

Abstract
Seventy‐five percent of all traumatic brain injuries are mild and do not cause readily visible abnormalities on routine medical imaging making it difficult to predict which individuals will develop unwanted clinical sequelae. Microglia are brain‐resident macrophages and early responders to brain insults. Their activation is associated with changes in morphology or expression of phenotypic markers including P2Y12 and major histocompatibility complex class II. Using a murine model of unrestrained mild closed head injury (mCHI), we used microglia as reporters of acute brain injury at sites of impact versus sites experiencing rotational stress 24 h post‐mCHI. Consistent with mild injury, a modest 20% reduction in P2Y12 expression was detected by quantitative real‐time PCR (qPCR) analysis but only in the impacted region of the cortex. Furthermore, neither an influx of blood‐derived immune cells nor changes in microglial expression of CD45, TREM1, TREM2, major histocompatibility complex class II or CD40 were detected. Using magnetic resonance imaging (MRI), small reductions in T2 weighted values were observed but only near the area of impact and without overt tissue damage (blood deposition, edema). Microglial morphology was quantified without cryosectioning artifacts using ScaleA 2 clarified brains from CX3CR1‐green fluorescence protein (GFP) mice. The cortex rostral to the mCHI impact site receives greater rotational stress but neither MRI nor molecular markers of microglial activation showed significant changes from shams in this region. However, microglia in this rostral region did display signs of morphologic activation equivalent to that observed in severe CHI. Thus, mCHI‐triggered rotational stress is sufficient to cause injuries undetectable by routine MRI that could result in altered microglial surveillance of brain homeostasis.
Acute changes in microglial morphology reveal brain responses to unrestrained mild traumatic brain injury
In areas subjected to rotational stress distant from impact site
In the absence of detectable changes in standard molecular indicators of brain damage, inflammation or microglial activation.
That might result in decreased surveillance of brain function and increased susceptibility to subsequent brain insults.
Keywords: confocal microscopy, traumatic brain injury, T2‐weighted imaging, neuroinflammation
Abbreviations used
- CNS
central nervous system
- CT
computed tomography
- GCS
glasgow coma score
- GFAP
glial fibrillary acidic protein
- GFP
green fluorescence protein
- iNOS
inducible nitric oxide
- mCHI
mild closed head injury
- MHC II
major histocompatibility complex class II
- MRI
magnetic resonance imaging
- MR
magnetic resonance
- PBS
phosphate buffered saline
- PE
phycoerythrin
- sCHI
severe closed head injury
- T2WI
T2 weighted images
- TBI
traumatic brain injury
Traumatic brain injury (TBI) can be caused by blunt force or blast‐induced trauma to the head and encompasses a heterogeneous array of injuries, all of which can range from mild to severe (Faul et al. 2015). Each year approximately 1.3 million individuals in the United States and an estimated 42 million worldwide sustain TBIs that are mild (mTBI) to moderate. The most common causes of mTBI in the general population are falls, motor vehicle accidents and contact sports. However, with changes in military strategies and military gear, survivable blast injuries are now frequent causes of mTBI among soldiers. Indeed, mTBI has been called the signature injury of the Afghanistan and Iraqi wars. In concussion or mild TBI, little or no overt brain damage is detected with standard magnetic resonance imaging (MRI) imaging methods used in clinical settings. Consequently, mTBI is likely underreported in annual incidence surveys because of the inability to detect damage in the emergency room as well the failure for those with minor injuries to seek medical help.
There is currently substantial debate on how to quantify TBI severity and category both accurately and in an unbiased manner. The Glasgow Coma Scale, which ranges from 3 (prolonged unconsciousness) to 15 (normal), is the method frequently used to differentiate the severity of TBI based on initial clinical responses (Gardner 2015). mTBI is defined as a TBI causing 0–30 min of unconsciousness, disorientation, post‐traumatic amnesia and/or neurologic deficits. mTBI corresponding to Glasgow coma score scores of 13–15 reveal little to no detectable brain abnormalities by MRI or CT imaging methodologies. Magnetic resonance imaging including T2‐weighted (T2; T2WI), is increasingly used for detection and diagnosis of moderate to severe TBI (sTBI). T2WI is a reflection of the unique relaxation properties within tissues, where for example, tissues with excess water (i.e. edema in stroke or TBI) will have longer or increased T2 values (relaxation time) (Obenaus et al. 2007). In contrast, T2 values can be reduced under certain circumstances, including the presence of blood, increased metabolic demands or reduced blood flow within the affected tissues (Choy et al. 2014). In contrast, mTBI typically reports no overt changes in T2 (Rutgers 2008).
Although mTBI often does not cause direct brain damage, it is not without long‐term clinical consequences. sTBI can cause brain swelling, blood deposition, axonal shearing and neuronal death whereas those with mTBI often develop persistent neurologic sequelae without overt brain damage (Obenaus 2015). For example, concussed individuals can report ‘feeling slowed down’, ‘feeling like “in a fog”’, ‘fatigued or low energy’, ‘difficulty concentrating’, often with an enduring headache for prolonged periods following mTBI (Gardner 2015). Failure to detect mTBI acutely after injury by common clinical imaging methodologies has hampered the ability to predict prognosis or define the progression of mTBI associated pathophysiology (Pacifico et al. 2015).
Here, we sought to utilize microglia, the brain's own surveillance mechanism, to identify cortical regions damaged by unrestrained mTBI close to the time of injury. Microglia are the tissue macrophage of the brain. Seminal studies by Davalos et al. (2005) and Nimmerjahn and Kirchhoff (2005) convincingly demonstrated that microglia continually survey all elements in their environment every 6 h with highly motile processes. Consistent with their roles in monitoring the homeostatic health of the CNS, microglia express a large array of receptors classified as the ‘sensesome’ that can detect ‘danger signals’ associated with tissue damage, neuronal activity, immune cell infiltration and blood‐derived molecules (Schmid et al. 2009; Vinet et al. 2012; Hickman et al. 2013; Butovsky et al. 2014). In their homeostatic state, microglia have small cell bodies with long fine processes that extend away from the soma. Upon robust activation, the microglial processes shorten and retract whereas the soma enlarges presumably to support activation‐induced production of immune or tissue repair molecules.
We hypothesized that injury induced changes in microglial morphology should parallel any brain abnormalities detected by MRI. At early times post‐injury, we hypothesized the greatest changes would be at the site of impact and would decline with increasing distance from impact. Because antibody‐mediated labeling of microglia often does not reliably detect the full extension of microglial processes, we chose to use heterozygous CX3CR1‐green fluorescence protein (GFP) mice in which the CX3CR1 promoter drives expression of green fluorescent protein (Jung et al. 2000, Davalos et al. 2005; Nimmerjahn and Kirchhoff 2005). Within the brain, CX3CR1‐GFP is specifically expressed by all microglia. The expressed GFP fills both the soma and the fine processes of microglia in all regions of the brain. To avoid tissue distortions associated with freezing and cryosectioning, we chose to quantify microglial process length and soma volume in brains chemically clarified using the ScaleA 2 method (Hama 2011).
Multiple models of TBI examining the pathologies ranging from mild to severe have been developed and well characterized (Mierzwa et al. 2014; Susarla et al. 2014; Roth et al. 2014; Rancan et al. 2004; Fenn et al. 2014; Loane et al. 2014; Mouzon et al. 2014; Ojo et al. 2015; Smith et al. 2013). Here, we chose to use an unrestrained closed head model for two reasons. First, removal of the skull such as for controlled cortical impacts was by itself sufficient to cause significant changes in the morphology of cortical microglia (Huang et al. 2013) and thus would confound detection of mTBI‐triggered changes. Second, most closed head injury models restrain the head causing the immobilized head to bear the full force of the impact device at the site of contact. In an unrestrained model with the mouse positioned upon a foam cushion, the head moves away from the contusion device both decreasing the force sustained at site of impact while also introducing large rotational stress caused by this same head movement.
In sum, we demonstrate that when the head is unrestrained, a single mTBI given directly above the right cortical hemisphere causes no observable overt edema and blood deposition as characterized by T2 weighted MRI. However, small but reproducible alterations of T2 values are detected at the impacted area. While microglia extend processes from their soma that survey their local brain region, the extension of their processes is not a simple perfect spherical halo surrounding around the cell body. Instead, the processes (and thus the brain region surveyed) may be aligned in a more oval shaped ‘halo’ with a short and a long axis of process extension away from soma. Here, we find that microglia are morphologically ‘activated’ as characterized by a decreased longest axis of processes in previously unsuspected injury sites, most notably the anterior cortex. These data suggest that acutely following injury, microglia may have altered surveillance of brain regions even at sites distant from direct impact. The consequences of altered surveillance are as yet unknown but may leave affected brain regions at greater risk of damage following subsequent insults.
Materials and methods
Animals
Adult C57Bl/6J and double transgenic CX3CR1‐GFP/PGRPs‐DsRed (Jung et al. 2001, Wang et al. 2011; Sakhon et al. 2015) male mice ages 6–8 weeks were housed in a temperature controlled animal facility on a 12‐h light/dark cycle. C57Bl/6J mice were randomly assigned to either Sham (n = 9) or mild TBI (n = 18) groups for MRI data analysis, whereas double transgenic CX3CR1‐GFP/PGRPs‐DsRed animals, expressing GFP in microglia (Jung et al. 2001) and DsRed in neutrophils (Wang et al. 2011, Sakhon et al. 2015), underwent sham (n = 3), mild (n = 3) or severe (n = 4) TBI for microglial morphology analysis. All protocols performed were in compliance with federal regulations and IACUC approved protocols at Loma Linda University and the University of California Riverside.
Traumatic brain injury
Mice were lightly anesthetized (isoflurane induction: 3%; maintenance: 1–2%) and placed on a foam bed (type E foam; Foam to Size Inc., Ashland, VA, USA) equipped with a nose cone. A midline incision was performed and the skin was retracted to expose the surface of the skull. A mild (1 mm displacement, 40 psi driving pressure, 250 dwell) or severe (4 mm displacement, 100 psi driving pressure, 250 ms dwell) closed head injury (CHI) was then induced to the right hemisphere immediately adjacent to the coronal and sagittal sutures by a pneumatically driven piston with a 4 mm diameter Delrin plastic tip. Following injury and the skull was inspected for skull fractures, which if present were cataloged. The skin was then sutured closed and animals were placed in a warm recovery chamber until they recovered from anesthesia before being returned to their cages. Sham controls underwent identical surgical procedures, but without a CHI.
Injury severity was confirmed and quantified using the following criteria: (i) Mild CHI: no visible skull fractures nor bleeding along skull sutures, no apnea immediately following CHI, no visible edema or blood on T2 images at 24 h post‐TBI. (ii) Severe CHI: visible skull fractures (~ 25% of animals) with/without the appearance of blood along skull sutures, apneic events present (~ 75% of mice), presence of edema and/or blood on T2WI at 24 h post‐CHI. In the mild group, all criteria had to be present, whereas in the severe group the presence of one or more criteria were sufficient.
Magnetic resonance imaging and analysis
Mice were anesthetized (isoflurane: 3% induction, 1% maintenance) for MRI data collection 1‐day post‐CHI or sham surgery. In vivo T2 weighted images (T2WI;TR/TE=2357.9 ms/10.2 ms, 20 × 1 mm slices) were collected with a 128 × 128 matrix on a 11.7T Bruker Avance instrument (Bruker Biospin, Billerica, MA, USA) (Fig. 1c). Quantitative T2 maps were computed from T2WIs using in‐house software written in Matlab (Mathworks, Natick, MA, USA) as previously described (Obenaus et al. 2007).
Figure 1.
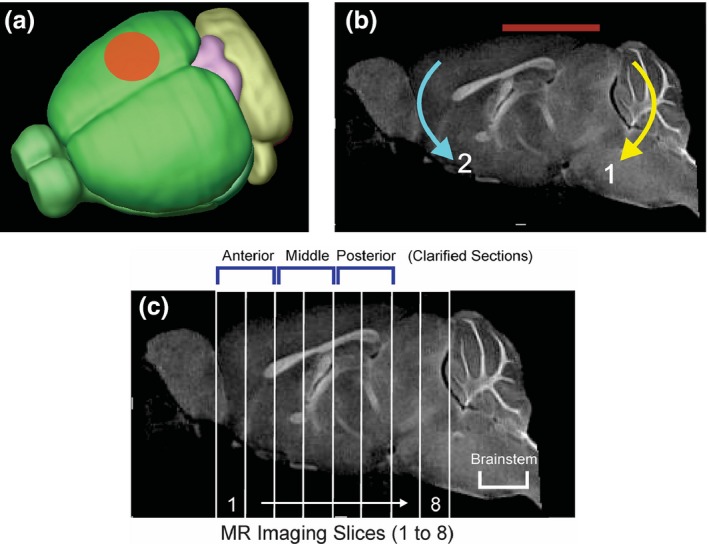
Closed Head Injury (CHI) model. (a) CHI was performed over the right hemisphere between bregma and lambda with a 4 mm diameter pneumatically driven piston. (b) The mouse was placed on a foam bed where the impactor displaced the cranium resulting in a rotational displacement of the brain (red line indicates impact zone). Initial impact causes rotation (1) followed by a secondary rotation (2). (c) A sagittal MR image illustrating the ~ 1 mm thick coronal sections from which T2‐weighted imaging (T2WI) analysis was performed. Six of the MRI slices corresponded to the three ~ 2 mm clarified brain sections: anterior (MRI sections 1–2), middle (MRI sections 3–4) and posterior (MRI sections 5–6) used for microglial morphologic analysis. In addition, the brainstem was also assayed for microglial morphology. The impact site was centered on MRI sections 4–6.
Ipsilateral and contralateral cortical regions of interest (ROI) were manually segmented on T2WIs from the anterior to posterior aspect of the brain from a total of eight MRI slices using Cheshire image processing software (Hayden Image/Processing Group, Waltham, MA, USA). ROIs were then transferred to quantitative T2 maps, where T2 values (ms) were extracted for each slice. Further MRI analysis aggregated three slices together to better represent routine clinical imaging and our clarified brain slices used for microglial analysis.
Brain clarification and imaging
Double transgenic CX3CR1‐GFP/PGRP‐DsRed mice were killed 1‐day post‐injury and transcardially perfused with 4% paraformaldehyde as previously described (Puntambekar et al. 2011, Huang et al. 2013). Brains were rapidly removed and post‐fixed in 4% paraformaldehyde overnight, followed by three 1 h washes in phosphate buffered saline. After the final wash, each brain was placed in a holding mold and cut into thick brain sections of ~ 2.2–2.6 mm based on distance from bregma: an anterior section from +3.4 bregma to +8 bregma, a middle section from +8 bregma to −1.2 bregma and a posterior section from −1.2 to 3.8 (Paximos Mouse Brain Atlas 3rd edition 2008). Anterior sections often retained attached olfactory bulb (that was not imaged) that increased total section thickness. This did not increase the thickness of the anterior cortex being imaged, as the olfactory bulb is not covered by cortical tissue. Each thick brain section was incubated in ScaleA 2 solution (4M Urea, 10% wt/vol glycerol, 0.1% Triton X‐100 pH 7.7) for 6 weeks to clarify brain tissues as previously described (Hama 2011). Following clarification, tissue sections were counter‐stained with 0.12 μg/mL Hoechst 3343 (Thermo Fischer Scientific, San Diego, CA, USA) in ScaleA2. All sections were stored in ScaleA2, in the dark at 4°C until confocal imaging was performed.
For imaging, all tissue sections were placed in 33 mm glass bottom microwell dishes (MatTek Corp., Ashland, MA, USA) containing ScaleA2. Images of the ipsilateral and contralateral cortices were obtained using a BD CARVII Confocal Imager (BD Biosystems, Franklin Lakes, NJ, USA) utilizing Metamorph Imaging Software (Ver. 7.7.0.0, Sunnyvale, CA, USA) on a Zeiss Axio Observer inverted microscope equipped with a long distance 40 × objective. Imaging of the cortices was initiated at the midline and progressed laterally for both the contralateral and ipsilateral cortices until the level of the corpus callosum was reached. Images were collected as 60 μm z‐stacks (0.59 μm/pixel thickness) that encompassed the entire microglial soma and processes.
Volocity image analysis
Confocal images (6–14 images per clarified brain slice, captured in 60 μm z‐stacks) underwent deconvolution (x = 0.2, y = 0.2, z = 0.59 μm/pixel) and contrast enhancement post‐processing using Volocity Imaging Software (Perkin Elmer, Waltham, MA, USA). Microglial cell length and cell body volume was visualized using the extended, 3D Opacity, and XYZ views in Volocity imaging software. Microglial process length (along the greatest diameter of each cell's processes surrounding the cell soma) was measured in Volocity using the longest axis function set at a threshold value of −20. This value was determined to be the most accurate value at which microglial processes were identified and distinguished from background. To quantify cell body volume, the Hoechst 3343 nuclear counter‐stain was used to identify and gate around microglial cell bodies. The cell volume function was then used to threshold images at an offset of 1.7 standard deviations, which was chosen based on visual evaluation of cell body identification from background and ability to distinguish individual cells.
RNA extraction and quantitative real‐time PCR (qPCR)
24 h post‐sham or mild closed head injury (mCHI) treatment (n = 5 per condition), mice were killed by CO2 inhalation. Brains were rapidly harvested. The right and left cortices were immediately removed and quartered, such that the posterior (back) right cortices encompassed the impact site whereas the anterior (front) right did not. The RNA was isolated from the two right hemisphere cortical quarters (front and back) of sham and mCHI treated mice. In brief, the cortical tissue was homogenized and total RNA was extracted using Trizol (Invitrogen, Carlsbad, CA, USA; Puntambekar et al. 2011).
Following RNA extraction, first strand cDNA was synthesized per the conditions outlined in the cDNA synthesis kit (GE healthcare, Pittsburgh, PA, USA; Puntambekar et al. 2011). qPCR was then performed as previously described using a CFX96 Real Time PCR Detection System (Bio‐Rad Laboratories, Hercules, CA, USA). The relative number of transcripts per hypoxanthine guanine phosphoribosyl transferase (HPRT) transcripts was determined using calibration standards for each of the tested molecules. Briefly, PCR products served as standards for calibration of quantitative PCR. These standards were diluted to obtain a standard curve of 50, 5, 0.5, 0.05, 0.005 and 0.0005 pg for qPCR analysis. To minimize experimental variations from one sample to another, the copy number per sample was normalized to the expression of HPRT. It was verified that the copy number of HPRT transcripts was of the same order of magnitude in all samples being compared. Primers for HPRT are CCCTCTGGTAGATTGTCGCTTA (forward) and AGATGCTGTTACTGATAGGAAATCGA (reverse), for inducible nitric oxide (iNOS) are GGCAGCCTGTGAGACCTTTG (forward) and GCATTGGAAGTGAAGCGTTTC (reverse), for glial fibrillary acidic protein (GFAP) are CAGCCCTGGCGTCGTGATTA (forward) and AGCAAGACGTTCAGTCCTGTC (reverse), and for P2Y12 are AGGCTTTGGGAACTTATGC (forward) and GGGTGGTATTGGCTGAGGTG (reverse).
Flow cytometric analysis of microglia and infiltrating immune cells
As previously described, microglia were isolated from the impacted right cortices of mice 24 h post‐injury (Carson et al. 1998; Schmid et al. 2009; Puntambekar et al. 2011). In brief, mice were killed by halothane inhalation, and the brains of the mice rapidly removed and mechanically dissociated. The cell suspension was separated on a discontinuous 1.03/1.088 percoll gradient and microglia/macrophages/infiltrating immune cells were collected from the interface as well as from the 1.03 Percoll fraction. Microglial activation was analyzed by flow cytometry using fluorescently conjugated antibodies (APC‐conjugated CD45, FITC‐conjugated FcR and phycoerythrin (PE)‐conjugated antibodies against Triggering Receptor Expressed on Myeloid cells‐1 (TREM1), TREM2, MHC class II, CD40 (BD Biosciences, San Diego, CA, USA).
Statistics
Sham and CHI animal MRI T2 values were compared using a two‐way analysis of variance (anova) to evaluate the rostro‐caudal changes in the ipsi‐ and contralateral cortices (SigmaPlot V11; Systat Software Inc, San Jose, CA, USA). Similarly, the aggregated MRI slices that aligned with clarified brain sections used for analysis of microglial morphology were analyzed using two‐way anova (SigmaPlot Software). Microglial morphology (length of longest axis, soma volume and volume/longest axis) in clarified brain sections from sham and CHI mice were analyzed using two‐way anova using Prism6 (GraphPad Software, San Diego, CA, USA). All data are presented as the Mean ± SEM. For all data, statistical significance was assigned at p < 0.05 with one star designating p < 0.05, two stars designating p < 0.001, three stars designating p < 0.0001.
Results
Mild traumatic brain injury results in MRI T2 values at the site of impact in the absence of apparent edema and blood deposition
Anesthetized mice were subjected to sham or mCHI on the right side of the exposed but intact skull (Fig. 1a). High‐speed video analysis demonstrated that in our model of unrestrained CHI, the posterior portion of the brain initially rotated downward followed by an anterior brain rotation (Fig. 1b and data not shown). Depending on the severity of injury, TBIs can trigger a rapid influx of blood‐derived macrophages and neutrophils into the brain. Numerous groups have demonstrated that immunolabeling of tissue sections is an unreliable method to distinguish brain‐resident microglia from acutely infiltrating macrophages. Even markers preferentially expressed by microglia such as P2Y12 or TREM2 may be down‐regulated in response to specific types of brain injury or inflammatory states making clear determination of cell type difficult by histologic analysis (Schmid et al. 2009; Amadio et al. 2014; Butovsky et al. 2014). Therefore, we distinguished brain‐resident microglia from blood‐derived macrophages in flow cytometric assays of brain cell suspensions based on their differential expression of CD45 (Carson et al. 1998; Puntambekar et al. 2011). Microglia are CD45lo in their homeostatic state but upon robust activation can acquire a CD45intermediate state. By contrast, acutely infiltrating macrophages display a CD45hi phenotype that is readily distinguishable from the lower levels of CD45 expressed by microglia (Carson et al. 1998; Puntambekar et al. 2011). Here, we find that CD45lo microglia were readily detected in the ipsilateral cortices of the sham (Fig. 2a) and mCHI (Fig. 2b) treated mice 24 h post‐treatment. CD45 and FcR levels were equivalent in microglia from both sham and mCHI treatment groups and no difference in the small numbers of CD45hi macrophages were detected in the two groups of brain cell suspensions (Fig. 2a–c). In addition, no difference in CD45 levels was detected between microglia in brain cell suspensions from sham‐treated and mCHI‐treated mice (Fig. 2c).
Figure 2.
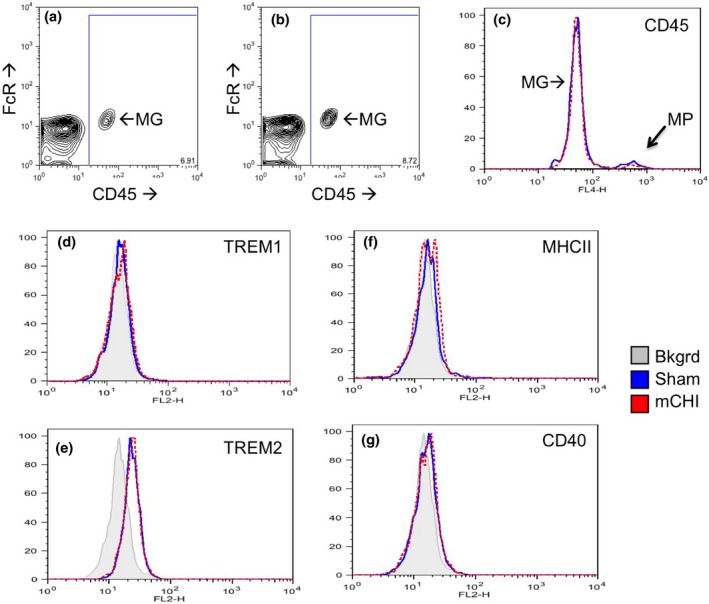
Microglial activation following mild closed head injury (mCHI) is not detected by flow cytometric analysis. Microglia (MG) from Sham‐treated mice are identified as CD45lo, FcR+ cells in brain cell suspensions from sham treated (Panel a) and mCHI treated (Panel b) right cortices. Panels (c–g) depict expression data from cells within the CD45 + gate (large boxes depicted in panels a and b). (c) Histogram analysis of CD45lo (microglia: MG) and CD45hi (macrophages: MP) confirms no difference in levels observed in cells from sham (blue line) versus mCHI treated cortices. Microglial expression of TREM1 (Panel D), TREM2 (Panel e), major histocompatibility complex class (MHC) class II (Panel f) and CD40 (Panel g) is compared in cells isolated from sham (blue solid line) and mCHI treated (red dashed line) cortices. Grey filled histogram depicts autofluorescence from unlabeled cells isolated from the right cortices of sham‐treated mice. Depicted flow cytometry is representative of three experiments.
We also examined microglial expression of molecules associated with exacerbating pro‐inflammatory responses (TREM1, Fig. 2d), with anti‐inflammatory tissue repair responses (TREM2, Fig. 2e), with general activation (MHC class II, Fig. 2f) or with neurotoxicity (CD40, Fig. 2g). Of all the molecules quantified by flow cytometry, only TREM2 was readily detected on microglia isolated from the right cortices of sham‐treated mice. We have previously demonstrated that microglia rapidly up‐regulate surface expression of TREM2 in the presence of Tumor Necrosis Factor (TNF), amyloid pathology or neuronal damage associated with neuronal axotomy and excitotoxity (Schmid et al. 2002, Carson et al. 2006, 2007, Schmid et al. 2009; Melchior et al. 2010). Therefore, we hypothesized that TREM2 induction would be a sensitive indicator of microglial detected brain injury. However, no difference in microglial expression of TREM2, TREM1, MHC class II or CD40 was detected from cells isolated from impacted or sham treated cortices. Taken together the MRI and flow cytometric data confirm the damage sustained in our model of unrestrained mCHI is mild.
As a potentially more sensitive analysis of injury and inflammation, we quantified cortical expression of iNOS (Fig. 3a), GFAP (Fig. 3b) and P2Y12 (Fig. 3c) as indicators of general inflammation, astrogliosis and microglial activation, respectively, by standard curve based qPCR analysis. For this set of experiments, the right cortices of sham and mCHi treated mice were divided into half with the front/rostral half being anterior to the impact zone (corresponding to MRI slices 1–3 in Fig. 1) and the back/caudal half encompassing the impact zone (corresponding to MRI slices 4–6 in Fig. 1). A wide variation in iNOS expression in the impacted ‘back’ cortices of mCHI treated mice was observed, with two mice exhibiting high levels of iNOS and three mice exhibiting very low levels of iNOS. By contrast, GFAP expression was unchanged between the sham and mCHI groups. This may be consistent with a mild injury leading to slight stochastic differences in injury responses. By contrast, a small but reproducible and statistically significant reduction in P2Y12 expression was detected in the back cortical region encompassing the impacted area of mCHI treated as compared to sham treated cortices.
Figure 3.
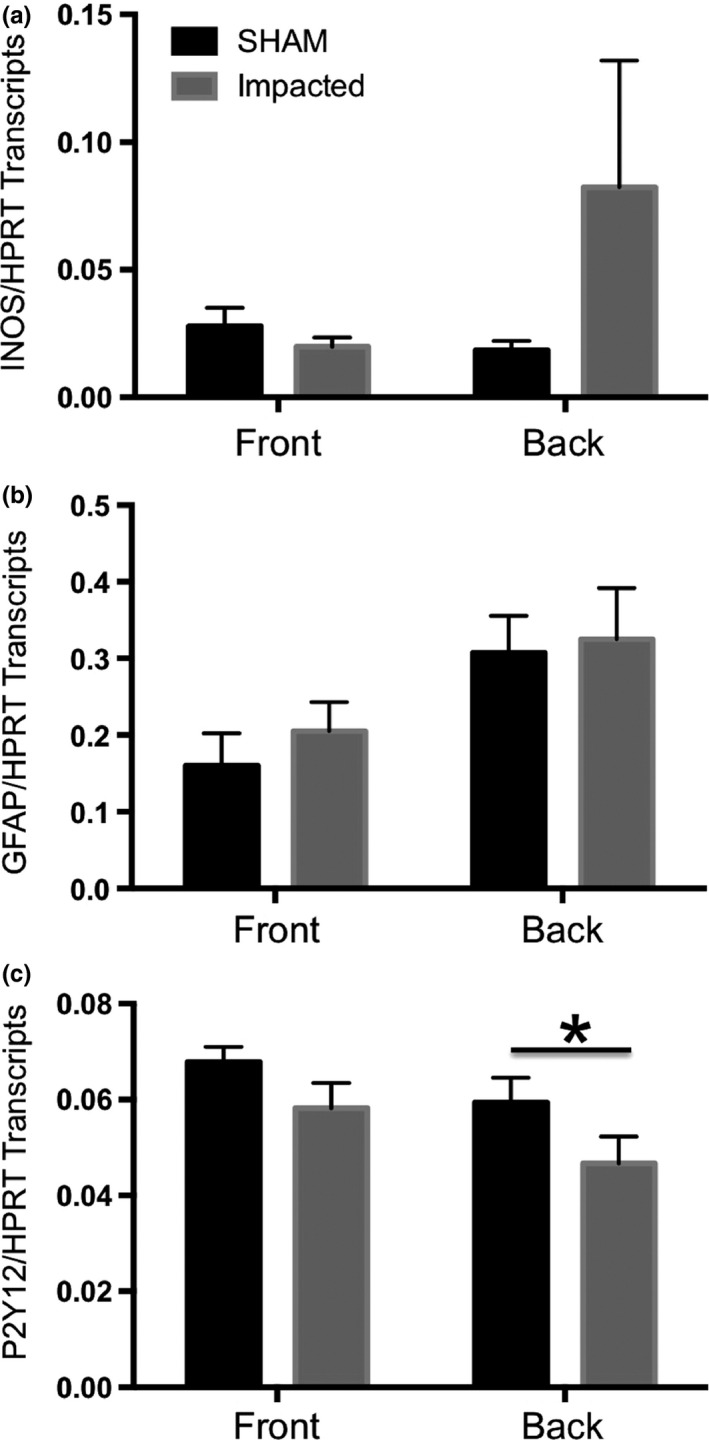
Standard curve based qPCR analysis of glial activation in the impacted cortex 24 h post‐treatment. Right cortices of treated mice were divided into two portions: the front, corresponding to the cortical regions rostral to the site of impact (corresponding to MRI sections 1–3 detailed in Fig. 1) and the back, encompassing the site of impact (corresponding to MRI sections 4–6 detailed in Fig. 1) The levels of inducible nitric oxide (iNOS) (Panel a), glial fibrillary acidic protein (GFAP) (Panel b) and P2Y12 (Panel c) transcripts were quantified in the right front cortices and the right back cortices from sham treated (black bars) and mild closed head injury (mCHI)‐treated mice (grey bars). A significant decrease in P2Y12 transcripts was detected in comparisons of transcript levels in the impact zone (back region) of cortices from mCHI versus sham‐treated mice (Student t‐test, *p < 0.05, n = 5 per treatment).
MRI was performed 24 h post‐CHI to assess tissue level alterations associated with mCHI as compared to sham‐treated mice. We have previously developed quantitative methods for detecting edema and blood deposition in MRI images (Bianchi et al. 2015; Donovan et al. 2014). T2WI was utilized to demonstrate the mild nature of our TBI model and lack of elevated T2 values that are reflective of edema. No evidence of edema was visualized in sham or mCHI tissue from T2WI, but quantitative T2 mapping revealed small but significant decrements in T2 values that could be visually observed on pseudo‐colored T2 maps (Fig. 4a). Compared to shams, no overt tissue level changes could be observed following CHI (Fig. 4a).
Figure 4.
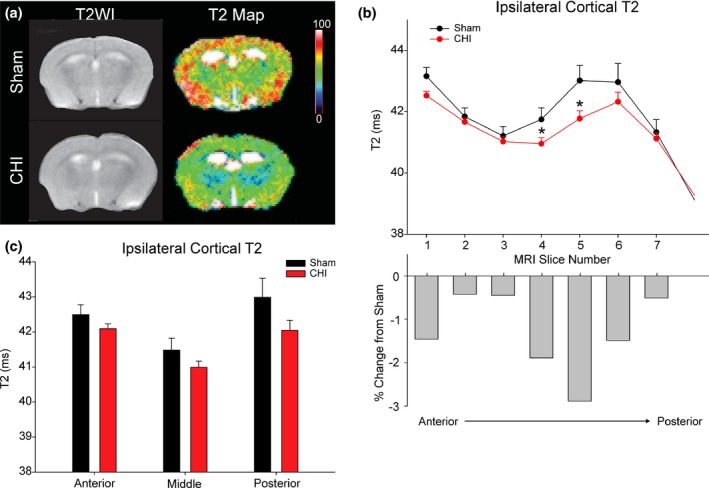
MRI imaging 24 h post‐mild closed head injury (mCHI). (a) Representative T2WIs (left panel within 4A) and their associated pseudo‐colored quantitative T2 maps (right panel within Fig. 4a). (Scale bar: 0–100 ms). (b) Ipsilateral anterior to posterior assessment of cortical T2 values revealed significant decreases in T2 values (slices 4–5) compared to shams (*p < 0.05). Percent change in T2 values between mCHI and sham treated mice is plotted for each MRI slice (impact zone centered on MRI sections 4–6). (c) The MRI T2 data were then binned to match the histological sections from which microglial assays were performed with decreases predominately in the posterior slice partially under the impact zone. Multi‐variate analysis of the aggregated data did not reach significance (posterior, p = 0.094).
Decrements in T2 values can be caused by multiple factors such as hypermetabolism or decreased/slowing blood flow but at minimum indicate TBI induced changes in tissue integrity. Changes in T2 values were quantified by extracting ROI from the ipsi‐ and contralateral cortices. T2 values from sham‐treated mice were found to not significantly differ between ipsi‐ and contralateral cortices (p = 0.631). In contrast, T2 values from the mCHI animals did exhibit a significant difference in T2 between the impacted and non‐impacted sides, (p = 0.048, data not shown). Therefore, T2 values were compared between the right cortices of sham and mCHI treated mice (Fig. 4b and c). No overall difference between sham and mCHI treated groups was detected when comparing T2 values across the entire rostral‐caudal extent (p = 0.089, Fig. 4b). However, post hoc testing revealed a significant difference between sham and mCHI treated mice at the site of impact (MRI slice 4–5). Reproducible, although modest decreases of 2–3% were observed in mCHI‐treated mice as compared to the same sites in sham‐treated mice (p < 0.05, Fig. 4b). When the T2 values were aggregated together to the same slice thickness corresponding to the clarified brain sections used for our analysis of microglial morphology, no significant differences in T2 values were found anterior to impact site. The subtle nature of the decrements in T2 at the site of injury are consistent with our goal of generating a mild model of CHI.
Unrestrained mild traumatic brain injury induces larger changes in microglial morphology at sites distant from the site of impact compared to the site of injury
Microglia are sensitive monitors of brain homeostasis with processes continually and actively surveying their environment (Davalos et al. 2005 and Nimmerjahn and Kirchhoff 2005). One of the earliest signs of microglial activation is a general shortening of processes and an increase in soma size. We used mice double transgenic for CX3CR1‐GFP (Jung et al. 2000) and PGRPs‐DsRed (Wang et al. 2011) to quantify microglial morphology in clarified brain sections as a function of CHI severity (Fig. 5). These mice heterozygously expressed CX3CR1‐GFP in which all brain‐resident microglia were visualized by the GFP reporter gene (Jung et al. 2000) and all neutrophils were visualized by PGRPs‐DsRed (Wang et al. 2011). DsRed+ neutrophils were not detected in any brain regions from mCHI mice at 1 day post‐injury (Figs 5 and 6) consistent with our failure to detect blood‐derived infiltrates in impacted cortices of mCHI‐treated mice (Fig. 2).
Figure 5.
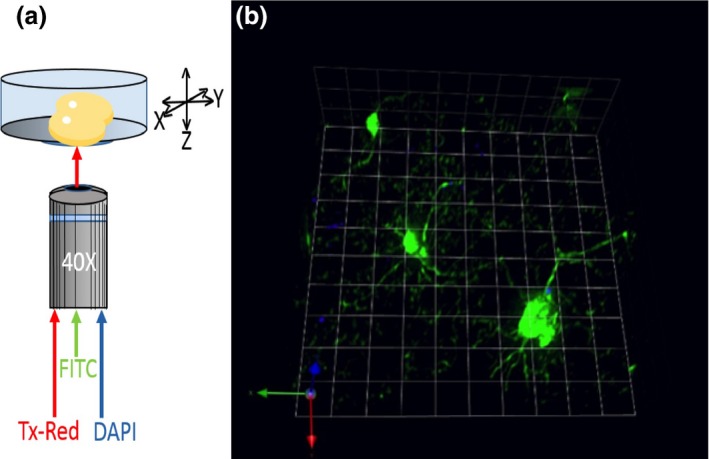
(a) Schema of clarified brain imaging while in the presence of ScaleA2. (b) Magnified 40 × image using 3D opacity function of Volocity Imaging Software (6.1.1). Image represents a complete 3D plane with CX3CR1‐GFP microglia and Hoescht 3343 positive nuclei.
Figure 6.
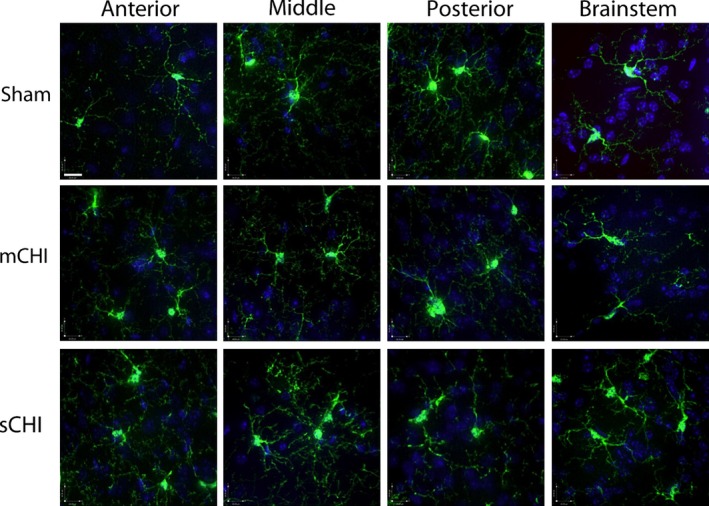
Representative extended focus confocal images from the ipsilateral rostral (anterior) to caudal (posterior) regions of the cortices (clarified brain regions) and from the brainstem from sham, mild closed head injury (mCHI), and sCHI treated mice (Scale bar = 20.00 μm).
GFP filled microglial processes did not extend away from the cell soma in a perfect sphere. Instead processes often extended away from the cell soma in an oval or flattened spherical array. This resulted in a long and short axis of process extension away from the cell soma. As a population, microglia also did not consistently align their longest process axis in a uniform orientation that could be reliably sampled in standard thin (5–25 μm) coronal or sagittal cryosections (Fig. 5b). Imaging of thick (~ 2 mm) clarified sections allowed imaging of multiple microglia regardless of orientation within the cortex. Because the MRI and flow cytometric data demonstrated that our mCHI model caused little apparent injury 24 h post‐treatment, for our morphometric analysis we also compared a severe model of CHI (sCHI) as a positive control for changes in the longest length (longest axis) of microglia processes (Fig. 7a and b), microglial soma volume (Fig. 7c and d) and the ratio of soma volume/longest axis of microglial processes (Fig. 7e and f).
Figure 7.
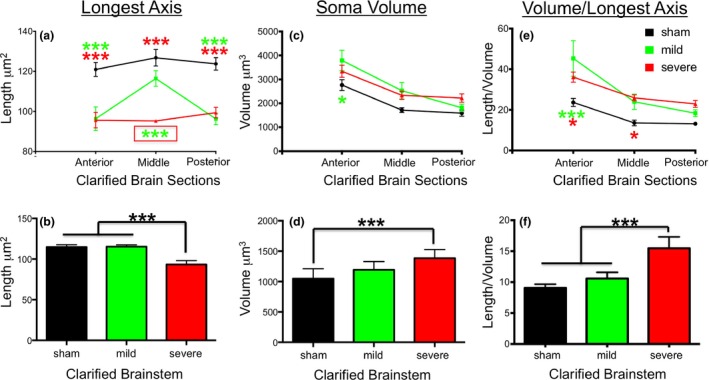
Quantification of microglia morphology by length of longest axis (a), soma volume (b) and the ratio of soma volume/longest axis (c) in clarified brain sections (anterior‐middle‐posterior) and in the brainstem (panels d–f). Sham treatment = black lines and bars; mCHI = green lines and bars; sCHI = red lines and bars. In panels (a–c), green and red stars indicate statistical difference of mild and severe CHI from sham treated mice. Boxed green stars in panel (a) indicate statistical difference of sCHI from both mCHI and sham treated mice. For all panels, three stars designates p < 0.001, one star designates p < 0.05 as analyzed by anova.
Analysis of the longest axis of microglial process length revealed that sCHI treatment significantly decreased microglial process length as compared to sham treated mice not only at the impact site but also in all brain regions examined including the brainstem (Fig. 7a and b). Surprisingly, we did not detect a significant change in microglia soma volume in the brain cortices of mice receiving sCHI (Fig. 7c). Thus, when comparing the ratio of microglia soma volume/longest axis of microglial process in mice receiving sCHI, only the impacted site and the rostral brain region showed significant differences from sham mice (Fig. 7e). However, perhaps because of the greater injury caused by rotational and/or ‘tethering’ stress effects to the brainstem during an unrestrained sCHI, microglia soma volume was increased in the brainstems of sCHI treated mice (Fig. 7d). This also resulted in a significant difference in microglia soma volume to longest axis length in the brainstem of mice receiving sCHI (Fig. 7f).
Analysis of the longest axis of microglial process length and microglial soma volume revealed a different pattern of activation in the cortices and brainstem of mice receiving a mCHI instead of sCHI. In mCHI‐treated mice, no statistically significant differences from sham‐treated mice were observed in microglial morphology in the clarified ‘middle’ brain section that partially overlapped with the impacted region of the cortex (Fig. 7a and c). By contrast, the longest axis of microglial process length was shortened to the same degree in mCHI‐treated mice as in sCHI mice not only at the posterior region partially encompassing the impact site (Fig. 7a). Shortened processes were also observed in the anterior brain region which was well away from the impact site but that did receive rotational stress (Fig. 1b and Fig. 7a). Changes in microglial soma volume in mice receiving mCHI as compared to sham mice were only observed in anterior brain region (Fig. 7c). No statistically significant differences in microglial morphology were observed in the brainstems of mCHI‐treated mice (Fig. 7b, d and e).
Discussion
In this study, we sought to establish a murine model with the same observable MRI pathology seen in human cases of mild traumatic brain injury comparable to the Glasgow coma scale of 13–15 (Faul et al. 2015). The model that we present reveals no overt damage in terms of edema or blood deposition after a mild unrestrained TBI allowing for a parallel observation between human and murine MRI pathology. Although our model parallels clinical observations, quantitative analysis of T2‐weighted values reveals a clear decrement, with changes reaching significance in comparison to sham animals at the impacted site. Because the observed changes are modest and highly localized to the impact site, they are readily lost to detection if larger MRI sections are grouped into thicker sections as might be used in clinical neuroimaging.
Microglia are long‐lived tissue macrophages that populate the brain early in post‐natal development and that are early responders to brain injury, disease and dysfunction (Carson et al. 2007). Previous reports have shown that various models of mild traumatic brain injury induce visible indications of microglial activation at the peri‐contusional lesion site such as soma volume enlargement and altered surveillance distance of microglial processes (Roth et al. 2014) as well as an up‐regulation in the CX3CR1 1 week post‐TBI (Rancan et al. 2004). In addition, recent studies demonstrate that a midline fluid percussion injury causing mild to moderate injury is accompanied by significant microglia activation as measured by changes in morphology, protein and mRNA 4–24 h post‐injury (Fenn et al. 2014). Furthermore, mTBI even results in persistent microglial activation in areas of diffuse axonal injury by 7 days and even months after injury (Fenn et al. 2014; Loane et al. 2014; Mouzon et al. 2014, Ojo et al. 2015).
While microglial activation, a key component of the inflammatory response in the brain, has been qualitatively reported (Bye et al. 2007; Morganti‐Kossmann et al. 2001), cortical morphologic responses of microglia to an unrestrained closed head injury with a rotational kinetic had not been previously described and quantified. Therefore, a key aspect that is different in our model is the rotational aspect that is not found in some weight drop models and in some fluid percussion injury models of mTBI. Our model potentially recapitulates many human injuries, in that, injury to one side of the brain along with rotational aspects might likely represent the kind of concussive injuries that are present in sports and other common causes of mTBI. We therefore speculate that the initial changes in microglial function that are associated with the altered morphology observed at 24 h post‐injury in our mCHI model likely sets the stage for the ongoing and long‐term inflammation seen in many other studies.
An important aspect of microglial biology is continual surveillance of the local environment. Here, we quantify microglial expression of activation markers as well as morphology acutely after injury in cortical regions experiencing impact versus rotational stress injuries. Our data suggest that whereas mCHI is sufficient to trigger canonical changes in microglial morphology associated with activation (soma enlargement and process retraction) in the anterior cortex distant from the impact site in a manner and degree similar to that following sCHI. The current data showing reduced process length in regions anterior to the impact that experienced rotational stress suggest these areas may be more vulnerable to future injury or insults because of decreased microglial surveillance and monitoring of brain function in these regions.
Our quantification of microglial morphology, and microglial activation by flow cytometry and qPCR also supports the growing evidence that morphological and molecular activation profiles are not on a linear spectrum of microglial responses (Butovsky et al. 2014; Vinet et al. 2012; Melchior et al. 2010). Rather these parameters are likely independent responses reflecting different mechanisms of action and possibly different activation subtypes. For example, while microglia may have a morphology similar to that of a homeostatic ramified cell, they may be already being activated following single or repeated insults (Conde and Streit 2006). Indeed, we have reported increased microglial responses to repeated mTBI in the cortical contusion injury model (Huang et al. 2013).
T2WI is a standard imaging method used clinically to evaluate the brain for the presence of tissue abnormalities. Previous experimental models of moderate‐severe TBI and stroke have demonstrated that T2WI correlates well with tissue pathology (Kochanek et al. 1995; Gerriets et al. 2004; Donovan et al. 2014). However, mild TBI results in subtle tissue changes that are not detectable visually on T2WI. Previous animal studies using quantitative T2 values have reported positive correlations between T2‐values and poor outcomes following moderate‐severe TBI (Irimia et al. 2011). T2 values can be affected by tissue pathology, including blood (iron) and edema, which typically manifest as low and high T2 values respectively. In addition, T2 values are sensitive to tissue iron levels and regions of high tissue metabolism can increase oxygen extraction from circulating blood with increased levels of deoxy‐hemoglobin resulting in decreased T2 values (Choy et al. 2014). Additional studies validating the neuroimaging changes in mild TBI are warranted.
Given the increasing concern of mild brain injury in sports and their potential for long‐term psychological and psychiatric sequelae there is an unmet need for non‐invasive imaging modalities that mirror the cellular events underlying the brain's response to injury. Our own work herein not only supports the need for improved imaging but also highlights the importance to examine the brain at sites distant from the impact site. The finding that the anterior brain is particularly sensitive to inflammatory activation, even distant from the injury, could suggest that ongoing and continued inflammation maybe a causative factor for long‐term disabilities reported clinically (Obenaus 2015).
Acknowledgments and conflict of interest disclosure
AH and VD contributed equally to these studies. AH was funded by a MarcU scholarship, VD is an IGERT fellow. Studies were supported by grants UCR PIC and NIH R01AG048099 (MJC) and DCMRP #DR080470 (AO).
All experiments were conducted in compliance with the ARRIVE guidelines. The authors have no conflict of interest to declare.
References
- Amadio S., Parisi C., Montilli C., Carrubba A. S., Apolloni S. and Volonté C. (2014) P2Y12 receptor on the verge of a neuroinflammatory breakdown. Mediators Inflamm. 2014, 975849. doi:10.1155/2014/975849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi A., Bhanu B. and Obenaus A. (2015) Dynamic low‐level context for the detection of mild traumatic brain injury. IEEE Trans. Biomed. Eng. 62, 145–153. [DOI] [PubMed] [Google Scholar]
- Butovsky O., Jedrychowski M. P., Moore C. S. et al (2014) Identification of a unique TGF‐β‐dependent molecular and functional signature in microglia. Nat. Neurosci. 17, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bye N., Habgood M. D., Callaway J. K., Malakooti N., Potter A., Kossmann T. and Morganti‐Kossmann M. C. (2007) Transient neuroprotection by minocycline following traumatic brain injury is associated with attenuated microglial activation but no changes in cell apoptosis or neutrophil infiltration. Exp. Neurol. 4, 220–233. [DOI] [PubMed] [Google Scholar]
- Carson M. J., Doose J. M., Melchior B., Schmid C. D. and Ploix C. C. (2007) CNS immune privilege: hiding in plain sight. Immunol. Rev. 213, 48–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson M. J., Reilly C. R., Sutcliffe J. G. and Lo D. (1998) Mature microglia resemble immature antigen presenting cells. Glia 22, 72–85. [DOI] [PubMed] [Google Scholar]
- Carson M. J., Thrash J. C. and Walter B. (2006) The cellular response in neuroinflammation: The role of leukocytes, microglia and astrocytes in neuronal death and survival. Clin. Neurosci. Res. 6, 237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy M., Dubé C. M., Patterson K., Barnes S. R., Maras P., Blood A. B., Hasso A. N., Obenaus A. and Baram T. Z. (2014) A novel, noninvasive, predictive epilepsy biomarker with clinical potential. J. Neurosci. 34, 8672–8684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde J. R. and Streit W. J. (2006) Microglia and the aging brain. J. Neuropathol. Exp. Neurol. 65, 199–203. [DOI] [PubMed] [Google Scholar]
- Davalos D., Grutzendler J., Yang G., Kim J. V., Zuo Y., Jung S., Littman D. R., Dustin M. L. and Gan W. B. (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758. [DOI] [PubMed] [Google Scholar]
- Donovan V., Bianchi A., Hartman R., Bhanu B., Carson M.J., Obenaus A. and (2014) Computational analysis reveals increased blood deposition following repeated mild traumatic grain injury. NeuroImage: Clinical. 1, 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul M. and Coronado V. (2015) Epidemiology of traumatic brain injury and book of clinical neurology. Handb Clin Neurol. 127, 3–13. [DOI] [PubMed] [Google Scholar]
- Faul M., Xu L., Wald M. M. and Coronado V. (2015) Traumatic Brain Injury in the United States: Emergency Visits Hospitalizations, and Deaths 2002‐2006. Atlanta, GA: CDC, National Center for Injury Prevention and Control. [Google Scholar]
- Fenn A. M., Gensel J. C., Huang Y., Popovich P. G., Lifshitz J. and Godbout J. P. (2014) Immune activation promotes depression 1 month after diffuse brain injury: a role for primed microglia. Biol. Psychiatry 76, 575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner R. C. (2015) Yaffe K Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol. Cell Neurosci. 66, 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerriets T., Stolz E., Walberer M., Müller C., Rottger C., Kluge A., Kaps M., Fisher M. and Bachmann G. (2004) Complications and pitfalls in rat stroke models for middle cerebral artery occlusion: a comparison between the suture and the macrosphere model using magnetic resonance angiography. Stroke 35, 2372–2377. [DOI] [PubMed] [Google Scholar]
- Hama H, Kurokawa H, Kawano H, Ando R, Shimogori T, Noda H, Fukami K, Sakaue‐Sawano A and Miyawaki A. (2011) Scale: a chemical approach for fluorescence imaging and reconstruction of transparent mouse brain. Nature Neurosci. 14, 1481–1488. [DOI] [PubMed] [Google Scholar]
- Hickman S. E., Kingery N. D., Ohsumi T. K., Borowsky M. L., Wang L. C., Means T. K. and El Khoury J. (2013) The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 16, 1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L., Coats J. S., Mohd‐Yusof A et al (2013) Tissue Vulnerability is increased following repetitive mild traumatic brain injury in the rat. Brain Res. 1499:109–120. [DOI] [PubMed] [Google Scholar]
- Irimia A., Chambers M. C., Alger J. R. et al (2011) Comparison of acute and chronic traumatic brain injury using semi‐automatic multimodal segmentation of MR volumes. J. Neurotrauma 28, 2287–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S., Aliberti J., Graemmel P., Sunshine M. J., Kreutzberg G. W., Sher A. and Littman D. R. (2000) Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 20, 4106–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochanek P. M., Marion D. W., Zhang W. et al (1995) Severe controlled cortical impact in rats: assessment of cerebral edema, blood flow, and contusion volume. J. Neurotrauma 12, 1015–1025. [DOI] [PubMed] [Google Scholar]
- Loane D. J., Stoica B. A., Tchantchou F., Kumar A., Barrett J. P., Akintola T., Xue F., Conn P. J. and Faden A. I. (2014) Novel mGluR5 positive allosteric modulator improves functional recovery, attenuates neurodegeneration, and alters microglial polarization after experimental traumatic brain injury. Neurotherapeutics 11, 857–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchior B., Garcia A. E., Hsiung B., Lo K. M., Doose J. M., Thrash J. C., Stalder A. K., Staufenbiel M., Neumann H. and Carson M. J. (2010) Dual induction of TREM2 and tolerance‐related transcript, Tmem176b, in amyloid transgenic mice: implications for vaccine‐based therapies for Alzheimer's disease. ASN Neuro. 2, 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mierzwa A. J., Sullivan G. M., Beer L. A., Ahn S. and Armstrong R. C. (2014) Comparison of cortical and white matter traumatic brain injury models reveals differential effects in the subventricular zone and divergent Sonic hedgehog signaling pathways in neuroblasts and oligodendrocyte progenitors. ASN Neuro. 6:pii: 1759091414551782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganti‐Kossman M. C., Rancan M., Otto V. I., Stahel P. F. and Kossmann T. (2001) Role of cerebral Inflammation after brain injury: a revisited concept. Shock 16, 165–177 [DOI] [PubMed] [Google Scholar]
- Mouzon B. C., Bachmeier C., Ferro A., Ojo J. O., Crynen G., Acker C. M., Davies P., Mullan M., Stewart W. and Crawford F. (2014) Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann. Neurol. 75, 241–254. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A. and Kirchhoff F. (2005) Helmchen F Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. [DOI] [PubMed] [Google Scholar]
- Obenaus A. (2015) Traumatic brain injury, in Encyclopedia of Mental Health, 2nd edition, Vol 4 (Friedman H. S. Editor in Chief), pp. 329–340. Academic Press, Waltham, MA. [Google Scholar]
- Obenaus A., Robbins M., Blanco G., Galloway N. R., Snissarenko E., Gillard E., Lee S. and Currás‐Collazo M. (2007) Multimodal magnetic resonance imaging alternations in two rat models of mild neurotrauma. J. Neurotrauma 24, 1147–1160. [DOI] [PubMed] [Google Scholar]
- Ojo J., Mouzon B. C. and Crawford F. (2015) Repetitive head trauma, chronic traumatic encephalopathy and tau: challenges in translating from mice to men. Exp. Neurol. pii: S0014–4886(15)30015–7. doi: 10.1016/j.expneurol.2015.06.003. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Pacifico A., Amyot F., Arciniegas D. et al (2015) A review of the effectiveness of neuroimaging modalities for the detection of traumatic brain injury. J. Neurotrauma 32(22):1693–1721. doi: 10.1089/neu.2013.3306. Epub 2015 Sep 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puntambekar S. S., Davis D. S., Hawel L., Crane J., Byus C. V. and Carson M. J. (2011) LPS‐induced CCR2 expression and macrophage influx into the murine central nervous system is polyamine‐dependent. Brain Behav. Immun. BBI 25, 883–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rancan M., Bye N., Otto V. I., Trentz O., Kossmann T., Frentzel S. and Morganti‐Kossmann M. C. (2004) The chemokine fractalkine in patients with severe traumatic brain injury and a mouse model of closed head injury. J. Cereb. Blood Flow Metab. 24, 1110–1118. [DOI] [PubMed] [Google Scholar]
- Roth T. L., Nayak D., Atanasijevic T., Koretsky A. P., Latour L. L. and McGavern D. B. (2014) Transcranial amelioration of inflammation and cell death after brain injury. Nat. Neurosci. 505, 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutgers D. R., Toulgoat F., Cazejust J., Fillard P., Lasjaunias P. and Ducreux D. (2008) White matter abnormalities in mild traumatic brain injury: a diffusion tensor imaging study. Am. J. Neuroradiol. 29(3), 514–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakhon O. S., Ross B., Gusti V., Pham A. J., Vu K. and Lo D. D. (2015) M cell‐derived vesicles suggest a unique pathway for trans‐epithelial antigen delivery. Tissue Barriers 3(1–2), e1004975. doi:10.1080/21688370.2015.1004975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid C. D., Melchior B., Masek K., Puntambeker S. S., Danielson P. E., Lo D. D., Sutcliffe J. G. and Carson M. J. (2009) Differential gene expression in LPS/IFNgamma activated microglia and macrophages: in vitro versus in vivo. J. Neurochem. 109 s1, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid C. D., Sautkulis L. N., Danielson P. E., Cooper J., Hasel K. W., Hilbush B. S., Sutcliffe J. G. and Carson M. J. (2002) Heterogeneous expression of the Triggering Receptor Expressed on Myeloid cells‐2 (TREM‐2) on adult murine microglia. J. Neurochem. 83, 1309–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C., Gentleman S. M., Leclercq P. D., Murray L. S., Griffin W. S., Graham D. I. and Nicoll J. A. (2013) The neuroinflammatory response in humans after traumatic brain injury. Neuropathol. Appl. Neurobiol. 39, 654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susarla B. T., Villapol S., Yi J. H., Geller H. M. and Symes A. J. (2014) Temporal patterns of cortical proliferation of glial cell populations after traumatic brain injury in mice. ASN Neuro. 6, 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinet J., Weering H. R., Heinrich A., Kälin R. E., Wegner A., Brouwer N. and Heppner F. L. (2012) Rooijen Nv, Boddeke HW, Biber K. J Neuroprotective function for ramified microglia in hippocampal excitotoxicity. J. Neuroinflammation 9, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Gusti V., Saraswati A. and Lo D. D. (2011) Convergent and divergent development among M cell lineages in mouse mucosal epithelium. J. Immunol. 187, 5277–5285. [DOI] [PMC free article] [PubMed] [Google Scholar]