

Abstract
Activation of the elongation factor 2 kinase (eEF2K) leads to the phosphorylation and inhibition of the elongation factor eEF2, reducing mRNA translation rates. Emerging evidence indicates that the regulation of factors involved in protein synthesis may be critical for controlling diverse biological processes including cancer progression. Here we show that inhibitors of the HIV aspartyl protease (HIV‐PIs), nelfinavir in particular, trigger a robust activation of eEF2K leading to the phosphorylation of eEF2. Beyond its anti‐viral effects, nelfinavir has antitumoral activity and promotes cell death. We show that nelfinavir‐resistant cells specifically evade eEF2 inhibition. Decreased cell viability induced by nelfinavir is impaired in cells lacking eEF2K. Moreover, nelfinavir‐mediated anti‐tumoral activity is severely compromised in eEF2K‐deficient engrafted tumors in vivo. Our findings imply that exacerbated activation of eEF2K is detrimental for tumor survival and describe a mechanism explaining the anti‐tumoral properties of HIV‐PIs.
Keywords: cancer, cell death, eEF2K, HIV‐protease inhibitors, mRNA translation
Subject Categories: Autophagy & Cell Death, Cancer, Signal Transduction
Introduction
Drug repositioning is emerging as a successful strategy that accounts for a significant share of newly US Food and Drug Administration (FDA)‐approved drugs in recent years 1, 2. The study of unanticipated drug effects in patients can uncover new pathways and mechanisms of biological interest that can contribute to the development of new therapeutics. Early studies in HIV patients treated with the HIV aspartyl protease inhibitors (HIV‐PIs) have suggested interesting off‐target actions of these molecules in cancer 3, 4, 5. The HIV‐PIs target the viral protease and are widely used to treat HIV. In 1997, nelfinavir (NFR) became one of the first HIV‐PI to be approved by the FDA for HIV treatment. In addition to its antiretroviral effects, this safe and orally available drug shows promising anti‐tumoral activity in mice and humans 6, 7, 8, 9, 10. Several phase I and phase II clinical trials are investigating the efficacy of NFR repositioning in cancer with encouraging initial results 11, 12, 13, 14, 15. NFR has been shown to affect multiple pathways regulating cellular homeostasis including the proteasome, the kinase AKT, and the unfolded protein response (UPR) 16. Recently, we found that NFR is a robust inducer of the integrated stress response (ISR), an adaptation response that promotes an ATF4‐dependant transcriptional program 17. While some of these pathways may contribute to its anti‐tumoral activity, none has been demonstrated to be essential and the molecular basis of NFR‐mediated anti‐tumoral effects remains unknown 16, 18, 19.
In this study, we generated clonal populations of cells with increased resistance to NFR‐mediated toxicity. These clones showed an unaltered activation of most NFR‐mediated responses, including those related to the ISR. However, among possible stress and survival pathways, we observed the downregulation of the eukaryotic translation elongation factor 2 kinase (eEF2K), suggesting that it could be a major player driving nelfinavir cytotoxic effects. Activation of eEF2K is one of the pathways that participate to the restoration of cellular homeostasis upon conditions of nutrient or energy depletion by decreasing translation rates at the stage of elongation 20, 21, 22, 23. Indeed, the eEF2K activity inhibits the translation elongation factor eEF2, which mediates GTP‐dependent translocation of the ribosome, thereby promoting peptide chain formation. In the tumoral context, increased activation of eEF2K downstream of mTORC1 inhibition by rapamycin has been linked to APC‐deficient adenoma growth arrest, suggesting that enhancing eEF2K activity can be beneficial in patients with colorectal cancer 24.
Here, we report that the anticancer molecule NFR triggered a robust eEF2K‐dependent eEF2 phosphorylation leading to decreased rates of translation elongation. We found that NFR‐mediated eEF2K activation decreased cell proliferation and promoted cell death. Consistent with these observations, we demonstrated in an in vivo model of engrafted tumors that NFR‐mediated anti‐tumoral activity is eEF2K dependent. Taken together, these data indicate that the eEF2K pathway can be therapeutically manipulated to drive a response that is detrimental for tumor survival.
Results
Nelfinavir resistance correlates with decreased eEF2K expression
Long‐term treatment of immortalized cells with NFR is toxic and triggers cell death 10. To get insight into the mechanisms involved, we treated HeLa cells with 10 μM of NFR and selected and characterized clones that survived and proliferated in the presence of the drug (Fig 1A). NFR concentration can reach up to 17 μM in the plasma of treated patients 25, 26 and around 10 μM in liver tissues of mice that receive a dose of NFR reproducing the plasma concentration measured in patients 17. Up to these concentrations of NFR, we can observe a significant increased viability in the resistant clones compared to the parental population (Fig 1B). We compared by RNA sequencing (RNA‐seq), parental cells treated with NFR and four clones maintained in the presence of the drug (Dataset EV1). We observed that many genes involved in ribonucleoprotein complex biogenesis and mRNA translation regulation were downregulated in the resistant clones compared to NFR‐treated parental cells. Among the translation regulating pathways significantly downregulated in the resistant clones, we noticed the decrease of the eukaryotic elongation factor 2 kinase (eEF2K) mRNA. This finding was confirmed by real‐time PCR (Fig 1C) and at the protein level by Western blot (Fig 1D). Then, we interrogated eEF2K expression in the clones following NFR withdraw. We found that in two out of three representative clones, eEF2K downregulation was stable and maintained for more than 3 weeks in the absence of NFR (Fig EV1). This indicated that decreased eEF2K expression was not a direct consequence of prolonged treatment with NFR, but could be the result of a selective advantage in a few cells that bypassed NFR‐mediated cell death. This hypothesis implied that eEF2K could be engaged by NFR to decrease viability.
Figure 1. Resistance to nelfinavir triggers eEF2K downregulation.
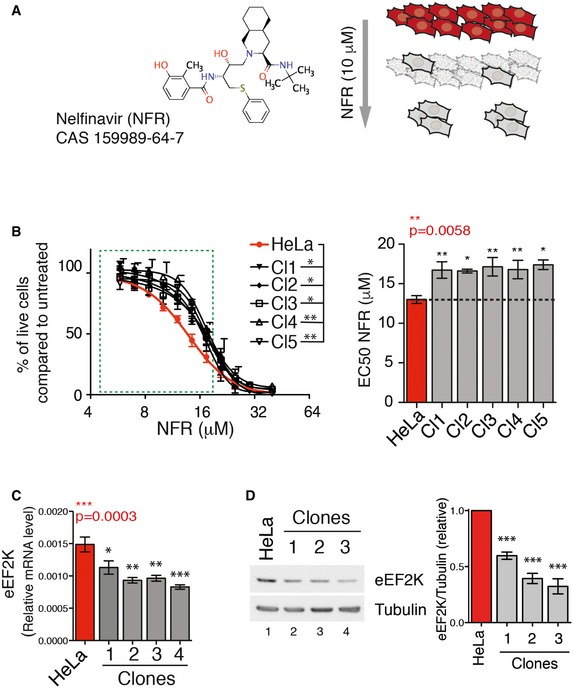
- Molecular structure of the HIV‐PI nelfinavir (NFR) and schematic representation of the protocol used to generate NFR‐resistant clones. HeLa cells were maintained with 10 μM NFR. After 15 days, clones proliferating in the constant presence of 10 μM NFR in the culture medium were selected and expanded for further analysis.
- Dose–response curves for viability of parental cells (in red) or selected clones (in black) upon treatment with NFR for 48 h. The green dashed box highlights the concentration in the physiologically relevant range. Curves are mean ± s.e.m. of three independent experiments performed in triplicate. P‐values were calculated using two‐way ANOVA between parental cells and each individual clone. Bar graph represents the EC50 (half‐maximal effective concentration) of the dose responses. Data are mean ± s.e.m. of three independent experiments performed in triplicate. P‐values were calculated using one‐way ANOVA (P‐value in red) followed by Dunnett's multiple comparison tests. *P‐value ≤ 0.05, **P‐value ≤ 0.01.
- The parental population and the NFR‐resistant clones were analyzed for eEF2K mRNA expression by real‐time PCR relative to β‐actin (mean and s.e.m. from three independent batches of cells are shown). P‐values were calculated using one‐way ANOVA followed by Dunnett's multiple comparison tests. *P‐value ≤ 0.05, **P‐value ≤ 0.01, ***P‐value ≤ 0.001.
- The parental population and representative NFR‐resistant clones were analyzed by Western blot for eEF2K total protein levels. Tubulin is used as a loading control. Histogram represents mean and s.e.m. of relative eEF2K levels quantified from Western blots of four different batches of cells collected on different dates. P‐values were calculated using one‐way ANOVA followed by Dunnett's multiple comparison tests. ***P‐value ≤ 0.001.
Source data are available online for this figure.
Figure EV1. eEF2K decreased level in NFR‐resistant HeLa clones.
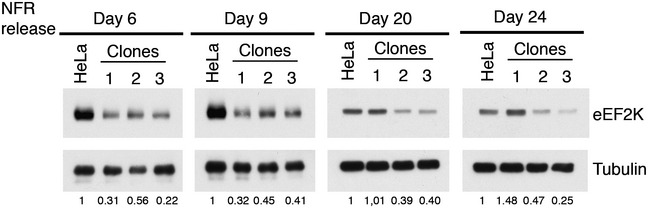
NFR was washed out from resistant clones' culture medium for the indicated time, and cells were harvested and analyzed for eEF2K protein level. Numbers below represent the relative eEF2K expression compared to untreated parental cell line and were obtained by Western blot quantification normalizing eEF2K on tubulin levels.
Nelfinavir triggers eEF2K to promote eEF2 phosphorylation
EEF2K controls the rate of translation elongation through the phosphorylation of the eukaryotic elongation factor 2 (eEF2) at threonine 56 (Thr56). We therefore tested whether NFR can activate eEF2K by monitoring eEF2 phosphorylation. Short‐term treatment with increasing concentrations of NFR triggered the phosphorylation of eEF2 in HeLa cells (Fig 2A). As expected, eEF2 phosphorylation was absent in eEF2K‐deficient mouse embryonic fibroblasts, demonstrating that NFR engages eEF2K to regulate eEF2 (Figs 2B and EV2A). EEF2K deficiency did not affect other pathways modulated by NFR 17, including the phosphorylation of the translation initiation factor eIF2α or the expression of ATF4 (Fig EV2A). Similarly impairing eIF2α phosphorylation and therefore the integrated stress response (ISR) did not affect NFR‐mediated eEF2 regulation (Fig EV2B), suggesting that these two pathways are engaged independently from one another. Then, we characterized these responses in the NFR‐resistant clones. NFR was withdrawn from the culture media for a few hours, and the NFR response was analyzed at 6 h of treatment. As expected, the clones showed a diminished expression of eEF2K protein that correlated with a reduced eEF2 phosphorylation in the presence of low doses of NFR (Figs 2C and EV3A). However, other pathways triggered by NFR such as the induction of ATF4 (Fig EV3B) were not affected in NFR‐resistant clones, which even expressed high level of NFR‐response markers such as CHOP or DNAJB9 (Fig EV3C).
Figure 2. HIV‐PIs induce eEF2K‐dependent eEF2 phosphorylation.
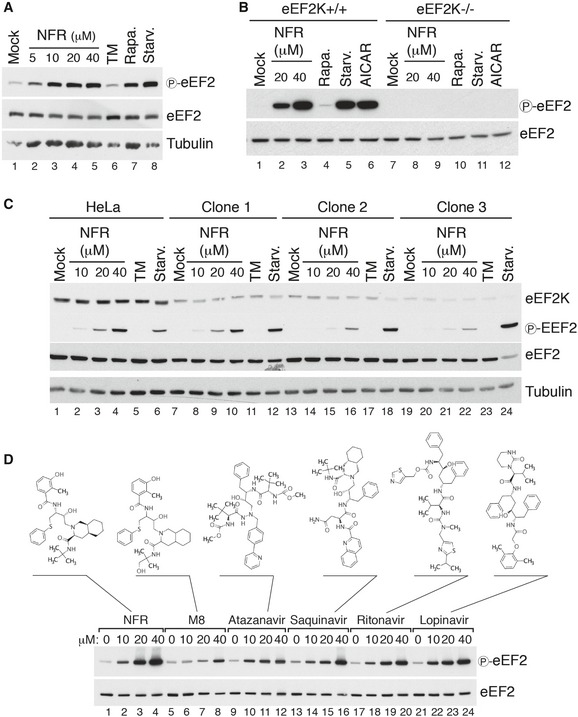
- Immunoblot analysis of eEF2 Thr56 phosphorylation (P‐eEF2) in HeLa cells treated for 6 h with increasing doses of NFR, and compared with 10 μg/ml tunicamycin (TM), 200 nM rapamycin (Rapa.), or 1‐h starvation (Starv.). Tubulin is used as a loading control.
- eEF2K WT and KO MEFs treated for 6 h with indicated concentrations of NFR, 200 nM rapamycin (Rapa.), 1‐h starvation (Starv.), or 1 mM of the AMPK activator AICAR, were analyzed by immunoblot with antibodies directed against total or phosphorylated eEF2 (Thr56).
- Three representative NFR‐resistant HeLa clones were analyzed by immunoblot for eEF2K expression level and eEF2 phosphorylation upon NFR treatment and compared to parental HeLa cells. For treatments, medium was replaced for 6 h with medium containing DMSO (Mock), increasing doses of NFR or 10 μg/ml tunicamycin (TM), or for 1 h with PBS for starvation (Starv.). Tubulin is used as a loading control.
- Immunoblot analysis of eEF2 Thr56 phosphorylation in HeLa cells treated for 6 h with increasing doses of different HIV‐PIs as indicated or the NFR metabolite M8 (hydroxy‐tert‐butylamide). Molecular structures of the different HIV‐PIs used are indicated.
Figure EV2. eEF2K deficiency does not impair NFR‐mediated ISR induction and vice versa.
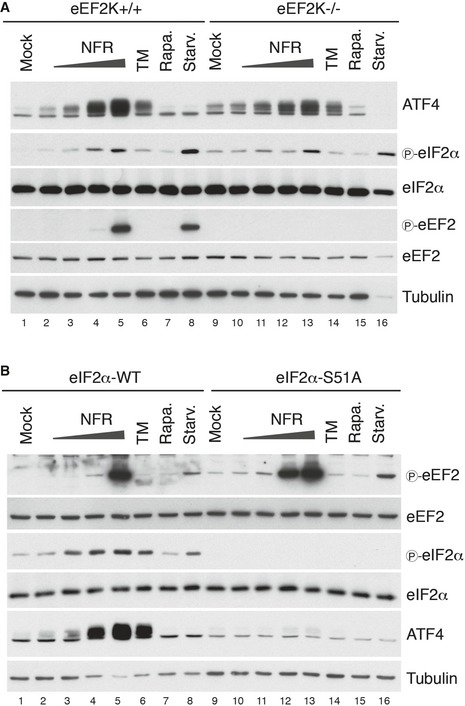
- Activation of the integrated stress response by NFR was not affected by eEF2K deficiency. ISR activation was measured in eEF2K−/− and eEF2K+/+ MEFs by immunoblotting of the translation factor ATF4 and the phosphorylation of the initiation factor eIF2α. NFR response (6 h of treatment) was compared with treatments using 10 μg/ml tunicamycin (TM), an inducer of the integrated stress response, 200 nM rapamycin (Rapa.), or 1 h of starvation in PBS (Starv.). Tubulin is used as a loading control.
- NFR mediates eEF2 phosphorylation independently from eIF2α phosphorylation, the effector of the ISR. NFR response was analyzed in eIF2α WT MEFs and cells unable to carry eIF2α phosphorylation on Ser51 (eIF2αS51A). Immunoblot analysis was performed to assess the phosphorylation of eEF2 and eIF2α as well as ATF4 expression after 6 h of treatment with indicated dose of NFR, 10 μg/ml tunicamycin (TM), 200 nM rapamycin (Rapa.), and after 1 h of starvation (Starv.). Tubulin is used as a loading control.
Figure EV3. ISR is not affected in NFR‐resistant clones and NFR triggers eEF2 phosphorylation independently of Akt.
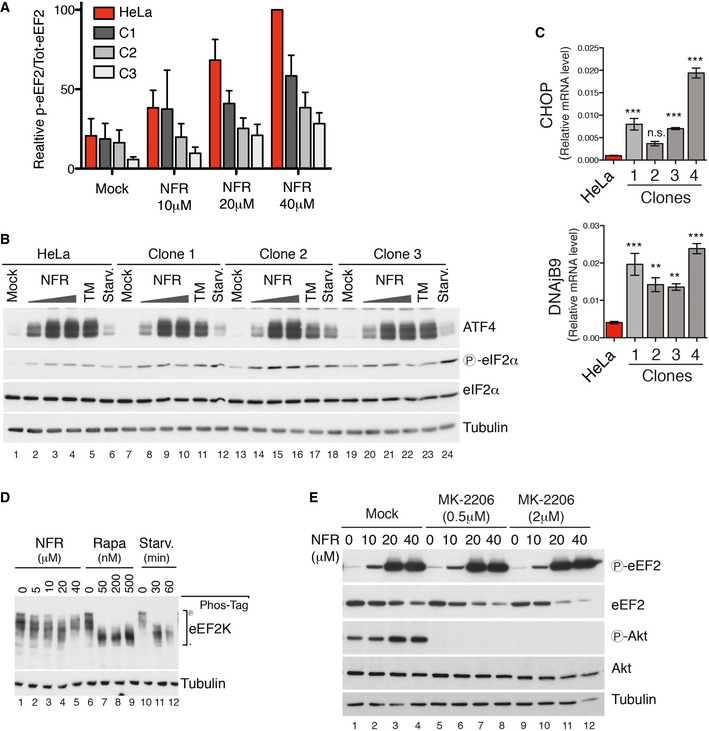
- Three representative NFR‐resistant HeLa clones were analyzed by immunoblot for eEF2 phosphorylation upon NFR and compared to parental HeLa cells (see Fig 2). Histogram shows the mean and s.e.m. of p‐eEF2/‐eEF2 ratio obtained from Western blot quantification of three independent experiments. Data were normalized using 40 μM NFR‐treated parental cell line as the maximum (100%) p‐eEF2 signal for each individual experiment.
- Immunoblot analysis of NFR‐mediated activation of ER‐stress markers in three NFR‐resistant HeLa clones compared to parental HeLa cells. EIF2α phosphorylation and expression of ATF4 are shown. For treatments, medium was replaced for 6 h with medium containing increasing doses of NFR or 10 mg/ml tunicamycin (TM), or for 1 h with PBS for starvation (Starv.). These data are representative of three experiments.
- NFR‐resistant clones and parental HeLa cells were analyzed for expression of the stress factors CHOP and DNAjB9 by real‐time PCR relative to β‐actin (mean and s.e.m. from three independent batches of cells are shown). P‐values were calculated using one‐way ANOVA followed by Dunnett's multiple comparison test; **P‐value ≤ 0.01, ***P‐value ≤ 0.001.
- eEF2K phosphorylation was measured using phos‐tag SDS–PAGE and specific antibodies in HeLa cells subjected to the indicated treatments. These data are representative of three experiments.
- MK‐2206‐mediated Akt inhibition does not affect the ability of NFR to induce eEF2 phosphorylation. MEFs were treated with the indicated concentrations of the potent Akt inhibitor MK‐2206 or left untreated. After 30 min, NFR was added for an additional 6 h. Immunoblot analysis was performed for phosphorylated and total Akt and eEF2 as indicated. Panel is representative of three independent experiments. Tubulin is used as a loading control.
Investigation of a panel of HIV‐PIs used in the clinic as well as hydroxy‐t‐butylamidenelfinavir (M8), the active NFR metabolite, showed that most HIV‐PIs trigger eEF2 phosphorylation (Fig 2D). Yet, we consistently found that among the HIV‐PIs, NFR is the most robust inducer of this pathway at relevant concentrations.
Nelfinavir does not inhibit the mTORC1 pathway to promote eEF2K activation
EEF2K activity is regulated by phosphorylation, which occurs at several sites downstream of specific signaling pathways. In particular, the mTORC1 downstream p70 S6 kinase negatively regulates eEFK by promoting its phosphorylation at inhibitory sites, thereby allowing translation to proceed 20, and AMP‐activated protein kinase (AMPK), a sensor of low energy status, has been shown to promote activator phosphorylation of eEF2K 27 (Fig 3A). Inhibition of mTORC1 with chemical inhibitors such as rapamycin or the starvation‐induced activation of AMPK impairs eEF2K phosphorylation leading to its activation and inhibition of eEF2 28, 29. Because many phosphorylation sites have been shown to regulate eEF2K 30, we separated cell extracts on a phos‐tag SDS–PAGE to get a comprehensive analysis of its phosphorylation status. No significant changes on the eEF2K phosphorylation pattern were detected in the presence of NFR, while rapamycin or starvation considerably affected the overall eEF2K phosphorylation (Fig EV3D). In line with these observations, the dephosphorylation of the mTORC1 effectors S6 ribosomal protein or 4EBP1, a hallmark of mTORC1 inhibition and treatment with rapamycin, was only minimally affected in the presence of NFR (Fig 3B, compare lanes 2–4 with 6–7). Importantly, this effect was not observed in eEF2K‐deficient cells (Fig 3B, compare lane 2–4 with lanes 14–16), suggesting that NFR only slightly affects the mTORC1 pathway downstream of eEF2K activation. Independence from mTORC1 was also confirmed by the observation that rapamycin, which does not trigger a strong eEF2 phosphorylation per se, did not affect NFR‐mediated eEF2 phosphorylation (Fig 3C). AMPK activation has been shown to trigger eEF2K activation both indirectly through mTORC1 inhibition and directly by phosphorylating eEF2K 27. Indeed, the AMPK agonist AICAR is a potent inducer of eEF2K‐dependent eEF2 phosphorylation (Figs 2B and 3B). Interestingly, we observed that concentrations above 20 μM of NFR triggered AMPK phosphorylation (Fig 3B, lane 3,4 and 15,16), suggesting that this pathway could be involved in mediating NFR responses. Yet, deletion of both AMPK isoforms α1 and α2 did not affect NFR‐induced eEF2 phosphorylation whereas it impaired AICAR response (Fig 3D). AKT has been shown to be a target of NFR 10, 31, 32; we therefore monitored AKT phosphorylation. Upon treatment with NFR, we did not observe decreased basal AKT phosphorylation (Fig 3B). Moreover, inhibition of AKT with the selective inhibitor MK‐2206 did not affect NFR‐mediated eEF2 phosphorylation (Fig EV3C). Similarly, inhibition of AKT phosphorylation upon treatment with the phosphoinositide 3‐kinase (PI3K) inhibitors 3‐methyladenine (3‐MA) and wortmannin did not affect NFR‐induced eEF2 phosphorylation (Appendix Fig S1). Altogether, these data demonstrate that NFR signals eEF2K activation independently of the eEF2K activating pathways such as mTORC1 inhibition, AMPK, or the ISR.
Figure 3. NFR‐mediated eEF2 phosphorylation is AMPK and mTOR independent.
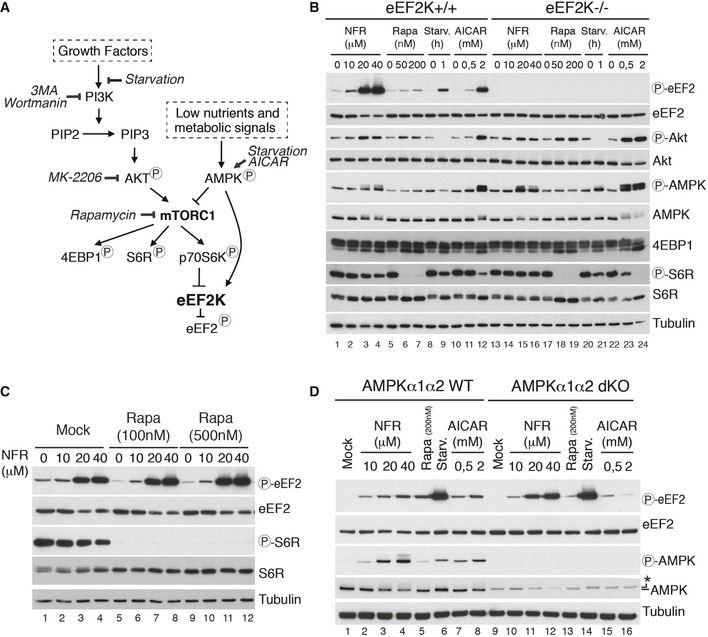
- Schematic representation of signaling pathways targeted by the inhibitors used in this study.
- eEF2K WT and KO MEFs treated for 6 h with the indicated concentrations of NFR, rapamycin (Rapa), AICAR, or starved for the indicated time in PBS (Starv.), were analyzed by immunoblot using the indicated antibodies. Tubulin is used as a loading control.
- Potent mTOR inhibition does not impair NFR‐mediated eEF2 phosphorylation. WT MEFs were treated with the indicated concentrations of rapamycin or with vehicle (Mock). After 30 min, indicated doses of NFR were added and cells were incubated for an additional 6 h and analyzed for phosphorylated S6R and eEF2. Tubulin is used as a loading control.
- NFR‐mediated eEF2 phosphorylation is not affected in AMPKα1α2 dKO. AMPKα1α2 WT and dKO MEFs treated for 6 h with indicated concentration of NFR, rapamycin (Rapa), AICAR, or starved for 1 h in PBS (Starv.), were analyzed by immunoblot with the indicated antibodies. Anti‐total AMPKα antibody gives an unspecific band (*) with a slightly higher molecular weight in dKO cells. Tubulin is used as a loading control.
eEF2K activation contributes to decreased translation rates
To quantify the role of eEF2K in NFR‐mediated translation inhibition, we measured global translation rates using 35S‐labeled methionine incorporation. We found that eEF2K deficiency partially rescued NFR‐mediated decreased translation rates without impacting on tunicamycin‐mediated regulation of translation (Fig 4A and B). In these cells, rapamycin triggered a relatively weak eEF2 phosphorylation (Fig 2B); thus, eEF2K did not significantly contribute to rapamycin‐mediated translation decrease (Fig 4B). Similar to eEF2K deficiency, impairing eIF2α phosphorylation partially restored methionine incorporation, indicating that both pathways contribute to NFR‐mediated reprogramming of mRNA translation (Fig 4B). We also examined the role of eEF2K in NFR‐mediated mRNA translation control by measuring polysomal distribution. Treatment with NFR resulted in a decrease of mRNA associated with polysomes and an increase of free ribosomes, overall reflecting a decrease of protein synthesis, whereas eEF2K deficiency reversed this effect (Fig 4C). On the contrary, treatment with the ER‐stress inducer tunicamycin triggered a decrease of the polysomal fraction that was unaffected by eEF2K deficiency but was rescued in the presence of the ISR inhibitor ISRIB or in cells unable to phosphorylate eIF2α (Fig EV4A and B). Inhibiting the ISR did not significantly affect the NFR‐mediated decrease in polysomes (Fig EV4A and B). Interestingly, while NFR clearly affected the polysomal profile in an eEF2K‐dependent manner, we did not observe the expected increase of the polysomal fraction that is predicted to accumulate upon elongation defects as observed in the presence of cycloheximide, a molecule that exerts its effect by interfering with the translocation step in protein synthesis (Fig EV4C). Instead, we observed a decreased polysomal fraction and increased free ribosomes signal. This indicates that specific eEF2K activation and eEF2 phosphorylation may affect additional steps in the translation program beyond its function during elongation. Previous reports have suggested that eEF2 could be involved in the splitting of the 80S ribosomes into subunits, a process required for the initiation steps of translation 33.
Figure 4. NFR regulates translation rates by phosphorylating eEF2.
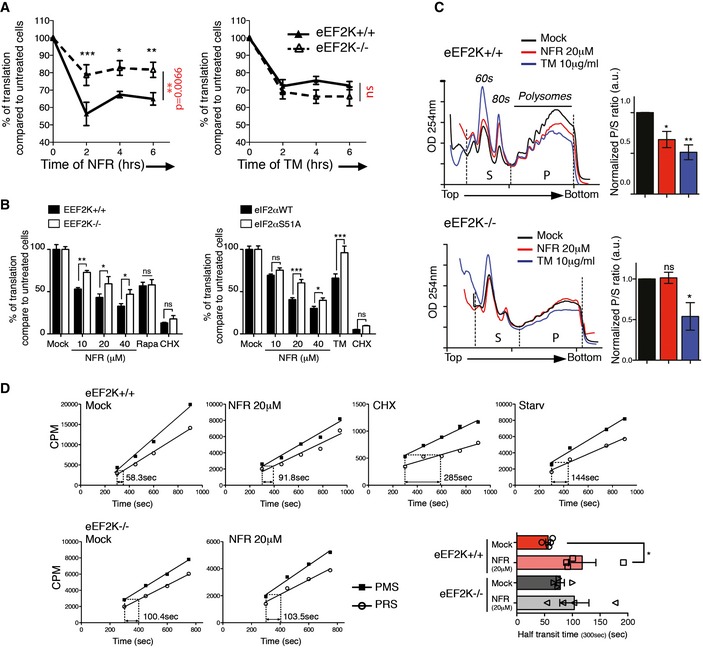
-
A, BEEF2K contributes to the decreased translation observed with NFR without impacting translation in the presence of TM. (A) Quantification of newly synthesized proteins at 0, 2, 4, and 6 h after 20 μM NFR (left panels) or 10 μg/ml TM (right panels) treatment in eEF2K+/+ and eEF2K−/− MEFs. Treated cells were labeled for 15 min with 35S‐methionine and visualized by SDS–PAGE and subsequent autoradiography. Autoradiographs from four different experiments (see also source data) were quantified, and results show percentage of translation compared to untreated cells. The mean and s.e.m. of four independent metabolic labeling experiments are shown. (B) eEF2K+/+, eEF2K−/−, eIF2αWT, and eIF2αS51A MEFs were treated for 6 h with indicated doses of NFR, 200 nM rapamycin, or 10 μg/ml TM, or for 30 min with 10 μg/ml CHX. 35S‐methionine incorporation was measured by liquid scintillation counting. Data are shown as the percentage of translation compared to untreated cells. *P‐value ≤ 0.05, **P‐value ≤ 0.01, ***P‐value ≤ 0.001 obtained using two‐way ANOVA (in red) followed by Bonferroni post‐test (in black).
-
CRepresentative polysome profiles of eEF2K WT and KO MEFs treated 6 h with NFR or TM. Area under the curve for sub‐polysomes (S) and polysomes (P) used to calculate the P/S ratio was indicated (see also source data). Bar graph represents the ratio normalized to untreated cells. OD254 nm is optical density at 254 nm. Data are mean ± s.e.m. of P/S ratio calculated from three independent experiments. P‐values were calculated using one‐way ANOVA followed by Bonferroni post‐test; *P‐value ≤ 0.05, **P‐value ≤ 0.01.
-
DThe ribosome half‐transit time in eEF2K+/+ and eEF2K−/− MEFs was determined as described in Materials and Methods. Incorporation of 35S‐methionine into total protein within the PMS and PRS was obtained by linear regression analysis. Presented graphs are representative of two (CHX 1 μg/ml for 30 min and 30‐min starvation in PBS in eEF2K+/+ cells) to four (NFR 20 μM for 6 h in eEF2K+/+ and eEF2K−/− cells) independent experiments. Indicated values represent the x displacement measurement (in time) between the PMS line at 300 s and the PRS line (see also source data). Histogram represents mean and s.e.m. of the ribosome half‐transit time from four independent experiments. P‐values were calculated using two‐tailed unpaired Student's t‐test; *P‐value ≤ 0.05.
Source data are available online for this figure.
Figure EV4. NFR‐mediated changes in polysomal profile are not dependent on eIF2α phosphorylation.
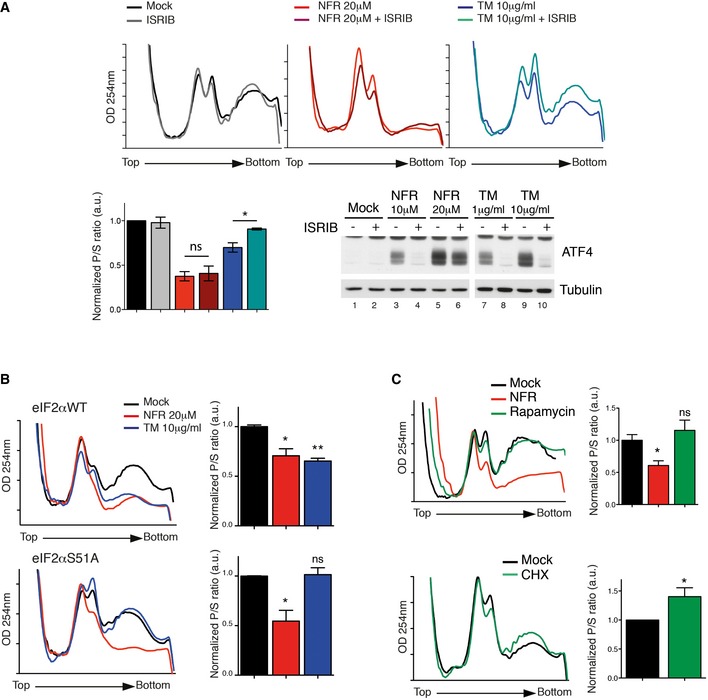
-
ARepresentative polysome profiles of WT MEFs treated 6 h with NFR or TM with or without 500 nM ISRIB. Bar graph represents the ratio of sub‐polysomes compared with polysomes (P/S). OD254 nm is optical density at 254 nm. Data are mean ± s.e.m. of P/S ratio calculated from two independent experiments. P‐values were calculated using two‐tailed unpaired Student's t‐tests comparing ratio +/− ISRIB; *P‐value ≤ 0.05. ISRIB efficiency at inhibiting NFR‐ or TM‐mediated ATF4 induction was tested by immunoblot.
-
B, CRepresentative polysome profiles of eIF2αWT and eIF2αS51A MEFs treated 6 h with NFR or TM (B) or WT MEFs treated for 6 h with 20 μM NFR or 200 nM rapamycin (C, upper panel) or 30 min with 1 μg/ml CHX (C, lower panel). Data are mean ± s.e.m. of P/S ratio calculated from 2 to 3 independent experiments. P‐values were calculated using one‐way ANOVA followed by Bonferroni post‐test (B and C, upper panel) or two‐tailed unpaired Student's t‐test (C, lower panel); *P‐value ≤ 0.05, **P‐value ≤ 0.01.
The observation that eEF2K deficiency restored the polysomal fraction clearly underlined the key role of this pathway in regulating translation by NFR. However, this observation is in apparent contradiction with the fact that eEF2K only partially restored overall protein synthesis (Fig 4A and B). A similar observation was made upon treatment with rapamycin (Figs 4B and EV4C). It is therefore possible that concomitant regulation of both the initiation and elongation machineries may mask the contribution of each mechanism on the polysomal traces. We therefore tested eEF2K contribution to elongation by examining NFR regulation of the ribosome half‐transit time 34. We compared the kinetics of 35S‐methionine incorporation into total protein isolated from the post‐mitochondrial supernatant (PMS) with the kinetics of radioactive amino acid incorporation measured in released polypeptides harvested from the post‐ribosomal supernatant (PRS). The time separating the PMS and PRS lines indicates the ribosome half‐transit time (Fig 4D). We measured that NFR significantly increased the ribosome half‐transit time. This effect on elongation was not observed in eEF2K‐deficient cells, indicating that eEF2K negatively regulates polypeptide elongation rates in the presence of NFR.
Overall, these experiments define eEF2K activation as a key pathway of NFR‐mediated protein synthesis regulation and indicate that eEF2K beyond its function as a regulator of elongation can affect polysomes by a mechanism that is yet to be identified.
eEF2K activation decreases proliferation and promotes cell death
The observation that loss of eEF2K may confer a fitness advantage to the cells in the presence of NFR (Fig 1B) prompted us to interrogate whether sustained eEF2K activation could affects cell growth and contribute to NFR‐mediated cell death. To test this hypothesis, we measured cell viability and proliferation in NFR‐treated eEF2K‐deficient MEFs and control cells. Consistently we found that NFR‐mediated decreased proliferation was affected by eEF2K deficiency in the presence of 10 μM NFR (Fig 5A). We performed genome editing by means of CRISPR/Cas9 to inactivate the eEF2K gene in additional cell lines (Appendix Fig S2A–C). Loss of eEF2K affected NFR‐mediated growth inhibition in all populations tested including those isolated from HeLa (Appendix Fig S2D), A549 (Appendix Fig S2E), and MCF7 cells (Appendix Fig S2F). Moreover, reconstitution of eEF2K−/− MEFs with a construct expressing the kinase (Appendix Fig S2G) restored NFR responses to the levels observed in wild‐type cells (Fig 5A). We also monitored eEF2K role in mediating NFR toxicity by analyzing cell viability by MTS assay upon increasing doses of NFR. We found that eEF2K deficiency decreased sensitivity to NFR (Fig 5B). This was mostly striking at physiological concentrations below 20 μM. As reported for the growth defect, reconstitution with eEF2K restored full NFR toxicity (Fig 5C). Similar findings were found in the eEF2K‐deficient cell lines tested (Fig 5D–F). Next we interrogated NFR‐mediated cell death by quantifying dying cells using AnnexinV and propidium iodide (PI) staining after 24 h of treatment. In line with the results obtained by monitoring NFR sensitivity, we found that eEF2K deficiency decreased NFR‐mediated cell death and that reconstitution of eEF2K‐deficient cells with eEF2K restored the response to NFR (Fig 5G and Appendix Fig S3).
Figure 5. NFR‐mediated eEF2K activation impairs cell proliferation and triggers cell death.
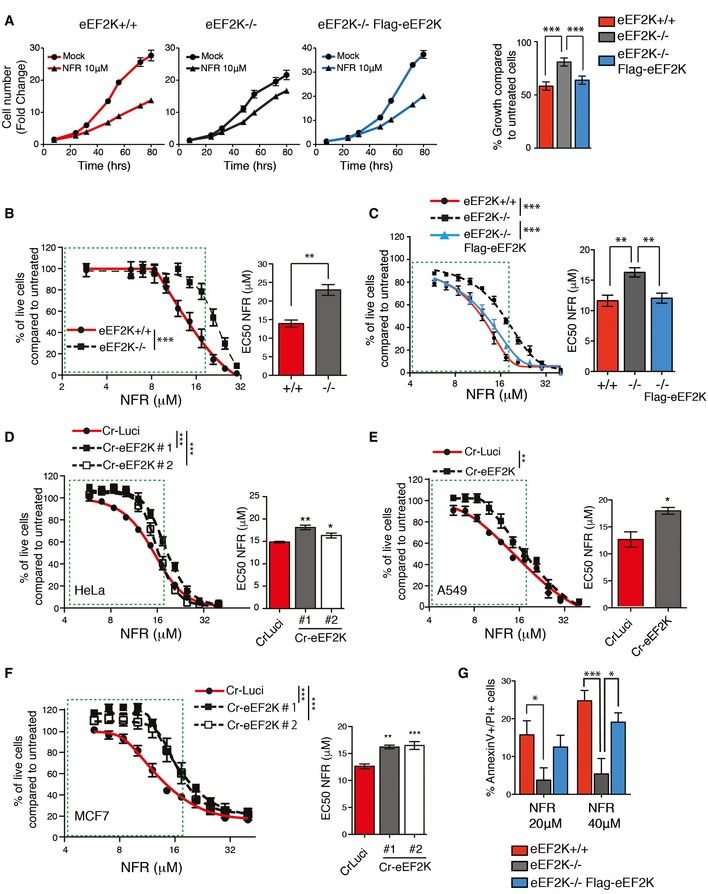
-
AeEF2K WT, KO, or KO reconstituted with human eEF2K (Flag‐eEF2K) MEFs were analyzed for cell growth at the indicated times. Fold change (means ± s.e.m. of triplicate) of the cell number just before treatment are shown. Histogram shows percentage of growth inhibition after 72 h of 10 μM NFR. P‐values were calculated using one‐way ANOVA with Bonferroni post‐test calculated from three independent experiments. ***P‐value ≤ 0.001.
-
B–FDose–response curves for cell viability after 48 h of NFR treatment measured using the MTS assay. Curves and bar graphs for EC50 are mean ± s.e.m. of three independent experiments performed in triplicate. For curves, P‐values were calculated using two‐way ANOVA. For the bar graphs, P‐values were calculated using two‐tailed unpaired Student's t‐tests (B, E), one‐way ANOVA with Bonferroni (C), or Dunnett's multiple comparison post‐tests (D and F). *P‐value ≤ 0.05, **P‐value ≤ 0.01, ***P‐value ≤ 0.001. (B, C) eEF2K−/− MEFs show a decreased susceptibly to NFR compared to eEF2K+/+ controls (B) whereas eEF2K reconstitution restores NFR sensitivity (C). (D‐F) HeLa (D), A549 (E), and MCF7 (F) clones with CRISPR/Cas9‐generated eEF2K deficiency (Cr‐eEF2K) show a decreased susceptibility to NFR compared to control cells (CrLuci).
-
GNFR‐mediated toxicity assessed using AnnexinV–PI staining and FACS analysis after 24 h of treatment. Histogram shows the percentage of dead cells (AV+/PI+). Data are mean ± s.e.m. of three independent experiments. P‐values were calculated using two‐way ANOVA with Bonferroni post‐test. *P‐value ≤ 0.05, ***P‐value ≤ 0.001.
EEF2K deficiency did not confer a promiscuous resistance to cell death as demonstrated by unaltered loss of viability in the presence of other compounds such as tunicamycin (TM) or the apoptosis‐inducing drug staurosporine (Appendix Fig S4). Other pathways including those related to the ISR are activated by NFR and could affect cell viability 7, 10, 17, 35. Cells deficient in key signaling components of the UPR pathway including PERK, ATF4, IRE1, and XBP1 and cells unable to phosphorylate eIF2α were tested for NFR sensitivity. Compared to control cells, no alteration in cell death was observed in these deficiencies (Fig EV5). Altogether, these observations demonstrate that sustained activation of eEF2K affects cell viability and contributes to cell death and growth inhibition in the presence of NFR, in particular at physiologically relevant concentrations of the drug, below 20 μM.
Figure EV5. UPR deficiency does not improve viability in the presence of NFR.
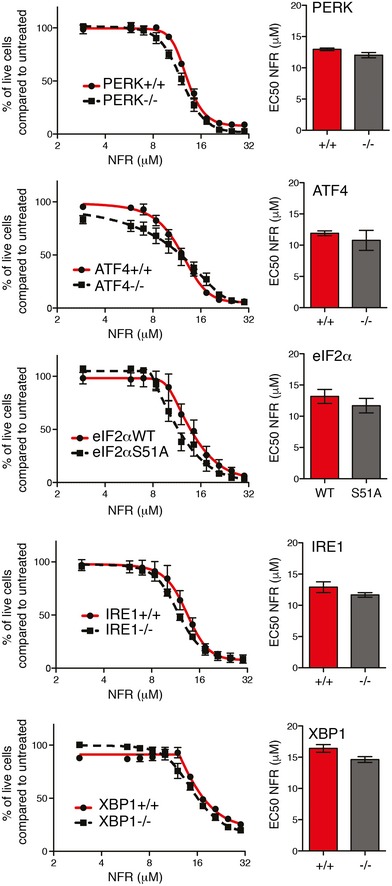
Dose–response curves for cell viability in MEFs unable to phosphorylate eIF2α (eIF2αS51A) or deficient for PERK, ATF4, IRE1, XBP1, and respective control cells treated with increasing doses of NFR for 48 h. Bar graph represents the EC50 (half‐maximal effective concentration) of the dose responses. Data are mean ± s.e.m. of three independent experiments performed in triplicate. Statistical significance was assessed using two‐way ANOVA for dose response and two‐tailed unpaired Student's t‐test for EC50. No significant differences were measured in the tested cell lines.
Pharmacological over‐activation of eEF2K reduces tumor growth
To determine the relevance of eEF2K in mediating NFR therapeutic effects in vivo, RasV12‐transformed proficient or eEF2K‐deficient MEFs were generated to transplant tumors into AGR129 (IFN‐α/β, IFN‐γ receptor, and RAG‐2 deficient) mice 36. Transformation did not significantly affect eEF2K activation (Appendix Fig S5A) or its role in mediating NFR susceptibility (Appendix Fig S5B). Mice were injected subcutaneously with eEF2K‐deficient cells on one flank and eEF2K‐proficient cells on the other flank. As reported previously in nu/nu immunocompromised mice 37, eEF2K deficiency did not affect overall tumor formation and growth in feed animals (Fig 6A). At day 6 post‐implantation, a daily treatment with NFR was started. This resulted in growth inhibition of WT tumor, but strikingly did not affect the growth of eEF2K‐deficient tumors (Fig 6A). As expected, NFR treatment increased eEF2 phosphorylation in postmortem tumor biopsies (Fig 6B and C), indicating that sustained eEF2K‐mediated eEF2 inhibition is a key factor contributing to the anticancer properties of NFR in vivo.
Figure 6. eEF2K is required for NFR‐mediated antitumoral activity.
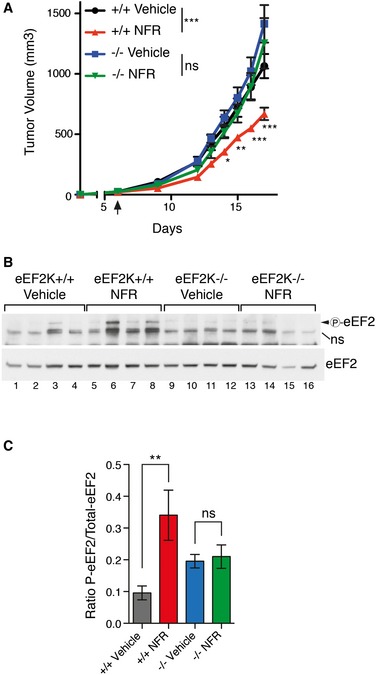
- Tumor volumes of eEF2K−/− and eEF2K+/+ RasV12 grafts implanted subcutaneously in AGR129 mice. Treatment with NFR or vehicle was initiated 6 days post‐implantation. Data are mean of tumor volume ± s.e.m. (n = 8 per group). P‐values were calculated using two‐way ANOVA followed by Bonferroni post‐test comparing WT tumor with vehicle or NFR treatment and KO tumors with vehicle or NFR treatment; *P‐value ≤ 0.05, **P‐value ≤ 0.01, ***P‐value ≤ 0.001. Experiment is representative of 2 performed in the same conditions.
- Immunoblots for total and phosphorylated eEF2 of tumor engraftment from (A). NS, non‐specific signal.
- Bar graph shows relative phosphorylation intensities of eEF2 compared with total eEF2 determined by densitometry analysis. Mean ± s.e.m. are shown (n = 4). P‐values were calculated using two‐way ANOVA followed by Bonferroni post‐test; **P‐value ≤ 0.01.
Source data are available online for this figure.
Discussion
We have previously shown that NFR triggers a transcriptional program that mostly rely on the ISR 17; however, here we found that this pathway is not a main player driving NFR‐mediated toxicity. Indeed, we showed that NFR‐resistant clones could survive and proliferate for weeks in the presence of the drug despite robust and sustained ISR activation. In addition, transgenic cells unable to activate ISR are not protected against NFR toxicity. In contrast, we found that NFR‐mediated toxicity relies on translation reprogramming rather than transcriptional responses. Mechanistically, this was mediated at least in part by activation of eEF2K, a stress response kinase that regulates translation programs. This pathway was downregulated in NFR‐resistant clones. Moreover, a decreased sensitivity to NFR and an impaired growth inhibition were observed in cells with eEF2K deficiency. In mice, we found that the anti‐tumoral effects of NFR are eEF2K dependent, demonstrating that over‐activation of this pathway within cancer cells can affect tumor growth. NFR ability to trigger eEF2K signaling was comparable in all human and mouse cell types tested, indicating that this pathway is a conserved hallmark of NFR‐mediated stress response. It is therefore reasonable to predict that this response could be part of the NFR anti‐tumoral effects observed in cancer patients 10, 11, 12.
The activation of eEF2K is part of an adaptation program that may help stressed cells, such as cancer cells, to cope with conditions of low nutriment and energy 38. This was shown, for example, in eEF2K‐deficient tumors that were found to be more sensitive to caloric restriction 37. Moreover, inhibition of eEF2K suppresses growth of Pten/p53‐deficient triple‐negative breast cancer (TNBC) with elevated Akt signaling xenografts in vivo 39. Downmodulation of eEF2K in breast cancer cell lines has also been reported to decrease the expression of oncogenes such as c‐Myc, thereby impacting on survival and growth 23. These findings clearly indicate that eEF2K activation is a hallmark of stressed cells within tumors. Accordingly, it was proposed that an AMPK‐eEF2K‐dependent reduction of global translation could contribute to preserving energy and conferring tolerance to stress in cancer cells 38, 40. Our observations that increased activation of eEF2K may signal cell death and decreased growth could be in apparent contradiction with its role as part of an adaptation program. However, deregulated or prolonged stress signals can also turn adaptation programs into programed cell death aimed at eliminating irremediably damaged cells. This scenario was demonstrated in the context of several stress pathways including during ER‐stress 41. It is therefore possible that eEF2K integrates signals that promote survival or death depending on the level or duration of its activation status. By increasing eEF2K activity, NFR may redirect the eEF2K program toward death. EEF2K activation observed in stressed tissues as part of the adaptation program may increase their sensitivity to the drug compared to healthy tissues. The status of eEF2K activation within the tumor may therefore influence NFR responsiveness and should be considered as a potential biomarker that could indicate NFR as a therapeutic choice.
The finding that pharmacological eEF2K activation could be detrimental for optimal tumor growth is also supported by a study investigating rapamycin effects in a mouse model of colon cancer that reported that rapamycin‐mediated APC‐deficient adenoma growth arrest required eEF2K 24. Rapamycin inhibits the mTORC1 complex, a master regulator that integrates the signals from nutrient and energy sensors with cell growth. Expression of many mRNA encoding oncogenes or growth‐promoting factors has been show to be translationally regulated downstream of mTOR activity 42. Interestingly, mTOR can modulate both initiation and elongation phase (respectively, through 4EBP‐1‐eIF4E and p70 S6K‐eEF2K‐eEF2 downstream effectors) of mRNA translation to facilitate cell proliferation. The discovery that mTOR inhibition is detrimental for tumor development in an eEF2K‐dependent manner raises the possibility that modifying the translational landscape by targeting elongation factors could be a promising strategy to decrease tumor growth. Nevertheless, therapeutic approaches aimed at targeting mTORC1 rely on a complex network of regulatory loops that affects its function impacting on cancer progression but also leading to increased numbers of potential side effects 43. In contrast, NFR does not significantly affect the mTORC1 signaling pathway and therefore represents a new valuable tool to study and specifically promote eEF2K activation in vitro, in mice as well as in patients. In this study, we show that eEF2K activating pathways such as mTORC1 inhibition, AMPK, or the ISR were not involved in NFR‐mediated eEF2K activation. Possible alternative pathway that could contribute to NFR‐mediated effects includes calcium ion influx, hypoxia, or ERK signaling, which have been proposed to regulate eEF2K 44. It is likely that the identification of NFR cellular targets and mechanism of action will shed new light on novel pathways that can drive eEF2K activation. These mechanisms are predicted to be complex and may involve NFR binding to multiple targets 19. Moreover, despite the fact that we do not observe eEF2K activation with the HIV‐PIs amprenavir, we cannot exclude that some aspects of the HIV‐PIs response could be related to the peptidometic nature of these molecules 45.
How increased eEF2K activation may affect proliferation and promote death is an important question that remains to be solved. It is possible that eEF2K activation may lead to exceed the acceptable threshold of overall translation inhibition or may specifically alter the repertoire of translated genes by preferentially affecting survival and proliferation factors. Malignant cells exhibit altered translational programming that is characterized by augmented activity of many components of the translation machinery, leading to increased overall protein synthesis and modulation of specific oncogenic networks 46. This was shown, for example, in mice engineered to carry only a single copy of the translation initiation factor eIF4E gene 47. These mice develop normally and show a functional translation machinery at basal. However, oncogenic transformation and tumorigenesis were affected by decreased eIF4E levels, indicating that tumors rely on maximal translation capacity 47. Therefore, therapeutic agents that target components of the translation machinery to decrease translation rates hold promise as broad activity anticancer drugs that could overcome intra‐tumor heterogeneity 48. NFR, by inhibiting eEF2, can decrease global rates of protein synthesis and possibly affect the translation of specific mRNAs that could promote tumor cell survival and proliferation. This activity likely contributes to the antitumoral effects and decreased cell growth observed upon eEF2K activation.
In conclusion, the data presented in this study highlight the importance of eEF2K as a therapeutic target in cancer and identify NFR, a well‐known drug with a relatively safe profile and oral bioavailability, as an anticancer treatment that over‐activates eEF2K to limit tumor viability. These data further suggest that in addition of being interesting targets for inhibition, stress responses and adaptation programs can be manipulated to provide increased signal that can lead to cell death and the elimination of stressed cancer tissues.
Materials and Methods
Cell culture and drug treatment
Each knockout or transgenic mouse embryonic fibroblast (MEF) cell line was compared to littermate control. EEF2K WT and KO MEFs were provided by A.G. Ryazanov. EIF2αWT and eIF2αS51A MEFs were from RJ. Kaufman (Sanford Burnham Medical Research Institute, La Jolla, CA, USA). IRE1−/−, XBP1−/−, PERK−/− MEFs, and their respective WT controls were from L. Glimcher (Weill Cornell Medical College, New York, USA). ATF4−/− and ATF4+/+ MEFs were provided by A. Bruhat (INRA, Saint‐Genes‐Champanelle, France). AMPKα1α2 WT and double KO MEFs were from B. Viollet (Institut Cochin, Université Paris Descartes, France). Cell lines have not been authenticated and are tested for mycoplasma once per year in the laboratory. Nelfinavir mesylate (CAS 159989‐65‐8) was from Axon Medchem, LGM pharma; ritonavir, atazanavir, lopinavir, and saquinavir were obtained from The NIH AIDS Reagent Program; nelfinavir hydroxy‐tert‐butylamide (M8) was from Santa Cruz (sc‐208088). Tunicamycin (TM), cycloheximide (CHX), AICAR (5‐amino‐1‐β‐D‐ribofuranosyl‐imidazole‐4‐carboxamide), MK‐2206, and rapamycin were from Enzo‐Life Sciences. Doxycycline, staurosporine, wortmannin and 3‐MA were from Sigma‐Aldrich. For starvation condition, cells were maintained for 1 h in PBS.
Immunoblot analysis
Every Western blot (WB) shown in the study is representative of at least three independent experiments performed in same conditions. Cells and tumor protein extracts were prepared with RIPA buffer (50 mM NaCl, 50 mM Tris pH 7.4, 1 mM EDTA, 0.1% SDS, 1% NP‐40, 1% sodium deoxycholate) supplemented with protease inhibitor cocktail (Roche), 10 mM Na3VO4, 50 mM NaF, 10 mM Na4P2O7, and 5 μM MG132 (Sigma‐Aldrich). The following antibodies were used for immunoblot analysis: anti‐EEF2K (Cell Signaling; #3692), anti‐phospho‐EEF2T56 (Cell Signaling; #2331), anti‐total‐EEF2 (Cell Signaling; #2332), anti‐phospho‐S6 ribosomal proteinS240/244 (Cell Signaling; #2215), anti‐total‐S6 ribosomal protein (Cell Signaling; #2317), anti‐phospho‐AktT308 (Cell Signaling; #9275), anti‐total‐Akt (Cell Signaling; #9272), anti‐phospho‐AMPKαT172 (Cell Signaling; #2535), anti‐total‐AMPKα (Cell Signaling; #2793S), anti‐4EBP1 (Cell Signaling; #9644), anti‐ATF4 (Santa Cruz; sc‐200), anti‐phospho‐eIF2α (Cell Signaling; #3597S), anti‐total‐eIF2α (Cell Signaling; #9722S), and anti‐tubulin (Adipogen; F2C). WB quantifications were performed using ImageJ software.
Polysomal profiling
Cells were treated with 20 μM NFR, 10 μg/ml tunicamycin or DMSO for 6 h. Culture medium was removed and cells were harvested using cold PBS supplemented with 5 mM EDTA and 100 μg/ml cycloheximide (CHX). After two washes in cold PBS containing 100 μg/ml CHX, pellets were resuspended with hypotonic buffer (1.5 mM KCl, 2.5 mM MgCl2, 5 mM Tris pH 7.4) supplemented with 0.1 μg/ml heparin, 100 μg/ml CHX, 20 U/ml RNasin, and protease inhibitors. Equal amount of lysis buffer (hypotonic buffer 2% DOC, 2% Triton X‐100, 2.5 mM DTT) was added, and subsequent cell lysates were vigorously vortexed, incubated 20 min on ice, and centrifuged at 15,000 g for 10 min at 4°C. Post‐nuclear lysates were layered on 10 ml 10–50% (w/v) sucrose gradients (50, 40, 30, 20, and 10% sucrose in 80 mM NaCl, 5 mM MgCl2, 20 mM Tris pH 7.4, 1 mM DTT, and 100 μg/ml CHX in DEPC H2O). Gradients were centrifuged at 30,000 rpm for 4 h at 4°C and separated through a live OD254 nm ultraviolet spectrometer. Comparison of polysomal (P) and sub‐polysomal (S) abundance was based on the measurement of area under the curve. All experiments were repeated at least two times in same conditions. P/S ratio was calculated for each condition and normalized to DMSO‐treated cells.
Metabolic labeling
Cells were treated for indicated times with NFR, 200 nM rapamycin, 1 μg/ml cycloheximide, or 10 μg/ml TM and incubated for the last 15 min with a mixture of L‐35S‐methionine/cysteine (5 μCi/ml). Cells were washed with PBS and lysed with RIPA buffer. Samples were analyzed by autoradiography after SDS–PAGE separation or collected on a glass fiber filter (GF/C, Whatman). Filters were washed twice with ice‐cold 10% TCA, once with 5% TCA, rinsed twice with ethanol, and air‐dried before being subjected to liquid scintillation counting. Values were normalized as percentage of mock‐treated cells.
Ribosome half‐transit time measurement
Ribosome half‐transit time measurements were assessed as in 37, 39 with the following modifications; 200,000 cells seeded for 24 h in 6‐well plates were treated for 6 h with 20 μM NFR; after 5.5 h of treatment, medium was replaced with labeling medium (DMEM, with 4,500 mg/l glucose and sodium bicarbonate, and without l‐methionine, l‐cystine, and l‐glutamine [Sigma‐Aldrich], supplemented with 10% FCS, 1% nonessential amino acids (Gibco Life Technologies™), and 2 mM l‐glutamine (AMIMED, Bioconcept)) for 30 min before addition of 1 μCi/well/ml of L‐35S‐methionine/cysteine. For cycloheximide treatment, CHX was added at 1 μg/ml to the labeling medium for 30 min. For starvation condition, cells were kept in PBS for 30 min before labeling. At indicated times (5, 7.5, 10, 12.5, and 15 min) after labeling, cells were washed twice in ice‐cold PBS containing 100 μg/ml cycloheximide and lysed by adding 250 μl of RSB lysis buffer (10 mM NaCl, 10 mM Tris–HCl pH 7.4, 15 mM MgCl2, 1% Triton X‐100, and 100 μg/ml heparin) containing protease inhibitors directly into the well. Cell lysate were harvested, vortexed, and incubated on ice for 20 min. Nuclei and mitochondria were cleared by centrifugation at 13,000 rpm for 10 min at 4°C; 250 μl post‐mitochondrial supernatant (PMS) was mixed with an equal volume of polysomal buffer (25 mM Tris–HCl pH 7.4, 10 mM MgCl2, 25 mM NaCl, 0.05% Triton X‐100, 0.14 M sucrose, 500 μg/ml heparin), and 250 μl was removed to measure incorporation of 35S‐methionine and cysteine into total protein (nascent and completed). Polysomes were pelleted by centrifugation of the remaining supernatant at 55,000 g for 1 h at 4°C in a Beckman TLA120 rotor, and 200 μl post‐ribosomal supernatant (PRS) was removed to measure the incorporation of 35S‐methionine and cysteine into completed proteins. Hundred microliters of PMS and PRS samples was collected on a glass fiber filter (GF/C, Whatman). Filters were washed twice with ice‐cold 10% TCA, once with 5% TCA, rinsed twice with ethanol, and air‐dried before being subjected to liquid scintillation counting. Incorporation of 35S‐methionine and cysteine into total protein within the PMS and PRS was obtained by linear regression analysis. Ribosome half‐transit time was calculated using the difference of × abscise values chosen for time of 300 s.
Cytotoxic assay
Cell viability was evaluated by using a 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfonphenyl)‐2H‐tetrazolium (MTS) assay (Promega, Madison, WI). All experiments were done in triplicate and were repeated at least three times. The inhibition of cells proliferation was expressed as the percentage of vehicle control cells. Dose–inhibition rate curves were plotted using a five‐parameter logistic equation. For all experiments, EC50 value (the 50% maximal inhibitory concentration) was calculated and mean ± s.e.m. of at least three independent experiments is shown.
Cell proliferation measurement
Thousand cells were seeded in 96‐well plates and let adhere for 24 h. Just before drug addition, cells were counted on a define area of each well using a SpectraMax imager technology in order to determine the number of cells at time 0. Cells were treated with vehicle or with 10 μM NFR, and each well was imaged again at indicated times. The fold change of cell number compared to time 0 was calculated for each individual well at indicated time. Curves represent the mean ± s.e.m. of three different wells treated or not with 10 μM NFR. The percentage of growth inhibition was determined using the ratio between growth of treated and untreated cells at 72 h of NFR treatment. For every cell line, curves showed one representative of three experiments performed in triplicate in the same conditions and histograms are mean ± s.e.m. of three independent experiments.
AnnexinV/PI staining and FACS analysis
AnnexinV/propidium iodide (PI) staining was performed using eBioscience kit (BMS500FI) according to the manufacturer's guidelines. Briefly, 2 × 105 cells plated in 12‐well plates were treated for 24 h with 0, 20, or 40 μM NFR. Cells and supernatant were harvested and centrifuged 5 min at 300 g. Cells were washed in ice‐cold PBS and resuspended in 50 μl of binding buffer containing 2.5 μl of AnnexinV‐FITC and incubated for 30 min on ice. Cells were washed once and resupended in 100 μl of binding buffer containing 10 μl of PI (20 μg/ml). Flow cytometric analysis was immediately performed using an Accuri flow cytometer instrument (BD Bioscience).
Mice
Animal experiments were approved by the Veterinary Office of the Canton de Vaud and the Animal Ethics Committee (authorization 2883). Immunocompromised AGR 129 (IFN‐α/β, IFN‐γ receptor, and RAG‐2 deficient) mice were provided by M. Gilliet (Department of Dermatology, CHUV, Lausanne) and housed at the University of Lausanne in accordance with local and national guidelines; 1 × 106 eEF2K WT‐HRasV12 and eEF2K−/− HRasV12 MEFs were, respectively, injected subcutaneously into the right and left flanks of 6–8‐week‐old female. Tumor growth was followed by measuring size of the tumor with a caliper. Tumor volume was calculated using the formula V = (L × l2)/2. When tumors reach 200 mm3, mice were randomly distributed into two homogenous groups and injected intraperitoneally daily either with vehicle (4% DMSO, 5% PEG, 5% Tween‐80 in saline) or with 100 mg/kg NFR. Experiments were repeated two times with 6–8 mice per group.
High‐throughput sequencing
For RNA sequencing, RNA was extracted using RNeasy mini kit (QIAGEN) from three independent plates of HeLa cells treated or not with 20 μM NFR and from four independent 10 μM NFR‐resistant HeLa cell clones. High‐throughput sequencing was performed at the Lausanne Genomics Technologies Facility (University of Lausanne) on the Illumina HiSeq 2500 using TruSeq SBS Kit v3 reagents. For the RNA‐seq analysis, we used a moderated t‐test from the R bioconductor package “limma” (R version 3.1.1, limma version 3.20.8). The “adjusted P‐value” corresponds to the P‐values corrected for multiple testing using the Benjamini–Hochberg method.
RT–PCR
SYBR Green fluorescent reagent and LightCycler 480 Real‐Time PCR System (Roche) were used for quantitative RT–PCR. Primer sequences used were as follows: EEF2K F, 5′‐CACCTGGAAGATATTGCCACC‐3′; R, 5′‐GCTTCGCCACGTAGTTGGA‐3′; EEF2 F, 5′‐AACTTCACGGTAGACCAGATCC‐3′; R, 5′‐TCGTCCTTCCGGGTATCAGTG‐3′; DNAjB9 F, 5′‐TCTTAGGTGTGCCAAAATCGG‐3′; R, 5′‐TGTCAGGGTGGTACTTCATGG‐3′; CHOP F, 5′‐GGAAACACAGTGGTCATTCCC‐3′; and FR, 5′‐CTGCTTGAGCCGTTCATTCTC‐3′. All RT–PCR were performed in experimental triplicate and repeated at least three times on independent samples.
Plasmid construction
LentiCRISPR‐v2 for EEF2K targeting: Optimized CRISPR target sequences were cloned into the lentiCRISPR‐v2 vector (Addgene #52961). The seed sequence was designed in the kinase domain of eEF2K as follows: CACCTGGAGCACTACATCGA. A sequence targeting luciferase was used as control sgRNA (CTTCGAAATGTCCGTTCGGT). p21‐FlagEEF2K for EEF2K reconstitution: Human EEF2K was amplified by PCR from pDONR223‐EEF2K (Addgene #23726) and sub‐cloned into lentiviral pINDUCER21‐plasmid (S. Elledge, Harvard Medical School).
Lentivirus production and cell line infection
Lentiviruses were produced as previously described 17. HeLa, A549, and MCF7 cells were infected with lentiCRISPR‐v2 viruses targeting luciferase or EEF2K. Positive populations were selected with 2 μg/ml puromycin. Clones were tested by WB for EEF2K protein level. EEF2K−/− MEFs were infected with p21‐FlagEEF2K viruses and GFP‐positive cells were FACS sorted 5 days after infection. Flag‐EEF2K expression was induced using doxycycline (1 μg/ml for 24 h). LZRS‐H‐rasV12 retroviruses used for EEF2K WT and KO MEF infections were provided by K. Lefort (GP Dotto laboratory, UNIL, Lausanne).
Statistical analysis
Statistical significance was ascertained by performing appropriate tests described in figure legends. Significant differences were indicated by *(P ≤ 0.05), **(P ≤ 0.01), or ***(P ≤ 0.001). For the RNA‐seq analysis, we used a moderated t‐test from the R bioconductor package “limma” (R version 3.1.1, limma version 3.20.8). The “adjusted P‐value” reported in the Dataset EV1 corresponds to the P‐values corrected for multiple testing using the Benjamini–Hochberg method, which controls for false discovery rate (FDR).
Author contributions
FM and ADG performed conceptualization; ADG and FM performed methodology; ADG, OD, RP, and LZ carried out investigation; FM and ADG were involved in writing the original draft; FM provided funding acquisition; AGR and MG provided resources; ADG and FM supervised the article.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 6
Acknowledgements
We thank L.H. Glimcher, A. Bruhat, R.J. Kaufman, B. Viollet, S. Elledge, C. Lefort, G.P. Dotto, and the NIH AIDS Reagent Program for sharing key reagents. We thank Jérome Lugrin, Etienne Meylan, Owen Sansom, and Margot Thome for critical reading of the manuscript. We thank Keith Harshman, Leonore Wigger, and the staff of the Lausanne Genomics Technologies Facility for high‐throughput sequencing. This project is supported by a grant from the European Research Council (ERC) starting grant (281996), a Human Frontier Science Program (HFSP) career development award (CDA00059/2011), and a grant from the Swiss National Science Foundation (31003A‐130476).
EMBO Reports (2016) 17: 1471–1484
References
- 1. Ashburn TT, Thor KB (2004) Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov 3: 673–683 [DOI] [PubMed] [Google Scholar]
- 2. Jin G, Wong ST (2014) Toward better drug repositioning: prioritizing and integrating existing methods into efficient pipelines. Drug Discov Today 19: 637–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lebbe C, Blum L, Pellet C, Blanchard G, Verola O, Morel P, Danne O, Calvo F (1998) Clinical and biological impact of antiretroviral therapy with protease inhibitors on HIV‐related Kaposi's sarcoma. AIDS 12: F45–F49 [DOI] [PubMed] [Google Scholar]
- 4. Krischer J, Rutschmann O, Hirschel B, Vollenweider‐Roten S, Saurat JH, Pechere M (1998) Regression of Kaposi's sarcoma during therapy with HIV‐1 protease inhibitors: a prospective pilot study. J Am Acad Dermatol 38: 594–598 [DOI] [PubMed] [Google Scholar]
- 5. Niehues T, Horneff G, Megahed M, Schroten H, Wahn V (1999) Complete regression of AIDS‐related Kaposi's sarcoma in a child treated with highly active antiretroviral therapy. AIDS 13: 1148–1149 [DOI] [PubMed] [Google Scholar]
- 6. Cuneo KC, Tu T, Geng L, Fu A, Hallahan DE, Willey CD (2007) HIV protease inhibitors enhance the efficacy of irradiation. Cancer Res 67: 4886–4893 [DOI] [PubMed] [Google Scholar]
- 7. Pyrko P, Kardosh A, Wang W, Xiong W, Schonthal AH, Chen TC (2007) HIV‐1 protease inhibitors nelfinavir and atazanavir induce malignant glioma death by triggering endoplasmic reticulum stress. Cancer Res 67: 10920–10928 [DOI] [PubMed] [Google Scholar]
- 8. Bernstein WB, Dennis PA (2008) Repositioning HIV protease inhibitors as cancer therapeutics. Curr Opin HIV AIDS 3: 666–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bruning A, Burger P, Vogel M, Rahmeh M, Gingelmaiers A, Friese K, Lenhard M, Burges A (2009) Nelfinavir induces the unfolded protein response in ovarian cancer cells, resulting in ER vacuolization, cell cycle retardation and apoptosis. Cancer Biol Ther 8: 226–232 [DOI] [PubMed] [Google Scholar]
- 10. Gills JJ, Lopiccolo J, Tsurutani J, Shoemaker RH, Best CJ, Abu‐Asab MS, Borojerdi J, Warfel NA, Gardner ER, Danish M et al (2007) Nelfinavir, A lead HIV protease inhibitor, is a broad‐spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo . Clin Cancer Res 13: 5183–5194 [DOI] [PubMed] [Google Scholar]
- 11. Chow WA, Jiang C, Guan M (2009) Anti‐HIV drugs for cancer therapeutics: back to the future? Lancet Oncol 10: 61–71 [DOI] [PubMed] [Google Scholar]
- 12. Blumenthal GM, Gills JJ, Ballas MS, Bernstein WB, Komiya T, Dechowdhury R, Morrow B, Root H, Chun G, Helsabeck C et al (2014) A phase I trial of the HIV protease inhibitor nelfinavir in adults with solid tumors. Oncotarget 5: 8161–8172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rengan R, Mick R, Pryma D, Rosen MA, Lin LL, Maity AM, Evans TL, Stevenson JP, Langer CJ, Kucharczuk J et al (2012) A phase I trial of the HIV protease inhibitor nelfinavir with concurrent chemoradiotherapy for unresectable stage IIIA/IIIB non‐small cell lung cancer: a report of toxicities and clinical response. J Thorac Oncol 7: 709–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brunner TB, Geiger M, Grabenbauer GG, Lang‐Welzenbach M, Mantoni TS, Cavallaro A, Sauer R, Hohenberger W, McKenna WG (2008) Phase I trial of the human immunodeficiency virus protease inhibitor nelfinavir and chemoradiation for locally advanced pancreatic cancer. J Clin Oncol 26: 2699–2706 [DOI] [PubMed] [Google Scholar]
- 15. Buijsen J, Lammering G, Jansen RL, Beets GL, Wals J, Sosef M, Den Boer MO, Leijtens J, Riedl RG, Theys J et al (2013) Phase I trial of the combination of the Akt inhibitor nelfinavir and chemoradiation for locally advanced rectal cancer. Radiother Oncol 107: 184–188 [DOI] [PubMed] [Google Scholar]
- 16. Koltai T (2015) Nelfinavir and other protease inhibitors in cancer: mechanisms involved in anticancer activity. F1000Res 4: 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De Gassart A, Bujisic B, Zaffalon L, Decosterd LA, Di Micco A, Frera G, Tallant R, Martinon F (2016) An inhibitor of HIV‐1 protease modulates constitutive eIF2alpha dephosphorylation to trigger a specific integrated stress response. Proc Natl Acad Sci USA 113: E117–E126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gantt S, Casper C, Ambinder RF (2013) Insights into the broad cellular effects of nelfinavir and the HIV protease inhibitors supporting their role in cancer treatment and prevention. Curr Opin Oncol 25: 495–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xie L, Evangelidis T, Xie L, Bourne PE (2011) Drug discovery using chemical systems biology: weak inhibition of multiple kinases may contribute to the anti‐cancer effect of nelfinavir. PLoS Comput Biol 7: e1002037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kenney JW, Moore CE, Wang X, Proud CG (2014) Eukaryotic elongation factor 2 kinase, an unusual enzyme with multiple roles. Adv Biol Regul 55: 15–27 [DOI] [PubMed] [Google Scholar]
- 21. Ryazanov AG, Ward MD, Mendola CE, Pavur KS, Dorovkov MV, Wiedmann M, Erdjument‐Bromage H, Tempst P, Parmer TG, Prostko CR et al (1997) Identification of a new class of protein kinases represented by eukaryotic elongation factor‐2 kinase. Proc Natl Acad Sci USA 94: 4884–4889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Proud CG (2015) Regulation and roles of elongation factor 2 kinase. Biochem Soc Trans 43: 328–332 [DOI] [PubMed] [Google Scholar]
- 23. Tekedereli I, Alpay SN, Tavares CD, Cobanoglu ZE, Kaoud TS, Sahin I, Sood AK, Lopez‐Berestein G, Dalby KN, Ozpolat B (2012) Targeted silencing of elongation factor 2 kinase suppresses growth and sensitizes tumors to doxorubicin in an orthotopic model of breast cancer. PLoS ONE 7: e41171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Faller WJ, Jackson TJ, Knight JR, Ridgway RA, Jamieson T, Karim SA, Jones C, Radulescu S, Huels DJ, Myant KB et al (2015) mTORC1‐mediated translational elongation limits intestinal tumour initiation and growth. Nature 517: 497–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hennessy M, Clarke S, Spiers JP, Kelleher D, Mulcahy F, Hoggard P, Back D, Barry M (2004) Intracellular accumulation of nelfinavir and its relationship to P‐glycoprotein expression and function in HIV‐infected patients. Antivir Ther 9: 115–122 [PubMed] [Google Scholar]
- 26. Ford J, Cornforth D, Hoggard PG, Cuthbertson Z, Meaden ER, Williams I, Johnson M, Daniels E, Hsyu P, Back DJ et al (2004) Intracellular and plasma pharmacokinetics of nelfinavir and M8 in HIV‐infected patients: relationship with P‐glycoprotein expression. Antivir Ther 9: 77–84 [PubMed] [Google Scholar]
- 27. Browne GJ, Finn SG, Proud CG (2004) Stimulation of the AMP‐activated protein kinase leads to activation of eukaryotic elongation factor 2 kinase and to its phosphorylation at a novel site, serine 398. J Biol Chem 279: 12220–12231 [DOI] [PubMed] [Google Scholar]
- 28. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30: 214–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Inoki K, Zhu T, Guan KL (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115: 577–590 [DOI] [PubMed] [Google Scholar]
- 30. Wang X, Regufe da Mota S, Liu R, Moore CE, Xie J, Lanucara F, Agarwala U, Pyr Dit Ruys S, Vertommen D, Rider MH et al (2014) Eukaryotic elongation factor 2 kinase activity is controlled by multiple inputs from oncogenic signaling. Mol Cell Biol 34: 4088–4103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang Y, Ikezoe T, Takeuchi T, Adachi Y, Ohtsuki Y, Takeuchi S, Koeffler HP, Taguchi H (2005) HIV‐1 protease inhibitor induces growth arrest and apoptosis of human prostate cancer LNCaP cells in vitro and in vivo in conjunction with blockade of androgen receptor STAT3 and AKT signaling. Cancer Sci 96: 425–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gupta AK, Cerniglia GJ, Mick R, McKenna WG, Muschel RJ (2005) HIV protease inhibitors block Akt signaling and radiosensitize tumor cells both in vitro and in vivo . Cancer Res 65: 8256–8265 [DOI] [PubMed] [Google Scholar]
- 33. Demeshkina N, Hirokawa G, Kaji A, Kaji H (2007) Novel activity of eukaryotic translocase, eEF2: dissociation of the 80S ribosome into subunits with ATP but not with GTP. Nucleic Acids Res 35: 4597–4607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fan H, Penman S (1970) Regulation of protein synthesis in mammalian cells. II. Inhibition of protein synthesis at the level of initiation during mitosis. J Mol Biol 50: 655–670 [DOI] [PubMed] [Google Scholar]
- 35. Gills JJ, Lopiccolo J, Dennis PA (2008) Nelfinavir, a new anti‐cancer drug with pleiotropic effects and many paths to autophagy. Autophagy 4: 107–109 [DOI] [PubMed] [Google Scholar]
- 36. Grob P, Schijns VE, van den Broek MF, Cox SP, Ackermann M, Suter M (1999) Role of the individual interferon systems and specific immunity in mice in controlling systemic dissemination of attenuated pseudorabies virus infection. J Virol 73: 4748–4754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Leprivier G, Remke M, Rotblat B, Dubuc A, Mateo AR, Kool M, Agnihotri S, El‐Naggar A, Yu B, Somasekharan SP et al (2013) The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell 153: 1064–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fu LL, Xie T, Zhang SY, Liu B (2014) Eukaryotic elongation factor‐2 kinase (eEF2K): a potential therapeutic target in cancer. Apoptosis 19: 1527–1531 [DOI] [PubMed] [Google Scholar]
- 39. Liu JC, Voisin V, Wang S, Wang DY, Jones RA, Datti A, Uehling D, Al‐awar R, Egan SE, Bader GD et al (2014) Combined deletion of Pten and p53 in mammary epithelium accelerates triple‐negative breast cancer with dependency on eEF2K. EMBO Mol Med 6: 1542–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Leprivier G, Rotblat B, Khan D, Jan E, Sorensen PH (2015) Stress‐mediated translational control in cancer cells. Biochim Biophys Acta 1849: 845–860 [DOI] [PubMed] [Google Scholar]
- 41. Maurel M, McGrath EP, Mnich K, Healy S, Chevet E, Samali A (2015) Controlling the unfolded protein response‐mediated life and death decisions in cancer. Semin Cancer Biol 33: 57–66 [DOI] [PubMed] [Google Scholar]
- 42. Pelletier J, Graff J, Ruggero D, Sonenberg N (2015) Targeting the eIF4F translation initiation complex: a critical nexus for cancer development. Cancer Res 75: 250–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Efeyan A, Sabatini DM (2010) mTOR and cancer: many loops in one pathway. Curr Opin Cell Biol 22: 169–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu R, Proud CG (2016) Eukaryotic elongation factor 2 kinase as a drug target in cancer, and in cardiovascular and neurodegenerative diseases. Acta Pharmacol Sin 37: 285–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Randolph JT, DeGoey DA (2004) Peptidomimetic inhibitors of HIV protease. Curr Top Med Chem 4: 1079–1095 [DOI] [PubMed] [Google Scholar]
- 46. Ruggero D (2013) Translational control in cancer etiology. Cold Spring Harbor Perspect Biol 5: a012336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Truitt ML, Conn CS, Shi Z, Pang X, Tokuyasu T, Coady AM, Seo Y, Barna M, Ruggero D (2015) Differential Requirements for eIF4E Dose in Normal Development and Cancer. Cell 162: 59–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, Topisirovic I (2015) Targeting the translation machinery in cancer. Nat Rev Drug Discov 14: 261–278 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 6