

ABSTRACT
Keratins are epithelial cell intermediate filament (IF) proteins that are expressed as pairs in a cell-differentiation-regulated manner. Hepatocytes express the keratin 8 and 18 pair (denoted K8/K18) of IFs, and a loss of K8 or K18, as in K8-null mice, leads to degradation of the keratin partner. We have previously reported that a K8/K18 loss in hepatocytes leads to altered cell surface lipid raft distribution and more efficient Fas receptor (FasR, also known as TNFRSF6)-mediated apoptosis. We demonstrate here that the absence of K8 or transgenic expression of the K8 G62C mutant in mouse hepatocytes reduces lipid raft size. Mechanistically, we find that the lipid raft size is dependent on acid sphingomyelinase (ASMase, also known as SMPD1) enzyme activity, which is reduced in absence of K8/K18. Notably, the reduction of ASMase activity appears to be caused by a less efficient redistribution of surface membrane PKCδ toward lysosomes. Moreover, we delineate the lipid raft volume range that is required for an optimal FasR-mediated apoptosis. Hence, K8/K18-dependent PKCδ- and ASMase-mediated modulation of lipid raft size can explain the more prominent FasR-mediated signaling resulting from K8/K18 loss. The fine-tuning of ASMase-mediated regulation of lipid rafts might provide a therapeutic target for death-receptor-related liver diseases.
KEY WORDS: Keratin, PKC, ASMase, Lipid raft, FasR, Hepatocyte
Summary: Hepatocytes express keratin 8 (K8) and 18 (K18) as their sole intermediate filaments. A loss of K8/K18 impacts on PKCδ- and ASMase-mediated modulation of lipid raft size and FasR-mediated apoptosis.
INTRODUCTION
Keratins, the intermediate filament (IF) proteins of epithelial cells, constitute the largest and most diverse family of cytoskeletal proteins and are grouped into type I (K9–K28) and type II (K1–K8 and K71–K80) classes (Schweizer et al., 2006). Epithelial cells express at least one type I and one type II keratin in a cell-differentiation-dependent manner (Coulombe and Wong, 2004). Structurally, like all IF proteins, keratins exhibit a conserved central α-helical (rod) domain flanked by N-terminal (head) and C-terminal (tail) globular domains (Coulombe and Omary, 2002; Herrmann and Aebi, 2004). The rod domain is responsible for the formation of the coiled-coil framework and provides the main driving force during the assembly of IF proteins. Notably, much of the IF protein diversity is determined by the N- and C-terminal domains (Coulombe and Omary, 2002), which are largely responsible for the spatiotemporal IF organization and their interactions with associated proteins, such as plectin (Coulombe and Wong, 2004; Green et al., 2005). In addition, these domains contain sites that can undergo posttranslational modifications in response to various stimuli, primarily phosphorylation sites that provide targets for several kinases, including mitogen-activated protein kinase (MAPK) and protein kinase C (PKC) isoforms (Snider and Omary, 2014). Keratin modulation of such signaling cascades, namely those implicating PKCδ, can occur through participation of a cytoskeletal linker (e.g. plectin) and a scaffolding protein (e.g. receptor of activated C kinase 1, RACK1) in spreading or migrating keratinocytes and hepatocytes (Bordeleau et al., 2010; Mashukova et al., 2009; Osmanagic-Myers and Wiche, 2004). In this context, keratin IFs can be part of a cytoskeleton-dependent signaling platform for PKCδ in epithelial cells, in response to surface receptor stimulation.
Ligand stimulation of surface receptors triggers the endocytosis of the ligand–receptor complexes and is frequently associated with lipid rafts (Schütze et al., 2008). These surface membrane microdomains are known to play key roles as signaling platforms for stimulatory cellular processes that require coordination between receptor trafficking and signaling (Futerman and Hannun, 2004; Holthuis et al., 2003). Structurally, lipid rafts are defined as dynamic membrane microdomains enriched in cholesterol and sphingolipids in which proteins and lipids are orderly assembled (Simons and Gerl, 2010). Functionally, small rafts coalesce together into domains ranging from 70 nm to 1 µm in diameter, depending on the stimulus and the cell type (Gupta and DeFranco, 2003) through additional specific lipid–lipid or lipid–protein interactions that also modulate their thickness (Castro et al., 2014; Lingwood and Simons, 2010; Silva et al., 2007). In addition, the lipid raft coalescence can be regulated by local accumulation of ceramide generated through the hydrolysis of a major constituent of the microdomains, sphingomyelin, by the acid sphingomyelinase enzyme (ASMase, also known as SMPD1) (Castro et al., 2014). Of note, owing to their rapid trans-bilayer movement (flip-flop), ceramide molecules are distributed between the outer and inner leaflets of the surface membrane. In this context, a variation in ASMase-dependent ceramide generation, in response to different stimuli, can modulate the performance of coalesced lipid rafts as signaling platforms (Silva et al., 2007; Zeidan and Hannun, 2010). This is particularly the case for the receptor FasR (also known as TNFRSF6) which, together with its ligand (FasL, also known as FASLG), constitutes one of the major regulators of cell death in mammalian cells (Castro et al., 2011). Indeed, FasR is recruited and clusters in lipid rafts after ligand binding, a ceramide-dependent process that is vital for efficient apoptosis triggering. Moreover, the activation of the ASMase and ceramide pathway with cisplatin induces signaling-mediated changes in cell morphology through rearrangement of fibrillar actin and redistribution of ezrin (Zeidan and Hannun, 2007; Zeidan et al., 2008). Furthermore, signaling-dependent activation of the ASMase constitutes a multiple-step process that involves its maturation in the lysosomal compartment and its translocation to the surface membrane through an indispensable PKCδ-dependent phosphorylation step (Jenkins et al., 2011; Liu et al., 2006; Zeidan and Hannun, 2007). As such, a PKC isoform is first activated by phosphorylation of specific domains, translocated to the surface membrane, and either downregulated through local ubiquitylation and proteasome-dependent degradation or internalized into early or late endosomal compartments en route for degradation into lysosomes (Gould and Newton, 2008; Lum et al., 2013). The work reported here examines the impact of keratin IFs on lipid raft size through modulation of PKC–ASMase activation in hepatocytes, a liver cell type particularly responsive to FasR stimulation (Marceau et al., 2007).
Hepatocytes exhibit typical simple epithelial cell features, and of particular note their IFs consist of the keratin 8 and 18 pair (denoted K8/K18) only (Omary et al., 2009). This means that, given the loss of one keratin leads to the degradation of the partner keratin after ubiquitylation (Ku and Omary, 2000), K8/K18-containing versus K8/K18-lacking hepatocytes provide unique cell models to address the roles of simple epithelium keratins at the molecular cell level (Marceau et al., 2004). For instance, K8-null mouse hepatocytes are more sensitive than wild-type (WT) mouse hepatocytes to FasR-induced apoptosis in vivo and in culture upon FasR stimulation (Gilbert et al., 2001; Marceau et al., 2007). From a disease perspective, point mutations in human KRT8 or KRT18 predispose to progression of acute or chronic liver disease progression in humans, thus acting as liver disease susceptibility genes (Ku et al., 2005; Strnad et al., 2010). These clinical associations in humans are supported by mouse genetic models, whereby overexpression of normal human K8 or a K8 G62C mutant (the amino acid number includes the first methionine before processing) in transgenic mouse liver predisposes hepatocytes to FasR-stimulated apoptosis in vivo (Ku et al., 2003, 2007). Mechanistically, we have shown previously that cultured K8-null hepatocytes are more sensitive than WT hepatocytes to FasR-induced apoptosis and experience a type II to type-I-like switch in FasR-activated death signaling as result of the K8/K18 IF loss (Gilbert et al., 2008). At the cellular level, absence of K8/K18 in K8-null hepatocytes leads to more efficient FasR-mediated apoptosis, in association with accelerated elaboration at surface membrane receptor signaling protein oligomerization transduction structures (SPOTS), prominent FasR cap formation and internalization, and modulation of the actin–ezrin interplay at lipid rafts (Gilbert et al., 2012). The results reveal the involvement of K8/K18 IFs in the regulation of lipid raft size through a modulation of PKCδ and ASMase activation. They further show that the enhanced FasR-stimulated apoptosis occurring in hepatocytes lacking K8/K18 requires an optimized lipid raft size.
RESULTS
K8/K18 IF modulation of lipid raft size in the surface membrane
In line with our previous work using hepatocytes isolated from WT and K8-null mouse livers and established as monolayer cultures (Gilbert et al., 2012), our first set of data derived from lipid raft labeling with Vybrant® Alexa-Fluor®-488-tagged GM1 confirmed that a K8/K18 IF loss leads to perturbation of the coalesced lipid raft distribution (Fig. 1A). Thereafter, 3D confocal images were acquired and then processed with Volocity® software to determine the three interrelated size parameters of the coalesced CTX-B-labeled-lipid rafts (CT-lipid rafts). As shown in Fig. 1B, the loss of K8/K18 IFs led to a 24% decrease in CT-lipid raft surface coverage in K8-null hepatocytes. In addition, considering that the CT-lipid rafts shown in Fig. 1A appeared smaller in K8-null compared to WT hepatocytes, we quantified their surface area. As shown in Fig. 1C, the CT-lipid raft average area surface for K8-null hepatocytes was 0.8 μm2 compared to 1.0 μm2 for WT hepatocytes, which represents a 20% reduction as result of the K8/K18 IF loss. Parallel measurements of the CT-lipid raft volumes for K8-null versus WT hepatocytes yielded comparable reductions as a result of the K8/K18 IF loss (Fig. 1D). Moreover, as a complementary raft visualization approach, hepatocyte transfection with a Lyn16–YFP vector, which is known to mark the inner leaflet of the plasma membrane (Sohn et al., 2008), confirmed that the fusion protein distribution correlated with CTX-B-labeling (Fig. S1A). Subsequent assessments of the Lyn16–YFP-tagged lipid raft volume revealed a reduction in K8-null hepatocytes (Fig. S1B), which matched the differential evaluations obtained with the CTX-B visualization tool. Taken together, the results indicate that K8/K18 IF loss resulting from the K8-null mutation leads to a decrease in lipid raft size.
Fig. 1.

K8/K18 IF modulation of three interrelated size parameters of CT-lipid rafts in hepatocytes. (A) Lateral (XY) and axial (XZ) fluorescence images of WT and K8-null hepatocyte lipid rafts labeled using Vybrant® Alexa Fluor® 488 (CTX-B labeling), showing that the lipid raft distribution is perturbed as result of the K8/K18 IF loss. (B) Percentage cell surface coverage by lipid rafts on WT and K8-null (K8n) hepatocytes, showing a decrease as result of the K8/K18 IF loss (n>74). (C) Surfaces of coalesced CT-lipid rafts on WT and K8-null (K8n) hepatocytes showing a reduction as result of the K8/K18 IF loss (n>74). (D) Volumes of coalesced CT-lipid rafts on WT and K8-null (K8n) hepatocytes showing a decrease as result of the K8/K18 IF loss (n>70). (E) Lateral (XY) and axial (XZ) fluorescence images of hK8 and K8G62C hepatocyte lipid rafts labeled using Vybrant® Alexa Fluor® 488 (CTX-B labeling), showing that the G62C mutation perturbs the lipid raft organization. (F) Percentage cell surface coverage by lipid rafts on hK8 and K8 G62C (G62C) hepatocytes, showing a decrease as result of the G62C mutation (n>40). (G) Surfaces of coalesced CT-lipid rafts on hK8 and K8 G62C (G62C) hepatocytes, showing a decrease as result of the G62C mutation (n>40). (H) Volumes of coalesced CT-lipid rafts on hK8 and K8 G62C (G62C) hepatocytes, showing a decrease as result of the G62C mutation (n>40). Quantitative results are mean±s.e.m. ***P<0.005 (t-test).
We then extended our raft size analysis to hepatocytes isolated from transgenic mice carrying a normal human K8 (hK8) gene or a human K8 gene bearing the K8 G62C point mutation (Ku et al., 2005; Strnad et al., 2006, 2010), and established in monolayer cultures, as above. Based on previous work performed at the organ level (Ku and Omary, 2006), we first looked at the IF network organization in these cultured hepatocytes. Confocal imaging of hK8 versus endogenous mouse K8 demonstrated that hK8 integrates perfectly into the endogenous K8/K18 IF network (Fig. S2A). In addition, although the K8 G62C expression created a well-organized IF network, it led to a reduced contribution of endogenous mouse K8 (Fig. S2A), confirming in culture the dominant effect of this mutation on IF assembly, as observed previously at the organ level (Ku et al., 2001). Moreover, given that K8 G62C transgenic mouse hepatocytes have been found to be more sensitive to FasR-mediated apoptosis in vivo (Ku and Omary, 2006), we extended the analysis to hepatocytes in culture, keeping in mind our previous data with WT versus K8-null hepatocytes in monolayer cultures (Gilbert et al., 2001, 2008). As expected, cultured K8 G62C hepatocytes were more sensitive to FasR-mediated apoptosis (Fig. S2B). In addition, they formed SPOTS faster at cell surface (Fig. S2C,D), in line with our recent data using K8-null hepatocytes (Gilbert et al., 2012). In this context, the next step was to assess whether the K8 G62C mutation affects the organization of the CT-lipid rafts. Confocal imaging showed that, as in the case of K8-null hepatocytes, CT-lipid raft distribution at the surface membrane was perturbed in K8 G62C hepatocytes compared to hK8 hepatocytes (Fig. 1E). Moreover, CT-lipid raft measurements yielded a 19% decrease in cell surface coverage (Fig. 1F), a 11% raft surface decrease (Fig. 1G) and a 16% raft volume decrease (Fig. 1H) in K8 G62C versus hK8 hepatocytes.
Taken together, these two sets of data indicate that K8/K18 IFs play a key role in maintaining the proper organization and size (i.e. surface and volume) of lipid raft microdomains.
K8/K18 IF modulation of lipid raft size is ASMase dependent
In conjunction with our previous findings that revealed no major reorganization in lipid-raft-specific proteins (e.g. caveolin) in K8-null hepatocytes (Gilbert et al., 2012), the above findings on the size of coalesced CT-lipid rafts led us to consider that the changes observed in K8-null hepatocytes were attributable to perturbations in their lipid components, namely variations in the level of ceramide generated by the ASMase-dependent hydrolysis of sphingomyelin (Silva et al., 2007). Accordingly, we used two inhibitors of the ASMase activity [desipramine (DPM) and imipramine (IPM)] to address its role in the modulation of CT-lipid raft size parameters. Hepatocytes were pretreated for 2 h with DPM (50 μM), IPM (50 μM) or with the cholesterol ‘solubilizer’ methyl-β-cyclodextrin (MBC; 2 mM) before labeling with CTX-B. As shown in Fig. 2A, confocal cell imaging readily indicated a lower CT-lipid raft staining intensity in presence of either DPM or IPM. Quantifications of cell surface coverage and raft surface revealed strong reductions in both WT and K8-null hepatocytes following DPM or IPM addition (Fig. 2B,C). As a control, the use of MBC to disrupt lipid rafts led to the expected drastic reduction in both cell types.
Fig. 2.

K8/K18 IF modulation of ASMase-dependent CT-lipid raft size. (A) Fluorescence images of the CT-lipid raft distributions at the surfaces of WT and K8-null hepatocytes, following a 2-h treatment with ASMase (50 µM DPM, 50 µM IPM) or lipid raft (2 mM MBC) inhibitors. The results show that the ASMase inhibition leads to a reduction in coalesced lipid raft size and distribution (CTRL, untreated control). (B) Percentage cell surface coverage by lipid rafts on WT and K8-null (K8n) hepatocytes following a 2-h treatment with ASMase or lipid raft inhibitors as in A, showing a strong reduction after these treatments (C, untreated control). n>25. (C) Surface of coalesced CT-lipid rafts on WT and K8-null (K8n) hepatocytes following a 2-h treatment with ASMase (or lipid raft inhibitors as in A, showing a surface reduction as result of the treatments (C, untreated control). n>25. (D) Volume of coalesced CT-lipid rafts on WT and K8-null hepatocyte surfaces following a 2-h treatment with ASMase or lipid raft inhibitors as in A, showing a reduction for both WT and K8-null hepatocytes (C, untreated control). n>25. Quantitative results are mean±s.e.m. **P<0.01; ***P<0.005 (t-test).
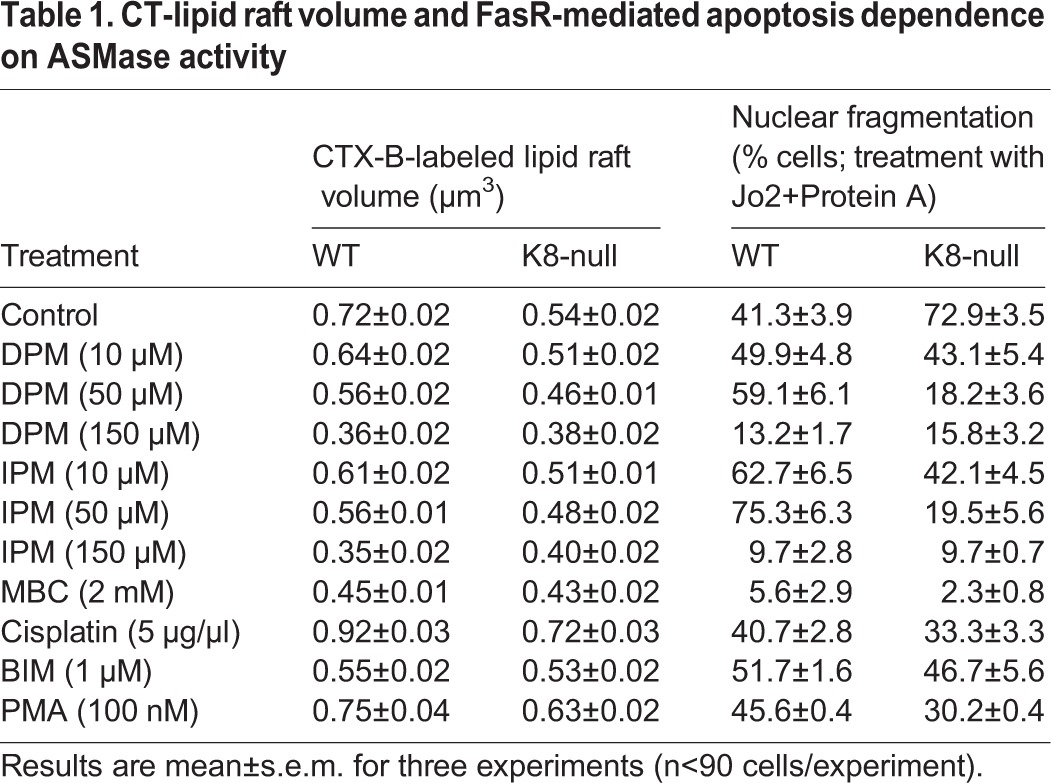
In the same way, a reduction was observed for CT-lipid raft volume in both WT and K8-null hepatocytes following DPM or IPM addition, with a higher effect in WT hepatocytes (22% with DPM and IPM, 15% with DPM alone and 11% with IPM alone in K8-null hepatocytes) (Fig. 2D). However, the addition of the lipid raft solubilizer MBC led to less volume reduction than expected, suggesting that the CT-lipid raft volume measurement tool was limited to a threshold volume detection, owing to the cell imaging system sensitivity or the fluorochrome used. To address this issue, we used a higher concentration of ASMase inhibitors (Fig. S3A–D; Table 1) and found that the minimal CT-lipid raft volume detected was 0.35 μm3 with IPM at 150 μM, with a distribution equivalent to the one obtained in control MBC-treated K8-null hepatocytes. Still, our assessments of volumes obtained with either ASMase inhibitor at 50 μM (Fig. 2D) yielded values that are well above this limit. Overall, the data indicate that the K8/K18-dependent modulation of coalesced lipid raft size takes place through a regulation of ASMase activation.
Table 1.
CT-lipid raft volume and FasR-mediated apoptosis dependence on ASMase activity
K8/K18 IF modulation of ASMase activation is PKC dependent
ASMase activation is a multi-step process that involves its maturation within lysosomes and its translocation to the surface membrane upon phosphorylation by PKCδ (Jenkins et al., 2011; Zeidan and Hannun, 2007; Zeidan et al., 2008). Thus, in search of an explanation for the reduced basal CT-lipid raft volume in K8-null hepatocytes, we first compared the ASMase distribution in WT versus K8-null hepatocytes. z-stack-projections (Fig. 3A) and xz views (Fig. 3B) of the ASMase in confocal images revealed its presence at the cell surface and also within the cytoplasmic space of both WT and K8-null hepatocytes. However, a slight decrease was evident at the surface of K8-null hepatocytes, suggesting a lower ASMase basal activity in these cells. Subsequent biochemical measurements of the ASMase activity in protein extracts from WT and K8-null hepatocytes demonstrated a 23% reduction in the basal ASMase activity in K8-null hepatocytes (Fig. 3C). Furthermore, PKC activation with cisplatin led to an increased ASMase targeting at the surface membrane in both cell types, as shown in the xz views (Fig. 3B). This increase in targeting matched the ASMase activity measurement resulting from the PKC activation with cisplatin or PMA in either WT or K8-null cells (Fig. 3C). Nonetheless, the differential ASMase activity between WT and K8-null cells was maintained. These data suggest that although the ASMase is functionally active in both WT and K8-null hepatocytes, its PKC-dependent activation process is affected by the K8/K18 loss.
Fig. 3.

K8/K18 IF modulation of ASMase localization and activity. (A) Immunofluorescence confocal imaging of the ASMase localization in WT and K8-null hepatocytes, revealing no major differences. (B) XZ views of ASMase immunofluorescence images with or without a 2-h treatment with cisplatin (Cis; 5 µg/ml) in WT and K8-null hepatocytes. In the absence of cisplatin, a slight decrease in ASMase localization is seen at the surface of hepatocytes lacking K8/K18 IFs, whereas an increased localization is observed at the surface of both cell types, following the cisplatin treatment. (C) ASMase activity assessments in WT and K8-null hepatocytes with or without a 2-h treatment with cisplatin (Cis; 5 µg/ml) or PMA (100 nM), showing a decrease in ASMase activity in hepatocytes lacking K8/K18 IFs, under either inhibitory conditions (C: untreated control). Results are mean±s.e.m. **P<0.01; ***P<0.005 (t-test).
K8/K18 IF modulation of lipid raft size occurs through PKC and ASMase activation
We next assessed whether the K8/K18 IF-dependent PKC and ASMase activation leads to CT-lipid raft size variations. WT and K8-null hepatocytes were stimulated for 2 h with PKC activators (cisplatin or PMA) or inhibitor (Bim), and lipid rafts were then labeled. Confocal imaging demonstrated significant increases in lipid raft size at the surface of both cell types after cisplatin or PMA treatment, and a reduction after Bim treatment (Fig. 4A). Subsequent volume measurements confirmed the above findings, with a 28% increase for WT and a 33% increase for K8-null hepatocytes, in response to cisplatin (Fig. 4B). However, in line with the ASMase activity, after cisplatin treatment, the change in CT-lipid raft volumes between WT and K8-null hepatocytes remained almost the same (22% versus 25%). PMA treatment yielded essentially the same results, but on a smaller scale (Fig. 4B). Notably, the same results were also obtained using deoxycholic acid (DCA), another ASMase activator (Gupta et al., 2004) (Fig. S3E,F). In contrast, PKC inhibition with Bim led to a strong reduction in volume for WT cells (24%), compared with only a slight decrease for K8-null hepatocytes (2%), suggesting that the absence of K8/K18 IFs alters proper PKC-dependent ASMase activation.
Fig. 4.
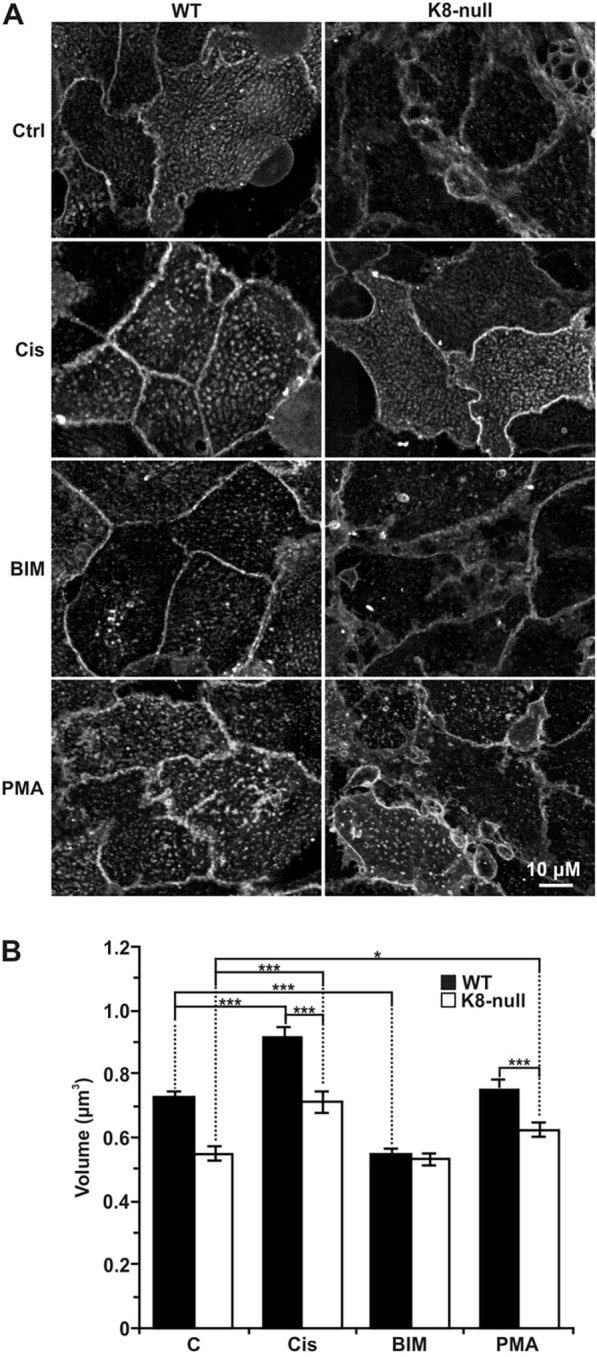
K8/K18 IF modulation of CT-lipid raft size through PKC activation. (A) Immunofluorescence confocal imaging of the CT-lipid raft distributions at the surfaces of WT and K8-null hepatocytes, following a 2-h treatment with two PKC activators [5 µg/ml cisplatin (Cis) or 100 nM PMA] or a PKC inhibitor (1 µM BIM). (B) Volume assessments of coalesced CT-lipid rafts on WT and K8-null hepatocytes following 2-h treatments with PKC activators or PKC inhibitor as in A, revealing that a PKC activation increases the lipid raft size, whereas a PKC inhibition reduces it in both cell types (n>25). Results are mean±s.e.m. *P<0.05; ***P<0.005 (t-test).
K8/K18 IF modulation of PKCδ translocation to the surface membrane versus internalization
Considering that different PKC isoforms can interact with K8/K18 IFs (Bordeleau et al., 2010; Mashukova et al., 2009; Omary et al., 1992), and that PKCδ modulates ASMase activation (Zeidan and Hannun, 2007), we first compared the surface membrane translocation of PKCα, PKCδ and PKCε, which is associated with their activations in the present K8-null hepatocyte model. At 52 h post seeding, WT and K8-null hepatocytes were transfected with a cDNA encoding EGFP-tagged PKCα, PKCδ and PKCε (Fig. S4A,B; Fig. 5A). At 48 h post transfection, cells were treated with PMA and then fixed at different times thereafter. In the absence of stimulation, PKCδ–EGFP was dispersed in the cytoplasm of both WT and K8-null hepatocytes (Fig. 5A). After PMA treatment, translocation of PKCδ–EGFP to the cell surface was observed in both cell types, except that it occurred at a much faster rate in K8-null hepatocytes, to reach a plateau at 15 min post treatment (Fig. 5B); in comparison, the translocation plateau was observed at 30 min in WT hepatocytes. This faster translocation toward the cell surface in K8-null hepatocytes fits with the higher PKCδ phosphorylation and activation observed at 5 min following PMA treatment (Fig. S1C). Notably, this increase in PKC translocation was specific to novel type PKCs, considering that a similar response was observed for PKCε–EGFP (Fig. S4B,C), and that no difference was revealed for PKCα–EGFP (Fig. 5C; Fig. S4A). Taken together, the results suggest that the K8/K18 IF loss leads to faster translocation kinetics for novel type PKCs, but that all PKCs still undergo full translocation in both cell types at 30 min post stimulation. Therefore, it appears that these slight variations in PKC cell surface translocation are not responsible for the major differences observed in ASMase activation and CT-lipid raft size.
Fig. 5.

K8/K18 IF modulation of PKCδ translocation to the surface membrane versus internalization. (A) Immunofluorescence confocal imaging of PKCδ–EGFP in transfected WT and K8-null hepatocytes treated with PMA (100 nM) and fixed at 1, 5 and 15 min post treatment. (B) Percentage assessments of WT or K8-null hepatocytes exhibiting a PKCδ–EGFP translocation at the surface membrane as a function of time after treatment with PMA (100 nM), showing a rapid translocation to the surface in both cell types, with even faster kinetics in hepatocytes lacking K8/K18 IFs (n>70). (C) Percentage assessments of WT or K8-null hepatocytes exhibiting a PKCα–EGFP translocation at the surface membrane as a function of time after a PMA treatment (100 nM), showing no difference in both cell types (n>60). (D) Lateral (XY) and axial (XZ) immunofluorescence confocal imaging of PKCδ–EGFP in transfected WT and K8-null hepatocytes treated with PMA (100 nM) and fixed at different time points, revealing less PKCδ–EGFP internalization in hepatocytes lacking K8/K18 IFs (CTRL: untreated control). Quantitative results are mean±s.e.m.
Alternatively, the downregulation of a PKC isoform, subsequent to its stimulation, has been shown to require its internalization through early and late endosomes and its degradation in lysosomes, a lipid-raft-linked process (Gould and Newton, 2008; Lum et al., 2013). Accordingly, we assessed the fate of PKCδ–EGFP transfected in WT and K8-null hepatocytes following a PMA treatment. As shown in Fig. 5D, PKCδ–EGFP internalization occurred as soon as 15 min after PMA treatment, and the aggregation of PKCδ–EGFP inside the cells continued for at least 60 min in WT hepatocytes, whereas in K8-null hepatocytes, the internalization was essentially absent at 15 min post treatment and barely detectable at 60 min.
Moreover, previous work by others has shown that PKCδ can localize to the lysosomal compartment after its internalization, where it associates with ASMase (Zeidan and Hannun, 2007). Thus, we examined the internalization of PKCδ in the presence or absence of K8/K18 IFs by transfecting WT and K8-null hepatocytes with PKCδ–EGFP in combination with labeling lysosomes with the LysoTracker® Red DND99. As shown in Fig. 6, at shortly after PMA stimulation, PKCδ–EGFP translocated to the surface membrane in WT hepatocytes, as described above (Fig. 5A,B), with no signal in the lysosomal area. Several emerging spots reflecting PKCδ internalization were seen at 15 min and became prominent at 60 min post PMA stimulation. In addition, some of the internalized PKCδ–EGFP colocalized with the lysosome tracker (yellow arrows), indicating that a proportion of the PKCδ–EGFP was moving toward the lysosomal compartment. By contrast, although a PKCδ–EGFP surface membrane translocation occurred in K8-null hepatocytes, only one spot reflecting a PKCδ and lysosome tracker colocalization was observed, even at 60 min post stimulation. Overall, these data indicate that in hepatocytes lacking K8/K18 IFs, the downregulation of PKCδ through the lysosomal compartment is decreased, leading to a reduced ASMase activation, which in turn decreases the lipid raft size.
Fig. 6.
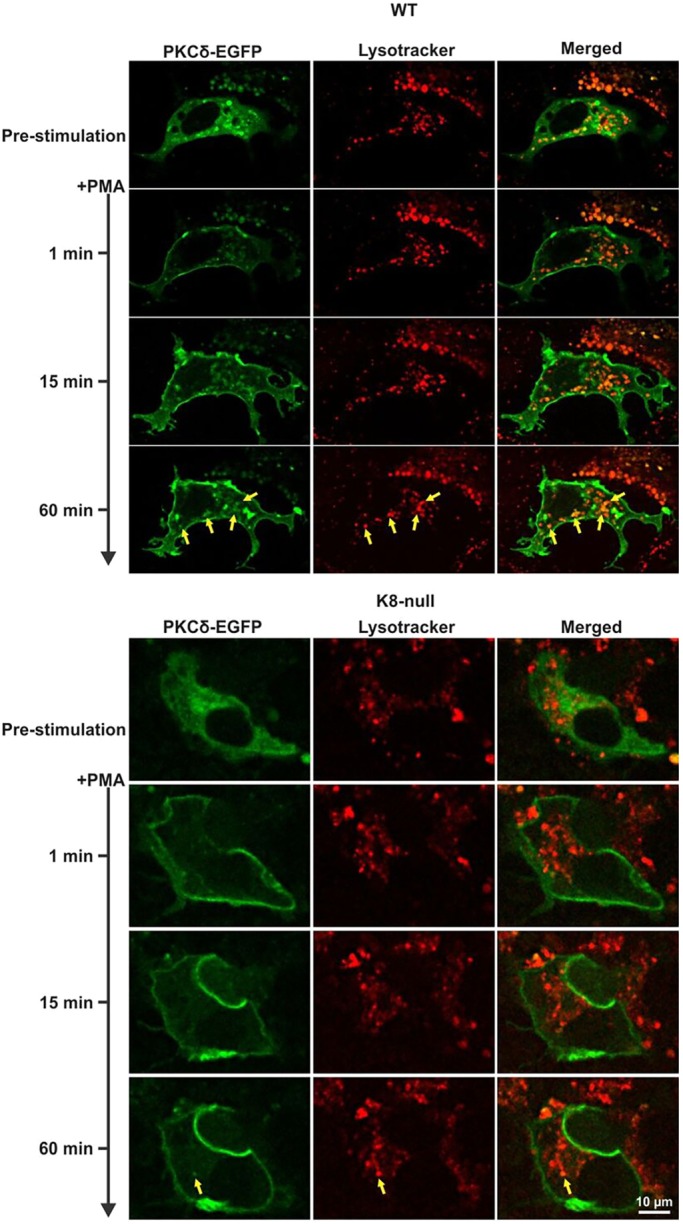
K8/K18 IF modulation of PKCδ localization in lysosomes. Live-cell confocal imaging of PKCδ–EGFP in transfected WT and K8-null hepatocytes along with Lysotracker-labeled lysosomes, following treatment with PMA (100 nM), showing less PKCδ–EGFP and lysosome colocalization in hepatocytes lacking K8/K18 IFs. Yellow arrows point to colocalizations.
The impact of lipid raft size on FasR-mediated apoptosis in hepatocytes
Given the above finding that a cisplatin treatment restored the lipid raft volume in K8-null hepatocytes to the basal level in WT hepatocytes, we wondered whether such a treatment was able to rescue FasR-mediated apoptosis. In line with our previous work (Gilbert et al., 2012, 2008), nuclear fragmentation measurements confirmed that K8-null hepatocytes are more sensitive to FasR-stimulation with Jo2 (antibody against mouse CD95) and Protein A (Fig. 7A). Moreover, the addition of cisplatin at the dose that rescued CT-lipid raft volumes restored K8-null hepatocyte apoptosis to the level of WT hepatocytes (Fig. 7A); this rescue was also observed following PMA stimulation. Notably, the addition of Bim led to an increase in apoptosis in WT hepatocytes, and a decrease in K8-null hepatocytes. Furthermore, to explore the effect of CT-lipid raft volume reduction, we examined ASMase inhibitor modulation of FasR-mediated apoptosis. As shown for Bim, the DPM- or IPM-mediated CT-lipid raft volume reduction led to an increased nuclear fragmentation in WT hepatocytes, but a reduction in K8-null hepatocytes (Fig. 7B). We thus suspected that an optimal CT-lipid raft size is required to maximize the apoptotic response.
Fig. 7.

K8/K18 IF-dependent lipid raft size as a modulatory parameter of FasR-mediated apoptosis. (A) Nuclear fragmentation assessments in response to a Jo2 (0.5 µg/ml)+Protein A (PA, 0.1 µg/ml) treatment of WT and K8-null hepatocyte for 7 h with or without a 1 h pre-treatment with PKC activators [5 µg/ml cisplatin (Cis) or 100 nM PMA] or PKC inhibitor (1 µM BIM), showing that PKC modulations affect FasR-dependent nuclear fragmentation (C, untreated control). (B) Nuclear fragmentation analysis following a Jo2 (0.5 µg/ml)+Protein A (PA, 0.1 µg/ml) treatment of WT and K8-null hepatocytes for 7 h without or with a 1 h pre-treatment with ASMase inhibitors (50 µM DPM or 50 µM IPM,), indicating that ASMase inhibition affects FasR-dependent nuclear fragmentation (C: untreated control). (C,D) Nuclear fragmentation assessments in response to a Jo2 (0.5 µg/ml)+Protein A (PA, 0.1 µg/ml) treatment for 7 h with or without a pre-treatment with the ASMase inhibitor DPM at different concentrations in WT (C) or K8-null (D) hepatocytes. (E) Scale representation of the optimal range of CT-lipid raft volume for seeing apoptosis, showing where WT and K8-null hepatocytes are positioned on the scale versus different treatment conditions. Quantitative results are mean±s.e.m. *P<0.05; **P<0.01; ***P<0.005 (t-test).
To examine this possibility, we used different concentrations of DPM or IPM and assessed FasR-mediated apoptosis versus the CT-lipid raft volume (Fig. 7C,D; Table 1). In WT hepatocytes, the reduction of CT-lipid raft volume with 10 or 50 μM DPM or IPM (Table 1; Fig. S3A,C) led to a clear increase in nuclear fragmentation (Fig. 7C, Table 1). However, when CT-lipid rafts were disrupted by using a higher dose of ASMase inhibitor (150 μM) or by a MBC treatment, a drastic reduction of apoptosis was observed; these findings were expected, given that FasR-mediated apoptosis has previously been shown to be lipid-raft dependent (Gilbert et al., 2012). In contrast, when K8-null hepatocytes were treated with 10 or 50 μM of DPM or IPM, their CT-lipid raft volume also decreased (Table 1; Fig. S3B,D), but in this case it led to a reduction in nuclear fragmentation (Fig. 7D). As for WT hepatocytes, when CT-lipid rafts were disrupted by a high DPM or IPM dose, or MBC treatment, apoptosis was almost absent. Compilation of all the volume versus nuclear fragmentation values (Table 1) leads to the conclusion that an optimal CT-lipid raft volume is between 0.54 and 0.61 μm3, because more than 50% of cells had fragmented nuclei within this volume range. Volumes above or below this range led to less than 50% of cells with fragmented nuclei (Fig. 7E). These data identify K8/K18 IF-dependent lipid raft size as a modulatory parameter of FasR-activation in mouse hepatocytes.
DISCUSSION
The results reported here uncover key mechanistic events underlying the involvement of the K8/K18 network as a cytoskeletal modulator of lipid raft size in hepatocytes. First, a K8/K18 loss leads to a perturbation in lipid raft organization, associated with a reduced CT-lipid raft size. Second, compared to hepatocytes expressing hK8, the K8 G62C mutation reduces CT-lipid raft size. Third, CT-lipid raft size depends on the ASMase activity and this activity is lower in hepatocytes lacking K8/K18 IFs. Fourth, K8/K18 loss downregulates PKCδ re-localization, in association with reduced ASMase activation. Finally, CT-lipid raft size needs to be within a particular range to optimize FasR-mediated apoptosis in hepatocytes. Overall, as schematically represented in our working model (Fig. S1D), the data indicate that K8/K18 IFs affect lipid raft size and FasR stimulation through a modulation of PKCδ relocalization and associated ASMase activation.
Our experimental approach relies on two methodological features. First, the ability of the cholera toxin homopentameric B-subunit (CTX-B) to specifically bind the GM1 ganglioside, which has been widely exploited to visualize coalesced lipid rafts at the surface membrane of a variety of cell types (Gupta and DeFranco, 2003; Pyenta et al., 2001). The cholera toxin binding to GM1 scaffolds five GM1 molecules together, meaning that the size measurements reported here should be viewed as CTX-B-enhanced lipid rafts. Moreover, Lyn16–YFP fusion protein expression in WT and K8-null hepatocytes, a well-known marker of lipid raft inner leaflet of the membrane (Sohn et al., 2008), also reveals a change in lipid raft size following a K8/K18 loss. This correlation between the two visualization conditions provides strong support for the conclusion that the values extracted from the deconvoluted CTX-B Alexa Fluor® 488 images reflect true variations in lipid raft size. Second, two hepatocyte models were used here to address the impact of K8/K18 IFs on lipid raft size in simple epithelial cells. The comparable results obtained with K8-null cells and a pathological K8 variant seen in humans lead us to the overall conclusion that the altered process of lipid raft organization is related to perturbation in K8/K18-dependent regulatory steps affecting the sphingolipid status (as discussed below) rather than a fault in the IF network per se. Moreover, the present results confirm in primary cultured hepatocytes a previous report revealing a notable similarity in IF staining patterns in mouse livers expressing hK8 and K8 G62C under basal conditions in vivo (Ku and Omary, 2006; Ku et al., 2003). Furthermore, the K8 G62C mutation is known to cause K8 cross-linking under oxidative conditions, through cysteine bridging, and disruption of K8 Ser74 phosphorylation by stress-activated kinases (SAKs). In addition, this K8 G62C-dependent inhibition of K8 Ser74 phosphorylation appears to be responsible for the increased susceptibility to FasR-mediated liver (hepatocyte) apoptosis (Ku and Omary, 2006). In this context, it would be of particular interest to determine the extent to which this K8/K18-dependent modulation of lipid raft size applies to other simple-type epithelial cells.
Sphingomyelin is the most abundant cellular sphingolipid and a key component of lipid rafts. Moreover, the ASMase site of action is also the lipid raft, where its activation results in the local generation of ceramides (Silva et al., 2007). Furthermore, in line with previous findings (Zeidan and Hannun, 2010), the present results confirm that ASMase inhibition or activation modulates dynamic features of lipid rafts. We find that a reduction in ASMase surface membrane localization is associated with a decrease in ASMase activity in K8/K18-lacking hepatocytes, meaning that reduced amounts of the different ceramides were produced in these cells. Given that ceramides are known to promote the formation of a large raft platform by the coalescence of small lipid raft structures, which in turn results in a spatial and temporal reorganization of receptors and signaling molecules (Castro et al., 2011), the reduction observed in ASMase activity might well explain the decrease in lipid raft size observed in K8/K18-lacking hepatocytes.
The present results extend our understanding of the signaling pathway upstream of ASMase that involves PKCδ, a member of the phospholipid-activated serine/threonine PKC family that has a role in a wide variety of fundamental cellular processes, including apoptosis (Zhao et al., 2012). For instance, overexpression of PKCδ results in the activation of death-signaling pathways, and in turn during apoptosis, PKCδ is proteolytically cleaved by caspase 3 to generate an activated catalytic fragment, which amplifies apoptosis cascades (Zhao et al., 2012). Mechanistically, as for other novel type PKCs, PKCδ can be activated by multiple factors, including the lipid second messenger diacyglycerol (DAG) and also agonists such as phorbol esters, which promote the membrane targeting required for its conformation-mediated activation (Leontieva and Black, 2004). The localization of activated PKCs is achieved through interactions with anchoring proteins, such as RACK1, which in turn can bind to plectin, a cytolinker that also possesses binding sites for IF proteins, actin and microtubule-associated proteins (Rezniczek et al., 2004). In addition, PKCδ signaling can be controlled by desensitization or internalization through clathrin-independent lipid-raft-mediated endocytic trafficking, with ultimate delivery to lysosomes for degradation (Lum et al., 2013). Remarkably, the present results demonstrate that efficient PKCδ desensitization through its delivery to lysosomes requires an intact K8/K18 network. In more specific terms, they reveal that a K8/K18 loss leads to less internalization of active PKCδ, which in turn results in a reduced ASMase activation. As to the ASMase-mediated downstream molecular steps that might regulate FasR-mediated apoptosis, previous work by others has established that the generated ceramide is a potent activator of PP2A, a regulator of ezrin phosphorylation status at the surface membrane (Zeidan et al., 2008). Notably, our previous work has indicated that a K8/K18 loss in hepatocytes alters the early FasR activation steps through perturbation of the ezrin–actin interplay at lipid rafts (Gilbert et al., 2012). Overall, as described in our working model (Fig. S1D), the work reported here uncovers an impact of K8/K18 on lipid raft size through a modulation of the PKCδ-mediated ASMase regulation. The connection to FasR-mediated apoptosis suggests, in addition, that a downstream interplay between ceramide, PP2A and ezrin also occurs at lipid rafts.
Intriguingly, hepatocytes isolated from ASMase-knockout mice exhibit resistance to FasR-mediated apoptosis, which can be overcome by the addition of C16 ceramide (Lin et al., 2000; Paris et al., 2001). On first pass, this observation seems difficult to reconcile with the increased apoptosis seen here in K8-null hepatocytes. We propose that the local production of ceramide by ASMase at the surface membrane generates variations in lipid raft volume that can, in turn, increase or decrease FasR-apoptosis efficiency. Indeed, when CT-lipid raft volumes are between 0.54 and 0.61 μm3 in both cell types, the FasR stimulation leads to over 50% apoptosis, whereas if the volume is above or below this range a reduction in hepatocyte death is observed (Fig. 4E). Hence, it appears that when the lipid raft is too large, FasR and its associated proteins are dispersed and have difficulty in assembling, whereas in lipid rafts that are too small, FasR has a lower probability of assembling with its associated proteins, and in either case the end result is a lower FasR-mediated apoptosis efficiency. In practical terms, we propose that the volume of the lipid raft needs to be within an optimal range to obtain an efficient FasR interaction with its associated proteins and to generate a maximum FasR-mediated apoptosis (Gajate and Mollinedo, 2005). This interpretation might help to reconcile many contradictions found in the literature on the role of ceramide in death-receptor-mediated cell death (Cock et al., 1998; De Maria et al., 1998; Lin et al., 2000; Paris et al., 2001). Moreover, the fact that keratin loss in hepatocytes generates a reduction of the lipid raft volume, which becomes optimal for FasR activation, explains at least in part the higher FasR-sensitivity previously observed in K8-null hepatocytes (Gilbert et al., 2001).
K8/K18 involvement in human liver disease has been highlighted by the discovery that single point mutations in K8 and K18 genes predispose to progression of acute or chronic liver disease (Omary et al., 2009; Strnad et al., 2010). For instance, overexpression of a human K8 G62C mutant in transgenic mouse liver predisposes to FasR-mediated apoptosis (Ku and Omary, 2006). The results reported here using hepatocytes isolated from K8 G62C mutant versus hK8 normal mouse liver demonstrate that the FasR-mediated apoptosis matches quite well with that observed with K8-null hepatocytes. Actually, this cell death event is reflected at the mechanistic level, where a single point mutation in K8 is sufficient to alter lipid raft volume in association with the FasR-mediated apoptosis. Moreover, considering that ASMase activation can be modulated rapidly by a variety of stimuli, including death receptor ligands (Zeidan and Hannun, 2010), we posit that the ASMase fine-tuning regulation of lipid raft volume might provide a therapeutic target for death-receptor-related liver diseases (Wang, 2014).
MATERIALS AND METHODS
Reagents
Isoflurane was purchased from Abbott Laboratories Ltd (Montreal, Canada). Hamster anti-mouse CD95 (Jo2) monoclonal antibody (#554255), hamster anti-mouse CD95 (Jo2) R-phycoerythrin-labeled monoclonal antibody (Jo2–PE; #554258) and EHS Matrigel Brand (#354234) were from BD Biosciences (Mississauga, Canada). Bisindolylmaleimide I (BIM; #203290) was from EMD Millipore (Etobicoke, Canada). Protein A (#P3838), cisplatin (#P4394), phorbol 12-myristate 13-acetate (PMA; #79346), methyl-β-cyclodextrin (MBC; #4555), desipramine (DPM; #D3900), imipramine (IPM; #I0899), deoxycholic acid (DCA; #30960) and all other reagents were from Sigma Chemical Co. (Mississauga, Canada). The fluorescent molecules Lysotracker Red DND-99 (#L7528) and Vybrant® Alexa Fluor® 488 (#V34403) were from Life Technologies (Carlsbad, CA). The antibodies used included: rat anti-mouse K8 (TROMA-1) monoclonal antibody (from Rolf Kemler, Freiburg, Germany); mouse anti-human K8 monoclonal antibody (TS1; #MS-997-P0) from ThermoFisher Scientific (Waltham, MA); rabbit anti-ASMase polyclonal antibody (H-181; #sc-11352) from Santa Cruz Biotechnology (Santa Cruz, CA); rabbit polyclonal anti-PKCδ (#2058) and anti-phospho-PKCδ(Thr-505) (#9374) from Cell Signaling Technology (Danvers, USA); mouse monoclonal anti-GAPDH (#10R-G109a) from Fitzgerald Industries Intl (Concord, USA); Alexa-Fluor®-488-conjugated anti-GFP (#A-21311), Alexa-Fluor®-488-conjugated goat anti-rabbit-IgG (#A11008), Alexa-Fluor®-488-conjugated goat anti-mouse-IgG (#A11001), Alexa-Fluor®-488-conjugated goat anti-rat-IgG (#A11006) (all H+L) and Alexa-Fluor®-594-conjugated goat anti-mouse-IgG (#A11005) antibodies from Life Technologies (Carlsbad, CA).
Hepatocyte isolation and culture
Hepatocytes were isolated from normal mice (WT), K8 knockout mice (K8-null) (Baribault et al., 1994) or transgenic mice expressing the normal human K8 gene (hK8) or the human K8 Gly62-to-Cys (G62C) (Ku and Omary, 2006) mutated gene according to a modified version of the two-step collagenase method as described previously (Marceau et al., 2004). The experiments using hepatocyte primary cultures were performed according to the guidelines of the Laval University Animal Care Committee (protocol #2012161). Hepatocytes were plated at a density of 1.2×105 cells/cm2 on fibronectin-coated dishes in Dulbecco's modified Eagle's medium with Ham's F12 (DMEM/F12) supplemented with sodium selenite (5 µg/l), insulin (5 mg/l), transferrin (5 mg/l), streptomycin (100 µg/ml) and penicillin (100 U/ml). After a 3-h attachment period, the culture medium was replaced by the same medium supplemented with dexamethasone (10−7 M) and EGF (20 ng/ml).
Hepatocyte treatment
At 24 h post seeding, the hepatocyte medium was changed to DMEM/F12 supplemented with selenious acid (5 µg/l), transferrin (5 mg/l), dexamethasone (10−7 M), matrigel (0.5 mg/ml), streptomycin (100 µg/ml) and penicillin (100 units/ml). For the inhibitor treatment, cells at 48 h post seeding were incubated with DPM, IPM, MBC or BIM (see figure legends for the specific concentrations) for 1 h at 37°C with 5% CO2 prior subsequent manipulations. For the inducer treatment, cells at 48 h post seeding were treated with PMA (100 nM), cisplatin (5 μg/ml) or DCA (50 μM) and incubated for 1 h at 37°C with 5% CO2 before subsequent manipulations. For apoptosis induction and nuclear fragmentation assessment, the apoptosis inducer Jo2 (0.5 µg/ml) alone or in combination with protein A (0.1 µg/ml) was added in the absence or presence of the above inductors or inhibitors and incubated for 7 h at 37°C with 5% CO2. The details on nuclear fragmentation assessment using the Acridine-Orange-based procedure were described previously (Gilbert et al., 2001).
Lipid raft visualization and size assessment
We visualized the distribution of coalesced lipid rafts at the surface membrane of cultured hepatocytes, making use of a fluorescent conjugate of CTX-B, which binds to the hepentasaccharide chain of ganglioside GM1, a constituent of the outer leaflet of the lipid raft (Aaronson and Blobel, 1975; Gilbert et al., 2012; Gupta and DeFranco, 2003). Briefly, at 48 h post seeding, hepatocytes were maintained for 10 min on ice before the addition of Vybrant® Alexa Fluor® 488 (CTX-B labeling) at 3 µg/ml followed by a 30-min incubation on ice. Cells were then washed twice with cold phosphate-buffered saline (PBS) and fixed with 2% formaldehyde for 10 min at room temperature (22°C). Images were acquired of 30 focal planes (z-stack), each separated from the adjacent plane by 0.2 µm, with a FV1000-Olympus confocal system, using the 488-nm excitation laser line and a 60×1.42 NA oil immersion objective.
Notably, the size of coalesced lipid rafts has been found to depend on the stimulus and the cell type (Gupta and DeFranco, 2003). It includes not only their surface but also their thickness (volume), which in turn depends on interplay between cholesterol, sphingolipids and transmembrane proteins (Lingwood and Simons, 2010). Here, hepatocytes were cultured in presence or absence of inhibitors or inducers for different time periods, and three size-related parameters of coalesced CTX-B-tagged lipid rafts were assessed, namely cell surface coverage (%), individual surface (µm2) and volume (µm3). Briefly, images were digitally deconvolved and combined to generate a three-dimensional reconstruction by using the Volocity® 3D image analysis software (PerkinElmer; Waltham, CA). Stacks were cropped so as to retain only the cell surface and to remove non-relevant staining below the membrane, followed by a second crop on single cells to visualize coalesced CTX-B-tagged lipid rafts at the cell surface. At that step, the resulting stacks were merged in one plane, prior to evaluations of the cell surface area and coalesced CTX-B-tagged lipid raft surfaces (µm2); visual examination allowed the exclusion of particles smaller than 0.3 µm2. The sum of these values divided by the total surface membrane area yielded the percentage of cell surface covered with CTX-B-tagged lipid rafts. For volume assessments, after cropping individual cells, a size threshold was established visually on one set of images and then applied for all the subsequent CT-lipid raft images; their volumes (µm3) were extracted using the above-mentioned evaluation tools, starting from the xyz view. Of note, only coalesced CT-lipid raft volumes between 0.1 and 3 µm3 were considered in order to readily exclude cell aggregates and small debris. For all measurements, the values of over 25 single cells were averaged.
Confocal cell imaging
For ASMase, cultured hepatocytes were fixed for 10 min at room temperature with 3.7% paraformaldehyde, blocked for 1 h at room temperature with 5% fetal bovine serum (FBS) in PBS and then permeabilized by treatment for 10 min at room temperature with 0.3% Triton X-100 in presence of 1% BSA in PBS. Then, labeling was performed using the anti-ASMase (1:100) antibody overnight at 4°C. Corresponding fluorescent-tagged secondary antibodies (1:100) were then added and incubation was continued for 60 min at room temperature. For PKC–EGFP, cultured hepatocytes were fixed for 10 min at room temperature with 2% paraformaldehyde, blocked for 1 h at room temperature with 5% FBS in PBS and then permeabilized for 10 min at room temperature with 0.3% Triton X-100 in the presence of 1% BSA in PBS. Then, labeling was performed using an Alexa-Fluor®-488-conjugated anti-EGFP- (1:100) incubated overnight at 4°C. For Lyn16–YFP, cultured hepatocytes were fixed by treatment for 10 min at room temperature with 2% paraformaldehyde. For mouse and human K8, cultured hepatocytes were fixed with 2% paraformaldehyde in PBS for 10 min at room temperature, and extracted with 100% methanol for 5 min at −20°C. The labeling was performed with anti-mouse K8 (1:100) or anti-human K8 (1:100) antibody overnight at 4°C. Appropriate fluorescent-tagged secondary antibodies (1:100) were then incubated for 60 min at room temperature. In some experiments, a 10-min incubation at room temperature with Hoechst 33258 (0.5 µg/ml in water) was performed to label the nuclei. Cells were then mounted on a slide with a drop of mounting medium [glycine (7.5 mg/ml), NaOH (3.5 mg/ml), NaCl (8.5 mg/ml), NaN3 (0.5 mg/ml) and glycerol (50% v/v) at pH 8.6]. Images were captured with a FV1000 Olympus confocal imaging system and analyzed with the Volocity® 3D image analysis software or Metamorph® imaging software (Molecular Device, Sunnyvale, CA).
ASMase activity assessment
The ASMase activity was assessed using a sphingomyelinase assay kit (#KA1374; Abnova, Taipei City, Taiwan), as described by the manufacturer. The ASMase samples were extracted from 6.6×106 hepatocytes for each of the conditions and experiments were performed in triplicates.
Hepatocyte transfection and imaging
At 24 h post seeding, the hepatocytes were placed in DMEM/F12 supplemented with sodium selenite (5 µg/l), insulin (5 mg/l), transferrin (5 mg/l), streptomycin (100 µg/ml), penicillin (100 U/ml), dexamethasone (10−7 M) and EGF (20 ng/ml). At 52 h post seeding, transfections of Lyn16–YFP (provided by Susan K. Pierce, NIH, Rockville, MD), human PKCα–EGFP, mouse PKCδ–EGFP or mouse PKCε–EGFP (provided by Dominique Joubert, Montpellier, France) were performed using Fugen®HD (Roche, Indianapolis, IN), according to manufacturer instructions using a 4:2 ratio. At 18 h post transfection, the cultures were washed twice with PBS pH 7.4 and then maintained in DMEM/F12 supplemented with selenious acid (5 μg/l), transferrin (5 mg/l), dexamethasone (10−7 M), matrigel (0.5 mg/ml), streptomycin (100 µg/ml) and penicillin (100 units/ml). At 42 h post transfection, cells were treated and then fixed. Thereafter, images were captured with the FV1000 Olympus confocal imaging system, using the 488- or 514-nm excitation laser line for GFP and YFP, respectively.
In one set of experiments, the lysosomes of PKCδ–EGFP-transfected WT and K8-null hepatocytes were labeled with Lysotracker Red DND-99 for 1 h in a 37°C and 5% CO2 environment. Thereafter, live-cell images were captured with the spinning disk system, using the 488- and 568-nm excitation laser lines, which in turn lead to emission spectra with maxima at 519 and 617 nm, respectively. Acquisitions were performed through 530/63 and 610/70 emission filters. Control experiments revealed no interfering bleed-through between the corresponding channels.
FasR SPOTS analysis
Hepatocytes were incubated for 10 min on ice after which the fluorescent-tagged Fas-agonist antibody Jo2–PE (0.5 µg/ml) coupled to protein A (0.1 µg/ml) was added for an additional 30 min on ice. Cells were then returned to 37°C for the indicated periods of time. The cells were fixed with 2% formaldehyde for 10 min at room temperature, and their surfaces were imaged using a FV1000 Olympus confocal system.
Western blotting
Protein samples (5 µg) were subjected to SDS-PAGE and transferred onto a PVDF membrane. The blots were incubated with the primary antibody (1:1000) and then with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (1:5000). The staining was revealed with the SuperSignal West Pico Kit (Pierce, Rockford, IL).
Acknowledgements
We thank H. Baribault for the original WT and K8-null mouse colonies, R. Kemler for the TROMA-1 hybridoma, S.K. Pierce for Lyn16–YFP cDNA, D. Joubert for EGFP-tagged PKC cDNAs and A. Anderson for proofreading the manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: S.G., N.M.; methodology: S.G., A.L., M.B.O.; writing – original draft preparation: S.G.; Writing – review and editing: N.M., S.G., M.B.O.; funding acquisition: N.M., M.B.O.
Funding
This work was supported by the Canadian Institutes of Health Research [grant number MOP 15529 to N.M.]; and the National Institutes of Health [grant number DK52951 to M.B.O.]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.171124.supplemental
References
- Aaronson R. P. and Blobel G. (1975). Isolation of nuclear pore complexes in association with a lamina. Proc. Natl. Acad. Sci. USA 72, 1007-1011. 10.1073/pnas.72.3.1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baribault H., Penner J., Iozzo R. V. and Wilson-Heiner M. (1994). Colorectal hyperplasia and inflammation in keratin 8-deficient FVB/N mice. Genes Dev. 8, 2964-2973. [DOI] [PubMed] [Google Scholar]
- Bordeleau F., Galarneau L., Gilbert S., Loranger A. and Marceau N. (2010). Keratin 8/18 modulation of protein kinase C-mediated integrin-dependent adhesion and migration of liver epithelial cells. Mol. Biol. Cell 21, 1698-1713. 10.1091/mbc.E09-05-0373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro B. M., de Almeida R. F., Goormaghtigh E., Fedorov A. and Prieto M. (2011). Organization and dynamics of Fas transmembrane domain in raft membranes and modulation by ceramide. Biophys. J. 101, 1632-1641. 10.1016/j.bpj.2011.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro B. M., Prieto M. and Silva L. C. (2014). Ceramide: a simple sphingolipid with unique biophysical properties. Prog. Lipid Res. 54, 53-67. 10.1016/j.plipres.2014.01.004 [DOI] [PubMed] [Google Scholar]
- Cock J. G. R. B.-d., Tepper A. D., de Vries E., van Blitterswijk W. J. and Borst J. (1998). CD95 (Fas/APO-1) induces ceramide formation and apoptosis in the absence of a functional acid sphingomyelinase. J. Biol. Chem. 273, 7560-7565. 10.1074/jbc.273.13.7560 [DOI] [PubMed] [Google Scholar]
- Coulombe P. A. and Omary M. B. (2002). ‘Hard’ and ‘soft’ principles defining the structure, function and regulation of keratin intermediate filaments. Curr. Opin. Cell Biol. 14, 110-122. 10.1016/S0955-0674(01)00301-5 [DOI] [PubMed] [Google Scholar]
- Coulombe P. A. and Wong P. (2004). Cytoplasmic intermediate filaments revealed as dynamic and multipurpose scaffolds. Nat. Cell Biol. 6, 699-706. 10.1038/ncb0804-699 [DOI] [PubMed] [Google Scholar]
- De Maria R., Rippo M. R., Schuchman E. H. and Testi R. (1998). Acidic sphingomyelinase (ASM) is necessary for fas-induced GD3 ganglioside accumulation and efficient apoptosis of lymphoid cells. J. Exp. Med. 187, 897-902. 10.1084/jem.187.6.897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futerman A. H. and Hannun Y. A. (2004). The complex life of simple sphingolipids. EMBO Rep. 5, 777-782. 10.1038/sj.embor.7400208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajate C. and Mollinedo F. (2005). Cytoskeleton-mediated death receptor and ligand concentration in lipid rafts forms apoptosis-promoting clusters in cancer chemotherapy. J. Biol. Chem. 280, 11641-11647. 10.1074/jbc.M411781200 [DOI] [PubMed] [Google Scholar]
- Gilbert S., Loranger A., Daigle N. and Marceau N. (2001). Simple epithelium keratins 8 and 18 provide resistance to Fas-mediated apoptosis. The protection occurs through a receptor-targeting modulation. J. Cell Biol. 154, 763-773. 10.1083/jcb.200102130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert S., Ruel A., Loranger A. and Marceau N. (2008). Switch in Fas-activated death signaling pathway as result of keratin 8/18-intermediate filament loss. Apoptosis 13, 1479-1493. 10.1007/s10495-008-0274-x [DOI] [PubMed] [Google Scholar]
- Gilbert S., Loranger A., Lavoie J. N. and Marceau N. (2012). Cytoskeleton keratin regulation of FasR signaling through modulation of actin/ezrin interplay at lipid rafts in hepatocytes. Apoptosis 17, 880-894. 10.1007/s10495-012-0733-2 [DOI] [PubMed] [Google Scholar]
- Gould C. M. and Newton A. C. (2008). The life and death of protein kinase C. Curr. Drug Targets 9, 614-625. 10.2174/138945008785132411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green K. J., Böhringer M., Gocken T. and Jones J. C. R. (2005). Intermediate filament associated proteins. Adv. Protein Chem. 70, 143-202. 10.1016/S0065-3233(05)70006-1 [DOI] [PubMed] [Google Scholar]
- Gupta N. and DeFranco A. L. (2003). Visualizing lipid raft dynamics and early signaling events during antigen receptor-mediated B-lymphocyte activation. Mol. Biol. Cell 14, 432-444. 10.1091/mbc.02-05-0078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S., Natarajan R., Payne S. G., Studer E. J., Spiegel S., Dent P. and Hylemon P. B. (2004). Deoxycholic acid activates the c-Jun N-terminal kinase pathway via FAS receptor activation in primary hepatocytes. Role of acidic sphingomyelinase-mediated ceramide generation in FAS receptor activation. J. Biol. Chem. 279, 5821-5828. 10.1074/jbc.M310979200 [DOI] [PubMed] [Google Scholar]
- Herrmann H. and Aebi U. (2004). Intermediate filaments: molecular structure, assembly mechanism, and integration into functionally distinct intracellular Scaffolds. Annu. Rev. Biochem. 73, 749-789. 10.1146/annurev.biochem.73.011303.073823 [DOI] [PubMed] [Google Scholar]
- Holthuis J. C. M., van Meer G. and Huitema K. (2003). Lipid microdomains, lipid translocation and the organization of intracellular membrane transport (Review). Mol. Membr. Biol. 20, 231-241. 10.1080/0988768031000100768 [DOI] [PubMed] [Google Scholar]
- Jenkins R. W., Idkowiak-Baldys J., Simbari F., Canals D., Roddy P., Riner C. D., Clarke C. J. and Hannun Y. A. (2011). A novel mechanism of lysosomal acid sphingomyelinase maturation: requirement for carboxyl-terminal proteolytic processing. J. Biol. Chem. 286, 3777-3788. 10.1074/jbc.M110.155234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N.-O. and Omary M. B. (2000). Keratins turn over by ubiquitination in a phosphorylation-modulated fashion. J. Cell Biol. 149, 547-552. 10.1083/jcb.149.3.547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N.-O. and Omary M. B. (2006). A disease- and phosphorylation-related nonmechanical function for keratin 8. J. Cell Biol. 174, 115-125. 10.1083/jcb.200602146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N.-O., Gish R., Wright T. L. and Omary M. B. (2001). Keratin 8 mutations in patients with cryptogenic liver disease. N. Engl. J. Med. 344, 1580-1587. 10.1056/NEJM200105243442103 [DOI] [PubMed] [Google Scholar]
- Ku N. O., Soetikno R. M. and Omary M. B. (2003). Keratin mutation in transgenic mice predisposes to Fas but not TNF-induced apoptosis and massive liver injury. Hepatology 37, 1006-1014. 10.1053/jhep.2003.50181 [DOI] [PubMed] [Google Scholar]
- Ku N.-O., Lim J. K., Krams S. M., Esquivel C. O., Keeffe E. B., Wright T. L., Parry D. A. D. and Omary M. B. (2005). Keratins as susceptibility genes for end-stage liver disease. Gastroenterology 129, 885-893. 10.1053/j.gastro.2005.06.065 [DOI] [PubMed] [Google Scholar]
- Ku N.-O., Strnad P., Zhong B.-H., Tao G.-Z. and Omary M. B. (2007). Keratins let liver live: Mutations predispose to liver disease and crosslinking generates Mallory-Denk bodies. Hepatology 46, 1639-1649. 10.1002/hep.21976 [DOI] [PubMed] [Google Scholar]
- Leontieva O. V. and Black J. D. (2004). Identification of two distinct pathways of protein kinase Calpha down-regulation in intestinal epithelial cells. J. Biol. Chem. 279, 5788-5801. 10.1074/jbc.M308375200 [DOI] [PubMed] [Google Scholar]
- Lin T., Genestier L., Pinkoski M. J., Castro A., Nicholas S., Mogil R., Paris F., Fuks Z., Schuchman E. H., Kolesnick R. N. et al. (2000). Role of acidic sphingomyelinase in Fas/CD95-mediated cell death. J. Biol. Chem. 275, 8657-8663. 10.1074/jbc.275.12.8657 [DOI] [PubMed] [Google Scholar]
- Lingwood D. and Simons K. (2010). Lipid rafts as a membrane-organizing principle. Science 327, 46-50. 10.1126/science.1174621 [DOI] [PubMed] [Google Scholar]
- Liu X., Zeidan Y. H., Elojeimy S., Holman D. H., El-Zawahry A. M., Guo G. W., Bielawska A., Bielawski J., Szulc Z., Rubinchik S. et al. (2006). Involvement of sphingolipids in apoptin-induced cell killing. Mol. Ther. 14, 627-636. 10.1016/j.ymthe.2006.07.001 [DOI] [PubMed] [Google Scholar]
- Lum M. A., Pundt K. E., Paluch B. E., Black A. R. and Black J. D. (2013). Agonist-induced down-regulation of endogenous protein kinase Calpha through an endolysosomal mechanism. J. Biol. Chem. 288, 13093-13109. 10.1074/jbc.M112.437061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marceau N., Gilbert S. and Loranger A. (2004). Uncovering the roles of intermediate filaments in apoptosis. Methods Cell Biol. 78, 95-129. 10.1016/S0091-679X(04)78005-X [DOI] [PubMed] [Google Scholar]
- Marceau N., Schutte B., Gilbert S., Loranger A., Henfling M. E. R., Broers J. L. V., Mathew J. and Ramaekers F. C. S. (2007). Dual roles of intermediate filaments in apoptosis. Exp. Cell Res. 313, 2265-2281. 10.1016/j.yexcr.2007.03.038 [DOI] [PubMed] [Google Scholar]
- Mashukova A., Oriolo A. S., Wald F. A., Casanova M. L., Kroger C., Magin T. M., Omary M. B. and Salas P. J. I. (2009). Rescue of atypical protein kinase C in epithelia by the cytoskeleton and Hsp70 family chaperones. J. Cell Sci. 122, 2491-2503. 10.1242/jcs.046979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omary M. B., Baxter G. T., Chou C. F., Riopel C. L., Lin W. Y. and Strulovici B. (1992). PKC epsilon-related kinase associates with and phosphorylates cytokeratin 8 and 18. J. Cell Biol. 117, 583-593. 10.1083/jcb.117.3.583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omary M. B., Ku N.-O., Strnad P. and Hanada S. (2009). Toward unraveling the complexity of simple epithelial keratins in human disease. J. Clin. Invest. 119, 1794-1805. 10.1172/JCI37762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmanagic-Myers S. and Wiche G. (2004). Plectin-RACK1 (receptor for activated C kinase 1) scaffolding: a novel mechanism to regulate protein kinase C activity. J. Biol. Chem. 279, 18701-18710. 10.1074/jbc.M312382200 [DOI] [PubMed] [Google Scholar]
- Paris F., Grassme H., Cremesti A., Zager J., Fong Y., Haimovitz-Friedman A., Fuks Z., Gulbins E. and Kolesnick R. (2001). Natural ceramide reverses Fas resistance of acid sphingomyelinase(-/-) hepatocytes. J. Biol. Chem. 276, 8297-8305. 10.1074/jbc.M008732200 [DOI] [PubMed] [Google Scholar]
- Pyenta P. S., Holowka D. and Baird B. (2001). Cross-correlation analysis of inner-leaflet-anchored green fluorescent protein co-redistributed with IgE receptors and outer leaflet lipid raft components. Biophys. J. 80, 2120-2132. 10.1016/S0006-3495(01)76185-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezniczek G. A., Janda L. and Wiche G. (2004). Plectin. Methods Cell Biol. 78, 721-755. 10.1016/S0091-679X(04)78025-5 [DOI] [PubMed] [Google Scholar]
- Schütze S., Tchikov V. and Schneider-Brachert W. (2008). Regulation of TNFR1 and CD95 signalling by receptor compartmentalization. Nat. Rev. Mol. Cell Biol. 9, 655-662. 10.1038/nrm2430 [DOI] [PubMed] [Google Scholar]
- Schweizer J., Bowden P. E., Coulombe P. A., Langbein L., Lane E. B., Magin T. M., Maltais L., Omary M. B., Parry D. A. D., Rogers M. A. et al. (2006). New consensus nomenclature for mammalian keratins. J. Cell Biol. 174, 169-174. 10.1083/jcb.200603161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva L. C., de Almeida R. F. M., Castro B. M., Fedorov A. and Prieto M. (2007). Ceramide-domain formation and collapse in lipid rafts: membrane reorganization by an apoptotic lipid. Biophys. J. 92, 502-516. 10.1529/biophysj.106.091876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K. and Gerl M. J. (2010). Revitalizing membrane rafts: new tools and insights. Nat. Rev. Mol. Cell Biol. 11, 688-699. 10.1038/nrm2977 [DOI] [PubMed] [Google Scholar]
- Snider N. T. and Omary M. B. (2014). Post-translational modifications of intermediate filament proteins: mechanisms and functions. Nat. Rev. Mol. Cell Biol. 15, 163-177. 10.1038/nrm3753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn H. W., Tolar P. and Pierce S. K. (2008). Membrane heterogeneities in the formation of B cell receptor–Lyn kinase microclusters and the immune synapse. J. Cell Biol. 182, 367-379. 10.1083/jcb.200802007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strnad P., Lienau T. C., Tao G.-Z., Lazzeroni L. C., Stickel F., Schuppan D. and Omary M. B. (2006). Keratin variants associate with progression of fibrosis during chronic hepatitis C infection. Hepatology 43, 1354-1363. 10.1002/hep.21211 [DOI] [PubMed] [Google Scholar]
- Strnad P., Zhou Q., Hanada S., Lazzeroni L. C., Zhong B. H., So P., Davern T. J., Lee W. M., Acute Liver Failure Study, G. and Omary M. B. (2010). Keratin variants predispose to acute liver failure and adverse outcome: race and ethnic associations. Gastroenterology 139, 828-835, 835 e1-3 10.1053/j.gastro.2010.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K. (2014). Molecular mechanisms of hepatic apoptosis. Cell Death Dis. 5, e996 10.1038/cddis.2013.499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidan Y. H. and Hannun Y. A. (2007). Activation of acid sphingomyelinase by protein kinase Cdelta-mediated phosphorylation. J. Biol. Chem. 282, 11549-11561. 10.1074/jbc.M609424200 [DOI] [PubMed] [Google Scholar]
- Zeidan Y. H. and Hannun Y. A. (2010). The acid sphingomyelinase/ceramide pathway: biomedical significance and mechanisms of regulation. Curr. Mol. Med. 10, 454-466. 10.2174/156652410791608225 [DOI] [PubMed] [Google Scholar]
- Zeidan Y. H., Jenkins R. W. and Hannun Y. A. (2008). Remodeling of cellular cytoskeleton by the acid sphingomyelinase/ceramide pathway. J. Cell Biol. 181, 335-350. 10.1083/jcb.200705060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M., Xia L. and Chen G.-Q. (2012). Protein kinase cdelta in apoptosis: a brief overview. Arch. Immunol. Ther. Exp. 60, 361-372. 10.1007/s00005-012-0188-8 [DOI] [PubMed] [Google Scholar]