

Abstract
Summary: Eukaryotic genomes are hierarchically organized into topologically associating domains (TADs). The computational identification of these domains and their associated properties critically depends on the choice of suitable parameters of TAD-calling algorithms. To reduce the element of trial-and-error in parameter selection, we have developed TADtool: an interactive plot to find robust TAD-calling parameters with immediate visual feedback. TADtool allows the direct export of TADs called with a chosen set of parameters for two of the most common TAD calling algorithms: directionality and insulation index. It can be used as an intuitive, standalone application or as a Python package for maximum flexibility.
Availability and implementation: TADtool is available as a Python package from GitHub (https://github.com/vaquerizaslab/tadtool) or can be installed directly via PyPI, the Python package index (tadtool).
Contact: kai.kruse@mpi-muenster.mpg.de, jmv@mpi-muenster.mpg.de
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Chromosome conformation capture (3C) studies (Dekker et al., 2002), specifically their high-throughput derivative Hi-C (Lieberman-Aiden et al., 2009), have revealed the hierarchical folding of eukaryotic genomes. Chromatin has been found to be typically organized into genomic regions with a strong enrichment of intra-chromosomal contacts (Dixon et al., 2012; Nora et al., 2012; Sexton et al., 2012; Hou et al., 2012). These regions, termed self- or topologically-associating domains (TADs), can range in size from hundreds to thousands of kilobases with smaller domains often being nested within larger ones (Fraser et al., 2015).
A number of algorithms to identify TADs computationally have been developed. Among the most popular and intuitive are the directionality index (Dixon et al., 2012) and insulation index (Crane et al., 2015). The former calculates upstream and downstream contact biases for each locus along the genome – TAD boundaries are then generally demarcated by strongly directionally biased loci. The latter uses a rectangular sliding window approach to sum up contacts within a given region surrounding each locus - as TADs are regions of increased contacts, they can easily be identified via contact count cutoffs.
The properties of identified domains critically depend on the parameter choice of the applied TAD-calling algorithms. For the directionality index, the window size around a locus for which biases are computed primarily determines the size of the identifiable TADs. The cutoff used for biases strongly influences the sensitivity and specificity of TAD identification. Changing the window size and contact cutoff in the insulation index algorithm has similar effects. Finding suitable, robust parameters for the data set of interest is often a multistep, iterative process of testing and recalculation. To reduce the element of trial-and-error and to facilitate the meaningful identification of self-associating domains, we have developed TADtool: an interactive plot that assists in choosing relevant TAD-calling parameters with immediate visual feedback.
2 Usage
TADtool is launched from the command line and requires two input files: a square normalized Hi-C matrix and a list of genomic regions that correspond to matrix bins. The user selects a genomic region of interest and can choose between the insulation and directionality index. After automatically calculating the values for the chosen TAD-calling algorithm with a wide range of window sizes, an interactive figure will be opened that shows: a Hi-C plot (Fig. 1A) , TADs as called with the current set of parameters (Fig. 1B), a line plot showing the respective index values for the current window size (Fig. 1C), and a heatmap summarizing index values for all calculated window sizes (Fig. 1D).
Fig. 1.
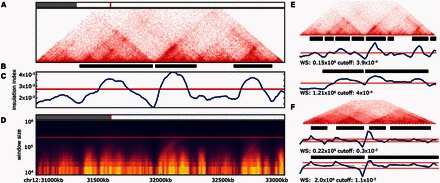
(A–D) TADtool window. (A) Hi-C plot, (B) TADs (black bars) called with current window size (WS: 2.5 × 105) and cutoff (2.7 × 10-3), (C) insulation index plot for current window size, (D) heatmap of insulation index for all window sizes. (E) Modifying TAD-calling parameters changes the identified TADs. (F) TADs called from Dixon et al. (2012) data with window size used in the original publication (bottom) and modified (top)
The figure supports synchronized zooming and panning, and the colour intensity of both heatmaps can be adjusted using sliders. By clicking the line plot and heatmap the user can interactively select the TAD-calling cutoff and window size, respectively – TAD regions will be updated automatically. Thus, with only a few clicks, a meaningful set of parameters for the chosen TAD-calling algorithm can be selected. TADs as called with the current parameters and other plot data can be directly saved to file using the provided buttons.
3 Discussion and conclusion
The usefulness of TADtool can easily be demonstrated on real-world data sets. Fig. 1A shows a 2Mb region on mouse chromosome 12 with TADs of varying sizes, some of them nested (lymphoblastoid Hi-C data at 10kb resolution from Rao et al., 2014). TADs naturally vary in size and intensity. By modifying parameters in TADtool (Fig. 1E), especially the window size, it is possible to deliberately call smaller (top) or larger (bottom) TADs.
Fig. 1F shows the same region in a lower-resolution embryonic stem cell Hi-C map (40 kb, Dixon et al., 2012), alongside directionality index plots for two different sets of parameters. In the original publication a window size of 2Mb was used (Fig. 1F, bottom). Similarly to the insulation index, by adjusting the window size of the directionality index we show that it is very easy to alternatively identify smaller, nested TADs (Fig. 1F, top).
Fraser et al. (2015) have used a similar approach to derive hierarchical TAD trees. With TADtool, the process of finding suitable window sizes and cutoffs matched to the nested TAD sizes is greatly simplified.
The chosen examples also illustrate that TAD calls can be very sensitive to parameter changes in certain ranges. TADtool is designed to find parameters that match the visual perception of how TADs should be called and, importantly, that produce stable TADs across a range of values. While this process is greatly simplified with this tool, it remains the user’s responsibility to verify TAD calls in their data using a range of parameter combinations and other, independent statistical means.
In summary, this tool speeds up and improves the process of finding meaningful parameters for two of the most popular TAD-calling algorithms. As such, TADtool is of great practical value for the genome organization community.
Supplementary Material
Acknowledgements
The authors thank all members of the Vaquerizas lab for useful input and feedback during the implementation of this project.
Funding
This work was supported by EpiGeneSys NoE, ZENCODE-ITN, Deutsche Forschungsgemeinschaft Cells-in-Motion Cluster of Excellence (EXC 1003–CiM), University of Münster and the Max Planck Society. C.B.H. was supported by a fellowship from the graduate school International Max Planck Research School–Molecular Biomedicine, Münster, Germany. B.H.-R. received support from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 643062 – ZENCODE-ITN.
Conflict of Interest: none declared.
References
- Crane E. et al. (2015) Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature, 523, 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J. et al. (2002) Capturing chromosome conformation. Science, 295, 1306–1311. [DOI] [PubMed] [Google Scholar]
- Dixon J.R. et al. (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature, 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser J. et al. (2015) Hierarchical folding and reorganization of chromosomes are linked to transcriptional changes in cellular differentiation. Mol. Syst. Biol., 11, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou C. et al. (2012) Gene density, transcription, and insulators contribute to the partition of the drosophila genome into physical domains. Mol. Cell, 48, 471–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E. et al. (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science, 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora E.P. et al. (2012) Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature, 485, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S.S.P. et al. (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton T. et al. (2012) Three-dimensional folding and functional organization principles of the Drosophila genome. Cell, 148, 458–472. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.