

Abstract
Mechanistic/mammalian target of rapamycin (mTOR) has emerged as a new potential therapeutic target for gastric cancer. Rapamycin and rapamycin analogs are undergoing clinical trials and have produced clinical responses in a subgroup of cancer patients. However, monotherapy with rapamycin at safe dosage fails to induce cell apoptosis and tumor regression which has hampered its clinical application. This has led to the exploration of more effective combinatorial regimens to enhance the effectiveness of rapamycin. In our present study, we have investigated the combination of rapamycin and a reactive oxygen species (ROS) inducer EF24 in gastric cancer. We show that rapamycin increases intracellular ROS levels and displays selective synergistic antitumor activity with EF24 in gastric cancer cells. This activity was mediated through the activation of c-Jun N terminal kinase and endoplasmic reticulum stress (ER) pathways in cancer cells. We also show that inhibiting ROS accumulation reverses ER stress and prevents apoptosis induced by the combination of rapamycin and EF24. These mechanisms were confirmed using human gastric cancer xenografts in immunodeficient mice. Taken together, our work provides a novel therapeutic strategy for the treatment of gastric cancer. The work reveals that ROS generation could be an important target for the development of new combination therapies for cancer treatment.
Abbreviations: 4E-BP-1, eukaryotic initiation factor 4E binding protein-1; ATF4, activating transcription factor 4; Bcl2, B-cell lymphoma 2; C/EBP, CAAT/enhancer binding protein; cdc-2, cyclin-dependent kinase 1 (cell division cycle protein 2); CHOP, CAAT/enhancer binding protein (C/EBP) homologous protein;; DCF, dichlorodihydrofluorescein; DCFH-DA, 2′, 7′-dichlorodihydrofluorescein diacetate; eIF2, eukaryotic initiation factor 2; ER, endoplasmic reticulum; FITC, fluorescein isothiocyanate; JC-1, cationic carbocyanine dye; HRP, horseradish peroxidase; JNK, c-Jun N terminal kinase; Ki-67, nuclear protein associated with cell proliferation; MDA, malondialdehyde; MDM-2, murine double minute 2; mTOR, mammalian target of rapamycin; mTORC, mTOR complex; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; NAC, N-acetyl cysteine; S6, S6 ribosomal protein; PARP, poly (ADP-ribose) polymerase; PERK, protein kinase RNA-like endoplasmic reticulum kinase; PI, propidium Iodide; PI3 kinase, phosphoinositide 3-kinase; ROS, reactive oxygen species
Keywords: Gastric cancer, Rapamycin, EF24, Oxidative stress, Cytotoxicity, Antitumor
Graphical abstract
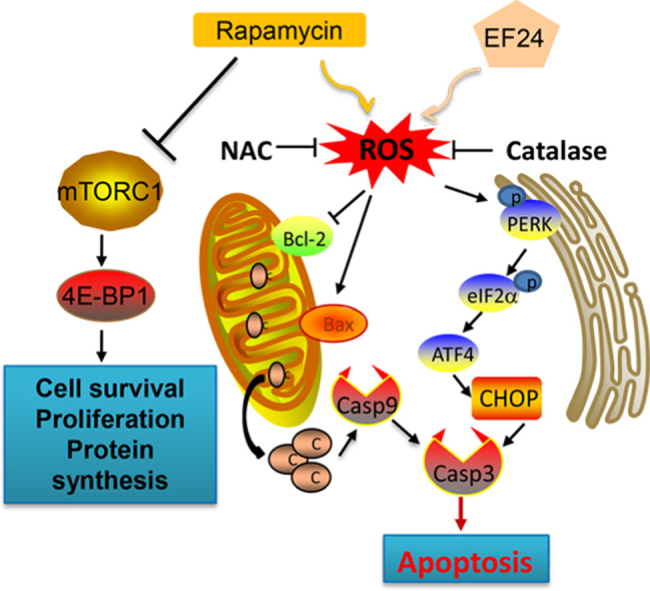
Highlights
-
•
Rapamycin displays synergistic antitumor activity with EF24 in gastric cancer cells.
-
•
EF24enhances rapamycin-induced ROS production.
-
•
Combination of EF24 and rapamycin activates ER stress and mitochondrial dysfunction.
-
•
EF24 amplifies the therapeutic effect of rapamycin in vivo.
1. Introduction
Gastric cancer is the second leading cause of cancer-related deaths worldwide [1]. Surgery is the only curative treatment for early-stage gastric cancer. However, most patients are asymptomatic in the early stages and more than half of the cases are diagnosed clinically with distant metastasis. Such metastatic cases are largely incurable with a 5-year survival rate of less than 10% [2], [3]. Adjuvantand targeted chemotherapy show remarkable benefits in reducing disease recurrence and increasing long-term survival [4]. However, severe side effects and complications including hematological and gastrointestinal toxicities of the current anticancer drugspose a major clinical challenge [5], [6]. Therefore, new drugs and/or new therapeutic combinations are needed for the treatment of patients with gastric cancer.
Rapamycin, a non-ATP-competitive inhibitor of mTOR complex 1, is a bacterial macrolide with antifungal and immunosuppressant activities [7], [8]. Rapamycin and its analogs have demonstrated efficacy in cancer treatment by inhibiting the mTOR pathway and inactivating the vital downstream kinases, p70S6 kinase and eukaryotic initiation factor 4Ebinding protein-1(4E-BP-1) [9]. Dephosphorylation of S6K1 and 4E-BP1 inhibits the expression of genes involved in cell cycle regulation and cell proliferation. Rapamycin and its analogs have been shown to exhibit anti-tumor activities in gastric cancer both in vitro and in animal models [10]. However, results of recent studies indicate that mTOR inhibitors lead to disease stabilization rather than regression for most cancer types [11]. It has, therefore, been suggested that mTOR-targeted therapy and other chemotherapy may be used in combination therapy to induce a cytotoxic response rather than a cytostatic response in cancer treatment.
We have recently shown that reactive oxygen species (ROS) production in cancer cells is one of the mechanisms underlying synergetic cytotoxicity seen with combination anti-tumor treatments [12], [13], [14]. ROS are a normal by product of numerous cellular processes, such as mitochondrial metabolism and protein folding [15]. Compared to normal cells, cancer cells have intrinsically higher levels of ROS and are under oxidative stress due to an imbalanced redox status [15], [16]. As a result, cancer cells are believed to be unable to cope with additional oxidative stress and become sensitive to agents that increase ROS levels [17]. Targeting ROS is an important therapeutic strategy for cancer as exemplified by cancer drugs such as trisenox [18], paclitaxel [19], and2-methoxyestradiol [20]. Our previous studies have found that auranofin and piperlongumine induce ROS-dependent synergistic cytotoxicity in gastric cancer cells [12]. Hence, novel ROS-based combination therapeutic strategies have been suggested for further improving the outcome of gastric cancer patients.
In the present study, we have examined a ROS-based combination therapy for gastric cancer utilizing rapamycin and a novel curcumin analog EF24. EF24 was identified as selectively toxic to cancer cells in ROS-dependent manner [13]. Our present study shows that EF24 sensitizes gastric cancer cells to rapamycin in vitro by triggering ROS-mediated c-jun N terminal kinase activation and inducing endoplasmic reticulum stress apoptotic pathways. Furthermore, we showed that rapamycin in combination with EF24 leads to a significant reduction in tumor growth in vivo. Together, these results suggest that rapamycin in combination with ROS inducers may provide an effective alternative for gastric cancer therapy.
2. Materials and methods
2.1. Cell culture and reagents
Rapamycin was purchased from Selleck Chemicals (Houston, TX). Curcumin analog EF24, N-acetylcysteine (NAC) and catalase were purchased from Sigma-Aldrich (St. Louis, MO). Human gastric cancer cell lines SGC-7901 and BGC-823, and normal human gastric epithelial cells GES-1 were purchased from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). All cell types were cultured in RPMI 1640 medium (Gibco, Eggenstein, Germany) supplemented with 10% heat-inactivated fetal bovine serum (Gibco), 100 units/mL penicillin, and 100 μg/mL streptomycin. Antibodies against Bcl-2, Bax, cleaved poly ADP ribose polymerase (PARP), murine double minute 2 (MDM2), CDC2, Cyclin B1, and GAPDH were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against p-mTOR(Ser2448), p-4EBP1(Thr37/46), CHOP (CCAAT/enhancer-binding protein homologous protein), ATF4 (activating transcription factor 4), p-JNK (Thr183/Tyr185), JNK, LC3A/B, Beclin-1, p-PERK (Thr981), Cleaved Caspase-3, Caspase-3, p-S6 (Ser240/244), and p-eIF2α and eIF2α (eukaryotic initiating factor 2) were purchased from Cell Signaling Technology (Danvers, MA). Secondary horseradish peroxidase (HRP)-conjugated antibodies were obtained from Santa Cruz Biotechnology. Fluorescein isothiocyanate(FITC)-AnnexinV apoptosis Detection Kit I and propidium iodide (PI) were purchased from BD Pharmingen (Franklin Lakes, NJ).
2.2. Cell viability assay
Cells were seeded in 96-well plates at the density of 8×103 per well and allowed to attach overnight in RPMI media containing serum. Rapamycin was dissolved in DMSO and diluted in 1640 medium to final concentrations of 0.3125, 0.625, 1.25, 2.5, 5, 10, 20, 40 and 80 μM. EF24 was dissolved in DMSO and diluted in 1640 medium to final concentrations of 0.1562,0.3125, 0.625, 1.25, 2.5, 5, 10 and 20 μM. Cells were then treated with increasing concentrations of rapamycin or EF24 as single treatments or in combination for 24 h before measuring viability by MTT assay.
2.3. Cell apoptosis analysis
Cells were treated with either rapamycin.EF24, or a combination of rapamycin and EF24 for 24 h. NAC or catalase pretreatment, where indicated, was carried out for 2 h. Cells were then harvested, labeled with FITC-conjugated Annexin V and PI, and analyzed by flow cytometry. Caspase9 activity was also determined in cell lysates using the Caspase 9 activity assay kit (Beyotime Institute of Biotechnology, Beijing, China). Activity levels were normalized by the protein concentration of the corresponding cell lysate and expressed as percentage of treated cells to that of control.
2.4. Measurement of reactive oxygen species generation
Cellular ROS levels were measured by flow cytometry as described previously [13], [14]. Briefly, 5×105 cells were plated on 60 mm dishes and allowed to attach overnight. Cells were exposed to either rapamycin, EF24, or a combination for the indicated time periods. Cells were then stained with 10 μM dichlorodihydrofluorescein diacetate (DCFH-DA;Beyotime Biotech, Nantong, China). DCFH-DA is oxidized to dichlorodihydrofluorescein (DCF) in the presence of ROS. DCF fluorescence was analyzed using FACSCalibur flow cytometry.
2.5. Western blotting analysis
Cell lysates were prepared and protein levels were determined by the Bradford Assay (Bio-Rad, Hercules, CA). Proteins were separated by 10% SDS-PAGE and transferred to poly-vinylidene difluoride transfer membranes. The blots were blocked for 2 h at room temperature with freshly prepared 5% nonfat milk in TBST and then incubated with specific primary antibodies over night at 4 °C. HRP-conjugated secondary antibodies and ECL substrate (Bio-Rad, Hercules, CA) were used for detection. The density of the immunoreactive bands was analyzed using Image J software (National Institute of Health, MD).
2.6. Electron microscopy
SGC-7901 cells were treated with vehicle control (DMSO, 3 μL) or a combination of rapamycin and EF24. NAC and catalase pretreatments were carried out for 2 h. Following treatment, cells were fixed with 2.5% glutaraldehyde overnight at 4 °C.The cells were then post-fixed in 1% OsO4 at room temperature for 60 min, stained with 1%uranyl acetate, dehydrated through graded acetone solutions, and embedded in epon. Areas containing cells were block-mounted and cut into 70 nm sections and examined with an electron microscope (H-7500, Hitachi, Ibaraki, Japan).
2.7. Evaluation of mitochondrial membrane potential (Δψm)
The synergistic effect of rapamycin and EF24 on mitochondrial membrane potential (Δψm) were examined by fluorescence microscope using JC-1 (Thermo Fisher). JC-1is a cationic carbocyanine dye that accumulates in the mitochondria. Upon changes to membrane potential, JC-1 leaks out into the cytosol. Cells were exposed to rapamycin and EF24 for 14 h. NAC and catalase pretreatments were carried out for 2 h. Fluorescence images were acquired by using Nikon epifluorescence microscope equipped with a digital camera (Nikon, Japan).
2.8. In vivo xenograft model
All animal experiments complied with the Wenzhou Medical University's Policy on the Care and Use of Laboratory Animals. Protocols for animal studies were approved by the Wenzhou Medical College Animal Policy and Welfare Committee (Approved documents:2012/APWC/0216). Protocols for animal studies also follow the National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978). Five-week-old athymic BALB/c nu/nu female mice (18–22 g) were obtained from Vital River Laboratories (Beijing, China). Animals were housed at a constant room temperature with a 12 h:12 h light/dark cycle and feda standard rodent diet. SGC-7901 cells were injected subcutaneously into the right flank (5×106 cells in 100 μL of PBS). Mice were treated with 5 mg/kgi.p. rapamycin once per day, 3 mg/kgi p. EF24 once per day, or with a combination of rapamycin and EF24 according to the same schedules. Treatment was initiated when tumors reached a volume of 40–50 mm3. The tumor volumes were determined by measuring length (l) and width (w) and calculating volume by using the formula: V=0.5×l×w2. At the end of study, animals were sacrificed, and the tumors were removed and weighed for use in proteins expression studies.
2.9. Malondialdehyde (MDA) assay
Tumors samples from mice were homogenized and sonicated. Tissue lysates were then centrifuged at 12,000×g for 10 min at 4 °C to collect the supernatant. Total protein content was determined by using the Bradford assay. Malondialdehyde (MDA) levels were measured by using Lipid Peroxidation MDA assay kit (Beyotime Institute of Biotechnology).
2.10. Statistical analysis
All experiments were assayed in triplicate (n=3). Data are expressed as means±SEM.The density of the immunoreactive bands was analyzed using Image Jcomputer software (NIH). All statistical analyses were performed using GraphPad Pro. Prism5.0 (GraphPad, San Diego, CA). Student's t-test and two-way ANOVA were employed to analyze the differences between sets of data. A p value <0.05 was considered statistically significant.
3. Results
3.1. High dose of rapamycin non-selectively decreases viability of human gastric cancer cells and normal cells
We first wanted to determine the effect of rapamycin on gastric cancer and normal gastric cell growth. We treated cells with different concentrations of rapamycin and performed viability test through the MTT assay. We show that rapamycin decreased viability of BGC-823, SGC-7901 and normal GES-1 cells with IC50 values of 26.1, 33.7 and 27.9 μM, respectively (Fig. 1A). These results show that rapamycin treatment non-selectively reduced viability of gastric cancer cells and normal cells at high concentrations. When used at lower concentrations (non-cytotoxic to normal GES1 cells), rapamycin showed no effect on gastric cancer cells. We next examined whether rapamycin reduced viability through inducing cell apoptosis. To do this, we stained cells with Annexin V/propidium iodide (PI) following rapamycin treatment. As shown in Fig. 1B and C, both of gastric cancer cell lines exhibited apoptosis after 24 h rapamycin treatment. This effect was dose-dependent with significant apoptosis evident at 20 µM rapamycin. To confirm these results, we determined the levels of apoptosis-related proteins in SGC-7901 and BGC-823cells. Our results show increased levels of Bax and cleaved poly ADP ribose polymerase (PARP), as well as decreased Bcl-2 in cells treated with rapamycin (Fig. 1D and Supplementary Fig. S1B).
Fig. 1.

Rapamycin causes cytotoxicity inhuman gastric cancer cells and normal cells. (A) Cytotoxic effect of rapamycin in human gastric cancer cells and normal GES-1 cells as assessed by cell viability. SGC-7901, BGC-823 and GES-1 cells were incubated with increasing doses of rapamycin (0.31–100 μM) for 24 h. Cell viability was determined by MTT assay. (B) Induction of apoptosis in human gastric cancer cells was determined by Annexin V/PI flow cytometry following treatment with rapamycin (10, 20 or 40 μM) for 24 h. (C) Quantification of apoptotic cells presented as percent total [*p<0.05, **p<0.01]. (D) Western blot analysis of apoptosis-related proteins in SGC-7901 and BGC-823 treated with rapamycin (10, 20 or 40 μM) for 20 h [Cle-PARP=cleaved PARP; GAPDH=loading control]. The densitometric quantification bar graphs are shown in Supplementary file. Representative data were shown from three independent experiments.
3.2. Curcumin analog EF24 increases the sensitivity of human gastric cancer cells to rapamycin by enhancing apoptosis
Cancer treatment is limited by our inability to maintain dose-intensification because of non-selective cytotoxicity. Therefore, current research efforts are focused on finding combination of therapies to yield enhanced cancer cell toxicity at low doses. Recent studies have shown that a novel curcumin analog EF24 sensitizes cancer cells to anti-cancer agents through multiple mechanisms [21], [22]. One such mechanism is production of ROS which has been shown in prostate and breast cancer cells [23]. Therefore, we tested whether EF24 enhances the effect of rapamycin on human gastric cancer cells. At a concentration of 0.5 μM, EF24 alone did not affect the viability of SGC-7901 and BGC-823 cells (Supplementary Fig. S1A). We then treated gastric cancer cells with increasing concentrations of rapamycin in combination with 0.5 µM EF24 and examined cell viability. Our results show that co-treatment with EF24 and rapamycin selectively enhances the cytotoxicity of rapamycin in gastric cancer cells and not normal cells (IC50=2.7 µM for SGC-7901, 8.3 μM for BGC-823, and 23.8 μM for GES-1; Fig. 2A and B). This remarkable finding prompted us to understand the mechanism underlying reduced viability following the combined treatment. We used low concentrations of rapamycin (2.5 and 5.0 μM) which still inhibit the mTOR signaling pathway (Fig. 2F and Supplementary Fig. S2B) but do not produce overt toxicity so as to mask the underlying mechanisms. In addition, western blot analysis showed that EF24 treatment alone had no effect on the mTOR signaling pathway (Supplementary Fig. S2C). We determined the pro-apoptotic effect of the combined treatment using Annexin V/PI staining and show significant enhancement in apoptosis in cells treated with EF24 and rapamycin (Fig. 2C and D). This was confirmed by cleavage of PARP and expression levels of Bcl-2 and Bax (Fig. 2E and Supplementary Fig. S2A). We next assessed cell cycle status as reduced cell viability seen in gastric cancer cells may be caused induction of apoptosis as well as cell cycle arrest. Indeed, we noted marked accumulation of gastric cancer cells in G2/M phase with a concomitant decrease in the number of cells in the S and G1 phase after treatment with rapamycin and EF24 (Supplementary Fig. S3A and B). Consistent with these results, protein levels of G2/M phase related factors murine double minute 2 (MDM-2), cyclin B1 and cell division cycle protein 2 (Cdc2) were reduced in gastric cancer cells treated with EF24 and rapamycin compared with rapamycin alone (Supplementary Fig. S3C). Furthermore, colony formation assays confirmed that EF24 enhanced the sensitivity of gastric cancer cells to rapamycin (Supplementary Fig. S3D). Collectively, our results indicate that EF24 sensitizes gastric cancer cells to rapamycin by enhancing apoptosis and reducing cell growth.
Fig. 2.

EF24 enhances the anti-tumor activity of rapamycin in gastric cancer cell lines. (A-B) SGC-7901,BGC-823 and GES-1 cells were pretreated with 0.5 μM EF24 and then exposed to increasing doses of rapamycin (0.31–100 μM) for 24 h. Cell viability was determined by MTT assay. (C) EF24 enhances rapamycin-induced apoptosis in SGC-7901 and BGC-823 cells as assessed by AnnexinV/PI staining. (D) Quantification of apoptotic cells [*p<0.05, **p<0.01]. (E) Western blot analysis of apoptosis-related proteins following EF24 and rapamycin combination treatment. (F) Western blot analysis of autophagy-associated proteins in SGC-7901 and BGC-823 treated with rapamycin. The densitometric quantification bar graphs are shown in Supplementary file. Representative data were shown from three independent experiments.
3.3. EF24enhances rapamycin-induced reactive oxygen species (ROS) production, apoptosis, and cell cycle arrest
As mentioned earlier, ROS production in cancer cells underlies synergetic cytotoxicity seen with select anti-tumor treatments [12], [24]. In addition, EF24 has been shown to increase ROS production in prostate and breast cancer cells [23]. Therefore, we explored whether ROS generation plays a role in the combined cytotoxic effects of rapamycin and EF24. Rapamycin alone increased ROS production in a time- and dose-dependent manner as assessed by the levels of dichlorodihydrofluorescein (DCF) (Fig. 3A, B, and S4A). Co-treatment of cells with rapamycin (2.5 and 5.0 μM) and 2 µM EF24 resulted in significant increases in ROS levels compared to rapamycin or EF24 treatments alone (Fig. 3C, D, and S4B). This increased DCF signals (ROS levels) were not seen when cells were pre-treated with ROS scavengers N-acetyl cysteine (NAC) or catalase as expected (Fig. 3C-D, and S4C-S4D). Since rapamycin is an inhibitor of mTOR complex 1, it is difficult to determine whether inhibition of mTOR passway could interfere with rapamycin-induced ROS production. We can, however, assess the mTOR pathway to get an insight. Rapamycin inhibited phosphorylation of mTOR and downstream protein 4EBP1 at 12 h after treatment (Supplementary Fig. S4E). Peak ROS levels are evident after only 1 h of rapamycin treatment (Fig. 3A) possibly indicating that inhibition of mTOR pathway is not associated with rapamycin-induced ROS accumulation. Furthermore, we found that inhibition of the mTOR pathway by rapamycin is not reversed by NAC pretreatment (Supplementary Fig. S4F). Scavenging ROS, however, did reverse apoptosis induced by the combined treatment in both SGC-7901 and BGC-823 cells (Fig. 4A and B). This was also evident through protein analysis for cleaved-PARP, Bcl-2 and Bax (Fig. 4C). In addition, blocking ROS prevented combined treatment-induced G2/M cell cycle arrest and down-regulation of cell cycle-related proteins MDM-2, cyclin B1 and Cdc2 in gastric cancer cells (Supplementary Fig. S5A–S5C). These results revealed a vital role of ROS in the combined effect of rapamycin and EF24. The results also suggest that the mechanism of ROS production may be independent of the mTOR signaling proteins, raising the question whether other targets contribute to rapamycin-induced oxidative stress.
Fig. 3.

EF24 enhances rapamycin-induced ROS generation. (A and B) Rapamycin-induced ROS generation was measured in SGC-7901 cells and BGC-823 cells by staining with DCFH-DA. (A) SGC-7901 and BGC-823 cells were treated with 10 μM rapamycin for different time periods. (B) Cells treated with different concentrations of rapamycin for 1 h. (C and D) EF24 enhances rapamycin-induced ROS generation in SGC-7901 (C) and BGC-823 (D) cells. Cells were treated with EF24, rapamycin, or a combination of both for 1 h. Intracellular ROS generation was inhibited with pretreatment of 5 mM NAC for 2 h. Right bar graphs show quantification of flow cytometry data [*p<0.05, **p<0.01]. Data were collected from three independent experiments.
Fig. 4.

Synergistic effect rapamycin and EF24 is dependent on ROS generation. (A) NAC or catalase pretreatment normalized the synergistic effect of rapamycin and EF24 in SGC-7901 and BGC-823 cells. Cells were pretreated with 5 mM NAC or 2000 U/mL catalase for 2 h before exposure to the combination treatment of rapamycin and EF24 for 24 h. Apoptotic cells were determined by AnnexinV/PI staining. (B) Quantification of Annexin V/PI flow cytometry data [*p<0.05, **p<0.01]. (C) Western blot analysis of apoptosis-related proteins in cells with NAC or catalase treatment prior to exposure to rapamycin and EF24. Data were collected from three independent experiments and representative images were shown.
3.4. Combination of EF24 and rapamycin activates endoplasmic reticulum stress and mitochondrial dysfunction
Increased ROS levels and perturbed intracellular redox status has been reported to increase the levels of unfolded proteins and induce endoplasmic reticulum (ER) stress response [25]. Therefore, we examined the expression of ER stress-related proteins, including CHOP (CCAAT/enhancer-binding protein homologous protein), ATF4 (activating transcription factor 4), eIF2α (eukaryotic initiating factor), and PERK (protein kinase RNA-like endoplasmic reticulum kinase). Here, we show that treatment of cells with rapamycin or EF24 alone slightly induced ER stress markers, whereas combined rapamycin and EF24 treatment dramatically activated ER-stress pathway as evident by induced levels of p-eIF2α, ATF4, p-PERK and CHOP (Fig. 5A and Supplementary Fig. S6A). In this system, both NAC and catalase pretreatment completely blocked the combined treatment-induced effects (Fig. 5B and Supplementary Fig. S6B). We next examined ER morphology in SGC-7901 cells exposed to the combined treatment through electron microscopy. Compared with control (DMSO-treated) SGC-7901 cells, the ER in SGC-7901 cells after 6 h of treatment with rapamycin and EF24 showed swelling (arrow, Fig. 5C). This morphological change was not observed with NAC pretreatment. These results suggest that treatment-induced ROS mediates ER stress response in gastric cancer cells.
Fig. 5.

EF24 enhances rapamycin-induced ER stress response by modulating ROS levels. (A) Western blot analysis of endoplasmic reticulum (ER) stress related proteins in cells following treatment with rapamycin and EF24 [p-eIF2α=phosphorylated eukaryotic initiation factor 2α; ATF4=activating transcription factor-4; p-PERK=phosphorylated protein kinase RNA-like endoplasmic reticulum kinase; and CHOP=CCAAT/enhancer-binding protein homologous protein]. (B) NAC and catalase reversed the combined treatment-induced ER stress response in cells as assessed by western blot analysis. (C) Effect of combined rapamycin and EF24 treatment on the morphology of endoplasmic reticulum in SGC-7901 cells. SGC-7901 cells were pre-treated 5 mM NAC for 2 h before exposure to 5 µM rapamycin in combination with 2 µM EF24 for 6 h [×10,000 or ×20,000]. The densitometric quantification bar graphs are shown in Supplementary file. Data were collected from three independent experiments and representative images were shown.
In addition to ER stress, mitochondrial dysfunction is central to the regulation of apoptosis. Loss of mitochondrial membrane potential (Δψm) is catastrophic for cells and leads to the release of cytochrome Cinto the cytosol [26]. We used fluorescence microscopy to confirm whether combined treatment-induced apoptosis was associated with disruption of mitochondrial homeostasis by using JC-1 dye. JC-1 is a cationic carbocyanine dye which accumulates in healthy mitochondria and leaks out when mitochondrial membrane integrity is compromised. Our results showed that the combined treatment of rapamycin and EF24 decreases the mitochondrial membranes potential in both SGC-7901 and BGC-823 cells (Fig. 6A). This mitochondrial deficit is also directly linked to ROS as pretreatment of cells with NAC prevented the decrease in membrane potential. Moreover, electron microscopy revealed that NAC attenuated the combined treatment-induced mitochondrial dysfunction including swelling and spheroid formation (Fig. 6B).
Fig. 6.

Combination of rapamycin and EF24 induces mitochondrial dysfunction by ROS/JNK signaling pathways. (A) Treatment of cells with combination of 5 µM rapamycin and 2 µM EF24 decreased mitochondrial membrane potential (Δψm). Cells were treated for 12 h and then stained with JC-1. Mitochondrial dysfunction was not evident when cells were pretreated with NAC or catalase [JC-1 accumulation in the mitochondria appear red, non-accumulating JC-1 as a monomer appear green; scale bar=20 μm]. (B) Effect of NAC on SGC-7901 mitochondrial morphology following combined rapamycin and EF24 treatment. (C) EF24 enhanced rapamycin-induced p-JNK levels. NAC or catalase pretreatment reversed the effects of 12-h combined rapamycin and EF24 in SGC-7901 and BGC-823 cells. The densitometric quantification bar graphs are shown in Supplementary file. Data were collected from three independent experiments and representative images were shown.
We next wanted to determine the potential signaling mechanism underlying the cellular and molecular changes we observed. Mitochondria-dependent cell apoptosis has been shown to involve an alteration of the Bcl-2 proteins by c-Jun N terminal kinase (JNK) [14], [27]. Our studies have shown that the combined treatment of rapamycin and EF24 induces ROS-dependent suppression of Bcl-2 and induction of Bax. Therefore, it is possible that the combined treatment induces cell apoptosis through JNK activation. Indeed, treatment of cells with rapamycin and EF24 increased the phosphorylation of JNK compared to single rapamycin or EF24 treatments, while scavenging ROS completely inhibited JNK phosphorylation induced by combined treatment (Fig. 6C and Supplementary Fig. S7A). Collectively these results indicate that combined treatment induced ROS-dependent JNK activation and mitochondrial dysfunction in gastric cancer cells.
3.5. EF24 amplifies the therapeutic effect of rapamycin in vivo
To confirm our promising combined treatment results, we evaluated the synergistic effect of rapamycin and EF24 in vivo by performing human gastric cancer xenografts in immunodeficient nude mice. As expected, treatment of mice bearing SGC-7901 tumors showed inhibited growth upon treatment with rapamycin, EF24, or a combination of the two (Fig. 7). Greatest inhibition of tumor growth was seen in mice treated with combined rapamycin and EF24 as illustrated by tumor weights (Fig. 7A) and tumor volumes (Fig. 7B–7C). No effect of any treatments was noted for body weights (Fig. 7D). Histopathological analyses of heart, kidney and liver tissues also revealed that combination of rapamycin with EF24 did not result in significant toxicity (Fig. S8). Lastly, we determined whether in vivo gastric tumor growth inhibition by rapamycin and EF24 also involves ROS generation and cell apoptosis. Tumor tissue lysates were subjected to western blot analysis to detect caspase-3 and PARP activation. Our results show significantly increased levels of p-eIF2α, and cleaved caspase 3 and PARP in mice treated with combined rapamycin and EF24 (Fig. 7E and G). We also found that combined treatment increased the level of lipid peroxidation product (MDA) in tumor tissues (Fig. 7F) indicating increased ROS production. Although the assay of lipid peroxidation product (MDA) is not specific for ROS level, it can be used as an indicator of oxidative stress [28]. The absent examinations for specific bio-markers of ROS level may be a limitation of this study. Furthermore, immunehistochemical staining for cell proliferation marker Ki-67 showed significantly reduced ki-67 positive cells in tumors treated with combined rapamycin and EF24 (Fig. 7G). Taken together, these results all indicated that EF24 can synergizes the therapeutic effect of rapamycin in vivo by elevating ROS levels and inducing cell apoptosis.
Fig. 7.

EF24 enhances the anti-tumor activity of rapamycin in human gastric cancer xenografts. SGC-7901 cells were injected in nude mice. Mice were then treated with rapamycin, EF24, or a combination of both. Figure showing tumor weight (A) and tumor volume (B and C) [*p<0.05, **p<0.01]. (D) No differences in body weights were noted for any of the treatments. (E) Western blot analysis of apoptosis-related protein in tumor specimens showing levels of p-eIF2α, and cleaved PARP and caspase3 (Randomly 2 mice were analyzed in each group). (F) The levels of oxidative stress marker MDA in the tumor tissue lysates [*p<0.05, **p<0.01]. (G) Tumor sections were stained with cell proliferation maker Ki-67 and apoptosis marker cleaved-caspase3 [scale bar=50 μm]. Data were collected from 5 mice in each group and representative images were shown.
4. Discussion
Despite improvements in detection and management, gastric cancer remains one of the leading causes of cancer death. As dysregulation of proteins in the mTOR pathway has been reported in many types of cancers, mTOR is an appealing therapeutic target. mTOR inhibitors including rapamycin and its analogs deforolimus, everolimus and temsirolimus are in clinical trials for treating multiple cancers [29] and some are already approved for metastatic renal cell carcinoma [30], [31]. However, monotherapy with mTOR inhibitors yields only modest therapeutic activity in advanced gastric cancer [32]. Emergence of drug resistance further hampers the clinical utility of rapamycin and its analogs [33]. Results from our present study as well as from others show that gastric cancer cells are resistant to low doses of rapamycin, while high doses of rapamycin cause cytotoxic effects in both cancer cells and normal cells. Therefore, combinatorial cancer therapies may provide a more synergistic anticancer effect and less systemic toxicity [34]. In the present study, we have studied the effect of combining EF24, a new curcumin analog, with rapamycin in human gastric cancer. Our findings demonstrate that EF24 sensitizes gastric cancer cells to rapamycin-induced selective growth inhibition and apoptosis induction. Furthermore, our study shows that EF24 mediates these effects through ROS-mediated ER-stress activation and mitochondrial dysfunction in gastric cancer cells.
ROS play a crucial role in tumorigenesis. Cancer cells exhibit high levels of ROS and higher antioxidant activities as compared to their normal counterparts [35]. Elevated ROS levels also render cancer cells more sensitive to agents that further increase ROS and oxidative stress [17]. Recent studies have also indicated that synergistic ROS-dependent cytotoxicity of combination therapies is cancer cell-specific [12]. Our previous studies show that EF24 inhibits thioredoxin reductase activity and induces ROS-mediated apoptosis in gastric cancer cells [13]. Therefore, we hypothesized that EF24 may represent an interesting target for combinatorial therapy. We found that rapamycin also induces the accumulation of intracellular ROS in a dose and time-dependent manner. Moreover, EF24 acted as a ROS inducer to enhance the anti-tumor activity of rapamycin in gastric cancer cells. To characterize the importance of ROS in our combined rapamycin and EF24 treatment, we used two ROS scavengers. Our findings demonstrate that NAC and catalase completely attenuate the synergistic anti-tumor effects of rapamycin and EF24 in gastric cancer cells. In addition, we found that ROS generation was independent of the mTOR signaling pathway as ROS scavengers failed to normalize mTOR suppression by rapamycin. Furthermore, time of ROS generation by rapamycin did not coincide with inhibition of the mTOR pathway. Further studies are necessary to identify the mechanism of ROS generation as well as to identify the direct redox-related targets of rapamycin.
In response to oxidative stress, accumulation of unfolded or misfolded proteins triggers a cellular adaptive procedure known as ER stress [36]. Normally, ER stress is designed to be protective as it shuts down protein synthesis and increases the production of molecular chaperones [37]. However, sustained ER stress leads to apoptosis mediated by CHOP [38]. Our findings showed that combined treatment with rapamycin and EF24 induced ER stress proteins p-PERK, p-eIF2α and ATF4. We also noted induction of ER stress-specific apoptotic cascade protein CHOP in gastric cancer cells. Importantly, scavenging of ROS by NAC and catalase abolished the ER-stress activation pathway by the combined treatments of rapamycin and EF24. This suggests that the anticancer effect of rapamycin and EF24 therapy is, at least, partially mediated by a ROS-dependent ER stress apoptotic pathway.
Excessive ROS production can lead to mitochondrial dysfunction, decrease the mitochondrial membrane potential, and release of cytochrome C [39]. Activated caspases subsequently induce proteolytic cleavage of PARP and finally result in cell apoptosis. Our study demonstrated that the combined use of rapamycin and EF24 results in a significant decrease of the mitochondrial membrane potential (Δψm) in SGC-7901 cells. Moreover, we showed that preventing ROS accumulation by NAC and catalase reverses rapamycin/EF24 treatment-induced mitochondrial dysfunction, indicating that ROS production may be the upstream regulator in mitochondrial deficits. Collectively, our results highlight that combined treatment-induced oxidative stress is linked to ER stress and mitochondrial dysfunction, which can amplify the synergistic anticancer effects of rapamycin and EF24 in gastric cancer cells.
In summary, we have reported that EF24 synergistically enhances the anticancer effects of rapamycin, highlighting a novel therapeutic avenue for gastric cancer. We found that EF24 enhances the anticancer activity of rapamycin in gastric cancer cells mainly via ROS-dependent ER stress and mitochondrial dysfunction and apoptosis induction. Furthermore, we verified the synergistic effect of rapamycin/EF24 combination on suppression of tumor growth in vivo using a xenograft tumor model. Taken together, we present evidence that combining rapamycin with EF24 can serve as a potential combination therapy for the treatment of human gastric cancer.
Conflict of interest disclosure statement
The authors disclose no potential conflicts of interest.
Acknowledgments
The work was supported by National Natural Science Foundation of China (81622043, 81503107, 81672305, and 81573657), Zhejiang Province Natural Science Funding of China (LY13H160022 and LY16H310011), and Wenzhou Science and Technology Project (2014Y0344).
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2016.09.006.
Contributor Information
Guang Liang, Email: wzmcliangguang@163.com.
Jiansong Ji, Email: jjstcty@sina.com.
Appendix A. Supplementary material
Supplementary material
.
References
- 1.Ferlay J., Shin H.R., Bray F., Forman D., Mathers C., Parkin D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer. 2010;127(12):2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Wagner A.D., Grothe W., Haerting J., Kleber G., Grothey A., Fleig W.E. Chemotherapy in advanced gastric cancer: a systematic review and meta-analysis based on aggregate data. J. Clin. Oncol. 2006;24(18):2903–2909. doi: 10.1200/JCO.2005.05.0245. [DOI] [PubMed] [Google Scholar]
- 3.Power D.G., Kelsen D.P., Shah M.A. Advanced gastric cancer – slow but steady progress. Cancer Treat. Rev. 2010;36(5):384–392. doi: 10.1016/j.ctrv.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 4.Waddell T., Verheij M., Allum W., Cunningham D., Cervantes A., Arnold D. Gastric cancer: ESMO-ESSO-ESTRO clinical practice guidelines for diagnosis, treatment and follow-up. Eur. J. Surg. Oncol.: J. Eur. Soc. Surg. Oncol. Br. Assoc. Surg. Oncol. 2014;40(5):584–591. doi: 10.1016/j.ejso.2013.09.020. [DOI] [PubMed] [Google Scholar]
- 5.Van Cutsem E., Moiseyenko V.M., Tjulandin S., Majlis A., Constenla M., Boni C., Rodrigues A., Fodor M., Chao Y., Voznyi E. Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 Study Group. J. Clin. Oncol. 2006;24(31):4991–4997. doi: 10.1200/JCO.2006.06.8429. [DOI] [PubMed] [Google Scholar]
- 6.Bouche O., Ychou M., Burtin P., Bedenne L., Ducreux M., Lebreton G., Baulieux J., Nordlinger B., Martin C., Seitz J.F. Adjuvant chemotherapy with 5-fluorouracil and cisplatin compared with surgery alone for gastric cancer: 7-year results of the FFCD randomized phase III trial (8801) Ann. Oncol.: Off. J. Eur. Soc. Med. Oncol. 2005;16(9):1488–1497. doi: 10.1093/annonc/mdi270. [DOI] [PubMed] [Google Scholar]
- 7.Vezina C., Kudelski A., Sehgal S.N. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. 1975;28(10):721–726. doi: 10.7164/antibiotics.28.721. [DOI] [PubMed] [Google Scholar]
- 8.Douros J., Suffness M. New antitumor substances of natural origin. Cancer Treat. Rev. 1981;8(1):63–87. doi: 10.1016/s0305-7372(81)80006-0. [DOI] [PubMed] [Google Scholar]
- 9.Meric F., Hunt K.K.: Translation initiation in cancer: a novel target for therapy. Mol. Cancer Ther. 2002;1(11):971–979. [PubMed] [Google Scholar]
- 10.Shigematsu H., Yoshida K., Sanada Y., Osada S., Takahashi T., Wada Y., Konishi K., Okada M., Fukushima M. Rapamycin enhances chemotherapy-induced cytotoxicity by inhibiting the expressions of TS and ERK in gastric cancer cells. Int. J. Cancer. 2010;126(11):2716–2725. doi: 10.1002/ijc.24990. [DOI] [PubMed] [Google Scholar]
- 11.Populo H., Lopes J.M., Soares P. The mTOR signalling pathway in human cancer. Int J. Mol. Sci. 2012;13(2):1886–1918. doi: 10.3390/ijms13021886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zou P., Chen M., Ji J., Chen W., Chen X., Ying S., Zhang J., Zhang Z., Liu Z., Yang S. Auranofin induces apoptosis by ROS-mediated ER stress and mitochondrial dysfunction and displayed synergistic lethality with piperlongumine in gastric cancer. Oncotarget. 2015;6(34):36505–36521. doi: 10.18632/oncotarget.5364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zou P., Xia Y., Chen W., Chen X., Ying S., Feng Z., Chen T., Ye Q., Wang Z., Qiu C. EF24 induces ROS-mediated apoptosis via targeting thioredoxin reductase 1 in gastric cancer cells. Oncotarget. 2016 doi: 10.18632/oncotarget.7633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zou P., Zhang J., Xia Y., Kanchana K., Guo G., Chen W., Huang Y., Wang Z., Yang S., Liang G. ROS generation mediates the anti-cancer effects of WZ35 via activating JNK and ER stress apoptotic pathways in gastric cancer. Oncotarget. 2015;6(8):5860–5876. doi: 10.18632/oncotarget.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cairns R.A., Harris I.S., Mak T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer. 2011;11(2):85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 16.DeNicola G.M., Karreth F.A., Humpton T.J., Gopinathan A., Wei C., Frese K., Mangal D., Yu K.H., Yeo C.J., Calhoun E.S. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475(7354):106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trachootham D., Alexandre J., Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat. Rev. Drug Discov. 2009;8(7):579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 18.Jeanne M., Lallemand-Breitenbach V., Ferhi O., Koken M., Le Bras M., Duffort S., Peres L., Berthier C., Soilihi H., Raught B. PML/RARA oxidation and arsenic binding initiate the antileukemia response of As2O3. Cancer Cell. 2010;18(1):88–98. doi: 10.1016/j.ccr.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 19.Alexandre J., Hu Y., Lu W., Pelicano H., Huang P. Novel action of paclitaxel against cancer cells: bystander effect mediated by reactive oxygen species. Cancer Res. 2007;67:3512–3517. doi: 10.1158/0008-5472.CAN-06-3914. [DOI] [PubMed] [Google Scholar]
- 20.Gao N., Rahmani M., Dent P., Grant S. 2-Methoxyestradiol-induced apoptosis in human leukemia cells proceeds through a reactive oxygen species and Akt-dependent process. Oncogene. 2005;24(23):3797–3809. doi: 10.1038/sj.onc.1208530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang Y., Zheng T., Song R., Wang J., Yin D., Wang L., Liu H., Tian L., Fang X., Meng X. Hypoxia-mediated sorafenib resistance can be overcome by EF24 through Von Hippel-Lindau tumor suppressor-dependent HIF-1alpha inhibition in hepatocellular carcinoma. Hepatology. 2013;57(5):1847–1857. doi: 10.1002/hep.26224. [DOI] [PubMed] [Google Scholar]
- 22.Selvendiran K., Tong L., Vishwanath S., Bratasz A., Trigg N.J., Kutala V.K., Hideg K., Kuppusamy P. EF24 induces G2/M arrest and apoptosis in cisplatin-resistant human ovarian cancer cells by increasing PTEN expression. J. Biol. Chem. 2007;282(39):28609–28618. doi: 10.1074/jbc.M703796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adams B.K., Cai J., Armstrong J., Herold M., Lu Y.J., Sun A., Snyder J.P., Liotta D.C., Jones D.P., Shoji M. EF24, a novel synthetic curcumin analog, induces apoptosis in cancer cells via a redox-dependent mechanism. Anticancer Drugs. 2005;16(3):263–275. doi: 10.1097/00001813-200503000-00005. [DOI] [PubMed] [Google Scholar]
- 24.Zou X., Liang J., Sun J., Hu X., Lei L., Wu D., Liu L. Allicin sensitizes hepatocellular cancer cells to anti-tumor activity of 5-fluorouracil through ROS-mediated mitochondrial pathway. J. Pharm. Sci. 2016 doi: 10.1016/j.jphs.2016.04.017. [DOI] [PubMed] [Google Scholar]
- 25.Chen W., Zou P., Zhao Z., Weng Q., Chen X., Ying S., Ye Q., Wang Z., Ji J., Liang G. Selective killing of gastric cancer cells by a small molecule via targeting TrxR1 and ROS-mediated ER stress activation. Oncotarget. 2016 doi: 10.18632/oncotarget.7565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weinberg S.E., Chandel N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015;11(1):9–15. doi: 10.1038/nchembio.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lei K., Davis R.J. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA. 2003;100(5):2432–2437. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gong K., Li W. Shikonin, a Chinese plant-derived naphthoquinone, induces apoptosis in hepatocellular carcinoma cells through reactive oxygen species: a potential new treatment for hepatocellular carcinoma. Free Radic. Biol. Med. 2011;51(12):2259–2271. doi: 10.1016/j.freeradbiomed.2011.09.018. [DOI] [PubMed] [Google Scholar]
- 29.Dancey J.E. Therapeutic targets: MTOR and related pathways. Cancer Biol. Ther. 2006;5(9):1065–1073. doi: 10.4161/cbt.5.9.3175. [DOI] [PubMed] [Google Scholar]
- 30.Hudes G., Carducci M., Tomczak P., Dutcher J., Figlin R., Kapoor A., Staroslawska E., Sosman J., McDermott D., Bodrogi I. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. New Engl. J. Med. 2007;356(22):2271–2281. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- 31.Motzer R.J., Escudier B., Oudard S., Hutson T.E., Porta C., Bracarda S., Grunwald V., Thompson J.A., Figlin R.A., Hollaender N. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372(9637):449–456. doi: 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- 32.Ohtsu A., Ajani J.A., Bai Y.X., Bang Y.J., Chung H.C., Pan H.M., Sahmoud T., Shen L., Yeh K.H., Chin K. Everolimus for previously treated advanced gastric cancer: results of the randomized, double-blind, phase III GRANITE-1 study. J. Clin. Oncol. 2013;31(31):3935–3943. doi: 10.1200/JCO.2012.48.3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carew J.S., Kelly K.R., Nawrocki S.T. Mechanisms of mTOR inhibitor resistance in cancer therapy. Target. Oncol. 2011;6(1):17–27. doi: 10.1007/s11523-011-0167-8. [DOI] [PubMed] [Google Scholar]
- 34.Chiarini F., Lonetti A., Teti G., Orsini E., Bressanin D., Cappellini A., Ricci F., Tazzari P.L., Ognibene A., Falconi M. A combination of temsirolimus, an allosteric mTOR inhibitor, with clofarabine as a new therapeutic option for patients with acute myeloid leukemia. Oncotarget. 2012;3(12):1615–1628. doi: 10.18632/oncotarget.762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szatrowski T.P., Nathan C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51(3):794–798. [PubMed] [Google Scholar]
- 36.Tabas I., Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011;13(3):184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walter P., Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 38.Rao R., Nalluri S., Fiskus W., Savoie A., Buckley K.M., Ha K., Balusu R., Joshi A., Coothankandaswamy V., Tao J. Role of CAAT/enhancer binding protein homologous protein in panobinostat-mediated potentiation of bortezomib-induced lethal endoplasmic reticulum stress in mantle cell lymphoma cells. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 2010;16(19):4742–4754. doi: 10.1158/1078-0432.CCR-10-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vrablic A.S., Albright C.D., Craciunescu C.N., Salganik R.I., Zeisel S.H. Altered mitochondrial function and overgeneration of reactive oxygen species precede the induction of apoptosis by 1-O-octadecyl-2-methyl-rac-glycero-3-phosphocholine in p53-defective hepatocytes. FASEB J. 2001;15(10):1739–1744. doi: 10.1096/fj.00-0300com. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material