

Abstract
Motivation: Recovering the gene structures is one of the important goals of genome assembly. In low-quality assemblies, and even some high-quality assemblies, certain gene regions are still incomplete; thus, novel scaffolding approaches are required to complete gene regions.
Results: We developed an efficient and fast genome scaffolding method called PEP_scaffolder, using proteins to scaffold genomes. The pipeline aims to recover protein-coding gene structures. We tested the method on human contigs; using human UniProt proteins as guides, the improvement on N50 size was 17% increase with an accuracy of ∼97%. PEP_scaffolder improved the proportion of fully covered proteins among all proteins, which was close to the proportion in the finished genome. The method provided a high accuracy of 91% using orthologs of distant species. Tested on simulated fly contigs, PEP_scaffolder outperformed other scaffolders, with the shortest running time and the highest accuracy.
Availability and Implementation: The software is freely available at http://www.fishbrowser.org/software/PEP_scaffolder/
Contact: lijt@cafs.ac.cn
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Identifying genes is a major goal of genome sequencing projects for downstream functional study and evolutionary analysis. Although large-insert libraries or long single-molecule reads improve the genome N50 size, it remains difficult to complete all gene structures. Thus, new scaffolding approaches are needed to recover the gene regions.
Many methods have been developed to increase the continuity of gene regions using transcripts or proteins as guides. Mortazavi et al. (2010) utilized paired RNA-seq data to scaffold Caenorhabditis nematode contigs. Nevertheless, the accuracy of this strategy and the improvement in the proportion of complete genes were not estimated. Previously, we developed L_RNA_scaffolder using long single-end RNA-seq reads for genome scaffolding, which was highly accurate (93.6%) (Xue et al., 2013). However, these strategies require RNA-Seq data, and genes that are not detected by RNA-seq will not be re-built. Different from using transcripts, ESPRIT (Dessimoz et al., 2011) and SWiPS (Li and Copley, 2013) used proteins to link contigs. Based on the predicted genes using AUGUSTUS (Stanke et al., 2006), ESPRIT identified split protein-coding regions and linked unassembled genomic segments. The accuracy of ESPRIT depends on that of predicted genes. The time-consuming nature of de novo gene prediction also limits its application. SWiPS determined coding contigs and exonic regions using tblastn and GeneWise (Birney et al., 2004), and then scaffolded contigs by optimization of the overall protein to contig mappings. SWiPS integrated multiple steps to refine the precise protein-contig mapping; consequently, it has long running time.
Herein, we present a novel and fast method to scaffold contigs using (homologous) proteins. The PEP_scaffolder has high scaffolding accuracy and is much faster than previous scaffolders. The improved proportion of fully covered genes is close to that of the finished genome.
2 Methods
The main steps of PEP_scaffolder are summarized as follows (Supplementary Figure S1 and Supplementary Methods 1). Initially, proteins are aligned to contigs using BLAT (Kent, 2002) and then the alignments are subjected to PEP_scaffolder. ‘Guide’ proteins are selected that have high-quality alignments above a certain minimal percent identity (MPI; Supplementary Methods 1) and are not fully covered under the minimal length coverage (MLC; Supplementary Methods 1). The longest alignment region in one block is then selected and all blocks are ordered following the alignment positions in the protein. The contigs corresponding to blocks are sorted and oriented following the block orders. If the interval between two blocks is shorter than the maximal intron length (MIL; Supplementary Methods 1), the connection between two contigs is retained. The optimal connection for each contig is selected and scaffolding paths are built by walking all optimal connections.
To assess performance and accuracy, we scaffolded 36 437 human contigs (N50 size of 148 715 bp) with different sources of human proteins. The accuracy of PEP_scaffolder was measured following the Genome Assembly Gold Standard Evaluations pipeline (Salzberg, et al., 2012) (Supplementary Methods 2). The N50 size and corrected N50 size were used as metrics to determine the scaffolding performance (Supplementary Methods 3). The genome coverage was measured to investigate its effect on scaffolding performance (Supplementary Methods 3). To evaluate the proportions of fully covered genes, we aligned human Swiss-Prot proteins to three assemblies including the contigs, the PEP_scaffolder assembly and the hg38 assembly (Speir et al., 2016), respectively. For each assembly, the proportion of fully covered proteins among all proteins was calculated (Supplementary Methods 4). To assess the accuracy of the method using non-human homologs, rodent and mammal proteins were used as guides to scaffold human contigs. Human, rodent and mammal proteins were downloaded from the UniProt database (Consortium, 2015). The contigs and hg38 assembly were obtained from NCBI GenBank (Benson et al., 2013).
We compared our method with SWiPS and ESPRIT. The Drosophila melanogaster genome from the Ensembl database (Cunningham et al., 2015) was fragmented into contigs of the same length (10 kb). Fly Ensembl proteins were used to scaffold the fly contigs (Supplementary Methods 5).
3 Results
The performance of PEP_scaffolder was measured using the N50 size. Using human Swiss-Prot proteins as test guides, the N50 sizes were saturated when the MPI was >0.9, the MLC >0.9 and the MIL >150 kb (Supplementary Figures S2, S3 and S4). Using these optimal parameters, the final N50 sizes were 171,032 bp (a 15% increase) and 168,047 bp (a 13% increase) for human Swiss-Prot proteins and TrEMBL proteins, respectively (Supplementary Tables S1 and S2). Using human Swiss-Prot and TrEMBL proteins as guides, the contig number was reduced from 36,437 to 30,550. The improvement on N50 size was 16.8% increase at an accuracy of 96.7%, which demonstrated the good performance of PEP_scaffolder. In a revised assembly where the scaffolds were split at error points, the corrected N50 size had an improvement of 16.1% increase.
The proportion of fully covered proteins in the PEP_scaffolder assembly (96.8%) was higher than the proportion in the contigs (82.8%) and nearly equal to that for hg38 (99.8%). In particular, the proportion of fully covered proteins over 500 amino acids showed a larger improvement (from 72.3 to 95.5%, a 32% increase) than did shorter proteins, indicating that PEP_scaffolder greatly improved the proportion of complete genes (Fig. 1a).
Fig. 1.
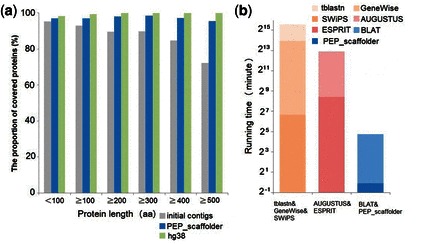
Evaluation of PEP_scaffolder performance. (a) Completeness of Swiss-Prot proteins in three human assemblies. Swiss-Prot proteins were aligned to three assemblies using BLAT, with a length coverage cutoff of 90%. (b) Running time for scaffolding the fly genome
PEP_scaffolder was able to scaffold human contigs using orthologs of distant species. Using rodent Swiss-Prot proteins, mammal Swiss-Prot proteins, rodent TrEMBL proteins and mammal TrEMBL proteins, we obtained improvements on N50 size of 6.84, 3.61, 10.84 and 20.84%, respectively (Supplementary Tables S1 and S2). The accuracy was as high as 90.82%, indicating that PEP_scaffolder could utilize orthologs to scaffold a target genome with high accuracy. More genome regions covered by proteins would generate longer scaffolds. To examine the improvement on N50 size with increasing numbers of proteins, we constructed multiple sets of proteins by combining human proteins, mammal proteins and rodent proteins together. The N50 size and corrected N50 size were improved to 182 433 and 176 257 bp (a 22.7% increase and 18.5% increase), respectively, using all proteins as guides (Supplementary Table S3), indicating that an enlarged proteome could increase the proportion of recovered genes.
We scaffolded fly contigs using SWiPS, ESPRIT and PEP_scaffolder, respectively. PEP_scaffolder produced the most connections (4191) with the highest accuracy (99.6%) and the shortest running time (27 minutes) (Fig. 1b and Supplementary Table S4), suggesting that PEP_scaffolder is superior to the other scaffolders.
4 Discussion
We demonstrated that PEP_scaffolder is an efficient and fast scaffolder that improves the proportion of complete genes. The performance of PEP_scaffolder could be improved in several ways. Protein variations between species might influence the accuracy of PEP_scaffolder. We observed higher accuracy using proteins from the target species compared with proteins from close species (Supplementary Tables S1 and S2). The annotations of Swiss-Prot proteins are created by manual analysis, whereas TrEMBL proteins are predicted automatically without manual annotation. Therefore, Swiss-Prot proteins are more credible than TrEMBL proteins. Our results showed that, using Swiss-Prot proteins, the accuracy of PEP_scaffolder was higher than using TrEMBL proteins (Supplementary Tables S1 and S2). To overcome the above limitations, we recommend that more supporting proteins are used to construct more accurate scaffolds (Supplementary Methods 6 and Supplementary Data). As shown in Supplementary Figure S6, scaffolding performance is significantly correlated with genome coverage. Therefore, increasing the number of (homologous) proteins would improve the performance. PEP_scaffolder could be useful for the genome analysis of non-model species, which lack high-quality genome assemblies.
Supplementary Material
Acknowledgement
We thank Drs. Yang I. Li and Richard R. Copley for providing the SWiPS software.
Funding
This study was supported by National Natural Science Foundation of China (31402353).
Conflict of Interest: none declared.
References
- Benson D.A. et al. (2013) GenBank. Nucleic Acids Res., 41, D36–D42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E. et al. (2004) GeneWise and Genomewise. Genome Res., 14, 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium T.U. (2015) UniProt: a hub for protein information. Nucleic Acids Res., 43, D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham F. et al. (2015) Ensembl 2015. Nucleic Acids Res., 43, D662–D669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessimoz C. et al. (2011) Comparative genomics approach to detecting split-coding regions in a low-coverage genome: lessons from the chimaera Callorhinchus milii (Holocephali, Chondrichthyes). Brief. Bioinformatics, 12, 474–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W.J. (2002) BLAT—The BLAST-Like Alignment Tool. Genome Res., 12, 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.I., Copley R.R. (2013) Scaffolding low quality genomes using orthologous protein sequences. Bioinformatics, 29, 160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A. et al. (2010) Scaffolding a Caenorhabditis nematode genome with RNA-seq. Genome Res., 20, 1740–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg S.L. et al. (2012) GAGE: A critical evaluation of genome assemblies and assembly algorithms. Genome Res., 22, 557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speir M.L. et al. (2016) The UCSC Genome Browser database: 2016 update. Nucleic Acids Res., 44, D717–D725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M. et al. (2006) AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res., 34, W435–W439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W. et al. (2013) L_RNA_scaffolder: scaffolding genomes with transcripts. BMC Genomics, 14, 604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.