

ABSTRACT
Axonal growth and targeting are fundamental to the organization of the nervous system, and require active engagement of the cytoskeleton. Polymerization and stabilization of axonal microtubules is central to axonal growth and maturation of neuronal connectivity. Studies have suggested that members of the tubulin polymerization promoting protein (TPPP, also known as P25α) family are involved in cellular process extension. However, no in vivo knockout data exists regarding its role in axonal growth during development. Here, we report the characterization of Ringmaker (Ringer; CG45057), the only Drosophila homolog of long p25α proteins. Immunohistochemical analyses indicate that Ringer expression is dynamically regulated in the embryonic central nervous system (CNS). ringer-null mutants show cell misplacement, and errors in axonal extension and targeting. Ultrastructural examination of ringer mutants revealed defective microtubule morphology and organization. Primary neuronal cultures of ringer mutants exhibit defective axonal extension, and Ringer expression in cells induced microtubule stabilization and bundling into rings. In vitro assays showed that Ringer directly affects tubulin, and promotes microtubule bundling and polymerization. Together, our studies uncover an essential function of Ringer in axonal extension and targeting through proper microtubule organization.
KEY WORDS: Nervous system, Axon growth, Cytoskeleton, Microtubules, Tubulin polymerization, Tubulin stabilization, TPPP
Summary: We report the characterization of Drosophila Ringmaker (Ringer), and demonstrate that Ringer regulates axonal growth by affecting microtubular organization during CNS development.
INTRODUCTION
The polarization of neurons during development gives rise to specialized processes in the form of dendrites and axons. The development of axons involves growth and guidance steps that essentially depend on the neuronal cytoskeleton (Lewis et al., 2013; Goldberg, 2003). A major constituent of the axonal cytoskeleton is the microtubule network, which provides structural support for the growing axon (de Forges et al., 2012; Dent et al., 2011). Microtubules form bundled parallel arrays along the axon and spread out at the growth cone with their plus end in the direction of growth (Witte and Bradke, 2008; Witte et al., 2008). Microtubule polymerization, bundling and stabilization are crucial for correct axonal extension and guidance (Lewis et al., 2013). These processes involve constant catastrophe and rescue events, and a variety of associated factors to maintain its dynamicity (Sept, 2007; Conde and Caceres, 2009). For instance, microtubule polymerization is aided by +TIPs, and in axons, bundling is accomplished through mediation of proteins like Tau (Prokop, 2013). A large number of microtubule-associated proteins have been identified (Prokop et al., 2013); however, many of those responsible for regulating the neuronal cytoskeleton to mediate axonal growth remain to be characterized.
Tubulin polymerization promoting proteins (TPPPs) are a superfamily of microtubule-associated proteins containing a common C-terminal p25α domain. Studies on TPPP (also known as TPPP1 and p25α), which is one of the three long p25α-containing paralogs in mammals (Orosz, 2012), have linked it to changes in microtubule dynamics such as the induction of double-walled microtubule formations and higher polymerization rates in vitro (Hlavanda et al., 2002). In in vivo settings, changes in postnatal TPPP expression have been correlated to pathologies like Parkinson's disease (Oláh et al., 2006). In samples from such affected individuals, TPPP colocalizes with α-synuclein aggregates in neurons, and cell studies have shown that increased TPPP stimulates α-synuclein aggregation into inclusions (Lindersson et al., 2005). TPPP loss has also been implicated in developmental disorders, as revealed by a high-resolution comparative genome hybridization that found a cohort of autistic children who exhibited TPPP deletions (Iourov et al., 2010). Further in vitro work has shown that reduction of TPPP levels in mammalian oligodendrocytes leads to defective differentiation and process extension, suggesting a role in growth of cellular processes during development. These studies, along with the identification of TPPPs as neuron outgrowth modifiers in a Drosophila primary neuron RNA interference (RNAi) screen (Sepp et al., 2008) and an in vivo screen in zebrafish (Aoki et al., 2014; Orosz, 2015) have led to the idea that TPPPs are involved in axonal growth. Although these previous studies indicate the involvement of TPPPs in developmental processes, no in vivo long-TPPP-knockout studies have addressed the endogenous role of these proteins at early stages. In addition, owing to potential paralog functional redundancy, developmental studies in mammalian systems might not fully address TPPP functions.
Here we report the identification and functional characterization of the only Drosophila long p25α homolog, named ringmaker (ringer; CG45057). Ringer displays a temporally dynamic expression in neurons and later in midline glia during ventral nerve cord (VNC) development. ringer-null mutants, generated through imprecise transposable element excision, show misplaced neurons and a variety of axonal phenotypes, including stalling and mistargeting. Ultrastructural analysis of nerve fibers from ringer mutants reveals defective microtubule organization and integrity. In vitro experiments and biochemical polymerization assays show that Ringer directly affects microtubule stabilization and polymerization, with cells overexpressing Ringer forming rings instead of regularly distributed microtubules. Together, our data demonstrate that Ringer is a major regulator of axonal microtubule organization, which is crucially required for proper axonal cytoskeletal architecture and growth during development.
RESULTS
Drosophila CG45057 locus encodes Ringer, a homolog of mammalian TPPP
The Drosophila CG45057 locus, named ringmaker (ringer), at cytological position 72E2 (Attrill et al., 2015) was uncovered through a deficiency screen aimed at identifying genes involved in VNC development (R.M. and M.B., unpublished). The ringer locus has four predicted splice variants that encode a polypeptide (Ringer) of 192 amino acids (Attrill et al., 2015; Wilson et al., 2008). To determine any molecular similarities between Ringer and its homologs in other species, we performed protein sequence alignment (Blosum62) (Altschul et al., 1997). These analyses revealed 39% identity and 54% similarity to mouse TPPP [expectation (E-) value 7×10−37], and 37% identity and 56% similarity to human TPPP (E-value 2×10−35). As shown in Fig. 1A, the most highly conserved region (red residues) is the C-terminus, which corresponds to the p25α domain (COBALT E-value 0.003) (Papadopoulos and Agarwala, 2007) (Fig. 1B). The mouse and human orthologs are 24 and 27 amino acids longer than Ringer, respectively (Fig. 1A,B) (Jensen et al., 2009). To determine the spatio-temporal expression of ringer during embryonic development, we performed in situ hybridization on stage-17 wild-type (WT) Canton S (+/+) embryos. Using antisense DIG-labeled probes, we observed prominent ringer mRNA expression in the embryonic central nervous system (CNS) at the midline of the VNC (Fig. 1C, arrow). The embryonic VNC midline is akin to the mammalian floor plate and constitutes a crucial developmental organizing center of the nervous system during development (Jacobs, 2000; Menne et al., 1997). Midline expression of ringer was consistent with that described in published data (Fisher et al., 2012) and was absent in sense probe controls (data not shown).
Fig. 1.

CG45057 (ringmaker) is dynamically expressed in the embryonic CNS. Drosophila CG45057, now ringmaker (ringer) encodes a 192-amino-acid polypeptide. (A) COBALT sequence alignment (E-value 0.003) of Drosophila Ringer (Dm, Drosophila melanogaster), mouse TPPP1 (Mm, Mus musculus) and human TPPP1 (Homo sapiens) shows Ringer has 54% and 56% sequence similarity to the mouse and human orthologs, respectively. Conserved areas are shown in red. (B) To-scale representation of p25α domains (lavender). LCR, low-complexity regions. (C) ringer mRNA expression in stage (S)17 WT (+/+) embryo CNS (arrow). (D) Ringer protein expression in the CNS midline (arrow), lateral CNS and PNS (arrowheads). Scale bar: 20 μm (C,D). (E) Immunoblots (IB) showing Ringer expression in embryo, larva and adult (23 kDa, arrowheads). Df−/−, homozygous embryos of Df(3L) BSC649, which have deletion of the ringer locus; Tub, Tubulin. (F) Midline Ringer expression is first observed in stage-13 neurons (F, arrowheads) and later in other neurons (a–e, arrowheads) and midline glia (c–e, arrows). (G) Immunostaining of stage-15 +/+ embryos with Ringer and ELAV showing Ringer in neurons (b, arrowhead). (H) Colocalization at stage 17 of Ringer and WRAP in midline glia (c, arrow). (I,K) At stage 13, Ringer colocalizes with FASII but not Eve-positive neurons (arrowhead). (J) At stage 14, other FASII neurons express Ringer (arrowheads). (L) Stage-15 Eve-positive RP2 motoneuron (arrowhead), and the aCC–pCC motoneuron–interneuron siblings (inset) also express Ringer. Arrow shows expression in glia. Scale bars: 20 μm (C,D); 10 μm (F–L).
To determine Ringer protein expression and subcellular localization, we generated polyclonal antibodies against the full-length Ringer protein (GENBank AY071358) (Benson et al., 2005). Antibody specificity was tested with embryos carrying Df(3L)BSC649, which deletes the entire ringer locus (Fig. 1E; Figs S1A and S2A). Immunohistochemical analyses of WT stage-17 embryos revealed that Ringer is prominently expressed in the VNC midline (Fig. 1D, arrow), with expression also observed in the surrounding CNS and peripheral nervous system (PNS) (Fig. 1D, arrowheads; Fig. S1C). Immunoblotting showed that, relative to Tubulin, Ringer had lower expression at embryonic stage 16 and the third instar larval stage but was abundant in adult fly heads (Fig. 1E, arrowhead). VNC midline embryonic developmental analyses revealed Ringer is initially detected in a pair of cells at stage 13 (Fig. 1Fa, arrowhead), at this stage, the pioneer midline axons initiate the formation of intersegmental connections (Kuzina et al., 2011). However, at later stages, expression was observed in other midline cells (Fig. 1Fb–Fd, arrowhead), peaking at stage 15 (Fig. 1Fc). Further analyses into the identity of these cells determined that at early stages, Ringer surrounds the neuronal nuclear marker ELAV (Fig. 1Gb, arrowhead) (Koushika et al., 1996) but not the nuclear lateral glia marker Repo (Fig. S1E,F), suggesting its presence in the neuronal soma (Fig. 1Ga–Gc, arrowhead). At stages 16 and 17, Ringer neuronal expression was still detected but was stronger in midline glia, which were identified by the midline-glia-specific marker Wrapper (WRAP) (Noordermeer et al., 1998; Wheeler et al., 2009) (Fig. 1Ha–Hc, arrows; Fig. S1B). Further midline neuronal analysis revealed that at stage 13, Ringer was expressed in MP1 neurons, as evidenced by its position and colocalization with the axonal marker FASII (Fig. 1Ia–Ic, arrowhead) and the transcription factor Even-Skipped (Eve) (Fig. 1Ka–Kc). At stage 14, in addition to MP1 expression, we observed expression in a subset of Eve-positive neurons (Skeath and Doe, 1998) (Fig. 1Ja–Jc and La–Lc, arrowheads). The RP2 motoneuron (Fig. 1Lb, arrowhead), and the aCC (motoneuron) and pCC (interneuron) siblings (Fig. 1Lb, arrowhead and inset) expressed Ringer until stage 16 when expression was reduced. Taken together, these data show that Ringer expression is dynamically regulated and coincides with stages that correspond with embryonic nervous system cell migration and development.
Ringer915 is a null mutant allele
To determine the effect of Ringer loss in vivo, we performed transposable-element mutagenesis using line w[1118];Mi-ET1-CG45057[MB04349] and a transposase source in Df(3L)BSC649 background (Fig. 2A) (Lin et al., 2014). We screened ∼4000 fly lines for imprecise excisions at the ringer locus by performing polymerase chain reaction (PCR) using genomic DNA from each line and primer combinations to identify deletions by shifts in band size (Fig. 2B). Lines carrying mutations were sequenced to obtain exact information on deleted regions. We obtained two independent lines with deletions in the ringer locus. The largest deletion started at the Minos element insertion site spanning 3734 base pairs and covered 80% of the ringer locus, including the p25α-domain-coding region (Fig. 2A). Genomic sequences that flank the deletion on the 5′ and the 3′ side are shown, with the ATG and the termination sites for all predicted polypeptides in the deleted segment (5′-CCTCCATTCGCTTTCCAGCGGTCCACTTGAGGTAGGTTGGGTTTATAATTGCGTTCCCCTCTCCAGGAAGTCCATTCTTTTCGAAAATGGCTATC- - - - -GTTTTCACAAGAATTTTGCTTGAATAAATACACTTTAAAATA-3′). This deletion allele is referred to as ringer915. Immunoblot of ringer915 homozygous adults revealed absence of the 23-kDa band corresponding to Ringer observed in control flies (Fig. 2C,D; Fig. S2). Immunostaining of ringer915 embryos at stage 15 (Fig. 2E,F) further confirmed absence of Ringer. Taken together, immunoblot and immunostaining analyses confirmed that ringer915 lacks Ringer and represents a null allele.
Fig. 2.

ringer-null mutants exhibit embryonic nervous system defects. (A) ringer-null mutant generation using Mi-ET1-CG45057[MB04349] and the extent of genomic deletion in ringer915 flies (red line). Exons are in dark blue. a-d indicate a subset of primer pairs used in PCR amplifications to determine genomic deletions. cpr72EA and pdh (gray) are neighboring genes. (B) Deletion size and location confirmation by using PCR analysis. (C,D) Immunoblots (IB) showing loss of 23-kDa Ringer in ringer915 flies (arrow). (E,F) Wild-type midline Ringer expression is absent in ringer915 embryos. (G,H) Viability analyses of ringer915 flies compared to +/+ flies indicates a decrease in mean survival at transitions from embryo to larva (G, *P=0.0211, Student's t-test) and larvae to adult (H, ****P<0.0001, Student's t-test) (n=210). (I,J) ELAV-specific immunostaining at stage 15 in +/+ (I) and ringer915 embryos (J) shows neuronal misplacement and bilateral symmetry errors in mutants (J, arrowhead indicates asymmetry, compare with arrow indicating symmetry in I). (K, L) SLIT-specific immunostaining in +/+ (K) and ringer915 (L) shows unaffected midline glia in ringer mutants. (M) Quantification of embryos with the neuronal misplacement phenotype as a percentage of the population. 71.7%±3.62 (mean±s.e.m.) ringer915 embryos showed changes at stage 16 compared to 7.02%±2.92 in WT (+/+) (n=100, P<0.0001, Student's t-test). Scale bars: 10 μm (E,F,I-L). Error bars are s.e.m.
Next, we determined the viability at crucial developmental stages because the ringer915 allele allowed eclosion of homozygous adult flies. To verify at which point ringer915 mutants were unable to progress through development, we calculated survival percentage within three experimental and control groups with a total of 210 embryos per genotype in three trials. Mean viability data indicated that only 69.9%±6.2 (mean±s.e.m.) of ringer915 embryos progressed to third instar larval stage, compared to WT, which had a mean survival of 93.7%±2.7 under identical conditions (P=0.0211) (Fig. 2G). Mean percent survival to adult stages was calculated by selecting a total of 210 larvae per genotype and following their development until eclosion. Only 34.7%±2.9 of mutant larva survived to adulthood, whereas 88.8%±2.7 WT larva eclosed as adults (Fig. 2H, P<0.0001). These data suggest that Ringer function affects viability at multiple stages of normal development.
ringer mutants display embryonic nervous system defects
Because we observed Ringer expression in the CNS, we next wanted to determine whether Ringer loss caused gross abnormalities in the VNC. ringer915 mutants were immunostained for the neuronal nuclear marker ELAV (Fig. 2J) and midline glial signaling molecule SLIT (Rothberg et al., 1988, 1990) (Fig. 2L). ringer mutants showed misplaced neurons (Fig. 2J, arrowhead) compared to the bilaterally symmetric placement in control embryos (Fig. 2I, arrow). Midline glial SLIT levels and distribution did not seem affected in ringer915 embryos (compare Fig. 2L with Fig. 2K). Quantification of neuronal misplacement in three independent experiments with a total of ∼100 embryos showed that an average 71.7%±3.62 (mean±s.e.m.) mutant embryos exhibited at least two segments with errors in bilateral symmetry of neurons at stage 16, in comparison to only 7.02%±2.92 observed in WT (P<0.0001) (Fig. 2M). These data suggest that loss of Ringer affects final placement of embryonic neuronal cells in the VNC midline.
Given the neuronal misplacement phenotype observed in ringer mutants, we wanted to determine whether loss of Ringer had any consequences on the CNS neuropile, and more specifically, in axonal development. To analyze stereotypical organization of CNS axonal tracks, we used Fasciclin II (FASII), an axon marker (Lin et al., 1994), and an antibody against horseradish peroxidase (HRP) as a neuronal membrane marker (Jan and Jan, 1982). Starting at stage 13, the stage at which Ringer is first observed, FASII-positive axons exhibit an overall collapse toward the midline (Fig. 3Ba, asterisk, compare with Fig. 3Aa) with instances of mistargeting (Fig. 3Ba, arrows), sometimes preventing axons from reaching the next segment (Fig. 3Ba, arrowhead). This trend continued in ringer915 flies through stages 14, 15 and 16 (Fig. 3Da,Fa,Ha, compare with control; Fig. 3Ca,Ea,Ga). At stage 16, we observed more pronounced guidance defects in which axons crossed the midline and extended posteriorly along FASII tracts (Fig. 3Ha, arrow, compared to 3Ga). To determine the phenotypic variability, we scored a total of 200 stage 16 embryos for presence of FASII phenotypes in three individual experiments per genotype. We found 74%±2.8 of ringer915 embryos had severe phenotypes (Fig. 3I), including 53%±4.5 of them presenting axonal collapse toward the midline, and 11.3%±2.4 exhibiting intersegmental axon breaks (Fig. 3J). Both phenotypes were observed in 9.7%±3.2 of embryos (Fig. 3J). Additionally, 20%±3.4 of ringer915 embryos showed mild axonal stalling, scored by monitoring the inability of the lateral FASII tract to reach the next segment by stage 16 (Fig. 3I). In contrast, 94.9%±0.5 control embryos did not present any of these phenotypes, and only 0.5%±0.3 presented stalling (Fig. 3I). The overall CNS structure, as highlighted with the antibody against HRP, also revealed defects in organization similar to those observed in FASII analysis (Fig. 3Bb,Db,Fb,Hb, arrowheads, compare to Fig. 3Ab,Cb,Eb,Gb). Additionally, to rule out any contributions of possible second site mutations created during mutagenesis, we quantified severe FASII phenotypes observed in ringer915/+ and ringer915/Df(3L)BSC649 embryos. Comparable to the WT population, which showed 5.133%±0.55 of embryos with a severe FASII phenotype, 9.59%±2.7 of ringer915/+ embryos exhibited severe FASII disruption (P=5931). Conversely, 65.67%±2.33 of ringer915 homozygous mutants exhibited a strong FASII phenotype comparable to the phenotype observed in 59.81±3.3% of the ringer915/Df(3L)BSC649 population (P=0.3857) (Fig. S1I,J), suggesting neuronal phenotypes originate owing to the disruption of the ringer locus. Taken together, these studies reveal that Ringer is required for proper axonal tract formation and overall architecture of the CNS midline during embryonic development.
Fig. 3.

Lack of ringer results in embryonic axonal growth and targeting defects. (A–H) Analysis of CNS axons at stages (S)13 through 16 using an anti-HRP antibody (Bb,Db,Fb,Hb compare to Ab,Cb,Eb,Gb) and staining of FASII reveals changes in the neuropil. Axons exhibit overall collapse toward the midline (Ba,Da,Fa,Ha, asterisks) with instances of mistargeting (Ba,Da,Fa,Ha, arrows) and intersegmental breaks (Ba,Da,Fa,Ha, arrowheads). (I) Quantification of embryos with specific phenotypes at stage 16 (n=200) of ringer915 and +/+ flies revealed severe phenotypes in 74%±2.8 (mean±s.e.m.) of ringer915 embryos, and 20%±3.4 showed mild axonal stalling (P<0.0001, ANOVA). (J) Breakdown of the severe phenotype in comparison to +/+. 53%±4.5 ringer915 exhibited axonal collapse, 11.3%±2.4 revealed axon breaks and 9.7%±2.92 exhibited both phenotypes. (K) Severe phenotypes were rescued by introduction of UAS-Ringer under elav-GAL4 in the mutant background. Scale bar: 10 μm (in Kb, applies to Aa–Hc and Ka). (L) Quantification of phenotypic rescue (n=300). At stage 14, phenotypes were observed in 21.75%±4 of rescue embryos (P<0.0001). By stage 16, 23.9%±0.5 of rescue embryos presented severe phenotypes compared to 74%±2.8 ringer915 (P<0.0001) and +/+ (P≤0.05) flies (ANOVA). There was no significant difference between rescue at stage 14 and 16 (P=0.7). (M) Breakdown of phenotypes observed in stage 16 rescue animals. Error bars are s.e.m. n.s., not significant; *P≤0.05, ****P<0.0001.
To determine whether nervous system re-expression of Ringer is sufficient to rescue axonal defects in ringer mutants, we generated UAS-ringer strains and expressed Ringer using elav-GAL4 in the ringer-mutant background (Fig. 3Ka,Kb) (Lin and Goodman, 1994). FASII axons in elav-GAL4,UAS-ringer;ringer915 embryos showed significant rescue compared to ringer mutants (Fig. 3Kb, arrows). For quantification of the rescue, 300 embryos were analyzed in three independent experiments per genotype. By stage 16, only 23.9%±0.5 of rescue embryos presented severe phenotypes compared to 74%±2.8 in ringer mutants (P<0.0001) and 5.1%±0.6 in WT (P≤0.05) (Fig. 3L,M). Because Ringer is expressed in both midline neurons and glia in the embryonic VNC, we addressed whether the observed neuronal defects were a result of loss of neuronal Ringer or a lack of Ringer in midline glia. To circumvent the contribution of midline glia, we analyzed the level of rescue of severe phenotypes at stage 14, before Ringer expression begins in midline glia. Phenotypic analysis of stage-14 elav-GAL4;UAS-ringer;ringer915 embryos revealed a significant rescue (P<0.0001) from 70.3%±1.3 of ringer915 embryos exhibiting a severe phenotype to 21.75%±4 (Fig. 3L). Only 6.1%±1.3 of control embryos showed a severe phenotype at stage 14 (Fig. 3L). There was no significant difference between rescue levels at stages 14 and 16, suggesting a minimal contribution of midline glia to the neuronal phenotype in ringer915 flies (Fig. 3L, P=0.7). These data suggest that Ringer might function in a cell autonomous manner in midline neurons and that midline glial Ringer is not essential for its axonal embryonic developmental function.
In addition to suggestions that the absence of TPPPs might lead to developmental disorders (Iourov et al., 2010; Lehotzky et al., 2010), reported cases of postnatal TPPP disruption leading to pathology are associated to in vivo increased protein expression (Oláh et al., 2011). To determine the effect of changes in Ringer protein levels in only a subpopulation of Ringer-expressing embryonic neurons, we used the available line eve-GAL4:UAS-tau-LacZ, which allowed to us to observe Eve-positive neuron soma and projections, as well as to drive other UAS transgenes under the same promoter. β-Galactosidase immunostaining of eve-GAL4:UAS-tau-LacZ lines (Fig. 4Ab) showed the proper arrangement of early stage-16 Ringer-expressing RP2, aCC and pCC neurons (Fig. 4Ab, arrowhead, and 4Ac, circles) with their corresponding projections (Fig. 4Ab, asterisk). Knockdown of Ringer with the UAS-ringer-RNAi line P{TRiP.HMS01740}attP40 (Ni et al., 2010) under control of eve-GAL4 led to a minimum of two segments with Eve-positive neuron placement defects in 67.25%±0.58 of the embryonic population (Fig. 4Bb–Bd,D), compared to 5%±3.8 in embryos expressing only LacZ in Eve-positive neurons (Fig. 4A,D) (P<0.0001). In the case of Ringer overexpression, we also observed neuronal misplacement (Fig. 4Cb–Cd, arrowhead) in 21.83%±1.94 of embryos (Fig. 4D, P=0.0082 compared to eve control), as well as axonal defects (Fig. 4Cb, asterisk, Fig. S3). Interestingly, in both knockdown and overexpression cases, phenotypes were stronger in RP2 neurons. The phenotypes observed in Eve-positive neurons were reflected in immunostaining of FASII, which showed disruption of axonal bundles (Fig. 4Aa,Ba,Ca, arrowhead) and position of the cell outline (Fig. 4Aa,Ba,Ca, arrows). Rescue experiments in which Ringer had been reintroduced into Eve-positive cells alone revealed a modest reduction of neuronal misplacement phenotypes to 44.29%±2.98 (Fig. 4D, P=0.0012 compared to RNAi flies, and P<0.0001 compared to WT flies, for each experiment, n=100). Taken together, these experiments suggest that a reduction or increase in Ringer in neurons causes neuron-specific deficits in the CNS midline and that optimal levels of Ringer are required for proper neural development.
Fig. 4.

Changes in Ringer levels affect neuronal position and process extension. (A–C) FASII- (a), β-Galactosidase- (b, B-Gal) and Ringer-specific (c) immunostaining of eve-GAL4:UAS-tau-LacZ embryos shows arrangement of early stage-16 RP2, aCC and pCC neurons and their projections. (B) Ringer expression changes in Eve-positive neurons lead to axonal (A–C; arrows, disruption of cell placement; asterisks, axonal defects) and soma defects (A–C). For each genotype, neuron soma placement is shown by circles in Ac,Bc,Cc, and midline glia are MG. FASII-specific immunostaining shows axonal bundle disruption (Aa–Ca). (D) Quantification of neuronal misplacement (n=∼100). 67.25%±0.58 (mean±s.e.m.) of knockdown (P<0.0001) and 21.83%±1.94 of overexpressing (P=0.0082) embryos exhibited misplacement compared to 5%±3.8 in WT (ANOVA). Rescue experiments exhibited a modest reduction to 44.29%±2.98 compared to knockdown (P=0.0012) and WT (P<0.0001) (ANOVA). (E,F) Primary neuronal cultures from larval CNS show that Ringer was also expressed in axons (arrow) and absent in mutant cultures. (E–I) Neuron identity was determined by staining of ELAV (Db,Eb) or with an anti-HRP antibody (Fb,Gb). Scale bars: 1 μm. (G–I) Immunostaining for acetylated tubulin. In ringer915 (H) and overexpressing embryos (I) microtubules appear disorganized (arrowheads) compared to +/+ (G). Arrowhead in Ringer overexpression image (I) shows Ringer accumulation at the axon tip. Scale bar: 5 μm. (J) Percent frequency distribution of FASII-positive primary neurons up to 30 μm in length. Quantification included neurons with axons up to 100 μm in length that were positive for FASII, anti-HRP antibody binding and acetylated tubulin. (K) After 24 h, the mean axon length for +/+ embryos was 13 μm±1.4 compared to 4.3 μm±0.4 in mutants (P 0.0002). Overexpression using elav-GAL4>UAS-ringer in the WT or in the mutant background resulted in mean axonal lengths of 5.3 μm±2.7 (P=0.0005) and 6.1 μm±0.4 (P=0.0010), respectively (ANOVA). n.s., not significant; *P≤0.05, **P≤0.01. Error bars are s.e.m.
Changes in Ringer expression affect neuronal process extension
Studies on mammalian TPPP have shown that p25α proteins have the ability to affect microtubule dynamics (DeBonis et al., 2015; Otzen et al., 2005; Tirián et al., 2003; To˝kési et al., 2010), as well as cellular projection extension (Lehotzky et al., 2010). We wanted to determine the consequences of Ringer changes on microtubules and how that affects axons. Because in vivo Tubulin distribution and single axon identification is complex owing to the presence of numerous neurons in a relatively small area, we addressed axonal extension aspects in primary neuronal cultures from control (Fig. 4G), ringer915 (Fig. 4H) and Ringer-overexpressing larval brains (Fig. 4I). We determined neuronal cell identity by immunostaining for the neuronal nuclear marker ELAV (Fig. 4Eb,Fb) or with the antibody against HRP (Fig. 4Gb,Hb). Analysis of WT CNS neurons showed that Ringer was expressed in the neuronal soma and within axonal projections (Fig. 4Ea, arrow) (Fig. 4Ec, see merged Ringer and ELAV). As expected, ringer-mutant primary neurons lacked Ringer immunoreactivity (Fig. 4Fa) (Fig. 4Fc, Ringer and ELAV merged image). Next, we immunostained control and ringer915 primary neurons with antibodies against acetylated Tubulin (Fig. 4Ga,Gc,Ha,Hc,Ia,Ic) to highlight axonal microtubules and HRP (Fig. 4Gb,Gc,Hb,Hc, blue) to label neuronal membranes. In WT neurons, acetylated microtubules exhibit uniform distribution along the axon, and in the cell body, microtubules form a prominent ring around the soma periphery (Fig. 4Ga,Gc, arrows). In contrast, ringer-mutant neurons had an irregular acetylated tubulin distribution within axons and asymmetric distribution at the soma (Fig. 4Ha,Hc, arrows), with tubulin more prominent toward the side proximal to the axon (Fig. 4Ha, asterisk). Conversely, increased Ringer expression in actin-GAL4>UAS-ringer neurons – which where identified using a combination of antibodies against FASII, mCherry or Ringer, and acetylated Tubulin – appeared to intensify distribution of acetylated microtubules throughout the cell (Fig. 4Ia). In addition, acetylated Tubulin accumulated at the growth cone area (Fig. 4Ib, arrowhead; Fig. 4Ic). These data suggest that changes in Ringer expression alter the distribution of acetylated microtubules in the axons.
As shown in Fig. 3A–H, loss of Ringer affects axonal development; therefore, we investigated whether lack of Ringer leads to defects in axonal extension in cultured neurons. We measured axon length specifically in FASII-positive neurons, which express Ringer, using acetylated tubulin and the anti-HRP antibody as markers for axon limits. We generated percent frequency distribution curves for axonal lengths measured in 500–700 WT control (Fig. 4J, blue), ringer915 (Fig. 4J, teal) and Ringer overexpression groups (Fig. 4J, elav-GAL4>UAS-ringer, magenta). Three independent experiments were performed under the same conditions 24 h after plating, and although we observed the formation of longer processes in these cultures, only FASII-axon lengths up to 100 μm were used for analysis. Axons in WT flies had a normal (P<0.06, D'Agostino–Pearson test) distribution, with most of the population within the range 0–50 μm in axon length and a maximum distribution at around 8 μm. ringer915 neurons had maximum distribution at shorter lengths, reaching 3–4 μm (Fig. 4J). The majority of elav-GAL4>UAS-ringer axons were 4–5 μm, with no significant difference between ringer mutants and Ringer-overexpressing neurons. The mean axonal length measured across experiments for the WT neuron population was 13 μm±1.4 (mean±s.e.m.), whereas in ringer mutants, mean axonal length was 4.3 μm±0.4 (P=0.0002, Fig. 4K). Ringer-overexpressing neurons exhibited a mean axonal length of 5.3 μm±2.7. In elav-GAL4>UAS-ringer;ringer915 neurons, mean axonal length increased to 6.1 μm±0.4 (P=0.0010), but axons did not reach full phenotypic rescue (Fig. 4K). In agreement with the primary culture studies, axonal stalling phenotypes were also observed when ringer under actin-GAL4 was overexpressed in vivo in embryos. We analyzed stage-13 through to stage-16 ringer-mutants using the anti-HRP antibody and FASII, which revealed that FASII-positive axons extended at a slower rate (Fig. S3 compare B,D,F to A,C,E). This was more evident at stage 16, with the formation of three FASII tracts, when overexpression led to a delay in the lateral tract in reaching the next segment. This stalling was quantified in three separate experiments following this phenotype at stage 16 in a total of 150 embryos. Compared to background controls, which only revealed a delay in 18.8%±2.3 (mean±s.e.m.) of embryos, 81%±1.6 of Ringer-overexpressing embryos exhibited axonal stalling (P<0.0001). Taken together, our cultured primary neuron and in vivo experiments suggest that too little or too much Ringer could prove detrimental to normal axonal outgrowth during development.
Ringer induces abnormal microtubule organization, and promotes microtubule bundling and polymerization in vitro
Phenotypic analysis of ringer mutants combined with primary culture analyses suggested that Ringer is involved in microtubule organization. We wanted to test whether Ringer expression in insect S2 cells affects their microtubule architecture. We generated UAS-ringer-mCherry, UAS-ringer and mCherry control constructs and expressed each under pMT-GAL4 in cultured S2 cells. Transfected cells were immunostained using antibodies against Ringer (Fig. 5A,B, asterisk), mCherry (Fig. 5C, red) and α-Tubulin (Fig. 5A–C, green). At lower levels of exogenous Ringer (Fig. 5A, asterisk), microtubules were sparse, failed to extend to the cell periphery, and exhibited an unusually high degree of curvature and looping (Fig. 5A, arrows) that was not observed in controls, which had normal microtubule architecture (Fig. 5A–C; Fig. S4). At higher levels of Ringer expression (Fig. 5B, asterisks), microtubules were bundled into compact rings that encircled the central cytoplasm and never extended into the cell periphery (Fig. 5B, arrowhead). We performed quantification of the microtubule phenotypes observed in mCherry–Ringer- or mCherry-expressing cells. Three replicates with n≥100 cells per treatment were then categorized into wild type, intermediate (sparse and curved) and strong (bundled rings) microtubule phenotypes. An average of 5.94%±0.55 (mean±s.e.m.) mCherry–Ringer cells exhibited wild-type microtubules compared to 90%±8.507 in control samples (P<0.0001). In 29.85%±2.99 of mCherry–Ringer-expressing cells, microtubules were thinly dispersed and curved compared to 10%±8.507 in control (P=0.1757). Owing to variability in the intermediate group, statistical analysis required data transformation. In the case of strong microtubule phenotypes, 64%±3.089 of mCherry–Ringer-expressing cells were identified with ring structures, whereas no cells with the strong phenotype were seen in controls (P<0.0001). These experiments show that Ringer expression affects microtubule distribution in single cells and that increased levels of Ringer might have more deleterious consequences on microtubule organization.
Fig. 5.
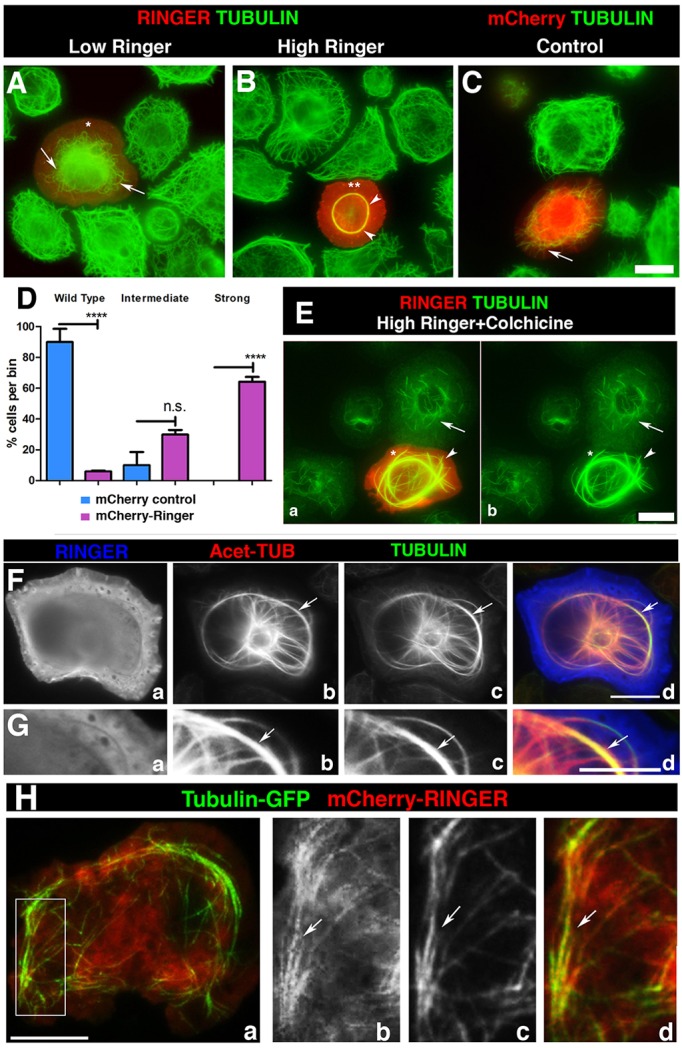
Ringer affects the morphology and stability of microtubules in cells. (A–C) mCherry-tagged Ringer (Ringer) and control mCherry alone (mCherry) was expressed in S2 cells, which were immunostained for Tubulin. Low Ringer expression (asterisk) leads to abnormal microtubule curvature and failure to grow into cell periphery (A, arrow). (B) At high levels of Ringer expression (asterisks), microtubules were incorporated into a central ring-like bundle (arrowheads) not seen in mCherry controls (C, arrow). (D) Quantification of microtubule phenotypes (n≥100). Individual cells were scored and categorized as wild-type, intermediate or strong microtubule phenotypes. 5.94%±0.55 (mean±s.e.m.) of mCherry–Ringer-expressing cells had wild-type microtubules compared to 90%±8.507 in control (P<0.0001, ANOVA). Intermediate phenotypes were observed in 29.85%±2.99 of mCherry–Ringer-expressing cells and 10%±8.507 in control (P=0.1757, ANOVA). Strong phenotypes were observed in 64%±3.089 of mCherry–Ringer-expressing cells, whereas no cells with the strong phenotype were seen in mCherry controls (P<0.0001, ANOVA). (E) Ringer-induced bundles were stabilized against depolymerization by colchicine (50 µM) (arrowhead) compared to controls (arrow). (F) Immunostaining in high-Ringer-expressing cells (Fa,Ga) for Tubulin (Fc,Gc) and acetylated Tubulin (Fb,Gb) shows that induced rings were acetylated microtubules. (H) S2 cells that had been transformed to coexpress GFP–Tubulin and mCherry–Ringer under pMT-GAL4 show partial colocalization of Ringer to microtubules (arrows) 10 s after the start of live imaging. Panels Hb–Hd are magnified from a section in Ha. Scale bars: 5 μm (A–C,E–H).
To further determine whether these ring structures are stabilized against microtubule depolymerization, we treated transfected cells with colchicine and examined its effect on Ringer-induced microtubule bundles. Consistent with a microtubule stabilizing activity, rings were resistant to high doses of colchicine (50 µM) (Fig. 5E, asterisk, arrowheads), a concentration that induced microtubule fragmentation and depolymerization in untransfected cells (Fig. 5Eb, arrow). We made further inquiries as to the nature of Ringer-induced microtubule rings (Fig. 5Fa,Ga, blue) with antibodies against acetylated Tubulin (T7451, Sigma) (Fig. 5Fb,Gb, red) and α-Tubulin (DMA1α, Sigma) (Fig. 5Fc,Gc, green). This immunostaining revealed that these rings were extensively acetylated (Fig. 5Fd,Gd, arrows) (mCherry control in Fig. S4), a post-translational modification widely observed in axonal microtubules. These effects of ectopic Ringer expression were also observed in HEK293 cells (Fig. S4), where tyrosinated microtubules remained unaffected. To examine Ringer localization in living S2 cells, we co-expressed mCherry–Ringer with GFP–actin (see Fig. S4 and Movie 1) or GFP–α-Tubulin (Fig. 5Ha–Hd; see Fig. S4 and Movie 2) to perform live imaging. We only imaged cells with low Ringer expression as high expression caused saturation and impaired imaging. As shown in Fig. 5H, a still at the 10-s timepoint revealed colocalization between Ringer (Fig. 5Hb, red) and microtubules (Fig. 5Ha, green, merged image; Fig. 5Hc and full cell image Fig. 5Hd, arrow). In contrast, S2 cells coexpressing mCherry–Ringer and GFP–actin showed no colocalization (Fig. S4). Taken together, these analyses indicate that Ringer regulates microtubule architecture and dynamics by inducing microtubule stabilization.
Because Ringer is able to affect microtubule organization in vivo and stabilize microtubules in cells, we wanted to know if purified Ringer is sufficient to induce changes in Tubulin dynamics. To test this, we expressed and purified recombinant GST–Ringer (Fig. 6A,B) and performed in vitro microtubule polymerization assays (Risinger et al., 2013; Shelanski et al., 1973) (Fig.6C). When purified Ringer was added in equimolar (18 μM) concentrations to purified Tubulin, optical density at 340 nm reached a maximum value of 0.40, with half maximum reached in 18 min. In contrast, buffer and GST-alone controls had maximum values of 0.06 and 0.04, respectively, and took 30 min to reach half maximum. As a positive control, we replicated these experiments in the presence of Taxol (Sigma) (Arnal and Wade, 1995; Schiff and Horwitz, 1980). Taxol was added at 10 μM, and Tubulin polymerization reached a density of 0.11 with half maximum reached after 2 min. Addition of GST–Ringer at 36 μM decreased the time for half maximum density to be reached to 11 min, although maximum density remained at ∼0.4, higher than that in the presence of Taxol. These biochemical data indicate that Ringer is sufficient to promote tubulin polymerization in vitro.
Fig. 6.

Ringer directly affects microtubule dynamics. (A,B) Representative silver-stain 12% gel (A) and anti-GST immunoblot (IB, B) showing purified proteins after the second round of purification for GST–Ringer (∼50 KDa, 2 μg) and GST alone (∼25 KDa, 1 μg) used for in vitro experiments. (C) In vitro tubulin polymerization assays, measured for optical densities (O.D.) at 340 nm and 37°C, showed purified GST–Ringer addition was sufficient to promote changes in microtubule polymerization rate and magnitude. (D–H) Representative images showing the ultrastructure of polymerization assay samples. Bottom panels in D–F are higher magnification images of those in the top panels. (D,E) Buffer and GST-only controls show unaided microtubule polymerization density. Single microtubules are marked with arrows (in Db,Eb,Fb,Gb,H). (F) Addition of Taxol increases microtubule density and promotes some bundling (arrowheads). (G,H) Addition of GST–Ringer to microtubules resulted in an increase of polymerization and had a microtubule-bundling effect (Ga,Gb,H, arrowheads). Scale bars: 10 μm (Da,Ea,Fa,Ga); 100 nm (Db,Eb,Fb,Gb,H).
The pronounced change in maximum optical density and time to reach the half maximum density after addition of Ringer during in vitro assays led us to use electron microscopy to directly evaluate the nature and extent of Tubulin changes. Buffer (Fig. 6Da,Db) and GST alone (Fig. 6Ea,Eb) samples exhibited background levels of polymerized microtubule density. Samples containing Taxol showed higher microtubule densities (Fig. 6Fa,Fb), with some lateral aggregation of microtubules (Fig. 6Fb). GST–Ringer samples, both at 36 μM (Fig. 6Ga,Gb) and 18 μM (Fig. 6H), showed a dramatic increase in polymerized microtubule density compared to controls (Fig. 6D,E). Addition of GST–Ringer to microtubules also promoted microtubule bundling, with aggregates reaching sizes of 820 nm in width (Fig. 6Gb compare with Fig. 6Fb, arrowheads). These data demonstrate that Ringer is sufficient to induce microtubule polymerization, bundling and stabilization, and indicate that in vivo Ringer has the potential to regulate proper microtubule assembly, architecture and distribution necessary for axonal growth during neuronal development.
Ringer is required for proper axonal microtubule organization in vivo
Our cell culture and in vitro experiments suggested that the phenotypes observed in vivo are the result of changes in axonal microtubules. To directly investigate whether Ringer loss contributes to structural changes in axonal microtubules, we analyzed larval segmental nerves. We used this system owing to their larger size compared to embryonic nerves. Immunohistochemical analysis showed that these neurons also expressed Ringer along their axon (Fig. 7Aa), which was absent in ringer mutants (Fig. 7Ba). Co-immunostaining for α-Tubulin showed that ringer915 nerves had disorganized microtubules within the axon (Fig. 7Bb, arrows). These wavy microtubule profiles were not observed in control axons (Fig. 7Ab,+/+). Next, we performed ultrastructural analysis of axons from control and ringer915 larvae to observe direct changes in microtubules. Imaging of transverse sections revealed that the axonal microtubule cross-section profile was compromised in ringer mutants (Fig. 7E–G, arrowheads). In WT axons, microtubules were seen as circular hollow rings with regular distribution within the axon (Fig. 7C, arrows; higher magnification in Fig. 7D, arrows). In ringer mutants, circular microtubule cross-sections appeared to be abnormally structured (Fig. 7F,G, arrowheads). Normal microtubule profiles were quantified in six WT and eight ringer915 mutant nerves from independent larvae and expressed as a percentage of the total microtubule population per axon. A minimum of 100 axons per genotype were counted. ringer mutants exhibited an average of 26.84%±2.8 (mean±s.e.m.) normal microtubule profiles compared to the 84.74%±2.69 observed in WT axons (Fig. 6H; P<0.0001). Ultrastructural analysis also revealed changes in microtubule distribution (Fig. 6G, arrowheads) consistent with those observed in immunostaining (Fig. 7A,B). WT axons exhibited characteristic uniform microtubule distribution, whereas ringer mutants had microtubule accumulations that were unevenly spaced (Fig. 7G, arrowheads). Taken together, these data demonstrate the Ringer is required for normal axonal microtubule assembly and organization during neuronal development.
Fig. 7.
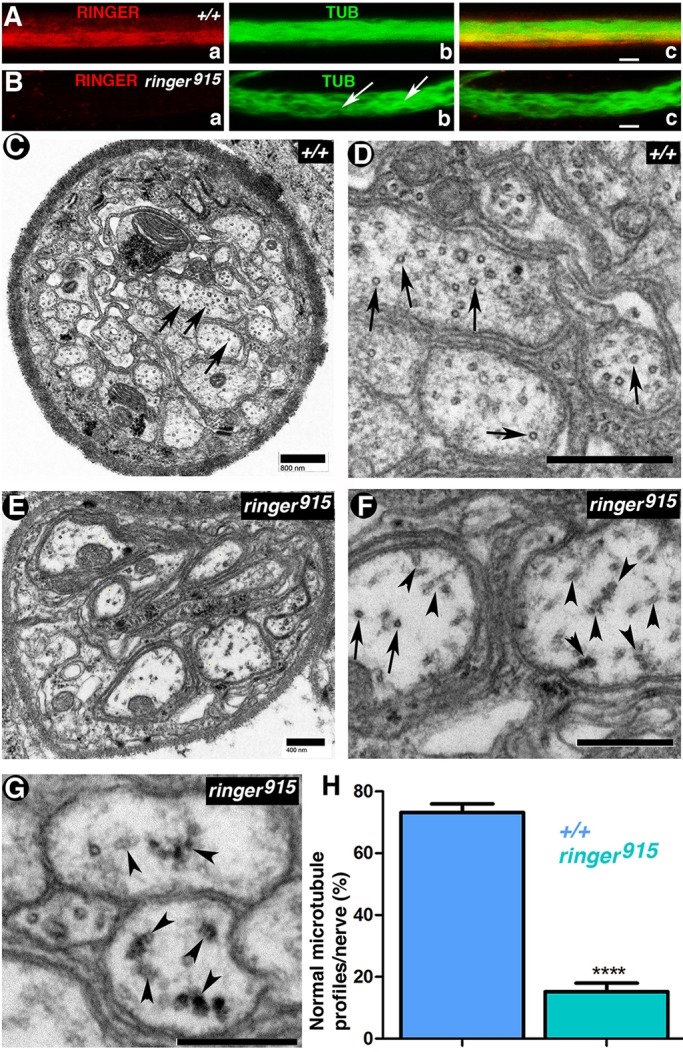
Lack of Ringer results in axonal microtubule defects. (A,B) Staining for α-Tubulin (TUB) and Ringer in +/+ and ringer915 third instar larval segmental nerves. Microtubules appear wavy and disorganized in mutants (Bb, arrows). Scale bars: 5 μm. (C–G) Representative transmission electron micrographs of +/+ (C,D) and ringer915-mutant segmental nerves (E–G). (C,D) In +/+ larval nerves, axonal microtubules (arrows) are normally observed as circular structures spread throughout the transverse face of the axon. Scale bars: 800 nm. (F,G) In ringer915 nerves, microtubule appearance (F,G, arrows) and distribution (F,G, arrowheads indicate instances of aberrant microtubule distribution) are compromised (F). F is a magnified image of an area of E. Scale bars: 400 nm. G shows a different example of microtubule accumulation in mutant nerves. Scale bar: 800 nm. (H) Quantification of the mean normal microtubule profiles per genotype as a percentage of the total per nerve shows that 84.74%±2.69 of +/+ (n=6) nerves exhibited a higher proportion of normal microtubules than the 26.84%±2.8 seen in ringer mutants (n=8) (****P<0.0001; Student's t-test).
DISCUSSION
Precise axon growth and guidance rely on microtubule polymerization, stabilization and bundling. These processes are central in establishing neuronal connectivity. Various proteins affecting microtubule dynamics have been characterized in the context of process extension (Lewis et al., 2013; Sánchez-Soriano et al., 2007). Proteins containing p25α domains are expressed in embryonic and postnatal brains (Skjoerringe et al., 2006), and are known to alter microtubule dynamics (Hlavanda et al., 2002). However, the majority of studies do not address their relevance during early development (Orosz, 2012). Using in vivo and in vitro studies, we address the previously uncharacterized function of Drosophila TPPP (Ringer), the only long p25α-containing protein in Drosophila, and its importance in neuronal development.
Neural development and Ringer expression
Through mRNA and protein localization, our work uncovers that Ringer is present in the nervous system and that its expression is variable and tightly modulated in the embryonic CNS midline. We also provide evidence that Ringer is necessary for correct nervous system development. Loss of Ringer results in soma misplacement, and defects in axonal extension and guidance in agreement with neuron-specific knockdown experiments showing similar defects. That loss of Ringer results in axonal disruption is strengthened by the findings of knockdown studies in vitro (Lehotzky et al., 2010) and in zebrafish (Aoki et al., 2014), which have shown TPPPs to have an effect on process extension. Similarly, ringer has been identified as a neuronal outgrowth modifier candidate (Sepp et al., 2008).
ringer mutants exhibit phenotypic variability. Initially, we supposed that these differences were due to a contribution of maternal Ringer, a suspicion arising from experiments involving deficiency lines (Fig. S1I,J). However, all ringer-mutant embryos analyzed were from homozygous stocks, which rules out this possibility. Phenotypic variance could also arise owing to compensation by other proteins. For instance, TPPP has been suggested to bundle microtubules in manner similar to that of Tau (DeBonis et al., 2015). In Drosophila neurons, Tau knockdown only shows exacerbated neuronal degeneration when combined with futsch mutations (Bolkan and Kretzschmar, 2014; da Cruz et al., 2005). We hypothesize that Ringer acts in a manner similar to Tau. Additionally, ringer-null mutants exhibit decreased organism viability. Lack of Ringer, as in the case of Tau, leads to reduced viability but not complete lethality (Bolkan and Kretzschmar, 2014).
Ringer and microtubule stabilization
Our studies determined that Ringer, like mammalian TPPPs, is able to regulate microtubule dynamics. This is evidenced in vivo by microtubule disruption at segmental nerves in ringer mutants and supported by our primary culture studies in which changes in Ringer translate into changes in acetylated tubulin. Ringer is likely to have a conserved stabilizing and bundling function similar to that of mammalian TPPPs (Hlavanda et al., 2002). Our cell culture experiments too suggest this, as they show Ringer can protect microtubules from depolymerization in addition to altering microtubule architecture, further underscoring a stabilizing function. Furthermore, our purified Ringer data show that no other external factors are necessary to induce changes in microtubule dynamics. Thus, our work demonstrates that Ringer alone is sufficient to induce higher rates of microtubule polymerization as well as bundling and stabilization.
Ringer-dependent microtubule changes in axonal extension and guidance
Our work provides evidence that Ringer regulates microtubule changes necessary for axonal development. Ringer is expressed along the axon in primary neurons, and at cellular margins and membrane-ruffle areas in S2 cells, a location concomitant with process growth (Ayala et al., 2007). Moreover, axon extension and growth cone advancement rely on microtubules (McCaig, 1989). Consequently, in ringer mutants exhibiting axonal stalling and breaks, phenotypes might be representative of lower microtubule polymerization rates that result from lack of Ringer function. This is supported by evidence that Ringer is necessary endogenously for proper axonal extension, and by micrographs showing axonal microtubule disruption in ringer mutants. Surprisingly, both ringer-mutant and -overexpressing neurons exhibit delayed axonal extension. Although defects observed in Ringer overexpression in vivo could be explained by the contribution of Ringer from surrounding cells, overexpression in primary neurons and in vivo Eve-positive neurons also results in soma placement and axonal phenotypes, revealing that there is a cell autonomous Ringer function.
The similar phenotypes produced by Ringer loss and gain of function appear counterintuitive. However, in vitro data show that Ringer has the ability to promote microtubule polymerization and bundling. Studies have revealed that modest microtubule overstabilization leads to an overall decrease in dynamics (Yvon et al., 1999). It is possible that Ringer overexpression stabilizes microtubules sufficiently to prevent axonal advancement, whereas in ringer mutants, axons delay advancement owing to lower tubulin polymerization. Additionally, Ringer loss might lead to depolymerization due to higher susceptibility to severing agents (McNally and Vale, 1993; Qiang et al., 2006). Perhaps there are Ringer concentration thresholds (Olah and Ovadi, 2014), post-translational modifications (Kleinnijenhuis et al., 2008) or other factors (Takahashi et al., 1991; To˝kési et al., 2010) that decide in favor of a specific function.
Conversely, the CNS axon mistargeting observed in ringer mutants might be an indirect result of delays in axonal extension. During embryogenesis, VNC midline neurons extend their axons as they migrate ventrally (Araujo and Tear, 2003). In mutants, axons from misplaced neurons might not extend at a normal rate, causing them to miss cues resulting in guidance defects. Additionally, axon guidance defects are repeatedly accompanied by severe neuronal misplacement, suggesting these two phenotypes are linked to migration errors. Alternatively, guidance phenotypes might result from a function of Ringer in growth cone directional movement through differential microtubule stabilization (Buck and Zheng, 2002). Thus, we postulate that during axonal development, Ringer regulates microtubule stabilization that is necessary for correct spatial distribution and polymerization to direct growth.
Interestingly, none of the phenotypic rescue experiments yielded a full recovery. Besides FASII embryonic phenotypic rescue, other attempts proved modest at best. We think that these differences are not due to changes in transgene expression but from diverging protein level requirements between systems. Moreover, our FASII rescue measurements were performed relative to the integrity of the combined neuronal connections, whereas single-cell measurements were made unhindered by environmental cues. Another possibility is that Ringer is necessary in surrounding cells, such as lateral glia, and that our antibodies are not robust enough to detect Ringer expression in such cells. If this is the case, elav-GAL4-mediated rescue, which expresses Ringer in all neurons and lateral glia at early stages (Berger et al., 2007), would be the only driver able to rescue phenotypes. Although our observations do not discard the notion of a function for Ringer in other cells that could influence development, they support the idea of an endogenous cell autonomous Ringer function in neurons.
In summary, our work has demonstrated that Ringer contributes to development in the regulation of axonal extension. We have shown that Ringer is sufficient to promote microtubule stabilization, bundling and polymerization and that its absence is likely to affect axonal microtubule dynamics, leading to extension delays, mistargeting and, consequently, abnormal neural development.
MATERIALS AND METHODS
Molecular constructs and in situ hybridization
ringer constructs were generated using cDNA (RE39465, GenBank) from Drosophila Genomics Resource Center (DGRC). For generation of antibodies against Ringer and of GST–Ringer, primers with flanking enzyme sites, and a 582-bp cDNA ringer fragment containing start and stop codons were subcloned into pET28(a+) and pGEX-4T-1, and expressed in Escherichia coli (BL21, Invitrogen). UAS-ringer expression constructs were generated by insertion of 1.2 Kb of ringer sequence, including 150 bp of the 5′UTR, into pUASTattB vector. In the UAS-mCherry-ringer construct, mCherry was inserted at the 5′ end, 4-bp upstream of the ringer start codon. Insect S2 cells and HEK293 cells were used to confirm expression of recombinant constructs before experiments were performed.
To In situ hybridization was performed as described previously in Kearney et al. (2004). In vitro transcription was performed with PCR product from the first 400 bp of the ringer locus as a template (1277073, Roche). A sense probe prepared under identical experimental conditions served as a negative control.
Generation of ringer mutants
To generate mutations in the CG45057 locus, Mi{ET1}CG45057MB04349 males were crossed to females carrying the transposase P{hsILMiT}2.4, both in the Df(3L)BSC649 background. Resulting progeny was heat-shocked at 37°C for 2 h until pupation. Mosaic F1 flies were crossed to yw;D/TM6,Tb to obtain males with single deletions. Progeny were tested for deletion size using PCR primers flanking the insertion site.
All stock lines were obtained from Bloomington Stock Center unless otherwise specified. Lines used: elavC155-GAL4, eve-GAL4:UAS-tau-LACZ and actin5c-GAL4, balanced over combinations of CyO,Twi-GFP; TM3,Ser,Twi-GFP or FM7,Kr-GFP or TM3,Ser,Kr-GFP. UAS-ringer and UAS-mCherry-ringer fly strains were generated using PhiC31 integrase-mediated transgenesis (GenetiVision, TX). Knockdown experiments with the corresponding controls were performed at 25°C using the available TRiP line TRiP.HMS01740}attP40. Canton S and WCS lines were used as wild-type controls.
Ringer antibody generation, immunostaining and immunoblotting
Primary antibodies against Ringer were generated in guinea pig and rat at Cocalico Biologicals, PA. His-tagged Ringer was expressed in E. coli BL21 for 3.5 h at 25°C after induction with 0.1 mM IPTG. His–Ringer Protein was purified with Talon Metal Affinity Resin beads (Clontech) using standard procedures. Antibodies were preabsorbed over ringer-mutant or -deficient embryos before their use in immunostaining procedures.
For immunoblotting and immunostaining, standard procedures were used (Banerjee et al., 2011). Primary antibodies used were: guinea pig anti-Ringer (1:3000), anti-FasII [Developmental Studies Hybridoma Bank (DSHB), 1:500], anti-ELAV (DSHB, 1:500), anti-Wrap (Wheeler et al., 2009), anti-Slit (DSHB, 1:250), anti-GFP (Life Technologies, 1:500), anti-β-Gal (Promega, 1:500), anti-acetylated-Tubulin (Sigma T7451 and T6793, 1:2000) and anti-α-Tubulin (DSHB, 1:50,000). To visualize HRP, we used Dylight-488-, -568- and -648-conjugated antibodies (Jackson ImmunoResearch, 1:400). Infrared-conjugated (LI-COR, 1:10,000) secondary antibodies and fluorescent Alexa-Fluor-488, -568 and -647 (Life Technologies, 1:400) secondary antibodies were used for immunoblotting and immunostaining, respectively.
Generation of larval primary neuronal cultures
Primary cultures from third instar larval brains from WT, homozygous ringer mutants and mCherry–Ringer-overexpressing stocks were generated and immunostained as described previously (Egger et al., 2013). Cultures were plated on polylysine-D coated glass-bottomed dishes (Corning, Biocoat) until fixation. Antibodies were used as listed above.
Insect S2 cell culture, transfection and immunofluorescence
Schneider (S2) cells were cultured in Schneider's medium (Invitrogen) supplemented with 10% heat-inactivated calf serum (HyClone Laboratories) and penicillin-streptomycin (Invitrogen). Cells were grown in 6-well plates and transfected with pMT-GAL4 and UAS-ringer (1 µg each) using FugeneHD (Promega) according to the manufacturer's protocol. On day 2, cells were induced with 100 µM copper sulfate for 24 h. On day 3, transfected cells were plated on to concanavalin-A-treated coverslips (MP Biologicals), washed for 2 min with PEM buffer (100 mM Pipes, pH 6.9; 1 mM EGTA; 1 mM MgCl2) and fixed for 20 min with 5% paraformaldehyde (EM Sciences) in PEM buffer. Some cells were treated with 50 µM colchicine (Sigma) for 2 h prior to fixation. After permeabilization with 0.1% Triton X-100 in PBS, cells were blocked with 5% normal goat serum and stained with anti-Ringer (1:1000) and anti-α-tubulin (1:1000) (DMA1α catalog no. T6199, Sigma) antibodies. After washing, Cy2 and Cy3 fluorescent secondaries (Jackson Immunologicals) were used. Cells were imaged on an inverted Nikon Eclipse Ti using a CoolSnap HQ CCD camera (Roeper Scientific) run by Nikon Elements.
For live imaging experiments, S2 cells were transfected with UAS-mCherry-ringer, pMT-GAL4 and pMT-GFP-α-tubulin 48 h prior to observation. Expression was induced with 100 μM copper sulfate 16–24 h before the experiment. Cells were seeded onto glass-bottomed dishes (MatTek) in serum-free Schneider's Drosophila medium supplemented with 100× antibiotic–antimycotic (Invitrogen) and allowed to attach for 1 h. Time-lapse imaging was performed on a motorized total internal reflection fluorescence (TIRF) system (Nikon) mounted on an inverted microscope (Ti; Nikon) equipped with a 100×1.49 objective lens, controlled by NIS Elements software. Images were captured with an Andor-Clara Interline camera (Andor Technology).
Microtubule polymerization and electron microscopy
For polymerization assays, GST and GST-tagged Ringer were expressed in E. coli (BL21) at 30°C. After centrifugation, bacteria were resuspended in TETN buffer (20 mM Tris, pH 8.0, 100 mM NaCl, 1 mM EDTA and 0.5% of Triton X-100) and sonicated. For purification, GST proteins were bound to glutathione beads (GE Healthcare) at 4°C overnight. Subsequent washes were done using high-salt TETN buffer. Elution was performed in 20 mM glutathione and 50 mM Tris-HCl, pH 8.0. The quality of purified proteins was assessed by Coomassie Blue staining, silver staining and immunoblotting (Fig. S2).
In vitro polymerization assays were performed as described previously (Risinger et al., 2013) using 18 μM 99% porcine Tubulin (BK006P, Cytoskeleton). A spectrophotometer running Softmax Pro was used to measure density. Experiments included GST–Ringer at 18 μM and 36 μM, GST at 18 μM and 36 μM, GST elution buffer only, Tubulin buffer only or 10 μM Paclitaxel. Immediately following assays, samples were processed for ultrastructure analysis, as described previously (Risinger et al., 2013).
Electron microscopy analysis of larval nerves was performed as previously described (Banerjee et al., 2006). Electron micrographs were obtained using a digital camera JEOL 1400 microscope system.
Imaging processing, quantification and statistical analysis
Imaging was performed with a Zeiss LSM710 confocal microscope. Identical parameters were set for control and mutant samples. Representative images are maximum intensity projections from 40X magnification confocal z-stacks with 0.4-µm intervals and 1× zoom for embryos, and 0.5 µm with 0.9× zoom for primary cultures. For quantification of embryos with specific phenotypes, a Zeiss Axioscop2 plus fluorescence microscope and AxioCam camera were used. ImageJ was used for axon length measurements. Images were processed with ImageJ and Adobe Photoshop.
All data are presented as the mean±s.e.m. Statistically significant differences were determined using Student t-test or one-way ANOVA. In cases in which variances were significantly different, data was transformed to y=log(y) for analysis. All statistical tests were performed using GraphPad Prism7. For significance: n.s. indicates P>0.05, * indicates a P≤0.05, ** indicates a P≤0.01 and *** indicates P≤0.001 and **** is P<0.0001.
Acknowledgements
ringer cDNA was obtained from the Drosophila Genomics Resource Center and fly stocks from the Bloomington Stock Center. Several antibodies were obtained from the Developmental Studies Hybridoma Bank (DSHB). We thank Barbara Hunt for electron microscopy sample processing, Raehum Paik (Mouse Engineering Laboratory) and Anna Taylor for assistance with reagents. We also thank Ben Eaton and Bhat laboratory members for valuable discussions.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
R.E.M. designed, performed and interpreted experiments, and prepared the manuscript. S.L.R. and A.L.R. performed and interpreted experiments. C.R. performed experiments. S.B. provided reagents and performed analysis. M.A.B. designed and interpreted experiments, and edited the manuscript.
Funding
This work was supported by the National Institutes of Health [grant number NS50356 (to M.A.B.)]; the Zachry Foundation; and funds from the State of Texas (M.A.B.). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.187294.supplemental
References
- Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W. and Lipman D. J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389-3402. 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki M., Segawa H., Naito M. and Okamoto H. (2014). Identification of possible downstream genes required for the extension of peripheral axons in primary sensory neurons. Biochem. Biophys. Res. Commun. 445, 357-362. 10.1016/j.bbrc.2014.01.193 [DOI] [PubMed] [Google Scholar]
- Araujo S. J. and Tear G. (2003). Axon guidance mechanisms and molecules: lessons from invertebrates. Nat. Rev. Neurosci. 4, 910-922. 10.1038/nrn1243 [DOI] [PubMed] [Google Scholar]
- Arnal I. and Wade R. H. (1995). How does taxol stabilize microtubules? Curr. Biol. 5, 900-908. 10.1016/S0960-9822(95)00180-1 [DOI] [PubMed] [Google Scholar]
- Attrill H., Falls K., Goodman J. L., Millburn G. H., Antonazzo G., Rey A. J., Marygold S. J. and Flybase Consortium. (2015). FlyBase: establishing a Gene Group resource for Drosophila melanogaster. Nucleic Acids Res. 44, D786-D792. 10.1093/nar/gkv1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala R., Shu T. and Tsai L.-H. (2007). Trekking across the Brain: the journey of neuronal migration. Cell 128, 29-43. 10.1016/j.cell.2006.12.021 [DOI] [PubMed] [Google Scholar]
- Banerjee S., Pillai A. M., Paik R., Li J. and Bhat M. A. (2006). Axonal ensheathment and septate junction formation in the peripheral nervous system of Drosophila. J. Neurosci. 26, 3319-3329. 10.1523/JNEUROSCI.5383-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S., Paik R., Mino R. E., Blauth K., Fisher E. S., Madden V. J., Fanning A. S. and Bhat M. A. (2011). A Laminin G-EGF-Laminin G module in Neurexin IV is essential for the apico-lateral localization of Contactin and organization of septate junctions. PLoS ONE 6, e25926 10.1371/journal.pone.0025926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson D. A., Karsch-Mizrachi I., Lipman D. J., Ostell J. and Wheeler D. L. (2005). GenBank. Nucleic Acids Res. 33, D34-D38. 10.1093/nar/gki063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger C., Renner S., Lüer K. and Technau G. M. (2007). The commonly used marker ELAV is transiently expressed in neuroblasts and glial cells in the Drosophila embryonic CNS. Dev. Dyn. 236, 3562-3568. 10.1002/dvdy.21372 [DOI] [PubMed] [Google Scholar]
- Bolkan B. J. and Kretzschmar D. (2014). Loss of tau results in defects in photoreceptor development and progressive neuronal degeneration in Drosophila. Dev. Neurobiol. 74, 1210-1225. 10.1002/dneu.22199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck K. B. and Zheng J. Q. (2002). Growth cone turning induced by direct local modification of microtubule dynamics. J. Neurosci. 22, 9358-9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde C. and Caceres A. (2009). Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 10, 319-332. 10.1038/nrn2631 [DOI] [PubMed] [Google Scholar]
- da Cruz A. B., Schwärzel M., Schulze S., Niyyati M., Heisenberg M. and Kretzschmar D. (2005). Disruption of the MAP1B-related protein FUTSCH leads to changes in the neuronal cytoskeleton, axonal transport defects, and progressive neurodegeneration in Drosophila. Mol. Biol. Cell 16, 2433-2442. 10.1091/mbc.E04-11-1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Forges H., Bouissou A. and Perez F. (2012). Interplay between microtubule dynamics and intracellular organization. Int. J. Biochem. Cell Biol. 44, 266-274. 10.1016/j.biocel.2011.11.009 [DOI] [PubMed] [Google Scholar]
- DeBonis S., Neumann E. and Skoufias D. A. (2015). Self protein-protein interactions are involved in TPPP/p25 mediated microtubule bundling. Sci. Rep. 5, 13242 10.1038/srep13242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent E. W., Gupton S. L. and Gertler F. B. (2011). The growth cone cytoskeleton in axon outgrowth and guidance. Cold Spring Harb. Perspect. Biol. 3, a001800 10.1101/cshperspect.a001800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger B., van Giesen L., Moraru M. and Sprecher S. G. (2013). In vitro imaging of primary neural cell culture from Drosophila. Nat. Protocols 8, 958-965. 10.1038/nprot.2013.052 [DOI] [PubMed] [Google Scholar]
- Fisher B., Weiszmann R., Frise E., Hammonds A., Tomancak P., Beaton A., Berman B., Quan E., Shu S., Lewis S. et al. (2012). BDGP insitu homepage.
- Goldberg J. L. (2003). How does an axon grow? Genes & Development. 17, 941-958. 10.1101/gad.1062303 [DOI] [PubMed] [Google Scholar]
- Hlavanda E., Kovacs J., Olah J., Orosz F., Medzihradszky K. F. and Ovadi J. (2002). Brain-specific p25 protein binds to tubulin and microtubules and induces aberrant microtubule assemblies at substoichiometric concentrations. Biochemistry 41, 8657-8664. 10.1021/bi020140g [DOI] [PubMed] [Google Scholar]
- Iourov I. Y., Vorsanova S. G., Saprina E. A. and Yurov Y. B. (2010). Identification of candidate genes of autism on the basis of molecular cytogenetic and in silico studies of the genome organization of chromosomal regions involved in unbalanced rearrangements. Russian J. Genet. 46, 1190-1193. 10.1134/S102279541010011X [DOI] [PubMed] [Google Scholar]
- Jacobs J. R. (2000). The midline glia of Drosophila: a molecular genetic model for the developmental functions of glia. Prog. Neurobiol. 62, 475-508. 10.1016/S0301-0082(00)00016-2 [DOI] [PubMed] [Google Scholar]
- Jan L. Y. and Jan Y. N. (1982). Antibodies to horseradish peroxidase as specific neuronal markers in Drosophila and in grasshopper embryos. Proc. Natl. Acad. Sci. USA 79, 2700-2704. 10.1073/pnas.79.8.2700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen L. J., Kuhn M., Stark M., Chaffron S., Creevey C., Muller J., Doerks T., Julien P., Roth A., Simonovic M. et al. (2009). STRING 8—a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 37, D412-D416. 10.1093/nar/gkn760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney J. B., Wheeler S. R., Estes P., Parente B. and Crews S. T. (2004). Gene expression profiling of the developing Drosophila CNS midline cells. Dev. Biol. 275, 473-492. 10.1016/j.ydbio.2004.08.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinnijenhuis A. J., Hedegaard C., Lundvig D., Sundbye S., Issinger O. G., Jensen O. N. and Jensen P. H. (2008). Identification of multiple post-translational modifications in the porcine brain specific p25α. J. Neurochem. 106, 925-933. 10.1111/j.1471-4159.2008.05437.x [DOI] [PubMed] [Google Scholar]
- Koushika S. P., Lisbin M. J. and White K. (1996). ELAV, a Drosophila neuron-specific protein, mediates the generation of an alternatively spliced neural protein isoform. Curr. Biol. 6, 1634-1641. 10.1016/S0960-9822(02)70787-2 [DOI] [PubMed] [Google Scholar]
- Kuzina I., Song J. K. and Giniger E. (2011). How Notch establishes longitudinal axon connections between successive segments of the Drosophila CNS. Development 138, 1839-1849. 10.1242/dev.062471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehotzky A., Lau P., To˝kési N., Muja N., Hudson L. D. and Ovádi J. (2010). Tubulin polymerization-promoting protein (TPPP/p25) is critical for oligodendrocyte differentiation. Glia 58, 157-168. 10.1002/glia.20909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis T. L., Courchet J. and Polleux F. (2013). Cellular and molecular mechanisms underlying axon formation, growth, and branching. J. Cell Biol. 202, 837-848. 10.1083/jcb.201305098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D. M. and Goodman C. S. (1994). Ectopic and increased expression of fasciclin II alters motoneuron growth cone guidance. Neuron 13, 507-523. 10.1016/0896-6273(94)90022-1 [DOI] [PubMed] [Google Scholar]
- Lin D. M., Fetter R. D., Kopczynski C., Grenningloh G. and Goodman C. S. (1994). Genetic analysis of Fasciclin II in Drosophila: defasciculation, refasciculation, and altered fasciculation. Neuron 13, 1055-1069. 10.1016/0896-6273(94)90045-0 [DOI] [PubMed] [Google Scholar]
- Lin S.-C., Chang Y.-Y. and Chan C.-C. (2014). Strategies for gene disruption in Drosophila. Cell Biosci. 4, 63 10.1186/2045-3701-4-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindersson E., Lundvig D., Petersen C., Madsen P., Nyengaard J. R., Højrup P., Moos T., Otzen D., Gai W.-P., Blumbergs P. C. et al. (2005). p25α stimulates α-synuclein aggregation and is co-localized with aggregated α-synuclein in α-synucleinopathies. J. Biol. Chem. 280, 5703-5715. 10.1074/jbc.M410409200 [DOI] [PubMed] [Google Scholar]
- McCaig C. D. (1989). Nerve growth in the absence of growth cone filopodia and the effects of a small applied electric field. J. Cell Sci. 93, 715-721. [DOI] [PubMed] [Google Scholar]
- McNally F. J. and Vale R. D. (1993). Identification of katanin, an ATPase that severs and disassembles stable microtubules. Cell 75, 419-429. 10.1016/0092-8674(93)90377-3 [DOI] [PubMed] [Google Scholar]
- Menne T. V., Luer K., Technau G. M. and Klambt C. (1997). CNS midline cells in Drosophila induce the differentiation of lateral neural cells. Development 124, 4949-4958. [DOI] [PubMed] [Google Scholar]
- Ni J. Q., Zhou R., Czech B., Liu L. P., Holderbaum L., Yang-Zhou D., Shim H. S., Handler D., Karpowicz P., Binari R. et al. (2010). A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat. Methods. 8, 405-407. 10.1038/nmeth.1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noordermeer J. N., Kopczynski C. C., Fetter R. D., Bland K. S., Chen W.-Y. and Goodman C. S. (1998). Wrapper, a novel member of the Ig superfamily, is expressed by midline Glia and is required for them to ensheath commissural axons in Drosophila. Neuron 21, 991-1001. 10.1016/S0896-6273(00)80618-2 [DOI] [PubMed] [Google Scholar]
- Olah J. and Ovadi J. (2014). Dual life of TPPP/p25 evolved in physiological and pathological conditions. Biochem. Soc. Trans. 42, 1762-1767. 10.1042/BST20140257 [DOI] [PubMed] [Google Scholar]
- Oláh J., To˝kési N., Vincze O., Horváth I., Lehotzky A., Erdei A., Szájli E., Medzihradszky K. F., Orosz F., Kovács G. G. et al. (2006). Interaction of TPPP/p25 protein with glyceraldehyde-3-phosphate dehydrogenase and their co-localization in Lewy bodies. FEBS Lett. 580, 5807-5814. 10.1016/j.febslet.2006.09.037 [DOI] [PubMed] [Google Scholar]
- Oláh J., Vincze O., Virók D., Simon D., Bozsó Z., To˝kési N., Horváth I., Hlavanda E., Kovács J., Magyar A. et al. (2011). Interactions of pathological hallmark proteins: tubulin polymerization promoting protein/p25, β-amyloid, and α-synuclein. J. Biol. Chem. 286, 34088-34100. 10.1074/jbc.M111.243907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orosz F. (2012). A new protein superfamily: TPPP-like proteins. PLoS ONE 7, e49276 10.1371/journal.pone.0049276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orosz F. (2015). On the tubulin polymerization promoting proteins of zebrafish. Biochem. Biophys. Res. Commun. 457, 267-272. 10.1016/j.bbrc.2014.12.099 [DOI] [PubMed] [Google Scholar]
- Otzen D. E., Lundvig D. M. S., Wimmer R., Nielsen L. H., Pedersen J. R. and Jensen P. H. (2005). p25α is flexible but natively folded and binds tubulin with oligomeric stoichiometry. Protein Sci. 14, 1396-1409. 10.1110/ps.041285605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos J. S. and Agarwala R. (2007). COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics 23, 1073-1079. 10.1093/bioinformatics/btm076 [DOI] [PubMed] [Google Scholar]
- Prokop A. (2013). The intricate relationship between microtubules and their associated motor proteins during axon growth and maintenance. Neural Dev. 8, 17 10.1186/1749-8104-8-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokop A., Beaven R., Qu Y. and Sánchez-Soriano N. (2013). Using fly genetics to dissect the cytoskeletal machinery of neurons during axonal growth and maintenance. J. Cell Sci. 126, 2331-2341. 10.1242/jcs.126912 [DOI] [PubMed] [Google Scholar]
- Qiang L., Yu W., Andreadis A., Luo M. and Baas P. W. (2006). Tau protects microtubules in the axon from severing by Katanin. J. Neurosci. 26, 3120-3129. 10.1523/JNEUROSCI.5392-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risinger A. L., Li J., Bennett M. J., Rohena C. C., Peng J., Schriemer D. C. and Mooberry S. L. (2013). Taccalonolide binding to tubulin imparts microtubule stability and potent in vivo activity. Cancer Res. 73, 6780-6792. 10.1158/0008-5472.CAN-13-1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothberg J. M., Hartley D. A., Walther Z. and Artavanis-Tsakonas S. (1988). slit: an EGF-homologous locus of D. melanogaster involved in the development of the embryonic central nervous system. Cell 55, 1047-1059. 10.1016/0092-8674(88)90249-8 [DOI] [PubMed] [Google Scholar]
- Rothberg J. M., Jacobs J. R., Goodman C. S. and Artavanis-Tsakonas S. (1990). slit: an extracellular protein necessary for development of midline glia and commissural axon pathways contains both EGF and LRR domains. Genes Dev. 4, 2169-2187. 10.1101/gad.4.12a.2169 [DOI] [PubMed] [Google Scholar]
- Sánchez-Soriano N., Tear G., Whitington P. and Prokop A. (2007). Drosophila as a genetic and cellular model for studies on axonal growth. Neural Dev. 2, 9 10.1186/1749-8104-2-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff P. B. and Horwitz S. B. (1980). Taxol stabilizes microtubules in mouse fibroblast cells. Proc. Natl. Acad. Sci. USA 77, 1561-1565. 10.1073/pnas.77.3.1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepp K. J., Hong P., Lizarraga S. B., Liu J. S., Mejia L. A., Walsh C. A. and Perrimon N. (2008). Identification of neural outgrowth genes using genome-wide RNAi. PLoS Genet. 4, e1000111 10.1371/journal.pgen.1000111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sept D. (2007). Microtubule polymerization: one step at a time. Curr. Biol 17, R764-R766. 10.1016/j.cub.2007.07.002 [DOI] [PubMed] [Google Scholar]
- Shelanski M. L., Gaskin F. and Cantor C. R. (1973). Microtubule assembly in the absence of added nucleotides. Proc. Natl. Acad. Sci. USA 70, 765-768. 10.1073/pnas.70.3.765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeath J. B. and Doe C. Q. (1998). Sanpodo and Notch act in opposition to Numb to distinguish sibling neuron fates in the Drosophila CNS. Development 125, 1857-1865. [DOI] [PubMed] [Google Scholar]
- Skjoerringe T., Lundvig D. M. S., Jensen P. H. and Moos T. (2006). P25α/Tubulin polymerization promoting protein expression by myelinating oligodendrocytes of the developing rat brain. J. Neurochem. 99, 333-342. 10.1111/j.1471-4159.2006.04073.x [DOI] [PubMed] [Google Scholar]
- Takahashi M., Tomizawa K., Ishiguro K., Sato K., Omori A., Sato S., Shiratsuchi A., Uchida T. and Imahori K. (1991). A novel brain-specific 25 kDa protein (p25) is phosphorylated by a Ser/Thr-Pro kinase (TPK II) from tau protein kinase fractions. FEBS Lett. 289, 37-43. 10.1016/0014-5793(91)80903-G [DOI] [PubMed] [Google Scholar]
- Tirián L., Hlavanda E., Oláh J., Horváth I., Orosz F., Szabó B., Kovács J., Szabad J. and Ovádi J. (2003). TPPP/p25 promotes tubulin assemblies and blocks mitotic spindle formation. Proc. Natl. Acad. Sci. USA 100, 13976-13981. 10.1073/pnas.2436331100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- To˝kési N., Lehotzky A., Horváth I., Szabó B., Oláh J., Lau P. and Ovádi J. (2010). TPPP/p25 promotes tubulin acetylation by inhibiting histone deacetylase 6. J. Biol. Chem. 285, 17896-17906. 10.1074/jbc.M109.096578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler S. R., Banerjee S., Blauth K., Rogers S. L., Bhat M. A. and Crews S. T. (2009). Neurexin IV and Wrapper interactions mediate Drosophila midline glial migration and axonal ensheathment. Development 136, 1147-1157. 10.1242/dev.030254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R. J., Goodman J. L. and Strelets V. B. and The FlyBase Consortium. (2008). FlyBase: integration and improvements to query tools. Nucleic Acids Res. 36, D588-D593. 10.1093/nar/gkm930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte H. and Bradke F. (2008). The role of the cytoskeleton during neuronal polarization. Curr. Opin. Neurobiol. 18, 479-487. 10.1016/j.conb.2008.09.019 [DOI] [PubMed] [Google Scholar]
- Witte H., Neukirchen D. and Bradke F. (2008). Microtubule stabilization specifies initial neuronal polarization. J. Cell Biol. 180, 619-632. 10.1083/jcb.200707042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvon A.-M. C., Wadsworth P. and Jordan M. A. (1999). Taxol suppresses dynamics of individual microtubules in living human tumor cells. Mol. Biol. Cell 10, 947-959. 10.1091/mbc.10.4.947 [DOI] [PMC free article] [PubMed] [Google Scholar]