

Abstract
Centrioles are essential for the formation of centrosomes and cilia. While numerical and/or structural centrosomes aberrations are implicated in cancer, mutations in centriolar and centrosomal proteins are genetically linked to ciliopathies, microcephaly, and dwarfism. The evolutionarily conserved mechanisms underlying centrosome biogenesis are centered on a set of key proteins, including Plk4, Sas‐6, and STIL, whose exact levels are critical to ensure accurate reproduction of centrioles during cell cycle progression. However, neither the intracellular levels of centrosomal proteins nor their stoichiometry within centrosomes is presently known. Here, we have used two complementary approaches, targeted proteomics and EGFP‐tagging of centrosomal proteins at endogenous loci, to measure protein abundance in cultured human cells and purified centrosomes. Our results provide a first assessment of the absolute and relative amounts of major components of the human centrosome. Specifically, they predict that human centriolar cartwheels comprise up to 16 stacked hubs and 1 molecule of STIL for every dimer of Sas‐6. This type of quantitative information will help guide future studies of the molecular basis of centrosome assembly and function.
Keywords: centriole, centrosome, fluorescence imaging, proteomics, selected reaction monitoring
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Cell Cycle; Post-translational Modifications, Proteolysis & Proteomics
Introduction
A thorough understanding of any cellular process will ultimately require precise quantitative information on the expression levels of endogenous proteins under physiological conditions. Moreover, information about the stoichiometry of proteins within multiprotein complexes will be indispensable for modeling approaches that aim at understanding the assembly and function of cellular organelles at systems level. Comprehensive studies on the proteomes of cells, tissues, or organisms have been reported (e.g. Addona et al, 2009; Nilsson et al, 2010; Beck et al, 2011; Nagaraj et al, 2011; for review, see Bensimon et al, 2012; Kulak et al, 2014; Wilhelm et al, 2014; Hein et al, 2015; Richards et al, 2015), but accurate quantitative information on proteins expressed at low levels remains scarce. As a consequence, published estimates for the abundance of specific proteins sometimes vary over several orders of magnitude.
Recent progress with targeted proteomics offers new opportunities for acquiring quantitative information about the absolute amounts of individual cellular proteins (Picotti et al, 2009; Gillette & Carr, 2013; Simicevic et al, 2013). In particular, selected reaction monitoring (SRM) allows the detection of specific proteins in complex mixtures with high sensitivity and over a broad dynamic range (Picotti et al, 2009; Bauer et al, 2014). In parallel, EGFP‐tagging of endogenous proteins through genetic engineering offers opportunities for quantifying fluorescence signals at specific subcellular locations (e.g. Wu & Pollard, 2005; Bodor et al, 2014). Here, we have used these methodologies for a quantitative analysis of key proteins implicated in the duplication and function of human centrosomes.
Centrosomes are the major microtubule organizing centers of animal cells and typically comprise two centrioles embedded in a matrix of pericentriolar proteins (PCM) (Gonczy, 2012; Conduit et al, 2015). Centrioles are microtubule‐based cylindrical structures that display evolutionarily conserved ninefold symmetry (Azimzadeh & Marshall, 2010; Hirono, 2014; Winey & O'Toole, 2014). Centrosomes are important for organelle positioning, cell shape, polarity, and motility, as well as chromosome segregation and cell division (Nigg & Stearns, 2011; Bornens, 2012). In addition to their role in nucleating centrosome assembly, fully mature centrioles also function as basal bodies for the formation of cilia and flagella (Ishikawa & Marshall, 2011; Bornens, 2012). Numerical and structural aberrations in centrioles and centrosomes have been implicated in human disease, including cancer, microcephaly, dwarfism, and ciliopathies (Nigg & Raff, 2009; Zyss & Gergely, 2009; Bettencourt‐Dias et al, 2011; Ishikawa & Marshall, 2011; Chavali et al, 2014; Jana et al, 2014; Gonczy, 2015).
In most proliferating cells, centrioles are duplicated exactly once per cell cycle (Nigg, 2007; Firat‐Karalar & Stearns, 2014). Critical for centriole formation is the assembly of a cartwheel structure onto which nine triplet microtubules are assembled (Gonczy, 2012; Hirono, 2014). The centriole‐associated PCM comprises more than 100 proteins, including many coiled coil proteins (Andersen et al, 2003; Jakobsen et al, 2011) that form a complex matrix (Fu & Glover, 2012; Lawo et al, 2012; Mennella et al, 2012; Sonnen et al, 2012). Additional prominent PCM components include γ‐tubulin ring complexes (γ‐TuRCs) crucial for microtubule nucleation (Kollman et al, 2011; Teixido‐Travesa et al, 2012), proteins important for tethering duplicated centrosomes (Nigg & Stearns, 2011; Agircan et al, 2014), as well as cell cycle regulators and signaling molecules (Arquint et al, 2014). The assembly of PCM components onto centrioles is regulated during cell cycle progression, with major expansion of the matrix occurring in preparation of bipolar spindle formation at the onset of mitosis (Woodruff et al, 2014; Conduit et al, 2015).
In proliferating human cells, one new (daughter) centriole forms at the G1/S transition in near‐orthogonal orientation close to the proximal end of each of the two G1‐phase (mother) centrioles. Centriole formation is initiated by the protein kinase Plk4 (also known as Sak; Bettencourt‐Dias et al, 2005; Habedanck et al, 2005), which undergoes activation through trans‐autophosphorylation at a T‐loop residue (Lopes et al, 2015). Steady‐state cellular levels of Plk4 are generally kept low through trans‐autophosphorylation of a DSG‐motif and subsequent degradation by SCF‐β‐TrCP (Guderian et al, 2010; Holland et al, 2010). Plk4 assembles at the surface of mother centrioles through binding to Cep152 and Cep192 (Cizmecioglu et al, 2010; Hatch et al, 2010; Kim et al, 2013; Sonnen et al, 2013; Park et al, 2014). However, at the site of daughter centriole formation, the STIL protein binds, activates, and stabilizes Plk4, and phosphorylation of STIL by Plk4 triggers the recruitment of Sas‐6 (Dzhindzhev et al, 2014; Ohta et al, 2014; Arquint et al, 2015; Klebba et al, 2015; Kratz et al, 2015; reviewed in Arquint & Nigg, 2016). Sas‐6 is a major component of the cartwheel and contributes to confer ninefold symmetry (van Breugel et al, 2011; Kitagawa et al, 2011; Guichard et al, 2013; Fong et al, 2014; Wang et al, 2015). The protein CPAP gets recruited to promote the assembly of microtubules onto the cartwheel (Tang et al, 2011a; Cottee et al, 2013; Sharma et al, 2016), and this step is likely assisted by Cep135 (the human homolog of Chlamydomonas Bld10) (Hirono, 2014). CPAP also cooperates with additional proteins, including CP110, in determining the length of nascent centrioles (Kohlmaier et al, 2009; Schmidt et al, 2009; Tang et al, 2009; Comartin et al, 2013; Lin et al, 2013; Sharma et al, 2016). Finally, centriole maturation is completed by the acquisition of subdistal and distal appendages (Paintrand et al, 1992; Tateishi et al, 2013).
Centriole biogenesis and maintenance of correct centrosome numbers in proliferating cells critically depend on the exact levels of the key duplication factors Plk4 (Bettencourt‐Dias et al, 2005; Habedanck et al, 2005), Sas‐6 (Leidel et al, 2005; Strnad et al, 2007), and STIL (Tang et al, 2011b; Arquint et al, 2012; Vulprecht et al, 2012). To fully understand the regulation of centriole duplication, it will thus be important to obtain quantitative data on the abundance of these proteins under physiological conditions. However, many centriolar and centrosomal proteins are anticipated to be expressed at very low levels, so that reliable measurement of their abundance represents a formidable task. Here, we have combined targeted proteomics (based on SRM) with EGFP‐tagging of proteins at endogenous loci (through homologous recombination) to determine the absolute amounts of a selected set of human centriolar and centrosomal proteins. Our data provide information about the abundance of regulatory and structural proteins and allow us to make specific predictions about the quantitative architecture of the human centrosome.
Results
Use of complementary approaches for measuring centrosome protein abundance
Most reported large‐scale proteomics studies do not fully address the challenges associated with determining accurate quantitative information for low abundance proteins. To measure the abundance of centrosomal proteins, both within cell lysates and at purified centrosomes, we therefore used a combination of approaches (see flowchart in Fig 1). First, we used SRM to measure the absolute abundance of selected proteins in whole lysates prepared from RPE‐1 cells as well as different transformed cell lines, including KE‐37 (Fig 2A). Then, SRM was used to determine the relative amounts of the same proteins associated with centrosomes purified from KE‐37 cells (Fig 2C). In addition, label‐free shotgun proteomics was used to assess the abundance of more than 70 proteins associated with the same centrosome preparations (Fig 3). In a complementary approach, EGFP‐tagging of endogenous alleles in RPE‐1 cells was used to quantify the localized abundance of γ‐tubulin, Sas‐6, and STIL at different cell cycle stages (Figs 4 and 5). Taken together, the results of this multipronged approach provide a comprehensive overview of the cellular and localized abundance of key centrosomal proteins.
Figure 1. Strategies for measuring abundance of centrosomal proteins.
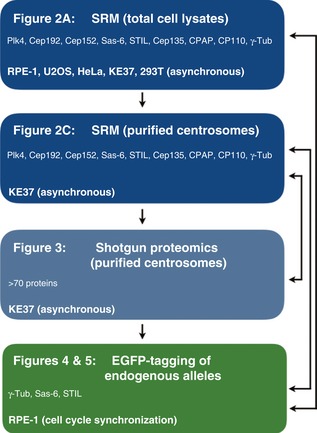
Schematic illustrates the use of different technologies for measuring the absolute or relative abundance of centrosomal proteins in total cell lysates or purified centrosomes. The flowchart refers to the main figures and also indicates the cell lines and cell cycle stages analyzed in each experiment. Arrows on the right emphasize the use of data from different experiments for comparison and calibration.
Figure 2. Abundance of centrosomal proteins in whole‐cell lysates and purified centrosomes.
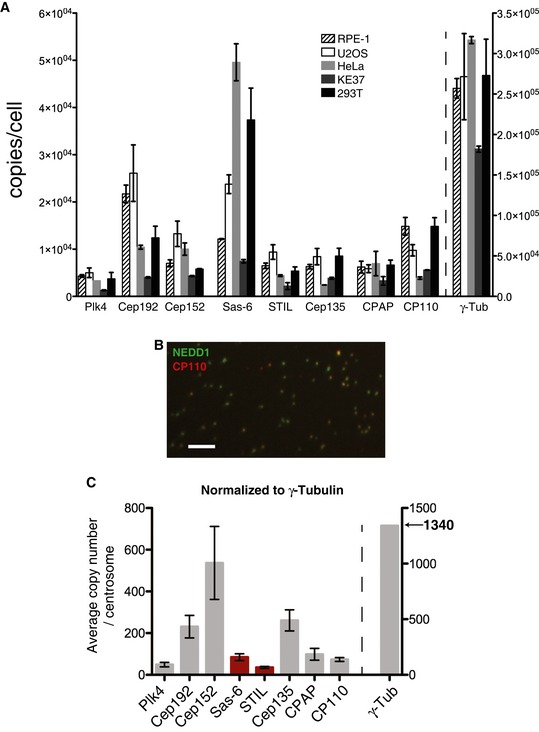
- The absolute abundance of centrosomal proteins in whole‐cell lysates, prepared from asynchronous growing cells, was determined using SRM mass spectrometry combined with stable isotope dilution. Copy numbers per cell were measured in three independent experiments. Because not all peptides could be detected in all experiments, histograms show average values determined in those two measurements in which most peptides could be measured reliably. Results presented are means ± SEM. RPE‐1 (non‐transformed) cells were compared with various tumor‐derived (U2OS, HeLa, KE37) or transformed cells (HEK293T). Note the different scale for γ‐tubulin on the y‐axis.
- Representative image showing centrosome preparation stained with antibodies to NEDD1 (green) and CP110 (red). Scale bar: 10 μm.
- Purified centrosomes from KE37 cells were analyzed by SRM with stable isotope dilution. Histogram a priori represents relative copy numbers per centrosome (results presented are means ± SEM). To predict protein abundance in absolute numbers (average number of copies/centrosome), data were normalized to 1,340 γ‐tubulin molecules, that is the average number of γ‐tubulin per centrosome as deduced by combining SRM and EGFP fluorescence measurements (Figs 4 and 5). Bars for Sas‐6 and STIL are marked in red to indicate that the corresponding values should be approximately doubled to correct for the fact that some 50% of purified centrosomes are derived from G1‐phase cells that mostly lack these two proteins. Note the different scale for γ‐tubulin on the y‐axis.
Figure 3. Label‐free quantification of centrosome preparations.
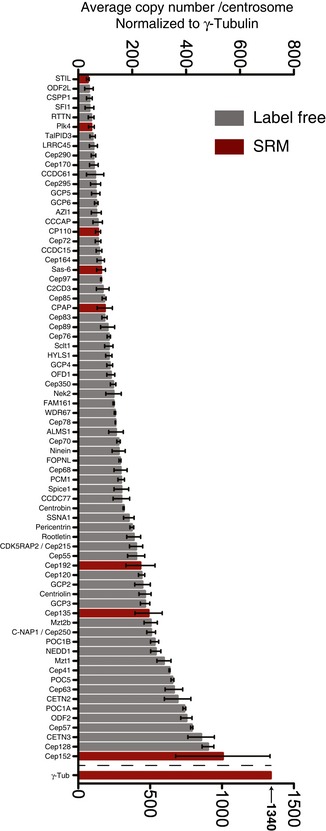
Centrosome preparations from KE37 cells were analyzed by shotgun proteomics and relative protein abundances determined using the iBAQ shotgun proteomics method. The iBAQ values were normalized with the trendline as obtained from the SRM measurements (Fig EV1E), and γ‐tubulin was normalized to 1,340 copies/centrosome. A subset of centrosomal proteins is shown as described in Azimzadeh et al (2012). Black bars represent the centrosome subset, and red bars represent the values obtained by SRM. Results presented are means ± SEM.
Figure 4. Determination of γ‐tubulin abundance at centrosomes.
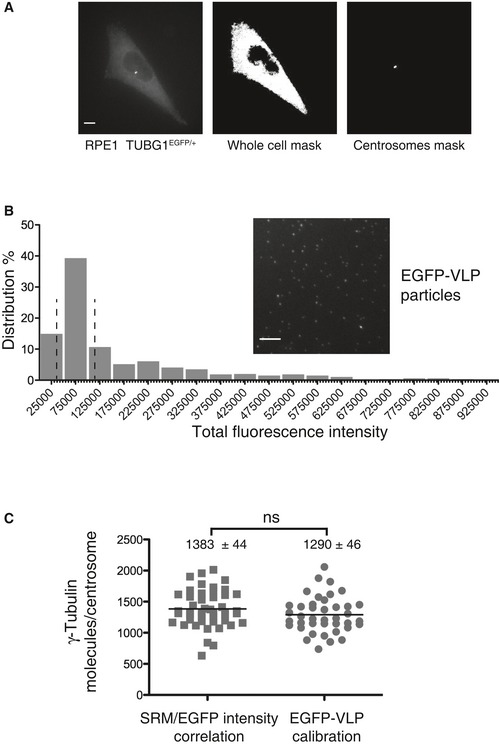
- Fluorescence analysis of RPE‐1 cells expressing endogenously tagged γ‐tubulin‐EGFP. Cells were synchronized in G2, using RO‐3306, to allow for a clear distinction of the two centrosomes present at this cell cycle stage. Panels illustrate sum projections of the stacks from an original picture (left), a whole‐cell mask (middle), and a centrosome mask (right); both masks are based on intensity thresholding. Scale bar: 10 μm.
- EGFP‐tagged viral‐like particles (GFP‐VLP2/6) are used as a reference for quantification of γ‐tubulin‐EGFP. The histogram shows the distribution of the total fluorescence intensity associated with single GFP‐VLP2/6; the two dashed lines delimit the population used for assigning an average fluorescence intensity value to represent the 120 GFP molecules associated with each VLP2/6 particle (Charpilienne et al, 2001). The inset shows a sum projection of EGFP‐VLPs fluorescence image. Scale bar: 10 μm.
- Scatter plots show the numbers of γ‐tubulin molecules per centrosome determined by calibrations using either SRM measurements (n = 46 cells) or EGFP‐VLP2/6 fluorescence (n = 40 cells). The black lines represent mean values; numbers ± SEM are indicated on top of each scatter plot. P‐values from t‐tests are indicated as follows: ***P < 0.001, **P = 0.001–0.01, *P = 0.01–0.05, and ns P > 0.05.
Figure 5. Abundance of centrosomal proteins through the cell cycle.
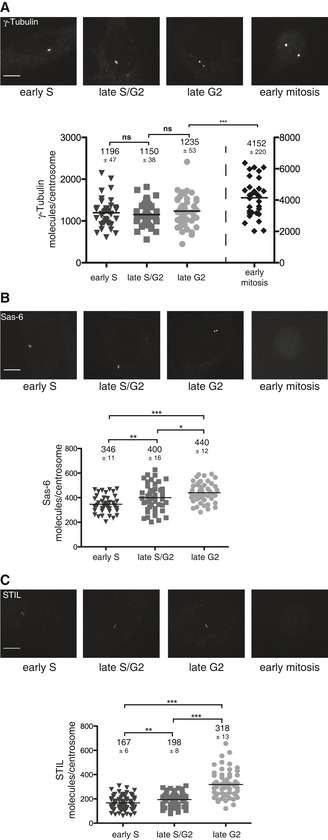
-
A–CTop panels: RPE‐1 cells expressing (endogenous) γ‐tubulin‐EGFP (A), Sas‐6 (B), or STIL (C) at the indicated cell cycle stages. Fluorescence images of representative cells are shown as maximum projections. Scale bars: 10 μm. Bottom panels: Fluorescence quantifications were performed using the EGFP‐VLP2/6 method for calibration. Scatter plots represent the number of γ‐tubulin (A), Sas‐6 (B), or STIL (C) molecules per centrosome at the indicated cell cycle stages. The black lines represent mean values; numbers ± SEM are indicated on top of each scatter plot. The following numbers of cells were analyzed in early S phase: γ‐tubulin: 45, Sas‐6: 43, STIL: 79; in late S/G2 phase: γ‐tubulin: 45, Sas‐6: 46, STIL: 68; in late G2 phase: γ‐tubulin: 48, Sas‐6: 48, STIL: 68; and in early mitosis: γ‐tubulin: 31. Note that fluorescence images were recorded using identical settings. P‐values from t‐tests are indicated as follows: ***P < 0.001, **P = 0.001–0.01, *P = 0.01–0.05, and ns P > 0.05.
Use of SRM to measure absolute protein abundance in cells
SRM relies on a three‐step process, the selection of precursor ions in a tandem mass spectrometer, the fragmentation of these ions in a collision chamber, and the monitoring of selected fragment ions, termed SRM transitions (Picotti & Aebersold, 2012). As an important preparatory step, the SRM workflow requires the identification of peptides, so‐called proteotypic peptides (PTPs), that are distinctive for a given protein and display high ionization efficiency, and the validation of optimized transitions specific for each PTP (Picotti & Aebersold, 2012). In pilot experiments, we identified the two most suited PTPs for each centrosomal protein to be quantified (Appendix Fig S1), as detailed in Appendix Fig S2, and acquired isotope‐labeled synthetic versions of these peptides (AQUA peptides; Gerber et al, 2003). After using these heavy reference peptides for optimizing collision energies for all selected SRM transitions, the linear dynamic range, reproducibility, and quantification limits of each SRM assay were thoroughly assessed through analyses of dilution series with whole human cell lysates (Bauer et al, 2014). Although wide dynamic ranges were observed for all assays (> 4 orders of magnitude), extract fractionation by Off‐Gel electrophoresis was required to further improve the sensitivity of SRM assays (Picotti et al, 2009) and allow confident detection and quantification of the least abundant target proteins. By comparing transition intensities of “heavy” reference peptides (spiked in known amounts into tryptic digests prior to any fractionation) and their corresponding “light” counterparts derived from endogenous proteins (Appendix Fig S3), the abundance of endogenous proteins in whole‐cell extracts could be accurately determined (Bauer et al, 2014).
Having established an efficient SRM workflow (Appendix Fig S2), the approach was applied to a core set of proteins implicated in the centrosome duplication cycle, many of which were expected to be expressed at low levels. The selected proteins include key regulators of centriole duplication and components of the centriolar cartwheel (Plk4, Sas‐6, STIL, Cep135), recruitment factors for Plk4 (Cep152 and Cep192), regulators of centriole length (CPAP, CP110), and the key PCM component required for microtubule nucleation (γ‐tubulin). We first used SRM to determine the absolute amounts of the above proteins in lysates prepared from different cells lines (Fig 2A and Table EV1). As expected (Holland et al, 2012), we found that Plk4 is a low abundance protein, ranging from 1,200 to 5,000 copies/cell in KE37 and U2OS cells, respectively. Considering that cultured human cells are estimated to harbor some 6–8 × 109 protein molecules per cell (Sims & Allbritton, 2007; Siwiak & Zielenkiewicz, 2013), this attests to the exquisite sensitivity of SRM and implies that we have detected one Plk4 polypeptide chain against a background of > 106 protein molecules. To the best of our knowledge, Plk4 has not previously been detected in any published whole‐cell proteomics study. In all cell lines analyzed, Cep152 and Cep192 are more abundant than Plk4 (Fig 2A), consistent with the notion that these proteins function as major Plk4 recruitment factors at centrioles (Cizmecioglu et al, 2010; Hatch et al, 2010; Kim et al, 2013; Sonnen et al, 2013; Park et al, 2014). The PCM component γ‐tubulin is much more abundant than any of the other proteins analyzed here, ranging from 180,000 to more than 300,000 copies per cell, depending on cell type. No correlation could be observed with malignant transformation. For example, Cep192 is more abundant in the non‐transformed RPE‐1 cells than in most cancer cells. Conversely, Sas‐6 is about 4 times more abundant in HeLa than in RPE‐1 cells. Of all proteins analyzed, Sas‐6 shows the largest variation in expression, differing by as much as 6.5‐fold between HeLa and KE37 cells. Although this observation may appear surprising, considering that Sas‐6 is a key structural component of the centriolar cartwheel (Gonczy, 2012; Hirono, 2014), it supports the view that recruitment of Sas‐6 to nascent centrioles is extensively controlled by posttranslational mechanisms (Dzhindzhev et al, 2014; Keller et al, 2014; Ohta et al, 2014; Arquint et al, 2015; Kratz et al, 2015). Similarly, although CPAP and CP110 have both been implicated in controlling the length of centrioles (Kohlmaier et al, 2009; Schmidt et al, 2009; Tang et al, 2009), their abundance in different cell lines shows no correlation, arguing that their functions at centrioles do not simply reflect protein concentration. Finally, we note that most centrosomal proteins were least abundant in the KE37 T‐lymphoblastoid line, which is commonly used for centrosome purification (Gosti‐Testu et al, 1986; Andersen et al, 2003). This most likely reflects the reduced cytoplasmic volume typical of lymphocytes, rather than lower protein concentrations.
Use of SRM to measure relative protein abundance in purified centrosomes
Having established absolute amounts of centrosomal proteins in a number of cell lines, we next used SRM to determine the abundance of proteins associated with centrosomes purified from KE37 cells (Fig 2B and C). Centrosome purification could readily be monitored by immunofluorescence microscopy (Fig 2B), but we cannot rigorously exclude selective loss or artefactual association of individual proteins during centrosome purification. Also, reliable measurement of the exact number of centrosomes entering the mass spectrometer proved difficult. Hence, our quantifications of protein abundance at purified centrosomes refer a priori to relative rather than absolute numbers of molecules per organelle. To predict absolute numbers, we used a value of 1,340 molecules of γ‐tubulin per interphase centrosome for calibration (Fig 2C). This value was derived from a comparison of whole‐cell SRM data with fluorescence measurements performed on RPE‐1 cells expressing one EGFP‐tagged allele of γ‐tubulin, as described in detail below (see Fig 4). The data compiled in Fig 2 indicate that no straightforward correlation exists between protein abundance in whole‐cell extracts (Fig 2A) and protein abundance at purified centrosomes (Fig 2C). This strengthens the notion that posttranslational mechanisms play an important role in determining the enrichment of individual proteins at centrosomes. For example, Cep192 and Cep152 are similarly abundant in total KE37 cell lysates (Fig 2A), but the number of centrosome‐associated Cep152 molecules exceeds that of Cep192 by a factor of more than 2 (Fig 2C). It is also remarkable that Cep135 is much more abundant at purified centrosomes than either STIL or Sas‐6 (Fig 2C). Although this result may appear surprising when considering the striking association of the Cep135 homolog Bld10 with the centriolar cartwheel in Chlamydomonas (Matsuura et al, 2004; Hiraki et al, 2007), it falls in line with the observation that the bulk of human Cep135 associates with the proximal ends of mother centrioles rather than the cartwheels of daughter centrioles (Sonnen et al, 2012). Finally, and most interestingly, our data predict that Sas‐6 is about twice as abundant as STIL. At face value, the data predict 85 and 36 molecules/centrosome for Sas‐6 and STIL, respectively. However, these numbers represent an underestimate, because ~50% of centrosomes purified from asynchronously growing KE37 cells are derived from G1‐phase cells. In contrast to most other proteins analyzed here, including the protein used for calibration (γ‐tubulin), both Sas‐6 and STIL show marked cycle regulation and very similar cell cycle profiles; as a consequence, they are barely detectable at most G1‐phase centrosomes (Strnad et al, 2007; Arquint & Nigg, 2014; Keller et al, 2014). Thus, an approximate doubling of the above numbers likely provides a better estimate of the absolute abundance of Sas‐6 and STIL on those centrosomes (S and G2 phases) that are positive for these proteins, resulting in 170 and 72 molecules for Sas‐6 and STIL, respectively. Importantly, our conclusion that Sas‐6 is about twice as abundant as STIL is valid regardless of the correction factor used to compensate for cell cycle effects.
Label‐free quantification of proteins in centrosome preparations
To obtain quantitative information about a broader range of centrosomal proteins, we subjected the above centrosome preparations to shotgun proteomics and estimated protein abundance using the iBAQ method (Schwanhausser et al, 2011; Arike et al, 2012; Ahrne et al, 2013). Figure 3 and Table EV2 summarize the results for 73 core centrosomal proteins [including 58/61 proteins described in Azimzadeh et al (2012)]. The results obtained by iBAQ and SRM were generally in excellent agreement (Fig EV1E), confirming that label‐free quantification provides useful, albeit approximate, information about protein abundance (Ahrne et al, 2013).
Figure EV1. Quantification of protein subsets within centrosome preparations.
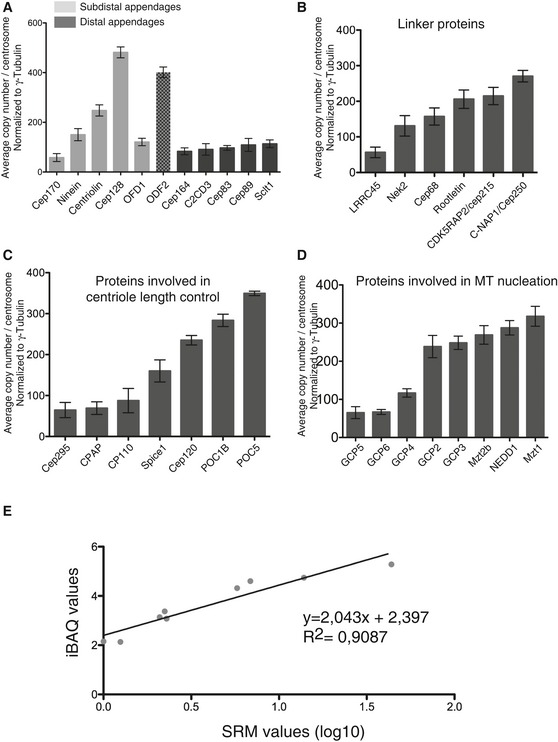
-
A–DHistograms show quantifications for subsets of centrosomal proteins; results presented are means ± SEM. Data are extracted from Fig 3 but arranged to facilitate comparisons of proteins with similar localizations and/or functions. Grouped together are distal and subdistal appendage proteins (A; note that ODF2 is believed to associate with both structures), centrosomal linker proteins (B), proteins involved in centriole length control (C), and core components involved in MT nucleation (D).
-
ECorrelation analysis shows excellent correspondence between values determined by iBAQ and SRM.
To facilitate comparisons of proteins with similar localizations and/or functions, information was extracted from the iBAQ and SRM quantification data for selected subsets of proteins (Fig EV1A–D), notably appendage proteins (A), centrosomal linker proteins (B), proteins involved in centriole length control (C), and core components involved in MT nucleation (D). These analyses reveal that most distal appendage proteins are of lower abundance than components of subdistal appendages; furthermore, while distal appendage proteins occur in nearly equal proportions, the abundance of subdistal appendage proteins differs by up to 10‐fold. With regard to centrosomal linker proteins, we were surprised to find that putative structural proteins are barely more abundant than Nek2, the kinase held responsible for their regulation. Concerning proteins implicated in the control of centriole length, our data indicate that POC5 and POC1B are 3–4 times as abundant as CPAP and CP110 and that Cep120 is more abundant than its putative interaction partners Spice1 and CPAP (Comartin et al, 2013), suggesting the existence of separate Cep120‐Spice1 and Cep120‐CPAP complexes. With regard to proteins implicated in MT nucleation, we find that GCP2 and GCP3, the core components of the γ‐tubulin small complex (γ‐TuSC), are of equal abundance, in line with structural data (Kollman et al, 2015). GCP5 and GCP6 are also present in stoichiometric amounts, but about three times less abundant than GCP2 or GCP3, and GCP4 is nearly twice as abundant as GCP5 and GCP6, but only half as abundant as GCP2 and GCP3. Finally, three proteins associated with the γ‐TuRC, Nedd1 (Haren et al, 2006; Luders et al, 2006) and Mozart 1, and Mozart 2 (Hutchins et al, 2010; Teixido‐Travesa et al, 2010) are similarly abundant as the GCP2 and GCP3 core components of γ‐TuSC. These data extend previous biochemical studies (Murphy et al, 2001; Choi et al, 2010) and support the notion that multiple γ‐TuSCs associate with GCP4, GCP5, and GCP6 into γ‐TuRCs (Kollman et al, 2011; Lin et al, 2015).
EGFP‐tagging of endogenous proteins to measure localized protein abundance
To obtain information about absolute numbers of proteins at centrosomes, we complemented the above‐mentioned mass spectrometry‐based approaches by an independent, fluorescence‐based quantification strategy. Specifically, we used adeno‐associated virus (AAV) for homologous recombination (Berdougo et al, 2009) to engineer EGFP reporter groups into one allele each of the endogenous genes coding for γ‐tubulin, Sas‐6, and STIL in RPE‐1 cells. Tagging of these centrosomal proteins did not detectably affect either cell growth or centriole numbers (Fig EV2). Following selection of clones expressing EGFP‐tagged transgenes (Figs EV3A, EV4A and EV5A), using either FACS (γ‐tubulin) or an excisable Neo resistance cassette (Sas‐6 and STIL), correct integration was ascertained by PCR. Western blotting demonstrated an approximately 50% reduction in the expression of the endogenous γ‐tubulin, Sas‐6, and STIL proteins (compared to parental RPE‐1 cells) and expression of the EGFP‐tagged counterparts at approximately equal levels (Figs EV3B, EV4B and EV5B). Moreover, fluorescence microscopy demonstrated centrosomal localization of all three EGFP‐tagged proteins, γ‐tubulin, Sas‐6 and STIL (Figs EV3C, EV4C and EV5C), and EGFP‐tagged γ‐tubulin and endogenous γ‐tubulin showed similar enrichments in centrosome fractions (Fig EV3D). We conclude that EGFP‐tagging through homologous recombination did not interfere with the expression or subcellular localizations of the corresponding proteins, supporting the assumption that EGFP signals recovered from tagged proteins represent roughly 50% of the corresponding protein at any given location.
Figure EV2. Cell cycle properties of EGFP‐tagged cell lines.
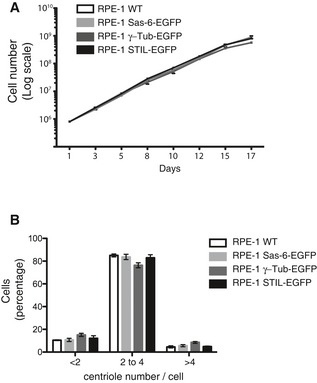
- EGFP‐tagging of centrosomal proteins does not detectably affect cell proliferation. The indicated cell lines were grown under standard conditions, and cell numbers counted at the indicated times. Note that all growth curves are nearly identical.
- EGFP‐tagging of centrosomal proteins does not detectably affect centriole number. Cells of the indicated genotype were grown on coverslips, and centriole numbers were counted after staining with anti‐CP110 antibodies (immunofluorescence microscopy). Histogram shows average of two experiments; results presented are means ± SEM. Note that the vast majorities of cells showed between 2 and 4 centrioles (as expected for G1‐ and G2‐phase cells, respectively), with no discernible differences between cell lines.
Figure EV3. Generation of RPE‐1 cells expressing endogenously tagged γ‐tubulin‐EGFP.
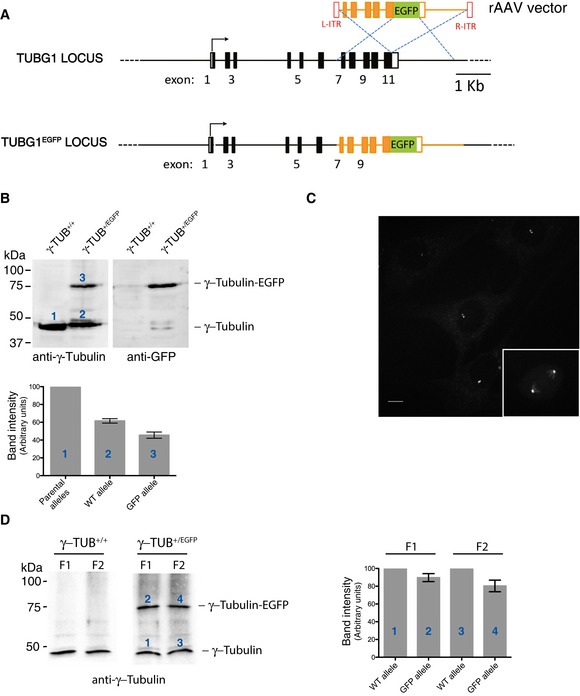
- Schematic representation of wild‐type (WT) TUBG1 locus and the targeted allele obtained after rAAV‐mediated homologous recombination. Black rectangles represent exons and black lines introns. After insertion of the EGFP coding sequence, cells expressing γ‐tubulin‐EGFP were selected by FACS.
- Western blot analysis of parental RPE‐1 and γ‐tubulin‐EGFP‐expressing RPE‐1 cells. Protein extracts were probed with anti‐γ‐tubulin (left panel) and anti‐GFP (right panel) antibodies. Histogram shows quantification of the numbered bands; average of two experiments. Results presented are means ± SEM. Band intensity for WT alleles was set to 100%.
- Maximum projection fluorescence microscopy image showing asynchronously growing RPE‐1 cells expressing γ‐tubulin‐EGFP. Inset shows mitotic cell (metaphase). Scale bar: 10 μm.
- Centrosomes were purified from parental RPE‐1 cells (left panel) or γ‐tubulin‐EGFP‐expressing RPE‐1 cells (right panel), using a previously described procedure (Blomberg‐Wirschell & Doxsey, 1998). In brief, starting with material derived from 2 × 15 cm plates, method 1 was employed with minor modifications. The first two centrosomal fractions (F1, F2; 0.5 ml each) were collected and concentrated by TCA precipitation prior to Western blot analysis. Membranes were probed by anti‐γ‐tubulin antibody. Histogram shows quantification of the numbered bands; average of two experiments. Results presented are means ± SEM. Band intensity for WT alleles was set to 100%. Note that both centrosome fractions (F1 and F2) contained WT γ‐tubulin and EGFP‐tagged γ‐tubulin in similar proportions.
Figure EV4. Generation of RPE‐1 cells expressing endogenously tagged STIL‐EGFP.
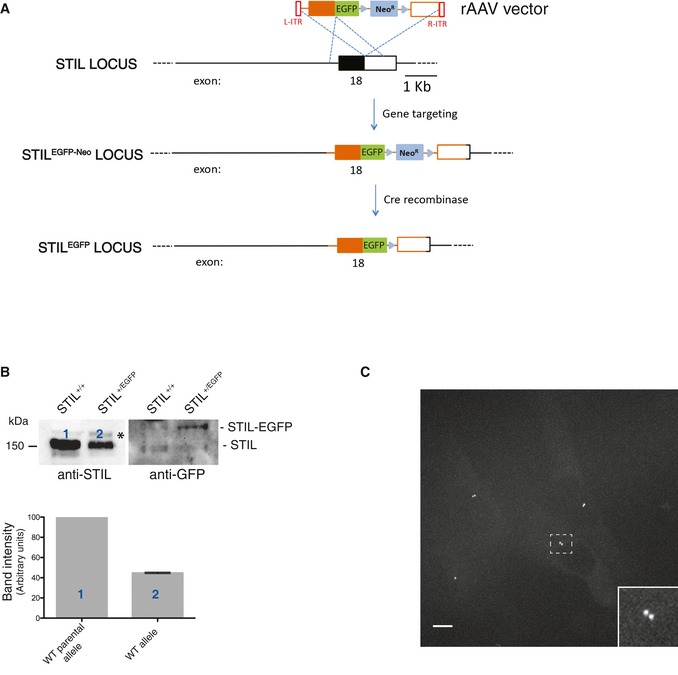
- Schematic representation of WT STIL locus and the targeted STIL allele obtained after rAAV‐mediated homologous recombination. Black rectangles represent exons and black lines introns. After insertion of the EGFP coding sequence and the G418 resistance cassette, cells were selected by addition of G418 to the medium. Then, the neomycin cassette was removed using an adenovirus‐expressed Cre recombinase (Vector Biolabs, Malvern, PA, USA), and clones were isolated by serial dilution (loss of resistance to G418 was confirmed by replica plating). Finally, positive cells were confirmed by fluorescence microscopy.
- Western blot analysis of parental RPE‐1 and STIL‐EGFP‐expressing RPE‐1 cells. Protein extracts were probed with anti‐STIL (left panel; asterisks denotes background band) and anti‐GFP (right panel) antibodies. Note that only the anti‐GFP antibody recognizes STIL‐EGFP; lack of reactivity by the anti‐STIL antibody almost certainly reflects interference of the EGFP tag with the epitope recognized by this antibody (this antibody was raised against a peptide corresponding to the C‐terminus of STIL (last 50 amino acids), and involvement of the C‐terminus in the fusion with EGFP makes interference plausible). Histogram shows quantification of the numbered bands; average of two experiments. Results presented are means ± SEM. Band intensity for WT alleles was set to 100%.
- Maximum projection fluorescence microscopy image showing asynchronously growing RPE‐1 cells expressing STIL‐EGFP. Scale bar: 10 μm. The inset shows an enlargement of the boxed interphase centrosome.
Figure EV5. Generation of RPE‐1 cells expressing endogenously tagged Sas‐6‐EGFP.
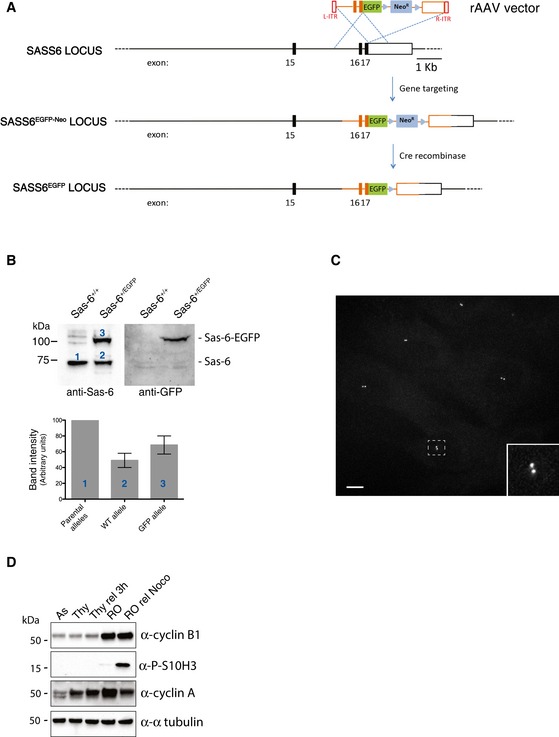
- Scheme of WT SASS6 locus and the targeted SASS6 allele obtained by rAAV‐mediated homologous recombination. Black rectangles represent exons and black lines introns. After insertion of the EGFP coding sequence and the G418 resistance cassette, cells were selected by addition of G418 in the medium. Then, the neomycin cassette was removed using an adenovirus‐expressed Cre recombinase (Vector Biolabs, Malvern, PA, USA), and clones were isolated by serial dilution (loss of resistance to G418 was confirmed by replica plating). Finally, positive cells were confirmed by fluorescence microscopy.
- Western blot analysis of parental RPE‐1 and Sas‐6‐EGFP‐expressing RPE‐1 cells. Protein extracts were probed with anti‐Sas‐6 (left panel) and anti‐GFP (right panel) antibodies. Histogram shows quantification of the numbered bands. Results presented are means ± SEM. Band intensity for WT alleles was set to 100%.
- Maximum projection fluorescence microscopy image showing asynchronously growing RPE‐1 cells expressing Sas‐6‐EGFP. Scale bar: 10 μm. The inset shows an enlargement of the boxed interphase centrosome.
- Efficacy of cell cycle synchronization was demonstrated by subjecting cell lysates to Western blotting with the indicated antibodies against cell cycle markers.
To quantify the number of EGFP‐tagged γ‐tubulin molecules at centrosomes, we first analyzed RPE‐1 cells that had been synchronized in G2; this allowed for a clear distinction of the two centrosomes present at this cell cycle stage. Quantification was then performed using two independent, complementary approaches. First, EGFP signals were measured in three‐dimensional space, using masks to integrate fluorescence intensities over the entire cells or the duplicated centrosomes only (Fig 4A). This analysis suggests that only a small fraction of the total cellular γ‐tubulin, only ~1%, is associated with a G2‐phase centrosome doublet (see Materials and Methods). Although this number may appear surprisingly small, we note that a previous cell fractionation study also concluded that at least 80% of γ‐tubulin exists in a cytosolic pool (Moudjou et al, 1996). Knowing from SRM measurements that the signal intensity measured for whole RPE‐1 cells corresponds to about 257,000 copies of γ‐tubulin, the relative signal intensity at G2 centrosome doublets implies that each of the two centrosomes harbors 1,383 (+/− 44) copies of γ‐tubulin (Fig 4C). To corroborate this estimate by a second, independent method for determining EGFP signal intensity at centrosomes, we used EGFP‐tagged virus‐like particles (VLPs) for calibration of fluorescence signals (Fig 4B). Taking into account that the signal emanating from a typical VLP particle represents 120 molecules (Charpilienne et al, 2001; Dundr et al, 2002), we calculate that centrosomes typically harbor about 1,290 (+/− 46) γ‐tubulin molecules. Thus, the two methods for EGFP quantification yield remarkably similar results and point to a mean of approximately 1,340 (+/− 32) copies of γ‐tubulin per interphase centrosome (Fig 4C). Hence, this number had been used for calibration of protein abundance at purified centrosomes (see above).
The availability of cell lines expressing endogenously EGFP‐tagged centrosomal proteins offered a straightforward approach for measuring centrosomal protein abundance as a function of cell cycle progression. Following synchronization of the three tagged RPE‐1 lines at different cell cycle stages, centrosome‐associated EGFP signals were quantified using the VLP particles for standardization. Levels of γ‐tubulin at centrosomes remained fairly constant at about 1,200 molecules during G1, S and G2 phases, but increased approximately 3.5‐fold at the G2 to M transition (Fig 5A). This is fully consistent with data demonstrating that γ‐tubulin is recruited to centrosomes in preparation of spindle formation at the onset of mitosis (Zheng et al, 1991; Lajoie‐Mazenc et al, 1994; Lane & Nigg, 1996; Khodjakov & Rieder, 1999). Similarly, the number of Sas‐6 molecules at centrosomes rose from ~346 in G1/S arrested cells to 440 in G2/M cells (Fig 5B) and those of STIL from 167 to 318 (Fig 5C). Neither Sas‐6‐EGFP nor STIL‐EGFP could be detected at centrosomes in mitotic cells (Fig 5B and C), consistent with ubiquitin‐dependent proteolytic degradation of both proteins (Strnad et al, 2007; Tang et al, 2011a; Arquint et al, 2012; Vulprecht et al, 2012; Arquint & Nigg, 2014).
Assuming that 25% of cells are typically in late G2 (Keller et al, 2014), a weighted average of the above numbers results in 383 and 212 molecules of Sas‐6 and STIL, respectively, per S‐ and G2‐phase centrosome. Because total cellular fluorescence of Sas‐6 and STIL was too weak to be measured, these values could not be corroborated by comparing EGFP signal intensity with SRM data. This notwithstanding, these results support our conclusion that centrosome‐associated Sas‐6 is about twice as abundant as STIL. Furthermore, they are reasonably similar to the values inferred for Sas‐6 (170 molecules) and STIL (72 molecules) on purified S‐ and G2‐phase KE37 centrosomes (after calibrating data to 1,340 molecules of γ‐tubulin; Fig 2C). Taking the average of the two approaches, our data indicate that S‐ and G2‐phase centrosomes harbor approximately 276 copies of Sas‐6 and about 142 copies of STIL. Together with corresponding estimates for many more centrosomal proteins (Fig 3), these numbers set the stage for approaching the assembly and architecture of this organelle from a quantitative perspective.
Discussion
To fully understand, and ultimately model, any complex biological process in mechanistic terms, it will be necessary to obtain quantitative information on the abundance and dynamics of all participant components. Although impressive progress has been made toward the characterization and quantification of comprehensive transcriptomes, proteomes, and interactomes, it often remains difficult to precisely quantify the abundance of individual proteins, particularly if these are expressed at low levels. Further compounding this challenge are frequent posttranslational modifications, and temporal changes in expression and/or subcellular localization. Most of the centrosomal proteins studied here are expected to be expressed at low levels, and information about their abundance remains scarce. In comprehensive proteomics studies, key regulators of centriole duplication, including Plk4, Sas‐6, and STIL, were barely detectable, if they were detected at all (Beck et al, 2011; Nagaraj et al, 2011). For example, one extensive mass spectrometry‐based analysis of U2OS cells concluded that Sas‐6 is expressed at fewer than 500 copies per cell (Beck et al, 2011). In striking contrast, an antibody‐based study, correlating immunofluorescence signal intensities with semi‐quantitative Western blotting in the same cell line, estimated Sas‐6 to be present at 185,000 copies in early S phase and 885,000 copies in late G2 phase (Keller et al, 2014). Such discrepancies in estimates, differing by several orders of magnitude, clearly document the need for quantitative approaches that are focused on specific proteins of interest.
SRM mass spectrometry in combination with stable isotope dilution is a powerful approach for protein quantification and offers major advantages over antibody‐based strategies (Addona et al, 2009). Not only does the method circumvent the need for antibody production, but it also provides information about absolute rather than relative amounts of protein. However, to reliably detect and quantify low abundance centrosomal proteins from cell extracts, we found that sample fractionation was inevitable. This resulted in large numbers of mass spectrometry runs and made it impractical to use SRM for monitoring expression profiles through the cell cycle. To partially alleviate these limitations and at the same time increase confidence in the accuracy of our estimates, we have combined SRM measurements with a second, independent approach centered on EGFP‐tagging of endogenous proteins. Microscopy‐based protein quantification through measurement of fluorescence intensity offers exquisite spatial and temporal resolution. Moreover, analysis of endogenous proteins tagged through homologous recombination minimizes concerns related to non‐physiological expression of tagged transgenes. However, possible interference of the fluorescent tag with protein function remains a concern. In addition, quantitative measurements of fluorescence signals are complicated by photo‐bleaching, quenching of signal intensity, and the difficulty of dependable standardization (Joglekar et al, 2008; Lawrimore et al, 2011; Coffman & Wu, 2012). Thus, we are reassured by the good concordance between results obtained through use of the two complementary approaches.
Plk4, the master regulator of centriole duplication, has generally been difficult to detect, leading to the assumption that this kinase is expressed “at low levels”. Our measurements now allow us to put a specific number to this term. Levels of Plk4 are remarkably similar in all cells analyzed, in line with the notion that human Plk4 is subject to tight autoregulation through proteolytic degradation (Guderian et al, 2010; Holland et al, 2010). STIL is expressed at similarly low levels as Plk4, but expression of Sas‐6 shows surprisingly high variation between cell lines. Considering that STIL and Sas‐6 are very low during most of G1 phase, an approximate doubling of the measured values likely provides a realistic estimate for S‐ and G2‐phase cells. For U2OS cells, we thus estimate that Sas‐6 levels may reach roughly 50,000 copies per S‐ and G2‐phase cell. This number is about 10 times lower than a previous estimate (Keller et al, 2014). We cannot definitively explain this discrepancy, but would surmise that a combination of mass spectrometry‐based SRM with EGFP‐tagging of endogenous centrosomal proteins is more likely to produce accurate results than a combination of immunofluorescence staining with semi‐quantitative Western blotting. In any case, it might in future be rewarding to explore whether deregulation of Sas‐6 may contribute to numerical and/or structural centrosome aberrations in human tumors.
As expected, the PCM component γ‐tubulin is expressed at more than 10 times higher levels than most other proteins analyzed here, ranging from 180,000 to 320,000 copies per cell, depending on cell line. These values are in good agreement with estimates reported in previous large‐scale proteomics studies (Nagaraj et al, 2011; Kulak et al, 2014; Hein et al, 2015), and this adds confidence to our use of γ‐tubulin data for calibration purposes. Also, we emphasize that although this calibration is critical for calculations of exact absolute copy numbers, any future correction of γ‐tubulin abundance would not affect relative numbers or any of the major conclusions.
As detailed in the Results section, our data make several predictions of biological relevance. These relate to the stoichiometry of γ‐TuRCs, the composition of subdistal and distal appendages, intracentriolar linker structures, as well as proteins implicated in centriole length control. Most intriguing are the implications of our quantitative data for cartwheel structure in human centrioles. First and foremost, our data predict that cartwheels comprise one molecule of STIL for every Sas‐6 dimer. It will be interesting to see whether future structural studies confirm or refute this prediction. Second, our data allow us to make a prediction on the number of stacked hubs in the cartwheels of human centrioles. Because visualization of cartwheels by electron microscopy has proven difficult in human cells, the number of stacks is unknown. Considering that each stacked hub is predicted to comprise 18 molecules (nine dimers) of Sas‐6 (van Breugel et al, 2011; Kitagawa et al, 2011), our prediction of 276 molecules of Sas‐6 per cartwheel‐containing centriole theoretically allows for the assembly of 15–16 stacks. This likely represents an upper limit, because not all Sas‐6 and STIL proteins are necessarily assembled into cartwheels at all times (Fong et al, 2014; Keller et al, 2014) and we also recognize that the number of stacks may differ between cell types. Nevertheless, it is interesting to compare our prediction with estimates of cartwheel thickness, albeit collected from centrioles of different organisms. Electron microscopy and cryo‐electron tomography suggest that cartwheels in most species display a thickness in the order of 100 nm (Dippell, 1968; Cavalier‐Smith, 1974; Nakazawa et al, 2007; Guichard et al, 2012, 2013; O'Toole & Dutcher, 2014). A similar value (60–150 nm) was reported in a recent study using 3D‐STORM imaging of stable Sas‐6‐positive structures, likely reflecting cartwheels, in detergent‐extracted human U2OS cells (Keller et al, 2014). Assuming that stacked hubs display a vertical periodicity of 8.5 nm (Kitagawa et al, 2011; Guichard et al, 2012, 2013; Li et al, 2012), a cartwheel thickness of 60–150 nm would thus allow for ~7–17 stacks. These numbers, albeit tentative, are encouragingly close to our estimate of 15–16 stacked hubs per cartwheel.
From a more general perspective, our study demonstrates that protein abundance at centrosomes does not scale with protein abundance in total cell lysates. This underscores the importance of posttranslational mechanisms for regulating centrosome assembly. Nevertheless, in view of the striking structural centrosome aberrations that are commonly observed in tumor cells (Lingle et al, 2002; Guo et al, 2007; Schnerch & Nigg, 2016), it may be rewarding to study whether massively enlarged centrosomes can be attributed to upregulation of critical matrix proteins (Woodruff et al, 2015). Recent evidence suggests that many non‐membrane‐bound organelles, including centrosomes, display properties of liquids (Hyman et al, 2014), and protein concentration is expected to influence these processes. In particular, coalescence of matrix molecules into droplet‐like assemblies may play a crucial role in the control of centrosome size (Brangwynne, 2013).
In conclusion, our study has addressed the challenges associated with extracting accurate quantitative information on key centrosomal proteins. It provides insight into the abundance of these proteins in human cells, as well their general stoichiometry within the human centrosome. This quantitative information provides a benchmark for validation of structural studies, and it will hopefully guide future biochemical and mechanistic studies. We anticipate that future modeling of centrosome assembly in time and space will require additional quantitative data, including detailed information on splice forms and proteins carrying specific posttranslational modifications. Acquisition of such data will almost certainly require major technological breakthroughs.
Materials and Methods
Cell culture and synchronization
Both wild‐type and genetically engineered TERT‐immortalized human retinal pigment epithelial (RPE‐1) cells were cultured in DMEM:F12 (Sigma‐Aldrich, St. Louis, MO, USA) supplemented with 10% FCS (Life technologies, CA, USA), 2 mM glutamine (Sigma‐Aldrich, St. Louis, MO, USA), 0.26% sodium bicarbonate (Sigma‐Aldrich, St. Louis, MO, USA), and Pen/Strep 100 μg/ml (Life technologies, Carlsbad, CA, USA). HeLa S3, U2OS, and HEK293T cells were grown in DMEM–Glutamax medium (Life technologies, Carlsbad, CA, USA), 10% FCS, and KE37 cells in DMEM medium containing 0.1% Pluronic solution and 10% FCS. Cells were maintained in a 37°C incubator with 5% CO2 and regularly tested for mycoplasma. Cell cycle synchronization was carried out as follows: to obtain an early S‐phase population, cells were treated for 18 h with 5 mM thymidine (Sigma‐Aldrich, MO, USA); a late S/G2 population was prepared by arresting cells for 18 h with 5 mM thymidine and then releasing them for 3 h into fresh medium; for a late G2 population, cells were treated for 18 h with 10 μM RO‐3306 (Merck Millipore, Darmstadt, Germany); for arrest in early mitosis, cells were arrested with 10 μM RO‐3306 for 18 h and then released for 2 h into 50 nM nocodazole (Sigma‐Aldrich, MO, USA).
Western blotting
Cells were washed in PBS and lysed in the following buffer: 50 mM Tris–HCl, pH 7.4, 0.5% IGEPAL (Sigma‐Aldrich, St. Louis, MO, USA), 150 mM NaCl, 1 mM DTT, 5% glycerol, 50 mM NaF, 1 mM PMSF, 25 mM β‐glycerophosphate, 1 mM vanadate, Complete Mini Protease Inhibitor Cocktail (Roche Diagnostics, Basel, Switzerland). Proteins were then resolved by SDS–PAGE and transferred onto a nitrocellulose membrane. Primary antibodies were directed against α‐tubulin (clone DM1A, 1:5,000; Sigma‐Aldrich, St. Louis, MO, USA), γ‐tubulin (clone GTU‐88, 1:5,000; Sigma‐Aldrich, St. Louis, MO, USA), GFP (ab290; 1:5,000; Abcam, Cambridge, UK), STIL (ab89314, 1:2,000; Abcam, Cambridge, UK), Sas‐6 (Kleylein‐Sohn et al, 2007), CP110 (1:2,000, Schmidt et al, 2009), cyclin A (1:2,000, Maridor et al, 1993), cyclin B1 (05‐373, 1:2,000, Merck Millipore, Darmstadt, Germany), and phospho‐histone H3/serine 10 (3377, 1:1,000, Cell Signaling Technology, Danvers, MA, USA); secondary antibodies were HRP‐conjugated anti‐mouse immunoglobulin (170‐6516, 1:3,000, Bio‐Rad, Hercules, CA, USA) or anti‐rabbit immunoglobulin (170‐6515, 1:3,000, Bio‐Rad, Hercules, CA, USA).
rAAV‐mediated gene targeting
Homology arms to human γ‐tubulin (TUBG1), human STIL (STIL), and human Sas‐6 (SASS6) genes were amplified from hTERT‐RPE‐1 genomic DNA using PfuUltra II (Agilent technologies, Santa Clara, CA, USA). Targeting constructs were obtained by four‐piece DNA ligation with a NotI‐digested pAAV vector backbone, the two homology arms and the coding sequence of EGFP (fused to a neomycin‐resistant cassette where indicated). For details, see Figs EV3, EV4 and EV5. Production of recombinant adenovirus‐associated virus (rAAV) particles and infection of cells were carried out as described previously (Berdougo et al, 2009; von Schubert et al, 2015). To establish the γ‐tubulin‐EGFP cell line, a FACSAria IIIu (BD Biosciences, San Jose, CA; USA) was used to isolate cells based on fluorescence. Specifically, a 514/30 nm bandpass filter was used against a 585/42 nm bandpass filter to discriminate between positive cells and background fluorescence. Cells were recovered on a 96‐well plate and positive cells screened by fluorescence microscopy. For isolation of cell lines harboring EGFP‐tagged STIL and Sas‐6, fluorescence signals were too weak to permit sorting by FACS. Thus, STIL‐EGFP and Sas‐6‐EGFP lines were generated using neomycin selection, as described previously (Kohli et al, 2004). In brief, neomycin‐resistant clones were screened by PCR on genomic DNA for correct integration. Then, the neomycin cassette was removed using an adenovirus‐expressed Cre recombinase (Vector Biolabs, Malvern, PA, USA), and clones were isolated by serial dilution (loss of resistance to G418 was confirmed by replica plating). Finally, positive cells were confirmed by fluorescence microscopy.
Microscopy and quantification of fluorescence
Cells were grown on μ‐Slide 8‐well slides (Ibidi, Martinsried, Germany) and imaged in DMEMGFP‐2 medium (Evrogen, Moscow Evrogen, Russia). All imaging was performed with a confocal spinning‐disk microscope (Intelligent Imaging Innovations, Denver, CO, USA) based on a Zeiss Axio Observer stand equipped with CSU‐X1 head (Yokogawa), a Photometric Evolve 512 back‐illuminated EMCCD camera, and a 63x/NA1.4 Plan‐Apochromat objective. For interphase cells, 50 Z sections of 0.27 μm were taken with 50 ms exposure time; for mitotic cells, 100 Z sections were taken to cover the whole cell. For quantification of fluorescence, two methods were used. In a first method, SRM data were used to calibrate signal intensities. For this approach, masks covering either a whole cell or a centrosome were generated in ImageJ based on intensity thresholding using the “3D objects counter” plugin. The integrated fluorescence intensity obtained after background subtraction for the whole cell was then correlated with the corresponding protein copy number obtained by SRM measurements, and the integrated fluorescence intensity obtained for the centrosome (1.076% of total fluorescence) allowed us to calculate the percentage of total protein associated with this structure. In a second method, fluorescence was calibrated by comparison to fluorescently labeled virus particles. To this end, GFP‐VLP2/6 virus‐like particles (stored in 20 mM PIPES, 10 mM CaCl2, 500 mM CsCl, pH 6.8) were diluted 900‐fold into 60 mM PIPES 27.3 mM Hepes, 10 mM CaCl2, pH 6.8), spun onto μ‐Slide 8‐well slides coated with poly‐L‐lysine (Ibidi, Martinsried, Germany), and imaged with the same settings as described above. The integrated fluorescence intensity was again obtained using “3D objects counter” from ImageJ. After background subtraction, the different values obtained from ~1,000 VLP were binned into a distribution histogram. A subpopulation, representing roughly 60% of all VLP measurements and falling within a twofold range of intensity, was then averaged. The resulting value was then attributed to the 120 VLP‐associated copies of GFP molecules and used for calibration of cellular and centrosomal GFP signals.
Mass spectrometry: SRM analysis of cells
The development of SRM assays is described in detail in Appendix Fig S1. For SRM analysis of whole‐cell lysates, cells were trypsinized, washed with PBS, and counted using a Scepter 2.0 cell counter (Merck Millipore, Darmstadt, Germany). After low‐speed centrifugation, cell pellets were lysed in lysis buffer [0.1 M ammonium bicarbonate, 8 M urea, 0.1% RapiGest (v/v)], subjected to three cycles of 30 s sonication and 5 min vortexing at 25°C. Following centrifugation at 20,000 g for 5 min at 4°C, supernatants were analyzed for protein content using the BCA assay (Pierce, Fisher Scientific, Perbio Science Switzerland SA, Lausanne, Switzerland). About 0.5–1 mg of protein was diluted with lysis buffer to a final concentration of 2.5 μg/μl. Samples were reduced in 5 mM TCEP (tris‐2‐carboxyethyl‐phosphine) at 37°C for 60 min, alkylated in 10 mM iodoacetamide at 25°C for 30 min in the dark, and incubated in 12.5 mM N‐acetylcysteine at 25°C for 10 min. Samples were then diluted 1:4 with 0.1 M ammonium bicarbonate and digested overnight with trypsin (Promega, Madison, WI, USA) at an enzyme‐to‐substrate ratio of 1:20 (w/w). The samples were supplemented with 250 fmol/mg of AQUA peptides (Thermo Scientific, Waltham, MA, USA), before digestion was stopped and RapiGest was cleaved with 0.5% trifluoroacetic acid and 50 mM HCl. After centrifugation at 20,000 g for 5 min at 4°C, peptides in the supernatant were desalted by solid‐phase extraction according to the manufacturer's instructions (C18 Sep‐Pak Vac columns, Waters, Baden‐Dättwil, Switzerland). Purified peptides were dried at 45°C under vacuum and resuspended in 200 μl of 10% ACN/90% water (v/v) and subjected to Off‐Gel electrophoresis (OGE) using 24‐cm strips with a pH range from 3 to 10 (3100 OFFGEL Fractionator, Agilent technologies, Santa Clara, CA, USA). The 24 OGE fractions were purified using microspin solid‐phase extraction C18 columns according to the manufacturer's instructions (The Nest Group, Southborough, MA, USA), dried at 45°C under vacuum, resuspended in an aqueous solution containing 5% ACN/0.15% formic acid, and subjected to SRM analysis in a TSQ Vantage mass spectrometer (Thermo Scientific, Waltham, MA, USA) as recently described (Bauer et al, 2014). Cycle time was set to 2 s resulting in a dwell time of 20 ms per transition. The transition lists with optimized collision energies for all peptides were reported previously (Bauer et al, 2014). Ratios between heavy‐labeled AQUA peptides and light endogenous peptides were determined using the Skyline software (MacLean B. et al, Bioinformatics, 2010; version 1.3). Notably, the transition intensities of peptides found across multiple fractions were summed before ratio calculation. For each protein, except Cep152, two peptides were used and average values determined; in case of Cep152, only one peptide (INEVLAAAK) recognized both long and short isoforms (Sonnen et al, 2013).
Mass spectrometry: SRM analysis of purified centrosomes
Centrosomes were purified as described previously (Bornens & Moudjou, 1999; Andersen et al, 2003). For analysis by immunofluorescence microscopy, 20 μl of the centrosomal fraction was diluted in 5 ml of 10 mM PIPES, pH 7.2 and spun onto coverslips at 23,600 g for 10 min at 4°C in a HB4 rotor. Coverslips were fixed in methanol at −20°C for 10 min, washed with PBS/0.1% Tween‐20, blocked for 30 min with PBS/0.1% Tween‐20/0.5% BSA, and incubated with anti‐centrosome antibodies diluted in PBS/0.1% Tween‐20/0.5% BSA for 90 min. After three washes with PBS/0.1% Tween‐20, the coverslips were mounted on glass slides and imaged using a 3i spinning‐disk confocal microscope. For analysis by mass spectrometry, 30–50 μl of the centrosomal fraction was supplemented with 10 M urea/0.1 M ammonium bicarbonate/0.1% RapiGest to a final concentration of 8 M urea. Samples were then subjected to three cycles of 30 s sonication and 5 min vortexing at 25°C, and lysates were digested with 1:500 (w/w) enzyme‐to‐substrate ratio endoproteinase LysC (Wako Diagnostics, Richmond, VA, USA) for 4 h at 37°C. Lysates were then diluted 1:4 with 0.1 M ammonium bicarbonate and digested with trypsin (Promega, Madison, WI, USA) 1:20 (w/w) enzyme‐to‐substrate ratio for 12 h at 37°C. The samples were supplemented with 10 fmol of AQUA peptides (Thermo Scientific, Waltham, MA, USA). The digestion was stopped, and RapiGest was cleaved with 0.5% trifluoroacetic acid and 50 mM HCl. After centrifugation at 20,000 g for 5 min at 4°C, peptides in the supernatant were desalted by solid‐phase extraction, purified using macro‐spin solid‐phase extraction C18 columns according to the manufacturer's instructions (The Nest Group, Southborough, MA, USA), dried at 45°C under vacuum, resuspended in an aqueous solution containing 5% ACN/0.15% formic acid, and subjected to SRM analysis in a TSQ Vantage mass spectrometer or shotgun analysis in a LTQ Velos mass spectrometer as described below (both Thermo Scientific, Waltham, MA, USA). Ratios between heavy‐labeled AQUA peptides and light endogenous peptides were determined using the Skyline software (MacLean et al, 2010).
Mass spectrometry: shotgun LC‐MS analysis of centrosome preparations
The centrosome preparations were analyzed by shotgun proteomics as described previously (Glatter et al, 2012) with minor modifications. The hybrid Orbitrap‐Velos mass spectrometer was interfaced to a nanoelectrospray ion source coupled online to an Easy‐nLC 1000 system (all Thermo Scientific, Waltham, MA, USA). About 1 μg of peptides was separated on a RP‐LC column (75 μm × 50 cm) packed in‐house with C18 resin (Reprosil‐AQ Pur, 1.9 μm; Dr. Maisch) using a linear gradient from 95% solvent A (98% water, 2% acetonitrile, 0.15% formic acid) and 5% solvent B (98% acetonitrile, 2% water, 0.15% formic acid) to 30% solvent B over 180 min at a flow rate of 0.2 μl/min. Each survey scan acquired in the Orbitrap at 60,000 FWHM was followed by 20 MS/MS scans of the most intense precursor ions in the linear ion trap. Preview mode was enabled, and dynamic exclusion was set for 60 s. Charge state screening was employed to select for ions with at least two charges and rejecting ions with undetermined charge state. The normalized collision energy was set to 32%, and one microscan was acquired for each spectrum. Notably, two LC‐MS analyses were carried out per sample.
For data analysis, the acquired raw files were imported into the Progenesis LC‐MS software (v4.0, Nonlinear Dynamics Limited), which was used to extract peptide precursor ion intensities across the samples applying the default parameters. The generated mgf‐files were searched using MASCOT (Matrix Science, version 2.4) against a decoy database (consisting of forward and reverse protein sequences) of the predicted SwissProt entries of Homo sapiens (www.ebi.ac.uk, release date 16/05/2012) and commonly observed contaminants (in total 41,250 sequences) generated using the SequenceReverser tool from the MaxQuant software (version 1.0.13.13). The search criteria were set as follows: Full tryptic specificity was required (cleavage after lysine or arginine residues, unless followed by proline); two missed cleavages were allowed; carbamidomethylation (C) was set as fixed modification; oxidation (M) was applied as variable modifications; mass tolerance of 10 ppm (precursor) and 0.6 Da (fragments). The database search results were filtered using the ion score to set the false discovery rate (FDR) to 1% on the peptide and protein level, respectively, based on the number of reverse protein sequence hits in the datasets. After normalizing the quantitative data of the technical replicates using the SafeQuant software tool (Glatter et al, 2012), the summed precursor intensities obtained from Progenesis for each centrosome preparation were aligned with the absolute protein concentrations determined by SRM analysis according to the iBAQ method (Schwanhausser et al, 2011), respectively. The resulting models were applied to estimate absolute protein levels for all proteins quantified in two centrosome preparations, and data were combined by calculating mean protein levels.
Mass spectrometry: accession codes
The raw mass spectrometric data used in this study and the Mascot analysis files are available via ProteomeXchange (accession code PXD003927).
Author contributions
MB performed all of the proteomics experiments, with assistance by AS; FC performed the fluorescence tagging of endogenous proteins in cell lines; EAN conceived the study and wrote the manuscript. All authors contributed to editing the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Acknowledgements
The authors thank Elena Nigg for expert technical assistance, Jonathon Pines and Prasad Jallepalli for reagents and advice concerning rAAV‐mediated gene tagging, and Didier Poncet (Institut de Biologie Integrative de la Cellule, UMR 9198, Gif sur Yvette, France) for the virus particles (GFP‐VLP2/6). We also thank all members of the Nigg laboratory as well as members of the FACS, proteomics, and imaging core facilities of the Biozentrum for assistance and helpful discussions. This work was supported by the Swiss National Science Foundation (310030B_149641) and the University of Basel.
The EMBO Journal (2016) 35: 2152–2166
References
- Addona TA, Abbatiello SE, Schilling B, Skates SJ, Mani DR, Bunk DM, Spiegelman CH, Zimmerman LJ, Ham AJ, Keshishian H, Hall SC, Allen S, Blackman RK, Borchers CH, Buck C, Cardasis HL, Cusack MP, Dodder NG, Gibson BW, Held JM et al (2009) Multi‐site assessment of the precision and reproducibility of multiple reaction monitoring‐based measurements of proteins in plasma. Nat Biotechnol 27: 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agircan FG, Schiebel E, Mardin BR (2014) Separate to operate: control of centrosome positioning and separation. Philos Trans R Soc Lond B Biol Sci 369: Article ID: 20130461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahrne E, Molzahn L, Glatter T, Schmidt A (2013) Critical assessment of proteome‐wide label‐free absolute abundance estimation strategies. Proteomics 13: 2567–2578 [DOI] [PubMed] [Google Scholar]
- Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M (2003) Proteomic characterization of the human centrosome by protein correlation profiling. Nature 426: 570–574 [DOI] [PubMed] [Google Scholar]
- Arike L, Valgepea K, Peil L, Nahku R, Adamberg K, Vilu R (2012) Comparison and applications of label‐free absolute proteome quantification methods on Escherichia coli . J Proteomics 75: 5437–5448 [DOI] [PubMed] [Google Scholar]
- Arquint C, Sonnen KF, Stierhof YD, Nigg EA (2012) Cell‐cycle‐regulated expression of STIL controls centriole number in human cells. J Cell Sci 125: 1342–1352 [DOI] [PubMed] [Google Scholar]
- Arquint C, Nigg EA (2014) STIL microcephaly mutations interfere with APC/C‐mediated degradation and cause centriole amplification. Curr Biol 24: 351–360 [DOI] [PubMed] [Google Scholar]
- Arquint C, Gabryjonczyk AM, Nigg EA (2014) Centrosomes as signalling centres. Philos Trans R Soc Lond B Biol Sci 369: 20130464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arquint C, Gabryjonczyk AM, Imseng S, Bohm R, Sauer E, Hiller S, Nigg EA, Maier T (2015) STIL binding to Polo‐box 3 of PLK4 regulates centriole duplication. Elife 4: e07888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arquint C, Nigg EA (2016) The PLK4–STIL–SAS‐6 module at the core of centriole duplication. Biochem Soc Trans doi: 10.1042/BST20160116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azimzadeh J, Marshall WF (2010) Building the centriole. Curr Biol 20: R816–R825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azimzadeh J, Wong ML, Downhour DM, Sanchez Alvarado A, Marshall WF (2012) Centrosome loss in the evolution of planarians. Science 335: 461–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M, Ahrne E, Baron AP, Glatter T, Fava LL, Santamaria A, Nigg EA, Schmidt A (2014) Evaluation of data‐dependent and ‐independent mass spectrometric workflows for sensitive quantification of proteins and phosphorylation sites. J Proteome Res 13: 5973–5988 [DOI] [PubMed] [Google Scholar]
- Beck M, Schmidt A, Malmstroem J, Claassen M, Ori A, Szymborska A, Herzog F, Rinner O, Ellenberg J, Aebersold R (2011) The quantitative proteome of a human cell line. Mol Syst Biol 7: 549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensimon A, Heck AJ, Aebersold R (2012) Mass spectrometry‐based proteomics and network biology. Annu Rev Biochem 81: 379–405 [DOI] [PubMed] [Google Scholar]
- Berdougo E, Terret ME, Jallepalli PV (2009) Functional dissection of mitotic regulators through gene targeting in human somatic cells. Methods Mol Biol 545: 21–37 [DOI] [PubMed] [Google Scholar]
- Bettencourt‐Dias M, Rodrigues‐Martins A, Carpenter L, Riparbelli M, Lehmann L, Gatt MK, Carmo N, Balloux F, Callaini G, Glover DM (2005) SAK/PLK4 is required for centriole duplication and flagella development. Curr Biol 15: 2199–2207 [DOI] [PubMed] [Google Scholar]
- Bettencourt‐Dias M, Hildebrandt F, Pellman D, Woods G, Godinho SA (2011) Centrosomes and cilia in human disease. Trends Genet 27: 307–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg‐Wirschell M, Doxsey SJ (1998) Rapid isolation of centrosomes. Methods Enzymol 298: 228–238 [DOI] [PubMed] [Google Scholar]
- Bodor DL, Mata JF, Sergeev M, David AF, Salimian KJ, Panchenko T, Cleveland DW, Black BE, Shah JV, Jansen LE (2014) The quantitative architecture of centromeric chromatin. Elife 3: e02137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornens M, Moudjou M (1999) Studying the composition and function of centrosomes in vertebrates. Methods Cell Biol 61: 13–34 [DOI] [PubMed] [Google Scholar]
- Bornens M (2012) The centrosome in cells and organisms. Science 335: 422–426 [DOI] [PubMed] [Google Scholar]
- Brangwynne CP (2013) Phase transitions and size scaling of membrane‐less organelles. J Cell Biol 203: 875–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Breugel M, Hirono M, Andreeva A, Yanagisawa HA, Yamaguchi S, Nakazawa Y, Morgner N, Petrovich M, Ebong IO, Robinson CV, Johnson CM, Veprintsev D, Zuber B (2011) Structures of SAS‐6 suggest its organization in centrioles. Science 331: 1196–1199 [DOI] [PubMed] [Google Scholar]
- Cavalier‐Smith T (1974) Basal body and flagellar development during the vegetative cell cycle and the sexual cycle of Chlamydomonas reinhardii . J Cell Sci 16: 529–556 [DOI] [PubMed] [Google Scholar]
- Charpilienne A, Nejmeddine M, Berois M, Parez N, Neumann E, Hewat E, Trugnan G, Cohen J (2001) Individual rotavirus‐like particles containing 120 molecules of fluorescent protein are visible in living cells. J Biol Chem 276: 29361–29367 [DOI] [PubMed] [Google Scholar]
- Chavali PL, Putz M, Gergely F (2014) Small organelle, big responsibility: the role of centrosomes in development and disease. Philos Trans R Soc Lond B Biol Sci 369: 20130468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YK, Liu P, Sze SK, Dai C, Qi RZ (2010) CDK5RAP2 stimulates microtubule nucleation by the gamma‐tubulin ring complex. J Cell Biol 191: 1089–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cizmecioglu O, Arnold M, Bahtz R, Settele F, Ehret L, Haselmann‐Weiss U, Antony C, Hoffmann I (2010) Cep152 acts as a scaffold for recruitment of Plk4 and CPAP to the centrosome. J Cell Biol 191: 731–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman VC, Wu JQ (2012) Counting protein molecules using quantitative fluorescence microscopy. Trends Biochem Sci 37: 499–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comartin D, Gupta GD, Fussner E, Coyaud E, Hasegan M, Archinti M, Cheung SW, Pinchev D, Lawo S, Raught B, Bazett‐Jones DP, Luders J, Pelletier L (2013) CEP120 and SPICE1 cooperate with CPAP in centriole elongation. Curr Biol 23: 1360–1366 [DOI] [PubMed] [Google Scholar]
- Conduit PT, Wainman A, Raff JW (2015) Centrosome function and assembly in animal cells. Nat Rev Mol Cell Biol 16: 611–624 [DOI] [PubMed] [Google Scholar]
- Cottee MA, Muschalik N, Wong YL, Johnson CM, Johnson S, Andreeva A, Oegema K, Lea SM, Raff JW, van Breugel M (2013) Crystal structures of the CPAP/STIL complex reveal its role in centriole assembly and human microcephaly. Elife 2: e01071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dippell RV (1968) The development of basal bodies in paramecium. Proc Natl Acad Sci USA 61: 461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dundr M, McNally JG, Cohen J, Misteli T (2002) Quantitation of GFP‐fusion proteins in single living cells. J Struct Biol 140: 92–99 [DOI] [PubMed] [Google Scholar]
- Dzhindzhev NS, Tzolovsky G, Lipinszki Z, Schneider S, Lattao R, Fu J, Debski J, Dadlez M, Glover DM (2014) Plk4 phosphorylates ana2 to trigger sas6 recruitment and procentriole formation. Curr Biol 24: 2526–2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firat‐Karalar EN, Stearns T (2014) The centriole duplication cycle. Philos Trans R Soc Lond B Biol Sci 369: 20130460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong CS, Kim M, Yang TT, Liao JC, Tsou MF (2014) SAS‐6 assembly templated by the lumen of cartwheel‐less centrioles precedes centriole duplication. Dev Cell 30: 238–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Glover DM (2012) Structured illumination of the interface between centriole and peri‐centriolar material. Open Biol 2: 120104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP (2003) Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci USA 100: 6940–6945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillette MA, Carr SA (2013) Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nat Methods 10: 28–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatter T, Ludwig C, Ahrne E, Aebersold R, Heck AJ, Schmidt A (2012) Large‐scale quantitative assessment of different in‐solution protein digestion protocols reveals superior cleavage efficiency of tandem Lys‐C/trypsin proteolysis over trypsin digestion. J Proteome Res 11: 5145–5156 [DOI] [PubMed] [Google Scholar]
- Gonczy P (2012) Towards a molecular architecture of centriole assembly. Nat Rev Mol Cell Biol 13: 425–435 [DOI] [PubMed] [Google Scholar]
- Gonczy P (2015) Centrosomes and cancer: revisiting a long‐standing relationship. Nat Rev Cancer 15: 639–652 [DOI] [PubMed] [Google Scholar]
- Gosti‐Testu F, Marty MC, Berges J, Maunoury R, Bornens M (1986) Identification of centrosomal proteins in a human lymphoblastic cell line. EMBO J 5: 2545–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guderian G, Westendorf J, Uldschmid A, Nigg EA (2010) Plk4 trans‐autophosphorylation regulates centriole number by controlling betaTrCP‐mediated degradation. J Cell Sci 123: 2163–2169 [DOI] [PubMed] [Google Scholar]
- Guichard P, Desfosses A, Maheshwari A, Hachet V, Dietrich C, Brune A, Ishikawa T, Sachse C, Gonczy P (2012) Cartwheel architecture of Trichonympha basal body. Science 337: 553 [DOI] [PubMed] [Google Scholar]
- Guichard P, Hachet V, Majubu N, Neves A, Demurtas D, Olieric N, Fluckiger I, Yamada A, Kihara K, Nishida Y, Moriya S, Steinmetz MO, Hongoh Y, Gonczy P (2013) Native architecture of the centriole proximal region reveals features underlying its 9‐fold radial symmetry. Curr Biol 23: 1620–1628 [DOI] [PubMed] [Google Scholar]
- Guo HQ, Gao M, Ma J, Xiao T, Zhao LL, Gao Y, Pan QJ (2007) Analysis of the cellular centrosome in fine‐needle aspirations of the breast. Breast Cancer Res 9: R48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA (2005) The Polo kinase Plk4 functions in centriole duplication. Nat Cell Biol 7: 1140–1146 [DOI] [PubMed] [Google Scholar]
- Haren L, Remy MH, Bazin I, Callebaut I, Wright M, Merdes A (2006) NEDD1‐dependent recruitment of the gamma‐tubulin ring complex to the centrosome is necessary for centriole duplication and spindle assembly. J Cell Biol 172: 505–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch EM, Kulukian A, Holland AJ, Cleveland DW, Stearns T (2010) Cep152 interacts with Plk4 and is required for centriole duplication. J Cell Biol 191: 721–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, Hyman AA, Mann M (2015) A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163: 712–723 [DOI] [PubMed] [Google Scholar]
- Hiraki M, Nakazawa Y, Kamiya R, Hirono M (2007) Bld10p constitutes the cartwheel‐spoke tip and stabilizes the 9‐fold symmetry of the centriole. Curr Biol 17: 1778–1783 [DOI] [PubMed] [Google Scholar]
- Hirono M (2014) Cartwheel assembly. Philos Trans R Soc Lond B Biol Sci 369: 20130458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland AJ, Lan W, Niessen S, Hoover H, Cleveland DW (2010) Polo‐like kinase 4 kinase activity limits centrosome overduplication by autoregulating its own stability. J Cell Biol 188: 191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland AJ, Fachinetti D, Zhu Q, Bauer M, Verma IM, Nigg EA, Cleveland DW (2012) The autoregulated instability of Polo‐like kinase 4 limits centrosome duplication to once per cell cycle. Genes Dev 26: 2684–2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchins JR, Toyoda Y, Hegemann B, Poser I, Heriche JK, Sykora MM, Augsburg M, Hudecz O, Buschhorn BA, Bulkescher J, Conrad C, Comartin D, Schleiffer A, Sarov M, Pozniakovsky A, Slabicki MM, Schloissnig S, Steinmacher I, Leuschner M, Ssykor A et al (2010) Systematic analysis of human protein complexes identifies chromosome segregation proteins. Science 328: 593–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman AA, Weber CA, Julicher F (2014) Liquid‐liquid phase separation in biology. Annu Rev Cell Dev Biol 30: 39–58 [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Marshall WF (2011) Ciliogenesis: building the cell's antenna. Nat Rev Mol Cell Biol 12: 222–234 [DOI] [PubMed] [Google Scholar]
- Jakobsen L, Vanselow K, Skogs M, Toyoda Y, Lundberg E, Poser I, Falkenby LG, Bennetzen M, Westendorf J, Nigg EA, Uhlen M, Hyman AA, Andersen JS (2011) Novel asymmetrically localizing components of human centrosomes identified by complementary proteomics methods. EMBO J 30: 1520–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana SC, Marteil G, Bettencourt‐Dias M (2014) Mapping molecules to structure: unveiling secrets of centriole and cilia assembly with near‐atomic resolution. Curr Opin Cell Biol 26: 96–106 [DOI] [PubMed] [Google Scholar]
- Joglekar AP, Salmon ED, Bloom KS (2008) Counting kinetochore protein numbers in budding yeast using genetically encoded fluorescent proteins. Methods Cell Biol 85: 127–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller D, Orpinell M, Olivier N, Wachsmuth M, Mahen R, Wyss R, Hachet V, Ellenberg J, Manley S, Gonczy P (2014) Mechanisms of HsSAS‐6 assembly promoting centriole formation in human cells. J Cell Biol 204: 697–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodjakov A, Rieder CL (1999) The sudden recruitment of gamma‐tubulin to the centrosome at the onset of mitosis and its dynamic exchange throughout the cell cycle, do not require microtubules. J Cell Biol 146: 585–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TS, Park JE, Shukla A, Choi S, Murugan RN, Lee JH, Ahn M, Rhee K, Bang JK, Kim BY, Loncarek J, Erikson RL, Lee KS (2013) Hierarchical recruitment of Plk4 and regulation of centriole biogenesis by two centrosomal scaffolds, Cep192 and Cep152. Proc Natl Acad Sci USA 110: E4849–E4857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa D, Vakonakis I, Olieric N, Hilbert M, Keller D, Olieric V, Bortfeld M, Erat MC, Fluckiger I, Gonczy P, Steinmetz MO (2011) Structural basis of the 9‐fold symmetry of centrioles. Cell 144: 364–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebba JE, Galletta BJ, Nye J, Plevock KM, Buster DW, Hollingsworth NA, Slep KC, Rusan NM, Rogers GC (2015) Two Polo‐like kinase 4 binding domains in Asterless perform distinct roles in regulating kinase stability. J Cell Biol 208: 401–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleylein‐Sohn J, Westendorf J, Le Clech M, Habedanck R, Stierhof YD, Nigg EA (2007) Plk4‐induced centriole biogenesis in human cells. Dev Cell 13: 190–202 [DOI] [PubMed] [Google Scholar]
- Kohli M, Rago C, Lengauer C, Kinzler KW, Vogelstein B (2004) Facile methods for generating human somatic cell gene knockouts using recombinant adeno‐associated viruses. Nucleic Acids Res 32: e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmaier G, Loncarek J, Meng X, McEwen BF, Mogensen MM, Spektor A, Dynlacht BD, Khodjakov A, Gonczy P (2009) Overly long centrioles and defective cell division upon excess of the SAS‐4‐related protein CPAP. Curr Biol 19: 1012–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollman JM, Merdes A, Mourey L, Agard DA (2011) Microtubule nucleation by gamma‐tubulin complexes. Nat Rev Mol Cell Biol 12: 709–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollman JM, Greenberg CH, Li S, Moritz M, Zelter A, Fong KK, Fernandez JJ, Sali A, Kilmartin J, Davis TN, Agard DA (2015) Ring closure activates yeast gammaTuRC for species‐specific microtubule nucleation. Nat Struct Mol Biol 22: 132–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratz AS, Barenz F, Richter KT, Hoffmann I (2015) Plk4‐dependent phosphorylation of STIL is required for centriole duplication. Biol Open 4: 370–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M (2014) Minimal, encapsulated proteomic‐sample processing applied to copy‐number estimation in eukaryotic cells. Nat Methods 11: 319–324 [DOI] [PubMed] [Google Scholar]
- Lajoie‐Mazenc I, Tollon Y, Detraves C, Julian M, Moisand A, Gueth‐Hallonet C, Debec A, Salles‐Passador I, Puget A, Mazarguil H, Raynaud‐Messina B, Wright M (1994) Recruitment of antigenic gamma‐tubulin during mitosis in animal cells: presence of gamma‐tubulin in the mitotic spindle. J Cell Sci 107(Pt 10): 2825–2837 [DOI] [PubMed] [Google Scholar]
- Lane HA, Nigg EA (1996) Antibody microinjection reveals an essential role for human polo‐like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J Cell Biol 135: 1701–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawo S, Hasegan M, Gupta GD, Pelletier L (2012) Subdiffraction imaging of centrosomes reveals higher‐order organizational features of pericentriolar material. Nat Cell Biol 14: 1148–1158 [DOI] [PubMed] [Google Scholar]
- Lawrimore J, Bloom KS, Salmon ED (2011) Point centromeres contain more than a single centromere‐specific Cse4 (CENP‐A) nucleosome. J Cell Biol 195: 573–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidel S, Delattre M, Cerutti L, Baumer K, Gonczy P (2005) SAS‐6 defines a protein family required for centrosome duplication in C. elegans and in human cells. Nat Cell Biol 7: 115–125 [DOI] [PubMed] [Google Scholar]
- Li S, Fernandez JJ, Marshall WF, Agard DA (2012) Three‐dimensional structure of basal body triplet revealed by electron cryo‐tomography. EMBO J 31: 552–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YN, Wu CT, Lin YC, Hsu WB, Tang CJ, Chang CW, Tang TK (2013) CEP120 interacts with CPAP and positively regulates centriole elongation. J Cell Biol 202: 211–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TC, Neuner A, Schiebel E (2015) Targeting of gamma‐tubulin complexes to microtubule organizing centers: conservation and divergence. Trends Cell Biol 25: 296–307 [DOI] [PubMed] [Google Scholar]
- Lingle WL, Barrett SL, Negron VC, D'Assoro AB, Boeneman K, Liu W, Whitehead CM, Reynolds C, Salisbury JL (2002) Centrosome amplification drives chromosomal instability in breast tumor development. Proc Natl Acad Sci USA 99: 1978–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes CA, Jana SC, Cunha‐Ferreira I, Zitouni S, Bento I, Duarte P, Gilberto S, Freixo F, Guerrero A, Francia M, Lince‐Faria M, Carneiro J, Bettencourt‐Dias M (2015) PLK4 trans‐autoactivation controls centriole biogenesis in space. Dev Cell 35: 222–235 [DOI] [PubMed] [Google Scholar]
- Luders J, Patel UK, Stearns T (2006) GCP‐WD is a gamma‐tubulin targeting factor required for centrosomal and chromatin‐mediated microtubule nucleation. Nat Cell Biol 8: 137–147 [DOI] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26: 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maridor G, Gallant P, Golsteyn R, Nigg EA (1993) Nuclear localization of vertebrate cyclin A correlates with its ability to form complexes with cdk catalytic subunits. J Cell Sci 106(Pt 2): 535–544 [DOI] [PubMed] [Google Scholar]
- Matsuura K, Lefebvre PA, Kamiya R, Hirono M (2004) Bld10p, a novel protein essential for basal body assembly in Chlamydomonas: localization to the cartwheel, the first ninefold symmetrical structure appearing during assembly. J Cell Biol 165: 663–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennella V, Keszthelyi B, McDonald KL, Chhun B, Kan F, Rogers GC, Huang B, Agard DA (2012) Subdiffraction‐resolution fluorescence microscopy reveals a domain of the centrosome critical for pericentriolar material organization. Nat Cell Biol 14: 1159–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moudjou M, Bordes N, Paintrand M, Bornens M (1996) gamma‐Tubulin in mammalian cells: the centrosomal and the cytosolic forms. J Cell Sci 109: 875–887 [DOI] [PubMed] [Google Scholar]
- Murphy SM, Preble AM, Patel UK, O'Connell KL, Dias DP, Moritz M, Agard D, Stults JT, Stearns T (2001) GCP5 and GCP6: two new members of the human gamma‐tubulin complex. Mol Biol Cell 12: 3340–3352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj N, Wisniewski JR, Geiger T, Cox J, Kircher M, Kelso J, Paabo S, Mann M (2011) Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol 7: 548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa Y, Hiraki M, Kamiya R, Hirono M (2007) SAS‐6 is a cartwheel protein that establishes the 9‐fold symmetry of the centriole. Curr Biol 17: 2169–2174 [DOI] [PubMed] [Google Scholar]
- Nigg EA (2007) Centrosome duplication: of rules and licenses. Trends Cell Biol 17: 215–221 [DOI] [PubMed] [Google Scholar]
- Nigg EA, Raff JW (2009) Centrioles, centrosomes, and cilia in health and disease. Cell 139: 663–678 [DOI] [PubMed] [Google Scholar]
- Nigg EA, Stearns T (2011) The centrosome cycle: centriole biogenesis, duplication and inherent asymmetries. Nat Cell Biol 13: 1154–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson T, Mann M, Aebersold R, Yates JR III, Bairoch A, Bergeron JJ (2010) Mass spectrometry in high‐throughput proteomics: ready for the big time. Nat Methods 7: 681–685 [DOI] [PubMed] [Google Scholar]
- Ohta M, Ashikawa T, Nozaki Y, Kozuka‐Hata H, Goto H, Inagaki M, Oyama M, Kitagawa D (2014) Direct interaction of Plk4 with STIL ensures formation of a single procentriole per parental centriole. Nat Commun 5: 5267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole ET, Dutcher SK (2014) Site‐specific basal body duplication in Chlamydomonas. Cytoskeleton 71: 108–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paintrand M, Moudjou M, Delacroix H, Bornens M (1992) Centrosome organization and centriole architecture: their sensitivity to divalent cations. J Struct Biol 108: 107–128 [DOI] [PubMed] [Google Scholar]
- Park SY, Park JE, Kim TS, Kim JH, Kwak MJ, Ku B, Tian L, Murugan RN, Ahn M, Komiya S, Hojo H, Kim NH, Kim BY, Bang JK, Erikson RL, Lee KW, Kim SJ, Oh BH, Yang W, Lee KS (2014) Molecular basis for unidirectional scaffold switching of human Plk4 in centriole biogenesis. Nat Struct Mol Biol 21: 696–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P, Bodenmiller B, Mueller LN, Domon B, Aebersold R (2009) Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell 138: 795–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P, Aebersold R (2012) Selected reaction monitoring‐based proteomics: workflows, potential, pitfalls and future directions. Nat Methods 9: 555–566 [DOI] [PubMed] [Google Scholar]
- Richards AL, Merrill AE, Coon JJ (2015) Proteome sequencing goes deep. Curr Opin Chem Biol 24: 11–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt TI, Kleylein‐Sohn J, Westendorf J, Le Clech M, Lavoie SB, Stierhof YD, Nigg EA (2009) Control of centriole length by CPAP and CP110. Curr Biol 19: 1005–1011 [DOI] [PubMed] [Google Scholar]
- Schnerch D, Nigg EA (2016) Structural centrosome aberrations favor proliferation by abrogating microtubule‐dependent tissue integrity of breast epithelial mammospheres. Oncogene 35: 2711–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schubert C, Cubizolles F, Bracher JM, Sliedrecht T, Kops GJ, Nigg EA (2015) Plk1 and Mps1 cooperatively regulate the spindle assembly checkpoint in human cells. Cell Rep 12: 66–78 [DOI] [PubMed] [Google Scholar]
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M (2011) Global quantification of mammalian gene expression control. Nature 473: 337–342 [DOI] [PubMed] [Google Scholar]
- Sharma A, Aher A, Dynes NJ, Frey D, Katrukha EA, Jaussi R, Grigoriev I, Croisier M, Kammerer RA, Akhmanova A, Gonczy P, Steinmetz MO (2016) Centriolar CPAP/SAS‐4 imparts slow processive microtubule growth. Dev Cell 37: 362–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simicevic J, Schmid AW, Gilardoni PA, Zoller B, Raghav SK, Krier I, Gubelmann C, Lisacek F, Naef F, Moniatte M, Deplancke B (2013) Absolute quantification of transcription factors during cellular differentiation using multiplexed targeted proteomics. Nat Methods 10: 570–576 [DOI] [PubMed] [Google Scholar]
- Sims CE, Allbritton NL (2007) Analysis of single mammalian cells on‐chip. Lab Chip 7: 423–440 [DOI] [PubMed] [Google Scholar]
- Siwiak M, Zielenkiewicz P (2013) Transimulation – protein biosynthesis web service. PLoS ONE 8: e73943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnen KF, Schermelleh L, Leonhardt H, Nigg EA (2012) 3D‐structured illumination microscopy provides novel insight into architecture of human centrosomes. Biol Open 1: 965–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnen KF, Gabryjonczyk AM, Anselm E, Stierhof YD, Nigg EA (2013) Human Cep192 and Cep152 cooperate in Plk4 recruitment and centriole duplication. J Cell Sci 126: 3223–3233 [DOI] [PubMed] [Google Scholar]
- Strnad P, Leidel S, Vinogradova T, Euteneuer U, Khodjakov A, Gonczy P (2007) Regulated HsSAS‐6 levels ensure formation of a single procentriole per centriole during the centrosome duplication cycle. Dev Cell 13: 203–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang CJ, Fu RH, Wu KS, Hsu WB, Tang TK (2009) CPAP is a cell‐cycle regulated protein that controls centriole length. Nat Cell Biol 11: 825–831 [DOI] [PubMed] [Google Scholar]
- Tang CJ, Lin SY, Hsu WB, Lin YN, Wu CT, Lin YC, Chang CW, Wu KS, Tang TK (2011a) The human microcephaly protein STIL interacts with CPAP and is required for procentriole formation. EMBO J 30: 4790–4804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YC, Williams BR, Siegel JJ, Amon A (2011b) Identification of aneuploidy‐selective antiproliferation compounds. Cell 144: 499–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateishi K, Yamazaki Y, Nishida T, Watanabe S, Kunimoto K, Ishikawa H, Tsukita S (2013) Two appendages homologous between basal bodies and centrioles are formed using distinct Odf2 domains. J Cell Biol 203: 417–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixido‐Travesa N, Villen J, Lacasa C, Bertran MT, Archinti M, Gygi SP, Caelles C, Roig J, Luders J (2010) The gammaTuRC revisited: a comparative analysis of interphase and mitotic human gammaTuRC redefines the set of core components and identifies the novel subunit GCP8. Mol Biol Cell 21: 3963–3972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixido‐Travesa N, Roig J, Luders J (2012) The where, when and how of microtubule nucleation – one ring to rule them all. J Cell Sci 125: 4445–4456 [DOI] [PubMed] [Google Scholar]
- Vulprecht J, David A, Tibelius A, Castiel A, Konotop G, Liu F, Bestvater F, Raab MS, Zentgraf H, Izraeli S, Kramer A (2012) STIL is required for centriole duplication in human cells. J Cell Sci 125: 1353–1362 [DOI] [PubMed] [Google Scholar]
- Wang WJ, Acehan D, Kao CH, Jane WN, Uryu K, Tsou MF (2015) De novo centriole formation in human cells is error‐prone and does not require SAS‐6 self‐assembly. Elife 4: e10586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm M, Schlegl J, Hahne H, Moghaddas Gholami A, Lieberenz M, Savitski MM, Ziegler E, Butzmann L, Gessulat S, Marx H, Mathieson T, Lemeer S, Schnatbaum K, Reimer U, Wenschuh H, Mollenhauer M, Slotta‐Huspenina J, Boese JH, Bantscheff M, Gerstmair A et al (2014) Mass‐spectrometry‐based draft of the human proteome. Nature 509: 582–587 [DOI] [PubMed] [Google Scholar]
- Winey M, O'Toole E (2014) Centriole structure. Philos Trans R Soc Lond B Biol Sci 369: 20130457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff JB, Wueseke O, Hyman AA (2014) Pericentriolar material structure and dynamics. Philos Trans R Soc Lond B Biol Sci 369: 20130459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff JB, Wueseke O, Viscardi V, Mahamid J, Ochoa SD, Bunkenborg J, Widlund PO, Pozniakovsky A, Zanin E, Bahmanyar S, Zinke A, Hong SH, Decker M, Baumeister W, Andersen JS, Oegema K, Hyman AA (2015) Centrosomes. Regulated assembly of a supramolecular centrosome scaffold in vitro . Science 348: 808–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JQ, Pollard TD (2005) Counting cytokinesis proteins globally and locally in fission yeast. Science 310: 310–314 [DOI] [PubMed] [Google Scholar]
- Zheng Y, Jung MK, Oakley BR (1991) Gamma‐tubulin is present in Drosophila melanogaster and Homo sapiens and is associated with the centrosome. Cell 65: 817–823 [DOI] [PubMed] [Google Scholar]
- Zyss D, Gergely F (2009) Centrosome function in cancer: guilty or innocent? Trends Cell Biol 19: 334–346 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File