

Abstract
In the context of the Human Toxome project, mass-spectroscopy-based metabolomics characterization of estrogen-stimulated MCF-7 cells was studied in order to support the untargeted deduction of pathways of toxicity. A targeted and untargeted approach using over-representation analysis (ORA), quantitative enrichment analysis (QEA), and pathway analysis (PA) and a metabolite network approach were compared. Any untargeted approach necessarily has some noise in the data owing to artifacts, outliers, and misidentified metabolites. Depending on the chemical analytical choices (sample extraction, chromatography, instrument and settings etc.) only a partial representation of all metabolites will be achieved, biased by both the analytical methods and the database used to identify the metabolites. Here, we show on the one hand that using a data analysis approach based exclusively on pathway annotations has the potential to miss much that is of interest and, in the case of misidentified metabolites, can produce perturbed pathways that are statistically significant yet uninformative for the biological sample at hand. On the other hand, a targeted approach, by narrowing its focus and minimizing (but not eliminating) misidentifications, renders the likelihood of a spurious pathway much smaller, but the limited number of metabolites also makes statistical significance harder to achieve.
To avoid an analysis dependent on pathways, we built a de novo network using all metabolites that were different at 24 hours with and without estrogen with a p-value less than .01 (53) in the STITCH database, which links metabolites based on known reactions in the main metabolic network pathways but also based on experimental evidence and text-mining. The resulting network contained a “connected component” of 43 metabolites, and helped identify non-endogenous metabolites as well as pathways not visible by annotation based approaches. Moreover, the most highly connected metabolites (energy metabolites such as pyruvate and alpha-ketoglutarate, as well as amino acids) showed only a modest change between proliferation with and without estrogen.
Here, we demonstrate that estrogen has subtle but potentially phenotypically important alterations in the acyl-carnitine fatty acids and acetyl-putrescine and succinoadenosine, in addition to likely subtle changes in key energy metabolites that, however, could not be verified consistently given the technical limitations of this approach. Finally, we show that a network-based approach combined with text-mining identifies pathways that would otherwise neither be considered statistically significant on their own nor be identified via ORA, QEA, or PA.
Introduction
A major challenge of 21st century life sciences is to make sense of the big data produced by the new technologies available. Biological pathways lend themselves as guidance to interpret such data, especially those derived from omics technologies. Metabolomics, i.e. which aims for a relatively complete analysis of all small molecules of a biological model, promises to be closest to phenotypical changes, as changes in gene expression and protein formation for example do not necessarily lead to altered functionality. Analyzing metabolites for altered phenotype is perhaps one of the oldest modalities employed by medicine—the tale goes that pre-modern doctors would test for diabetes mellitus by seeing if ants were attracted to a patient’s urine, a crude but effective and accurate assay for a biomarker of disease(King and Rubin, 2003). Nonetheless, metabolomics—defined as measuring the concentration of the low molecular weight (<1,500 Da) molecules in a system of interest—has yet to join transcriptomics and proteomics as an essential part of systems biology. In part, this may be because metabolomics presents many technical and analytical challenges in comparison to transcriptomics. While metabolomics is ultimately very close to the phenotype, this turns out to be a double- edged sword, as it means that metabolomics can be extraordinarily sensitive to slight changes in experimental parameters, and it requires a scrupulous commitment to protocol as virtually any small change can introduce artifacts (Bouhifd et al., 2015b). Metabolomics also presents a challenge in terms of analytical chemistry in comparison to transcriptomics or even proteomics, which is comparatively straight-forward from an analytical perspective, as both technologies targets a single class of compounds that are comparatively easy to isolate and more stable. The universe of metabolites consists of chemicals with a vast range of properties—over 2000 metabolites total, consisting of polar and neutral lipids, amino acids, redox metabolites, and sugars (Wishart, 2011)—and the different biochemical properties precludes coverage with any one platform, e.g. HPLC will have different coverage than gas chromatography, positive or negative polarity will ionize different metabolites, etc. Therefore, while untargeted metabolomics attempts to catch “all” the metabolites, the choice of platform will likely privilege some over others. This is important to keep in mind for any analysis based on pathways, as metabolites that are invisible to a specific platform but are heavily represented on a pathway of interest may skew the result, i.e. cells treated with estrogen may have steroid-specific pathways up-regulated, but if a technology does not adequately capture large, non-polar compounds, any impact on that pathway may be difficult to see unless a combination of analytical platforms is used.
Lastly, metabolomics, unlike transcriptomics, does not produce a list of unambiguously identified “features”. Instead, it depends on several intricate steps of data analysis to go from a chromatograms to a list of metabolites with concentrations, including peak alignment, deconvolution, adequate identification of ions, isotopes, and possible adduct modifications (water, sodium, or other small molecules that may be bound to or lost from the compound and therefore reflected in the m/z), and lastly accurate metabolite identification, which is dependent not only on all of the above steps, but also on the accuracy and metabolite coverage provided by the database used for compound identification. Finally, this last step is an immensely time consuming step, seriously compromising the use of untargeted metabolomics for high-throughput screening. In this respect, metabolomics has yet gain the maturity that transcriptomics, which in the past decade has achieved broad consensus on data analysis.
As part of the Mapping the Human Toxome project (Bouhifd et al., 2015a; Bouhifd et al., 2014), where concepts and tools for the elucidation of molecular pathways of toxicity (Hartung and McBride, 2011; Kleensang et al., 2014) are being developed using the test case of estrogen endocrine disruption, we examined the effect of 17B-estradiol on the metabolism of MCF-7 cells with both a targeted and untargeted approach using over-representation analysis (ORA), quantitative enrichment analysis (QEA), and pathway analysis (PA) and a network approach. Here, we show that QEA and PA must be used with caution when applied to untargeted metabolomics, given the noise typically present in such data. On the other hand, a network approach both helped identify pathways that would otherwise be invisible, eliminated incorrectly identified metabolites, and allowed visualization of the metabolic perturbations that respects the intrinsic interconnectedness of metabolites. Finally, we show how estradiol specifically perturbs several metabolic pathways: our analysis indicates that carnitine-fatty acid derivative pathway appears regulated by estrogen, as are polyamine degradation and production, and succinoadenosine.
Materials and Methods
Cell Culture
MCF-7 cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA no. HTB-22, lot number 59388743) and were grown using an optimized protocol, modified from the protocol used in the ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods) validation study for the MCF-7 cell proliferation test method (ICCVAM, 2003), which was positively evaluated by the US validation body of the National Toxicology Program (ICCVAM, 2006b). Vials received from ATCC at passage 147 were thawed and expanded for 8 passages to provide a sufficient available stock, and then frozen and stored in liquid nitrogen. MCF-7 cells were maintained in complete growth medium composed of DMEM-F12 (GIBCO, Life Technologies, Grand Island, NY, USA, no. 11309 ) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Norcross, GA, USA, no. S11150), nonessential amino acids (GIBCO, Life Technologies, no. 11140), 10μg/mL bovine insulin (Akron Biotech, Boca Raton, FL, USA, no. AK8213) and 0.01 mg/ml gentamicin (Invitrogen, Life Technologies, no. 15710) in BPA-free culture flasks. Cultures were fed every 2–3 days and passaged when 70–80% confluent. To control for genetic drift, MCF-7 cells from the initial thawed vial were only used in experiments for up to 10 passages. For quality control, MCF-7 cells were analyzed for karyotyping and mycoplasma. Comparative genomic hybridization (CGH) (Kleensang et al., 2015) as well as regular morphological assessment were also performed on the MCF-7 cells.
Treatment of cell cultures
MCF-7 cells were seeded at a density of 300,000 cells/well in 6-well plates and allowed to grow for 72 hours in complete growth media. After 72 hours, cells were rinsed with PBS and placed in treatment media composed of DMEM/F12 supplemented with 5% dextran charcoal stripped fetal bovine serum (DCC, Gemini Bio-products, Sacramento, CA, US, no. 100–119), nonessential amino acids, 6 ng/mL bovine insulin and gentamicin for 48 hours. Cells were then exposed to 1 nm/L 17β estradiol (E2, Sigma Aldrich, St. Louis, MO, USA, no. E8875) or vehicle control dimethylsulfoxide (DMSO, Sigma Aldrich, no. D8418) in fresh treatment media for 4 and 24 hours.
Untargeted metabolomics analysis
After E2 treatment, the cell culture media was removed by gentle vacuum suction and the remaining cells were washed 2 times with 1 mL of pre-warmed PBS. Any residue of PBS is removed from the wells. A solution of 700uL dry-ice cold 80:20 (v/v) methanol/water was immediately added, and the cells were scraped and collected in a 1.5 ml Eppendorf tube. The wells were washed again with an additional 700uL solution of methanol/water and this solution was combined with the previous one. The solution was vortexed for 1 min and then stored at −80°C for 2 h to allow for protein precipitation. For metabolite extraction from the cell lysate, tubes were placed on dry ice for 15 min and centrifuged at 14 000 × g for 5 min at 4°C. The supernatant was transferred to a new 1.5 ml tube and placed on dry ice. Then, 300 μl of 80 : 20 methanol/water was added to the pellet and a second extraction was performed. The combined supernatants were evaporated overnight to dryness at room temperature in a Speedvac concentrator (Savant, Thermo Fisher Scientific, Waltham, MA, USA). The dried samples were reconstituted with 60 μL of 60% methanol with 0.1% formic acid and clarified by centrifugation at 14 000 × g for 5 min. The clarified samples were transferred to HPLC vials for LC-MS measurements.
Chromatographic separations were performed using an Agilent 1260 high-performance liquid chromatography system with a well-plate autosampler (Agilent, Santa Clara, CA, USA). For aqueous normal phase (ANP) separation, a Cogent Diamond Hydride (MicroSol, Eatontown, NJ, USA) column (150 × 2.1 mm i.d., 4 μm particle size, 100 Å pore size) was used for separation of metabolites. The LC parameters were as follows: autosampler temperature, 4°C; injection volume, 5 μL; column temperature, 35°C; and flow rate, 0.4 mL/min. The solvents and optimized gradient conditions for LC were: Solvent A, 50% methanol/50% water/0.05% formic acid; Solvent B, 90% acetonitrile with 5 mM ammonium acetate; elution gradient: 0 min 100% B; 20–25 min 40% B; post-run time for equilibration, 10 min in 100% B. The LC system was coupled directly to the Q-TOF mass spectrometer. A blank injection was run after every three samples and a QC sample was run after every five samples to identify the sample carryover and check for stability.
A 6520 accurate-mass Q-TOF LC-MS system (Agilent) equipped with a dual electrospray (ESI) ion source was operated in negative-ion mode for metabolic profiling. The optimized ESI Q-TOF parameters for MS experiments were: ion polarity, negative; gas temperature, 325°C; drying gas, 10 l/min; nebulizer pressure, 45 psig; capillary voltage, 4000 V; fragmentor, 140 V; skimmer, 65 V; mass range, 70–1100 m/z; acquisition rate, 1.5 spectra/s; instrument state, extended dynamic range (1700 m/z, 2 GHz). Spectra were internally mass-calibrated in real-time by continuous infusion of a reference mass solution (standards with known mass at specific concentrations, which are introduced into the ion-source throughout the sample run to perform dynamic calibration) using an isocratic pump connected to a dual sprayer feeding into an electrospray ionization source. Data were acquired with MassHunter Acquisition software from Agilent.
For the data processing and chemometric analysis of the LC-MS untargeted data, the acquired raw data files (.d files) were first checked for quality in MassHunter Qualitative Analysis software (Agilent, version 6.0). Reproducibility of chromatograms was visually inspected by overlaying the Total Ion Chromatograms (TICs) of all samples. Data files that exhibit outlier peaks; i.e. replicates with very dissimilar chromatograms, were excluded for further processing. The raw data files were then converted to mzXML using ProteoWizard 3.0 (Kessner et al., 2008). Raw LC-MS data were analyzed by the MZmine 2 software (Pluskal et al., 2010)for chromatogram deconvolution, peak detection and alignment. The optimized parameters of the data processing are given in supplementary material S1. The putative identification was achieved by online searching for the accurate m/z values of the peaks against HMDB and KEGG databases (Kanehisa and Goto, 2000; Wishart et al., 2007). Those peaks were manually inspected for the quality of the EIC (extracted ion chromatograms) and also for remaining duplicate compounds names.
Targeted metabolomics analysis
The cell culture and treatment were the same as described above. The metabolite extraction was made in a similar manner as for the untargeted analysis using an 80:20 methanol/water solution with some minor modifications; after the PBS wash, a quick final rinse with deionized water was added to remove excess salts. The metabolism was then quenched using a similar solution as previously (dry-ice cold 80:20 MeOH/water) and the cells scraped and transferred in cold methanol and stored at −80°C. In addition, a bead-beating homogenization of the pellet was applied. The dried samples were reconstituted in 70% acetonitrile + 0.2% ammonium hydride for both positive and negative mode. The LC-MS platform for metabolite profiling was described previously (Chen et al., 2012). For the targeted metabolite identification method the assignment of structural identities to metabolites was based exclusively on a match of retention times and accurate masses to an in-house database, established by Dr. Gross lab at Cornell University, based on a match of retention times and accurate masses.
Targeted metabolomic confirmation by LC-MS/MS
Targeted metabolomics confirmation analysis was performed on an Agilent 6490 triple quadropole LC-MS/MS system with iFunnel and Jet-Stream® technology (AgilentTechnologies, Santa Clara, CA) equipped with an Agilent 1260 infinity pump and autosampler. Chromatographic separation was performed on a Diamond Hydride column (150mm x 2.1 mm i.d., 4μm particle size, Microsolv, Eatontown, NJ). The LC parameters were as follows: autosampler temperature 4°C; injection volume 4 μL; column temperature 35°C; and flow rate 0.4 ml/min. The solvents and optimized gradient conditions for LC were: Solvent A, water with 5mM ammonium acetate, pH=7.2; Solvent B, 90% acetonitrile with 10mM ammonium acetate, pH=6.5; elution gradient: 0 min 95% B; 15–20 min 25% B; post-run time for equilibration, 5 min in 95% B. MS was operated in positive/negative polarity switching mode (unit resolution) with all analytes monitored by multiple reaction monitoring (MRM). Compound identity was confirmed by comparison to the retention times of pure standards. The optimized operating ESI conditions were: gas temperature 230°C (nitrogen); gas flow 15 L/min; nebulizer pressure 40 psi; sheath gas temperature 350°C and sheath gas flow 12 L/min. Capillary voltages were optimized to 4000V in positive mode with nozzle voltages of 2000 V. All data processing was performed with the Mass Hunter Quantitative Analysis software package.
Data analysis
Over-representation analysis using only the metabolite identifiers was performed with Metaboanalyst, which tests all metabolites with database identifiers against human KEGG pathways for over-representation. Metabolite Set Enrichment Analysis, the metabolomics equivalent of Gene Set Enrichment Analysis/quantitative enrichment analysis was performed using the entire dataset to look for statistically significant perturbations along KEGG Pathways was performed via Metaboanalyst. Pathway Impact Analysis, which focuses on perturbations that are considered topologically important, was performed with Metaboanalyst (Xia et al., 2009; Xia et al., 2015) against all human pathways available. In all cases, the background was assumed to be all possible metabolites. Impala was also used for over-representation analysis. (Cavill et al., 2011; Kamburov et al., 2011).
Network analysis
All metabolites statistically significantly different at 24 hours with control vs. treated with a p-value less than .01 were used to create a network in STITCH Version 4 (Kuhn et al., 2008) with a medium stringency of .40 and no additional proteins. All unconnected metabolites were discarded. The network was plotted in Cystoscape (Shannon et al., 2003) using Neighborhood Connectivity map.
Results
Initially, we attempted to characterize the metabolomic effects of estradiol after 24 hours treatment with an untargeted approach. To examine reproducibility between two experiments, we performed identical experiments (hereafter called EXP1 and EXP2) one week apart with an otherwise identical protocol and cells harvested at passages 158 and 160, respectively.
For an untargeted approach, there are several approaches that could be taken to initially identify the metabolites. For our untargeted metabolomics workflow, only putative identification of the metabolites is possible, i.e. an estimate of the identity (formula, name or both) based on algorithms for feature extraction and annotation. However, the multiple possible metabolites for each identified feature means that few metabolites can be identified definitively based on mass alone, and even with retention time there is still ambiguity in the assigned identity. Even with a clear mass the possibility of adducts, contaminants, and inaccurate peak calling translates into considerable ambiguity in any attempt to identify metabolites, making this step a major bottleneck in analysis of metabolomics data. Several methods and algorithms have been developed to meet this challenge, as extensively reviewed by Katajamaa and Orešič (Katajamaa and Oresic, 2007). Based on studies of different methods in terms of alignment, deconvolution and accuracy of identification and ease of use it was found that MZmine is highly suitable for LC-MS datasets, and typically allows for more than 80% accuracy (Niu et al., 2014). Mzmine, a freely available and widely used software for metabolomics analysis, was used for the untargeted method. The algorithm allows for chromatogram deconvolution, peak detection and alignment. One of the most valuable features of the software is its performance in handling missing values. Usually, after peak alignment, the resulting peak list may contain missing peaks that affect the successive analysis. A missing value does not necessarily imply that the peak is not present in the raw data, but could equally be a misidentification in one of the algorithm steps, which MZmine can fill in.
Most importantly, we verified the performance of the method in terms of accuracy of metabolite putative identification using a publicly available dataset (URL: http://www.ebi.ac.uk/metabolights under accession number MTBLS67). The main result is the high accuracy of the putative identification, with 80% extracted and accurately annotated using our method, i.e. same m/z, RT, formula and name, 7% were assigned the same formula but a different compound name, 9% were assigned a different formula and only 4% were not found, although we have to caution that the accuracy may be different for our dataset due to differences in sample preparation, etc. The detail of the performance assessment of the metabolite putative identification method is provided in Supplementary Material S2.
After metabolite identification, Metaboanalyst was used for both over-representation analysis (ORA), Quantitative Set Enrichment Analysis (QEA), and Pathway Analysis (PA) (Xia and Wishart, 2010). ORA analyzes whether, for a given list of differentially expressed metabolites, one particular pathway is over-represented, that is to say there are more hits on that pathway than would be expected by chance compared to an expected value based on the size of the pathway and assuming a hypergeometric distribution, after correcting for a false discovery rate (FDR). Critically, ORA is dependent only looks at group selected on the basis of an inferential statistics and does not look at the magnitude of the fold-change of the given metabolite. The second approach, QEA, is based on the “global test” algorithm (Goeman et al., 2004), commonly used for microarray experiments, to perform enrichment analysis directly from expression data; like ORA it looks for more hits on a pathway than by chance, but it additionally takes into consideration the magnitude of the expression values; there a pathway can be significant because of a few highly changed genes or metabolites on a pathway, or alternatively, multiple smaller-scale changes in genes or metabolites on a pathway. “Global test” was originally created to examine associations between gene sets and clinical outcomes, but it has been used extensively for microarray data and adapted for multiclass and continuous phenotypes. It uses a generalized linear model to compute a ‘Q-stat’ for each metabolite set, using the average of the squared covariance between the expression level of the phenotype label. QEA includes appropriate methods to adjust for the multiple testing problems that occur during enrichment analysis (e.g. Benjamini and Hochberg FDR). PA attempts to improve on QEA by taking into consideration the topology of the pathway - in other words, it looks for changes in central, highly connected nodes when considering the likelihood that given pathway is perturbed (Aittokallio et al., 2003).
Our results indicate that all of the above approaches must be used with considerable caution when analyzing untargeted metabolomic data. Initially, we examined differentially expressed metabolites determined via Over-Representation Analysis, using all metabolites different at 24 h vs. control with an uncorrected p-value less than .05, which show identical results for both data sets - only one fairly uninformative, overly general pathway (“Protein biosynthesis”) [Table 1] that was statistically significant after correcting for multiple hypothesis testing; it is likely that given the requirement for multiple hits on a given pathway to achieve statistical significance, ORA is of limited usefulness for smaller pathways (i.e. glutathione biosynthesis, with only 10 compounds annotated to that pathway). Finally, ORA will necessarily be sensitive to the background of identified metabolites – in this case, the main pathway identified by using all the metabolites identified in the experiment was also protein biosynthesis. The lists of all metabolites used in the analysis are provided in supplementary materials. S5 lists all metabolic features generated using the untargeted method for EXP1 and EXP2. S6 contains all metabolic features generated using the targeted method. S7 provides the results of the two confirmation experiments.
Table 1.
Untargeted metabolomics analyzed by Over-Representation Analysis from 24 hour 1nM estradiol-treated MCF-7 vs. control in EXP1 and EXP2
| Total Metabolites on Pathway | Expected Metabolites | Actual Metabolites | Raw p | Holm p | FDR | |
|---|---|---|---|---|---|---|
| PROTEIN BIOSYNTHESIS | 19 | 1.24 | 9 | 6.31E-07 | 5.05E-05 | 5.05E-05 |
| AMMONIA RECYCLING | 18 | 1.18 | 5 | 0.00444 | 0.351 | 0.178 |
| UREA CYCLE | 20 | 1.31 | 5 | 0.00727 | 0.567 | 0.194 |
After metabolite identification, Metaboanalyst was used for Over-Representation Analysis. Only protein biosynthesis was identified as a statistically significant pathway after correction for multiple hypothesis testing, likely reflecting the large number of amino acids, although the amino acids had relatively modest fold-change.
Since both QEA and PA offer a more sensitive measurement of pathway perturbation, as expected they both identified more pathways with higher statistical power. However, the top three pathways identified by QEA were all based on fewer than three - and often as few as one - metabolite, meaning such an analysis can be easily misled by a single misidentification or outlier in the data [ Figure 1, Table 2, Supplementary material S3]. Moreover, there was minimal overlap between the two data sets from the two experiments, EXP1 and EXP2 in terms of the pathways identified.
Figure 1. Untargeted Metabolomics analyzed by Quantitative Enrichment Analysis from 24 hour 1nM estradiol-treated MCF-7 cells vs. control in EXP1 and EXP2.


After metabolite identification, Metaboanalyst was used for Quantitative Enrichment Analysis. Using a Holmes adjusted p-value of .05, there were more pathways unique to each experiment than in common when QEA was used to analyze the data, despite being identical experiments.
Table 2.
Pathways identified by Quantitative Enrichment Analysis of untargeted metabolomics of 24 hour 1nM estradiol-treated MCF-7 cells vs. control in EXP1 and EXP2
| Pathways unique to EXP1 | Pathways unique to EXP2 | Pathways in Common |
|---|---|---|
| Beta oxidation of very long chain fatty acids | Betaine metabolism | Cysteine metabolism |
| Fructose and mannose degradation | Biotin metabolism | Folate and pterine biosynthesis |
| Glutathione metabolism | Catecholamine biosynthesis | Methionine metabolism |
| Glycerolipid metabolism | Phenylalanine and tyrosine metabolism | Sphingolipid metabolism |
| Glycine, serine and threonine metabolism | Propanoate metabolism | |
| Glycolysis | Protein biosynthesis | |
| Tyrosine metabolism | ||
| Valine, leucine and isoleucine degradation |
Similarly, PA analysis identified only five pathways in common between EXP1 and EXP2, and the top pathway in both (“methane metabolism”) datasets is based on three metabolites (Dihydroxyacetone, Glycine, and Serine) - reflecting the extent to which pathways used for such approaches typically build upon conserved consensus pathways initially identified in bacteria or yeast and then extrapolated to human data (Popescu and Yona, 2005); in this case, methane metabolism is clearly non-informative.
Quantitative Enrichment Analysis (QEA)
Pathway Analysis (PA)
Any untargeted approach necessarily has some noise in the data owing to artifacts, outliers, and misidentified metabolites. In the case of both QEA and PA, it is quite possible that the noise intrinsic to untargeted metabolomics creates the appearance of a statistically significant perturbation in a pathway, and any such analysis must be treated as more exploratory than definitive, especially if the pathway is identified by a solitary metabolite.
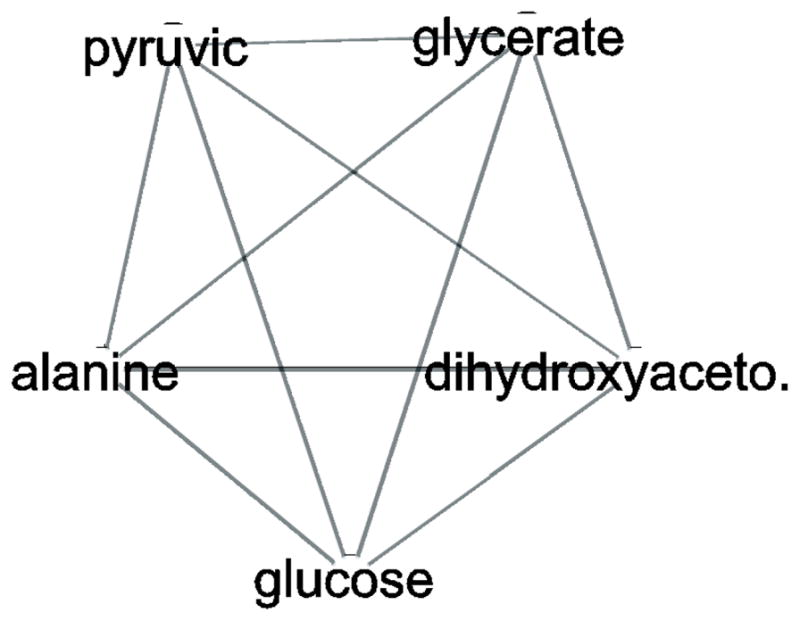
Interestingly, using a network approach, the data appears more similar than the identified pathways would imply some overlap – for example, the dihydroxyacetone subnetwork consisted of pyruvic acid and alanine in EXP1 and the same metabolites, plus glycerate and glucose, in EXP2. Therefore, while dihydroxyacetone is annotated to both the methane and glycerolipid pathways, a network approach points to a more likely role in carbohydrate metabolism, although given the putative nature of metabolite identification, this hypothesis would need confirmation from a more precise approach.
We next tried a targeted approach based on metabolite identification with known standards and retention times. While this targeted method usually allows for detection of a smaller number of metabolites, the accuracy of identification is greatly improved, even though identification is only more certain, not definitive. The targeted approach identified 38 metabolites statistically significantly with a p-value of less than .05 (without correction for multiple hypothesis testing) and 53 with a p-value less than .01. When the statistically significant metabolites were analyzed via ORA no significant pathways were found (the signal was weak in both QEA and Pathway Impact analysis, but it was statistically significant). However, only 36 of the 53 metabolites that were statistically significant after estrogen treatment were annotated to pathways even when using Impala (Cavill et al., 2011; Kamburov et al., 2011) which looks at not only KEGG pathways but also HMDB, SMPD, and Wikipathways. This indicates that even with a targeted approach and all possible databases, a substantial part of the signal was simply lost.
To build our de novo network, we used all metabolites that were different at 24 hours with and without estrogen with a p-value less than .01 (53) in the STITCH database, which links metabolites based on known reactions in the main metabolic network pathways but also based on experimental evidence and text-mining; we selected a medium stringency level (.40).
The resulting network contained a “connected component” of 43 metabolites. Of the metabolites excluded, several were clearly non-endogenous (e.g. diethanolamine, geranyl pyrophosphate); one advantage of this approach is that while it does not completely eliminate spurious metabolites, it helps identify them, as they tend to be unconnected to the larger network. However, one metabolite (succinoadenosine) was unconnected although it is both endogenous and was later verified by in-house standards. Figure 4 shows the network plotted both by the fold change difference (node size) and p-value (node color). On the whole, the most highly connected metabolites (energy metabolites such as pyruvate (pyruvic acid) and alpha-ketoglutarate, as well as amino acids) showed only a modest change between proliferation with and without estrogen. The relatively subtle changes at such key nodes may indicate that PA, by focusing on highly connected nodes considered key points in pathways, may miss the most phenotypically relevant changes, as flux through such points is possibly so tightly controlled that profound changes are unlikely in the absence of targeted enzyme inhibition.
Figure 4. Metabolic network, plotted with Circular Neighborhood Connectivity layout, of targeted metabolomics of 24 hour 1nM estradiol-treated MCF-7 cells.

Targeted metabolomicss was carried out at Cornell university. All metabolites statistically significantly different with a p-value less than .01 were used to create a network in STITCH Version 4 with a medium stringency of .40 and no additional proteins. All unconnected metabolites were discarded. The network was plotted in Cystoscape using Neighborhood Connectivity map. Node color = absolute value of the difference between the fold change of 0 vs 24 h with estrogen and without estrogen (i.e. if a metabolite increased 2.5-fold with estrogen from 0 to 24 hours, and 1.5 without, Absolute Fold Change would equal to 1, darker is equivalent to a higher change); node size is equivalent to “degree” (the number of connected metabolites).
The most marked differences in terms of fold-change between proliferating cells with and without estrogen were found in metabolites that were relatively “unconnected” – hypoxanthine (4 connected metabolites), purine (1 connected metabolite), acetyl-methionine (1 connected metabolite); only glutathione (11 connected metabolites) was both markedly different and highly connected.
Nonetheless, the use of the network approach suggests some connections that might otherwise be invisible - for example, the glutathione subnetwork (S4) (all first degree neighbors of glutathione) shows several metabolites not annotated to the glutathione pathway but connected either by experimental data or text-mining. Fumarate, for example, is not annotated to the glutathione pathway but is known to bind directly to glutathione and produce succinated glutathione which thus acts to both deplete NADPH and increase ROS (Sullivan et al., 2013) and is considered an onco-metabolite. However, succinyl-glutathione levels would likely have been invisible in most experiments that used HMDB to identify metabolites, and certainly in most pathway analyses, as it is not in HMDB as a metabolite and is not annotated to any pathways.
Moreover, the network helped identify several clear “pathways” not visible when focusing on annotated pathways. Although fatty acid beta-oxidation was identified in QEA as a pathway, the carnitine and fatty acid acyl carnitine derivatives formed a small cluster (Figure 5) that would not be apparent via pathway-dependent data analysis, as the carnitine derivatives were, with the exception of propionyl-L-carnitine, not annotated to pathways. In this instance, the network links were added via text-mining.
Figure 5. Carnitine subnetwork cluster in targeted metabolomics of 24 hour 1nM estradiol-treated MCF-7 cells.

Carnitine and carnitine derivatives were linked in the network, but were not connected via pathway databases but instead via text-mining (using simple PubMed co-ocurrence); only propionyl-L-carnitine was connected to carnitine via known reactions in the pathway databases. In addition to the carnitine derivatives, carnitine was a “hub” in the network and connected to many other metabolites (names not shown).
Finally, to verify the altered metabolites, we analyzed 20 of the statistically significant metabolites using pure chemical standards in estrogen treated vs. untreated. 20 metabolites were analyzed using an optimized targeted method (Table 4): Putrescine, Hypoxanthine, Deoxycytidine, Taurine, Purine, Leucine, Pyruvate, α-Ketoglutarate, Octanoyl L-Carnitine, Glycerate, NAA, Proline, Glutathione (reduced), Succinoadenosine, Valeryl L-Carnitine, Butyryl L-Carnitine, Propionyl L-Carnitine, Cystathionine, L-Carnitine and N-acetylputrescine.
Table 4.
Coefficient of variation of repeat MS/MS confirmation with metabolomics of 20 of the statistically significant metabolites following 1nM estradiol treatment for 24h of MCF-7 cells
| Cornell | In house 1 | In house 2 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| CTL | 1nM | FC direction | CTL | 1nM | FC direction | CTL | 1nM | FC direction | |
| Putrescine | 19% | 12% | ↑ | 13% | 11% | ↓ | 4% | 12% | ↑ |
| Hypoxanthine | 27% | 50% | ↓ | 17% | 16% | ↓ | 52% | 48% | ↑ |
| Deoxycytidine | 26% | 25% | ↑ | 20% | 19% | ↑ | 26% | 23% | ↑ |
| Taurine | 19% | 18% | ↑ | 6% | 5% | ↓ | 2% | 3% | ↓ |
| Purine | 34% | 34% | ↓ | 9% | 11% | ↑ | 11% | 6% | ↑ |
| Leucine | 20% | 13% | ↑ | 10% | 9% | ↑ | 18% | 10% | ↑ |
| Pyruvate | 24% | 25% | ↑ | 22% | 27% | ↓ | 16% | 13% | ↑ |
| α-Ketoglutarate | 17% | 19% | ↑ | 17% | 12% | ↑ | 15% | 13% | ↑ |
| Octanoyl L-Carnitine | 52% | 52% | ↑ | 41% | 27% | ↓ | 58% | 43% | ↓ |
| Glycerate | 34% | 33% | ↑ | 11% | 9% | ↑ | 5% | 4% | ↓ |
| NAA | 18% | 19% | ↑ | 15% | 11% | ↑ | 22% | 22% | ↓ |
| Proline | 41% | 27% | ↑ | 33% | 22% | ↑ | 3% | 2% | ↑ |
| Glutathione (reduced) | 42% | 64% | ↑ | 28% | 26% | ↑ | 23% | 25% | ↓ |
| Succinoadenosine | 17% | 19% | ↑ | 20% | 16% | ↑ | 3% | 7% | ↑ |
| Valeryl L-Carnitine | 42% | 28% | ↑ | 23% | 13% | ↑ | 12% | 12% | ↑ |
| Butyryl L-Carnitine | 39% | 29% | ↑ | 14% | 16% | ↑ | 27% | 27% | ↑ |
| Propionyl L-Carnitine | 51% | 37% | ↑ | 4% | 4% | ↑ | 6% | 9% | ↑ |
| Cystathionine | 57% | 54% | ↑ | 12% | 8% | ↑ | 4% | 5% | ↑ |
| L-Carnitine | 28% | 29% | ↑ | 5% | 4% | ↑ | 5% | 5% | ↓ |
| N-acetylputrescine | 7% | 7% | ↑ | 18% | 12% | ↑ | 7% | 7% | ↑ |
Comparison three repeat experiments using targeted metabolomics with standards. The same experiment was carried out three times and targeted metabolomics was carried out first at Cornell and then restricted to 20 of the significant metabolites in Johns Hopkins.
6 replicates were analyzed for each condition, controls and estrogen treated cells, for a total of 12 samples, and we repeated the experiment twice. The abundances spanned over a 3-fold range with an average coefficient of variation of 11% and 17% for estrogen treated samples and controls, respectively. However, the coefficient of variation was markedly different for each metabolite and our results indicate that it needs to be considered carefully when interpreting results.
We compared the abundances of the analysis based on standards to the previous targeted data. For each metabolite, we first calculated the fold-change observed in each experiment between controls and treated samples and then we plotted these fold-changes for each compound (Figure 6) For the 20 metabolites analyzed, the carnitine pathways were, with one exception, fairly consistent in the direction in the (i.e. either increase or decrease) and somewhat less consistent in terms of change magnitudes although the dynamic range was quite different – fold-changes observed were between −1.2 and −5.4 for Cornell and the targeted analysis, respectively. However, several of the metabolites showed a statistically significant difference in only one of the experiments using standards. Generally speaking, these tended to be very low-abundance metabolites - suggesting a technical bias due to the limit of detection with this approach. Moreover, the coefficient of variation of these compounds tended to be the highest, (e.g. 34% for purine) and the observed fold-changes obtained were very small and therefore inconclusive; while these may be affected by estrogen treatment, it would take a more targeted analysis specific for that metabolite (or flux analysis) to confirm the change.
Figure 6. Reproducibility of fold-changes of 20 metabolites in three experiments using targeted metabolomics of 20 metabolites following 1nM estradiol treatment for 24h of MCF-7 cells.

The three experiments with targeted metabolomics shown in Figure 6 analyzed first at Cornell and then restricted to 20 of the significant metabolites in Johns Hopkins are shown with regard to fold-change in metabolite.
Discussion
Often, metabolomic (like transcriptomic) data are analyzed based on inferential statistics, which generates a relatively small list of metabolites differentially expressed. The problems of relying on inferential statistics alone for high-dimensional data are well-known: the stringency of false-discovery corrections tends to lose much of the signal, and a key advantage of high-dimensional data - the ability to see each gene, protein, or metabolite acting in concert with others - is often lost with such an analysis. This is especially likely to diminish the signal in metabolomics data since the tight control of most metabolic pathways entails that most metabolites will fluctuate within a fairly narrow range – instead of dramatic fold-changes in one or two key metabolites, it is more likely to see a modest increase or decrease at key points in a given pathway.
This has led to the development of other approaches such as Over-Representation Analysis (ORA), Gene and/or Metabolite Set Enrichment Analysis (GSEA/MSEA), and finally Pathway Analysis (PA), all of which seek to interpret data in the context of known pathways. This approach, however, is only as useful as the quality of the databases and the reproducibility of the data used - for transcriptomics this may cover a significant portion of the data, but it likely leaves relevant gaps for toxicology, as shown in our previous work (Maertens et al., 2015) and (Pendse et al., in preparation). Metabolomics, on the other hand, has a relatively underdeveloped database infrastructure, and owing to inconsistent or incomplete pathway annotation may miss key information.
Turning to the sources that focus on pathways and attempt to provide a comprehensive map—Recon/EHMNM, HumanCyc, KEGG, and Reactome—there is remarkably little overlap (Consensus and Conflict Database1): These databases differ in size—from a low of 970 metabolites in Recon1 (reflecting that it is based on manual curation) to a high of 2,676 metabolites for the EHMN (which is based in part on automated annotations) (Stobbe et al., 2013). However, somewhat worryingly, there is a striking lack of agreement amongst the databases in terms of commonality—the five main databases agree only on 402 metabolites (9% of the total metabolites in the different databases) and a full 3,107 of metabolites are present in only one database (Stobbe et al., 2013). This can be attributed to several reasons. One, the databases may have different levels of granularity—for example, a reaction may include all associated molecules (including “currency molecules,” such as ATP and NADH) in one database, but another database may focus only on the main players. Two, various databases were started with different aims in mind and use different identifiers—the lack of database interoperability makes it exceedingly difficult to translate chemical identifiers from one database to another, because of the lack of efficient ID conversion tools, the complexity of chemical nomenclature, and the difficulty in using structural-based IDs such as InChI and SMILES for database indexing.
This problem is aggravated when approaches such as ORA and QEA (Quantitative Enrichment Analysis) are used in metabolomics without necessarily taking into consideration some of the problems of metabolomic data. While microarrays offer a data set where all the discrete features are labeled unambiguously at the outset and all such features have an assigned value, in the case of metabolomic data, due to the nature of the technology, no experiment can possibly identify a complete set of metabolites; of those identified as discrete, not all can be assigned an identity precisely - peak identification may have been incorrect, ions may have been incorrectly identified, or metabolites may be confused with compounds with the same weight. Finally, of those assigned identities, not all are annotated with pathway information. Moreover, the missing metabolites may not be missing at random, but may reflect a chemical class. As a consequence, at the same time ORA and QEA have a substantial loss of information (because of the incomplete pathway mappings), they also have the potential to be inaccurate due to a small number of misidentified metabolites. Additionally, all annotation-based statistical tests are predicated on an accurate assumption of the “background”—that is, the total number of pathways and metabolites possible. Generally, the assumed background is the number of total metabolites in pathways, although in fact even the best untargeted approaches will be able to identify only a fraction of any given pathway. Therefore, there are potentially two sources of error—non-random, missing data or an incorrect assumption about background size. Because of this, ORA and QEA can lead to inconsistent or misleading results owing both to errors in identification or a lack of annotations in the database. Finally, it is important to keep in mind that a linear pathway is an artificial construct—all pathways are abstracted from a broader, global cellular network and therefore are, at some level, an oversimplification. Focusing on discrete pathways for statistical analysis may miss a key aspect of a shift that is taking place within a global metabolic network, rather than a pathway.
While the estrogen-receptor signaling pathway is relatively well known, the full phenotypic consequences of estrogen or estrogen-like substances have yet to be fully explored. Here, we demonstrate that estrogen has subtle but potentially phenotypically important alterations in the acyl-carnitine fatty acids and acetyl-putrescine, in addition to likely subtle changes in key energy metabolites that, however, could not be verified consistently given the technical limitations of this approach. Our findings are consistent with a separate metabolomics study of ER-negative and positive breast cancer, which also found that butrylcarnitine – one of the carnitine-associated fatty acids in our network verified via standards – was highly correlated with a proliferation index (Tang et al., 2014).
Interestingly, in the same study hypoxanthine was correlated with receptor status (Tang et al., 2014). Hypoxanthine was markedly decreased in estrogen-treated cells vs. controls in the targeted analysis and confirmed via analysis with one data set using standards but not the other. Estrogen has been reported to decrease both protein levels and enzyme activity of xanthine dehydrogenase/xanthine oxidase (XDH/XO) in MCF-7 cells (Taibi et al., 2008). Loss of XO activity has been linked to aggressive breast cancer growth both in vivo and in vitro (Fini et al., 2008). The phenotypic significance is unclear but offers an intriguing hypothesis that would require further exploration with an approach tailored to this goal.
Succinoadenosine, which was identified in the Cornell experiment and confirmed in both experiments with in-house standards, is a metabolite that is very much off the map, as it was not annotated to pathways and was unconnected in the STITCH network. Succinoadenosine is mostly found in the urine of patients with ADSL (Adenylosuccinate Lyase) deficiency (Zikanova et al., 2005). The only other disease context that reports an increase in succinodenosine is fumarase deficiency (Zeman et al., 2000), which suggests two possibilities for the elevated levels detected in estrogen-treated cells. One, estrogen suppressed ADSL levels, or two, ADSL inhibited fumarase such that succinoadenisone levels increased.
Acetyl-putrescine, which was consistently altered in both the targeted and in-house standards, and showed one of the largest fold-change differences, offers another interesting hypothesis. Both SAT1 and SAT2 are predicted to acetylate spermidine although with lower affinity than other polyamines (Hamosh et al., 2005). SAT2 is thought not be implicated in polyamine production or degradation in vivo and therefore the most likely candidate is SAT1. Transgenic SAT1 overexpression in mice is known to induce an increase in acetylspermidine as well as acetyl-putrescine with significant phenotypic consequences – including infertility in females. SAT1 does not have an estrogen response element and does not appear to be regulated by estrogen, but instead by the PMF1 and NRF2 mediated pathway (Wang et al., 1999).
While this paper shows that for this dataset, a network analysis offers insights unavailable in an approach that depends upon pathway annotations, the network built from this model must be treated with substantial caution. Because the overall effects were subtle, we used a less stringent p-value to build the network. While the metabolites that were verified with standards are unambiguously identified, for the remainder the identification remains putative. Although the network approach likely eliminated obvious non-endogenous metabolites, several metabolites were either misidentified as they are not endogenous or are unlikely to be in these cells (e.g. proline betaine). However, such an approach – especially if combined with chemical similarity network – offers one way forward to improve the percentage of accurately identified metabolites.
Metabolomics remains a field with both substantial technical challenges (most critically, the data analysis bottleneck of accurate metabolite identification) as well as bioinformatics challenges. To begin with, metabolomics has yet to standardize and optimize sample preparation techniques (washing, quenching, protein separation) to minimize the bias introduced by the methodology chosen. As was the case with transcriptomics, there needs to be a substantial investment in the necessary tools to allow the improvement of analytical and data processing methods, and the time-consuming but necessary task of curating databases, pathways, and annotations. Here, we show that using a data analysis approach based exclusively on pathway annotations will at best miss much that is of interest and at worst will produce perturbed pathways that are statistically significant yet uninformative for the biological sample at hand (e.g. methane metabolism). Any pathway large enough to achieve statistical significance with ORA in an untargeted approach is equally likely to be a pathway so broad that it is uninformative. While both QEA and PA seem like they would offer an improvement as they are taking into consideration prior knowledge about the pathways (QEA) or pathway topology (PA), the noise intrinsic to an untargeted approach is at least as likely to produce artifacts as it is to produce a meaningful result, since both methodologies can use as few as one metabolite on any given pathway to achieve statistical significance. Given the intrinsic inter-relatedness of metabolic pathways, many metabolites appear on multiple pathways – e.g. methionine appears on 5 pathways in KEGG, 17 on HumanCyc – the likelihood of a misleading positive hit increases, and the experiments will then give the appearance of profound problems in reproducibility.
On the other hand, a targeted approach, by narrowing its focus and minimizing (but not eliminating) misidentifications, renders the likelihood of a spurious pathway much smaller, but the limited number of metabolites also makes statistical significance harder to achieve. Furthermore, the lack of annotations for several metabolites entails that such an approach will miss much that is of interest, and not necessarily provide the most useful information about any given perturbation.
A network based approach to data analysis – and specifically, one that exploits not just database reactions but text-mining as well – offers a chance to “connect the dots” (in this case, several acyl-carnitine derivatives) that would otherwise neither be considered statistically significant on their own (as each carnitine derivative was only modestly changed) nor be identified via ORA, QEA, or PA. Moreover, a network approach respects the intrinsic interconnectedness of the metabolites; instead of the statistical problems associated with analyzing pathways with several metabolites (e.g. amino acids and common energy metabolites) appearing in several different pathways, a metabolite can be seen in the context of its connectivity to the broader map.
Metabolomics bears considerable promise for toxicology to assess the actual phenotypical changes in a reasonably fast and affordable manner with chemical-analytical tools of a high level of standardization (Bouhifd et al., 2013; Ramirez et al., 2014). The challenge arises when we want to move from signatures of altered metabolites to underlying adverse outcome pathways or pathways of toxicity, which at the same time promise to link to the respective mode of action and separate thereby the signal from the noise. This is an indispensible step moving to a systems toxicology approach modeling the perturbations of the organism by a toxicant (Hartung et al., 2012). Nonetheless, a key message is that metabolomics experiments in some respects resemble the famous blind men trying to independently ascertain the nature of an elephant – any one experiment is going to be limited by the technical biases of the platform chosen, the noise intrinsic to the biological system and the data analysis, and the limitations of existing databases to interpret the data – both to identify metabolites and to identify perturbed pathways. Any one of our experiments taken independently would have led to at the very least a limited understanding of the biology, and at worst an artifact published as a striking new finding. As is true for all – omics technologies, but especially for metabolomics, both caution regarding conclusions and confirmation of hypotheses are of critical importance. Finally, for metabolomics to offer sufficient reliability to help illuminate Pathways of Toxicity, it will require climbing the steep hill that once faced transcriptomics: a community-wide commitment to trouble-shooting experimental protocols, improving and standardizing data processing, and slowly boot-strapping our way from the known pathways to a truly global understanding of the cellular metabolic network.
Supplementary Material
S1: Metabolite putative identification workflow parameters
S2: Performance of the metabolite putative identification method
S3: Quantitative Enrichment Analysis
S4 Network first degree neighbors glutathione
S5 List of all metabolic features generated using the untargeted method
S6 List of all metabolic features generated using the targeted method
S7 Metabolite confirmation results
Figure 2. Untargeted metabolomics analyzed by Pathway Analysis of 24 hour 1nM estradiol-treated MCF-7 cells vs. control in EXP1 and EXP2.
After metabolite identification, Metaboanalyst was used for Pathway Analysis. Using a Holm-adjusted p-value of 0.05, only 5 pathways were common to both experiments while another 18 pathways were present in only one experiment, 6 in EXP1 and 12 in EXP2 using pathway impact analysis. Only one pathway was similar in both experiments using Quantitative Enrichment Analysis and Pathway Analysis.
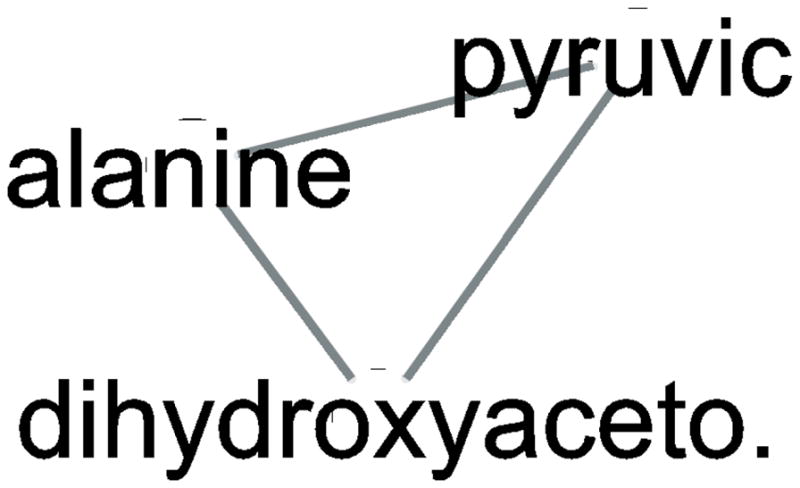
Figure 3. Dihydoxyacetone subnetwork, for untargeted metabolomics analyzed by Pathway Analysis of 24 hour 1nM estradiol-treated MCF-7 cells (A) EXP1 and (B) EXP2.
Dihydroxyacetone and all linked metabolites. While dihyxdroxyacetone was annotated to the methane pathway, a network analysis connected it to alanine and pyruvic acid in both experiments.
Table 3.
Pathways identified by Pathway Analysis of untargeted metabolomics of 24 hour 1nM estradiol-treated MCF-7 cells vs. control in EXP1 and EXP2
| Pathways unique to EXP1 | Pathways unique to EXP2 | Pathways in Common |
|---|---|---|
| Pentose phosphate pathway | Biotin metabolism | Methane metabolism |
| Folate biosynthesis | Propanoate metabolism | Sphingolipid metabolism |
| N-Glycan biosynthesis | Valine, leucine and isoleucine degradation | Glycerolipid metabolism |
| Cysteine and methionine metabolism | Valine, leucine and isoleucine biosynthesis | Sulfur metabolism |
| Tryptophan metabolism | Ubiquinone and other terpenoid- quinone biosynthesis | Arginine and proline metabolism |
| Glutathione metabolism | Tyrosine metabolism | |
| Thiamine metabolism | ||
| Phenylalanine metabolism | ||
| Phenylalanine, tyrosine and tryptophan biosynthesis | ||
| Nitrogen metabolism | ||
| Ascorbate and aldarate metabolism | ||
| Aminoacyl-tRNA biosynthesis |
Pathway Analysis found only three common five common pathways between the two experiments; despite being restricted to human pathways, the top pathway identified, methane metabolism, is incorrect.
Acknowledgments
This work was supported by an NIH Transformation Research Grant, “Mapping the Human Toxome by Systems Toxicology” (RO1 ES 020750). We are most grateful of the collaboration and discussions, which made this work possible, to the following individuals and their coworkers: Dr. Melvin E. Andersen, Dr. Patrick D. McMullen and Salil Pendse at The Hamner Institute, Research Triangle Park, NC, USA, Dr. Kim Boekelheide and Dr. Marguerite Vantangoli at Brown University, Pathology & Laboratory Medicine, Providence, RI, USA, Dr. Kevin M. Crofton and Dr. Russell Thomas at EPA, National Center for Computational Toxicology, Research Triangle Park, NC, USA, Dr. Albert J. Fornace Jr. and Henghong Li at Georgetown University Medical Center, Washington, DC, USA, as well as Dr. Carolina Livi and Dr. Michael Rosenberg at Agilent Inc., Santa Clara, CA, USA. The authors thank Dr. Steven Gross and his team at Cornell University for performing the targeted metabolomics analysis.
Footnotes
http://www.molgenis.org/c2cards/molgenis.do (last accessed 27 Sep 2015)
References
- Aittokallio T, Kurki M, Nevalainen O, Nikula T, West A, Lahesmaa R. Computational strategies for analyzing data in gene expression microarray experiments. J Bioinform Comput Biol. 2003;1:541–586. doi: 10.1142/s0219720003000319. [DOI] [PubMed] [Google Scholar]
- Bouhifd M, Andersen ME, Baghdikian C, Boekelheide K, Crofton KM, Fornace AJ, Jr, Kleensang A, Li H, Livi C, Maertens A, McMullen PD, Rosenberg M, Thomas R, Vantangoli M, Yager JD, Zhao L, Hartung T. The human toxome project. ALTEX. 2015a;32:112–124. doi: 10.14573/altex.1502091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhifd M, Beger R, Flynn T, Guo L, Harris G, Hogberg H, Kaddurah-Daouk R, Kamp H, Kleensang A, Maertens A, Odwin-DaCosta S, Pamies D, Robertson D, Smirnova L, Sun J, Zhao L, Hartung T. Quality Assurance of Metabolomics. ALTEX. 2015b doi: 10.14573/altex.1509161. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhifd M, Hogberg HT, Kleensang A, Maertens A, Zhao L, Hartung T. Mapping the human toxome by systems toxicology. Basic Clin Pharmacol Toxicol. 2014;115:24–31. doi: 10.1111/bcpt.12198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavill R, Kamburov A, Ellis JK, Athersuch TJ, Blagrove MS, Herwig R, Ebbels TM, Keun HC. Consensus-phenotype integration of transcriptomic and metabolomic data implies a role for metabolism in the chemosensitivity of tumour cells. PLoS Comput Biol. 2011;7:e1001113. doi: 10.1371/journal.pcbi.1001113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Park HC, Goligorsky MS, Chander P, Fischer SM, Gross SS. Untargeted plasma metabolite profiling reveals the broad systemic consequences of xanthine oxidoreductase inactivation in mice. PLoS ONE. 2012;7:e37149. doi: 10.1371/journal.pone.0037149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fini MA, Orchard-Webb D, Kosmider B, Amon JD, Kelland R, Shibao G, Wright RM. Migratory Activity of Human Breast Cancer Cells Is Modulated by Differential Expression of Xanthine Oxidoreductase. Journal of Cellular Biochemistry. 2008;105:1008–1026. doi: 10.1002/jcb.21901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeman JJ, van de Geer SA, de Kort F, van Houwelingen HC. A global test for groups of genes: testing association with a clinical outcome. Bioinformatics. 2004;20:93–99. doi: 10.1093/bioinformatics/btg382. [DOI] [PubMed] [Google Scholar]
- Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic acids research. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung T, McBride M. Food for Thought ... on mapping the human toxome. ALTEX. 2011;28:83–93. doi: 10.14573/altex.2011.2.083. [DOI] [PubMed] [Google Scholar]
- ICCVAM. Interagency Coordinating Committee on the Validation of Alternative Methods Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays. 2003 NIH Publication No. 03-4503. [Google Scholar]
- Kamburov A, Cavill R, Ebbels TM, Herwig R, Keun HC. Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics. 2011;27:2917–2918. doi: 10.1093/bioinformatics/btr499. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katajamaa M, Oresic M. Data processing for mass spectrometry-based metabolomics. J Chromatogr A. 2007;1158:318–328. doi: 10.1016/j.chroma.2007.04.021. [DOI] [PubMed] [Google Scholar]
- Kessner D, Chambers M, Burke R, Agus D, Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24:2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King K, Rubin G. A history of diabetes: from antiquity to discovering insulin. British journal of nursing. 2003:12. doi: 10.12968/bjon.2003.12.18.11775. [DOI] [PubMed] [Google Scholar]
- Kleensang A, Maertens A, Rosenberg M, Fitzpatrick S, Lamb J, Auerbach S, Brennan R, Crofton KM, Gordon B, Fornace AJ, Jr, Gaido K, Gerhold D, Haw R, Henney A, Ma’ayan A, McBride M, Monti S, Ochs MF, Pandey A, Sharan R, Stierum R, Tugendreich S, Willett C, Wittwehr C, Xia J, Patton GW, Arvidson K, Bouhifd M, Hogberg HT, Luechtefeld T, Smirnova L, Zhao L, Adeleye Y, Kanehisa M, Carmichael P, Andersen ME, Hartung T. t4 workshop report: Pathways of Toxicity. ALTEX. 2014;31:53–61. doi: 10.14573/altex.1309261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleensang A, Vantangoli M, Andersen ME, Boekelheide K, Bouhifd M, Fornace AJ, Jr, Maertens A, Rosenberg M, Yager JD, Hartung T. Irreprodu-cell-bility: why genotyping cells is necessary, but not necessarily sufficient. 2015 in preparation. [Google Scholar]
- Kuhn M, von Mering C, Campillos M, Jensen LJ, Bork P. STITCH: interaction networks of chemicals and proteins. Nucleic Acids Res. 2008;36:D684–688. doi: 10.1093/nar/gkm795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertens A, Luechtefeld T, Kleensang A, Hartung T. MPTP’s pathway of toxicity indicates central role of transcription factor SP1. Arch Toxicol. 2015;89:743–755. doi: 10.1007/s00204-015-1509-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu W, Knight E, Xia Q, McGarvey BD. Comparative evaluation of eight software programs for alignment of gas chromatography-mass spectrometry chromatograms in metabolomics experiments. J Chromatogr A. 2014;1374:199–206. doi: 10.1016/j.chroma.2014.11.005. [DOI] [PubMed] [Google Scholar]
- Pluskal T, Castillo S, Villar-Briones A, Oresic M. MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics. 2010;11:395. doi: 10.1186/1471-2105-11-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu L, Yona G. Automation of gene assignments to metabolic pathways using high-throughput expression data. BMC Bioinformatics. 2005:6. doi: 10.1186/1471-2105-6-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LB, Martinez-Garcia E, Nguyen H, Mullen AR, Dufour E, Sudarshan S, Licht JD, Deberardinis RJ, Chandel NS. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell. 2013;51:236–248. doi: 10.1016/j.molcel.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taibi G, Di Gaudio F, Nicotra CM. Xanthine dehydrogenase processes retinol to retinoic acid in human mammary epithelial cells. J Enzyme Inhib Med Chem. 2008;23:317–327. doi: 10.1080/14756360701584539. [DOI] [PubMed] [Google Scholar]
- Tang X, Lin CC, Spasojevic I, Iversen ES, Chi JT, Marks JR. A joint analysis of metabolomics and genetics of breast cancer. Breast Cancer Res. 2014;16:415. doi: 10.1186/s13058-014-0415-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Devereux W, Stewart TM, Casero RA., Jr Cloning and characterization of human polyamine-modulated factor-1, a transcriptional cofactor that regulates the transcription of the spermidine/spermine N(1)-acetyltransferase gene. J Biol Chem. 1999;274:22095–22101. doi: 10.1074/jbc.274.31.22095. [DOI] [PubMed] [Google Scholar]
- Wishart DS. Advances in metabolite identification. Bioanalysis. 2011;3:1769–1782. doi: 10.4155/bio.11.155. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Tzur D, Knox C, Eisner R, Guo AC, Young N, Cheng D, Jewell K, Arndt D, Sawhney S, Fung C, Nikolai L, Lewis M, Coutouly MA, Forsythe I, Tang P, Shrivastava S, Jeroncic K, Stothard P, Amegbey G, Block D, Hau DD, Wagner J, Miniaci J, Clements M, Gebremedhin M, Guo N, Zhang Y, Duggan GE, Macinnis GD, Weljie AM, Dowlatabadi R, Bamforth F, Clive D, Greiner R, Li L, Marrie T, Sykes BD, Vogel HJ, Querengesser L. HMDB: the Human Metabolome Database. Nucleic Acids Res. 2007;35:D521–526. doi: 10.1093/nar/gkl923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Psychogios N, Young N, Wishart DS. MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009;37:W652–660. doi: 10.1093/nar/gkp356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Sinelnikov IV, Han B, Wishart DS. MetaboAnalyst 3.0--making metabolomics more meaningful. Nucleic Acids Res. 2015;43:W251–257. doi: 10.1093/nar/gkv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Wishart DS. MSEA: a web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 2010;38:W71–77. doi: 10.1093/nar/gkq329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman J, Krijt J, Stratilova L, Hansikova H, Wenchich L, Kmoch S, Chrastina P, Houstek J. Abnormalities in succinylpurines in fumarase deficiency: Possible role in pathogenesis of CNS impairment. Journal of Inherited Metabolic Disease. 2000;23:371–374. doi: 10.1023/a:1005639516342. [DOI] [PubMed] [Google Scholar]
- Zikanova M, Krijt J, Hartmannova H, Kmoch S. Preparation of 5-amino-4-imidazole-N-succinocarboxamide ribotide, 5-amino-4-imidazole-N-succinocarboxamide riboside and succinyladenosine, compounds usable in diagnosis and research of adenylosuccinate lyase deficiency. Journal of Inherited Metabolic Disease. 2005;28:493–499. doi: 10.1007/s10545-005-0493-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1: Metabolite putative identification workflow parameters
S2: Performance of the metabolite putative identification method
S3: Quantitative Enrichment Analysis
S4 Network first degree neighbors glutathione
S5 List of all metabolic features generated using the untargeted method
S6 List of all metabolic features generated using the targeted method
S7 Metabolite confirmation results