

Abstract
In response to diverse stress stimuli, eukaryotic cells activate a common adaptive pathway, termed the integrated stress response (ISR), to restore cellular homeostasis. The core event in this pathway is the phosphorylation of eukaryotic translation initiation factor 2 alpha (eIF2α) by one of four members of the eIF2α kinase family, which leads to a decrease in global protein synthesis and the induction of selected genes, including the transcription factor ATF4, that together promote cellular recovery. The gene expression program activated by the ISR optimizes the cellular response to stress and is dependent on the cellular context, as well as on the nature and intensity of the stress stimuli. Although the ISR is primarily a pro‐survival, homeostatic program, exposure to severe stress can drive signaling toward cell death. Here, we review current understanding of the ISR signaling and how it regulates cell fate under diverse types of stress.
Keywords: activating transcription factor 4, eIF2α kinase, eukaryotic translation initiation factor 2 alpha, integrated stress response
Subject Categories: Autophagy & Cell Death, Signal Transduction
Glossary
- β‐TrCP
F‐box protein β‐transducin repeat‐containing protein
- 5′UTR
5′ untranslated region
- AARE
Amino acid response element
- Akt
Protein kinase B
- ALS
Amyotrophic lateral sclerosis
- AP‐1
Activator protein 1
- APAF‐1
Apoptotic peptidase‐activating factor 1
- ASNS
Asparagine synthetase
- ATF/CREB
Activating transcription factor/cyclic AMP response element binding protein
- ATF
Activating transcription factor
- Atg
Autophagy‐related protein
- BBC3
p53 upregulated modulator of apoptosis
- BCL‐2
B‐cell lymphoma 2
- Becn1
Beclin‐1
- BIM (BCL2L11)
BCL‐2 interacting mediator of cell death
- BNIP3L
BCL2/adenovirus E1B 19‐kDa protein‐interacting protein 3‐like
- bZIP
Basic leucine zipper domain
- C/EBPβ
CCAAT/enhancer‐binding protein beta
- CARE
C/EBP‐ATF response element
- CDS
Coding sequence
- CH1
Constant heavy chain domain 1
- CHOP
C/EBP homologous protein
- CK1
Casein kinase 1
- CK2
Casein kinase 2
- CLOCK
Circadian locomotor output cycles protein kaput
- CMT
Charcot–Marie–Tooth disease
- CReP
Constitutive repressor of eIF2α phosphorylation
- DR5
Death receptor 5
- eIF
Eukaryotic translation initiation factor
- ER
Endoplasmic reticulum
- ERO1α
Endoplasmic oxidoreductin‐1‐like protein
- FOXO
Forkhead box O
- Gabarap
Gamma‐aminobutyric acid receptor‐associated protein
- Gabarapl2
Gamma‐aminobutyric acid receptor‐associated protein‐like 2
- GADD34
Growth arrest and DNA damage‐inducible protein GADD34
- GCN2
General control non‐depressible protein 2
- GRP78
78‐kDa glucose‐regulated protein
- HIV
Human immunodeficiency virus
- HRI
Heme‐regulated eIF2α kinase
- HSP70 and 90
Heat shock proteins 70 and 90
- HSV
Herpes simplex virus
- IAPs
Inhibitor of apoptosis proteins
- IBTKα
α‐isoform of inhibitor of Bruton's tyrosine kinase
- IRE1
Inositol‐requiring enzyme‐1
- IRES
Internal ribosome entry site
- ISRIB
Integrated stress response inhibitor
- ISR
Integrated stress response
- Map1lc3b (LC3)
Microtubule‐associated proteins 1 light chain 3 beta
- MCL‐1
Myeloid cell leukemia‐1
- MEFs
Mouse embryonic fibroblasts
- mTORC1
mTOR complex 1
- mTOR
Mammalian target of rapamycin
- NAIP
Neuronal apoptosis inhibitory protein
- NBR1
Neighbor of BRCA1 gene 1
- NF‐κB
Nuclear factor kappa light chain enhancer of activated B cells
- NRF2
Nuclear factor erythroid 2‐related factor 2
- NSRE‐1
Nutrient‐sensing response elements 1
- NSRE‐2
Nutrient‐sensing response elements 2
- NUPR1
Nuclear protein 1
- OLA1
Obg‐like ATPase 1
- p300
Histone acetyltransferase p300
- PDX1
Pancreatic and duodenal homeobox 1
- PERK
PKR‐like ER kinase
- PHD1
Prolyl hydroxylase 1
- PHD3
Prolyl hydroxylase 3
- PI3K
Phosphatidylinositol‐3 kinase
- PIC
Pre‐initiation complex
- PKA
Protein kinase A
- PKR
Double‐stranded RNA‐dependent protein kinase
- PMAIP1
Phorbol‐12‐myristate‐13‐acetate‐induced protein 1
- PP1c
Protein phosphatase 1 catalytic subunit
- PP1
Protein phosphatase 1
- PPP1R15A
Protein phosphatase 1 regulatory subunit 15A
- PPP1R15B
Protein phosphatase 1 regulatory subunit 15B
- REDD1 (Ddit4)
Regulated in development and DNA damage response 1
- Rpl7
60S ribosomal protein L7
- RSK2
Ribosomal protein S6 kinase alpha‐2
- SCFβTrCP
SCF E3 ubiquitin ligase complex
- Siah 1/2
E3 ubiquitin protein ligases 1 and 2
- Sqstm1
Sequestosome 1
- SyK
Spleen‐associated tyrosine kinase
- TFE3
Transcription factor E3
- TFEB
Transcription factor EB
- TNFRSF10B
TNF receptor superfamily member 10b
- TRAIL
TNF‐related apoptosis‐inducing ligand
- TRIB3, SKIP3
Tribbles homolog 3
- TXNIP
Thioredoxin‐interacting protein
- uORF
Upstream open reading frame
- UPR
Unfolded protein response
- VEGF
Vascular endothelial growth factor
- WARS
Tryptophanyl‐tRNA synthetase
- XBP1
X‐box binding protein 1
- XIAP
X chromosome‐linked inhibitor of apoptosis
- ZIPK
Zipper‐interacting protein kinase
Introduction
The integrated stress response (ISR) is an elaborate signaling pathway present in eukaryotic cells, which is activated in response to a range of physiological changes and different pathological conditions 1, 2, 3, 4, 5. Such stresses commonly include cell extrinsic factors such as hypoxia, amino acid deprivation, glucose deprivation, and viral infection 2, 4, 5, 6, 7, 8. However, cell intrinsic stresses such as endoplasmic reticulum (ER) stress, caused by the accumulation of unfolded proteins in the ER, can also activate the ISR 9. Furthermore, in the context of cancer biology, the ISR can be triggered by oncogene activation 10, 11. The common point of convergence for all the stress stimuli that activate ISR is phosphorylation of the alpha subunit of eukaryotic translation initiation factor 2 (eIF2α) on serine 51 1. In mammalian cells, this is catalyzed by a family of four serine/threonine (S/T) eIF2α kinases that are activated by distinct stress stimuli 12. eIF2α phosphorylation causes a reduction in global protein synthesis while allowing the translation of selected genes including activating transcription factor 4 (ATF4), aiding cell survival and recovery 13. However, if the cellular stress is severe, either in intensity or in duration, it will overwhelm the capacity of the adaptive response to resolve it and additional components become activated to execute cell death. Dephosphorylation of eIF2α signals termination of the ISR and return to normal protein synthesis (Fig 1) 14, 15, 16. It is likely that the duration and level of eIF2α phosphorylation, as well as ATF4 regulation and its interactions with other proteins, determine the ultimate ISR outcome resulting from different environmental and physiological stresses 16, 17, 18.
Figure 1. Integrated stress response signaling.

ER stress, viral infection, and other cellular stress signals activate PERK, PKR, HRI, and GCN2 kinases that converge on phosphorylation of eIF2α, the core of ISR. This leads to global attenuation of Cap‐dependent translation while concomitantly initiates the preferential translation of ISR‐specific mRNAs, such as ATF4. ATF4 is the main effector of the ISR. It forms homo‐ and heterodimers that bind to DNA targets to control the expression of genes involved in cellular adaptation. Termination of the ISR is regulated by the constitutively expressed CReP and stress‐inducible phosphatase GADD34 that dephosphorylate eIF2α. For more details, see main text and Table 1. Arrows denote activation or induction, while blunt lines indicate inhibition.
Here, we outline current knowledge of the molecular mechanisms of the ISR, highlighting the importance of eIF2α phosphorylation/dephosphorylation and how signaling events downstream of ATF4 ultimately regulate the cell responses to the particular stress insult. We also discuss the ISR pathway in the context of cell survival and cell death in mammalian cells, and describe pharmacological modifications of the pathway that may have therapeutic value.
Activation of the integrated stress response
The eIF2α kinases act as early responders to disturbances in cellular homeostasis. There are four members of the family: PKR‐like ER kinase (PERK) 19, double‐stranded RNA‐dependent protein kinase (PKR), heme‐regulated eIF2α kinase (HRI), and general control non‐derepressible 2 (GCN2) (Fig 1) 5, 12. All four eIF2α kinases share extensive homology in their kinase catalytic domains, but possess distinct regulatory domains 9, 20, 21, 22, 23, 24. Each eIF2α kinase dimerizes and autophosphorylates for full activation 25. However, each kinase responds to distinct environmental and physiological stresses (Fig 1), which reflects their unique regulatory mechanisms 12.
For example, PERK, found in metazoans, is located in the endoplasmic reticulum (ER) membrane where its luminal domain is normally bound by 78‐kDa glucose‐regulated protein (GRP78). ER stress (e.g. due to the accumulation of unfolded proteins in the ER, or perturbations in cellular energy, calcium homeostasis, or redox status) causes the activation of PERK. Two models for the activation of PERK by ER stress have been put forward 26, 27. The classical model proposes that upon the accumulation of misfolded or unfolded proteins in the ER lumen, GRP78 becomes dissociated from PERK, leading to its autophosphorylation and activation 9, 22, 28, 29. However, recent research suggests an alternative mechanism whereby PERK may be activated directly by binding of unfolded or misfolded proteins to its luminal domain 26, 30. While there is somewhat limited direct evidence for the latter model of PERK activation, it is supported by evidence from another ER sensor of unfolded proteins, inositol‐requiring enzyme‐1 (IRE1), whose ER luminal domain exhibits strong sequence homology with that of PERK and which in yeast has been shown to be directly activated by unfolded or misfolded proteins 26, 30. PERK activation in response to glucose deprivation has been reported in neuronal cells and in pancreatic β cells 31, 32 where it was shown that it is the ATP depletion that leads to the activation of PERK through the inhibition of sarcoplasmic/ER Ca2+‐ATPase pump. It is also interesting to note that in cancer cells, oncogene activation can trigger the ISR through PERK. For example, amplified HRAS induces PERK/eIF2α/ATF4 and cellular senescence in a premalignant model of melanoma 10 and increased activity of proto‐oncogene MYC elicits the activation of the PERK/eIF2α/ATF4 resulting in cellular transformation and tumorigenesis 11.
GCN2 is highly conserved from yeast to humans 33. Mechanistic insights into GCN2 regulation, largely derived from studies in yeast, indicate that GCN2 binds to deacylated transfer RNAs (tRNAs) via a histidyl‐tRNA synthetase‐related domain to become active in response to amino acid deprivation 34. GCN2 activation has also been reported in response to a prolonged glucose deprivation in tumor cells, although this is probably an indirect effect resulting from the consumption of amino acids as an alternative energy source in the absence of glucose 7. GCN2 can also be activated by UV light in mouse embryonic fibroblast (MEF) cells and human keratinocytes 35, 36. The mechanism of GCN2 activation by UV light is unclear and at least two models have been suggested, including UV‐induced cross‐linking between tRNA and GCN2, or the rapid consumption of arginine due to UV activation of nitric oxide synthetase 33, 35, 36.
The mammalian PKR is activated mainly by double‐stranded RNA (dsRNA) during viral infection 37, 38. Dimerization of PKR via its C‐terminal kinase domain results in autophosphorylation at T446 and the subsequent functional activation of the kinase 39, 40, 41. Thus, the presence of dsRNA during viral infection results in PKR activation and the subsequent inhibition of viral and host protein synthesis through eIF2α phosphorylation 37, 42, 43. Interestingly, other stresses such as oxidative and ER stress 44, 45, growth factor deprivation, cytokine or bacterial infection 46, 47, ribotoxic stress 48, stress granules 49, and heparin 50 have also been shown to stimulate PKR, but in a dsRNA‐independent manner. Interestingly, PKR can also be stimulated by caspase activity in the early stage of apoptosis, indicating a role for the inhibition of protein synthesis in this cell death program 51.
Unlike the other eIF2α kinases that have a broad tissue distribution, HRI is mainly expressed in erythroid cells where it is involved in erythrocyte differentiation during erythropoiesis 52. HRI couples the translation of globin mRNAs with the availability of heme for the production of hemoglobin and thus protects erythroid cells against the accumulation of toxic globin aggregates in iron deficiency 52, 53, 54, 55. Similar to the other eIF2α kinases, HRI activation requires dimerization and autophosphorylation of its kinase domain 56, 57. Regulation of HRI kinase activity by heme is mediated by the two heme‐binding domains that are located in the N‐terminus and the kinase insertion domain 56, 57. Binding of heme to the N‐terminus is stable and it co‐purifies with HRI, whereas heme binding to the kinase insertion domain is reversible and inhibits HRI kinase activity 54, 56. Heme inhibits HRI kinase activity in vitro and in vivo by promoting disulfide bond formation between HRI monomers, keeping them in an inactive dimer conformation 52, 58, 59. However, in the absence of heme, non‐covalent interactions between HRI molecules occurs, resulting in an active HRI dimer 52, 58, 59. HRI can also be activated by other stresses including arsenite‐induced oxidative stress, heat shock, osmotic stress, 26S proteasome inhibition, and nitric oxide 52, 60, 61, 62, 63. Interestingly, activation of HRI by these diverse stresses is independent of heme and occurs with the aid of heat shock proteins HSP90 and HSP70; however, the exact mechanism of HRI activation remains to be investigated 63.
The eIF2α kinases have overlapping functions and, as such, can act cooperatively to specifically tune cellular responses to a wide variety of stressors, hereafter referred as cellular stress, unless otherwise stated. For example, GCN2 contributes to ER stress‐induced eIF2α phosphorylation in Perk −/− MEF cells 64. Conversely, PERK can compensate for Gcn2 loss in a genetically engineered mouse model of soft tissue sarcoma 65 and in a Gcn2‐knockout 5XFAD mouse model of Alzheimer's disease 66, as well as for Hri loss in mouse hepatocytes 58. Further support for signaling redundancy between PERK and GCN2 comes from experiments on HeLa cells stressed by protein overload where knockdown of either PERK or GCN2 was compensated by a rapid upregulation of the other kinase 67. Additionally, both PERK and GCN2 become activated during nucleofection 68 and together with PKR can regulate host response to viral infection 8, 69, 70. All eIF2α kinases become activated in response to oxidative stress 2, 55, 71, 72. PERK and PKR kinases activate the ISR to effectively manage heat stress and to limit the aggregation and accumulation of the denatured proteins in the ER of human endothelial cells and MEFs, respectively 73, 74. Possibly, in such cases when eIF2α kinases act cooperatively, the cellular response is determined not only by ISR activation but also by the activation of other specific substrates of eIF2α kinases. Recently, it has been reported that in in vitro models of several types of tumors, upon ER stress, amino acid starvation, and oxidative stress, the ISR can be also modulated by OLA1, a GTPase that inhibits de novo formation of the eIF2 ternary complex, providing a secondary mechanism for the inhibition of global mRNA translation while permitting ATF4 protein synthesis 75.
Termination of the ISR
Dephosphorylation of eIF2α is central to ISR signal termination to restore protein synthesis and normal cell functioning 15. It is mediated by protein phosphatase 1 (PP1) complex that recruits a PP1 catalytic subunit (PP1c) and one of the two regulatory subunits. In mammals, phosphatase activity is regulated by either PPP1R15A (also known as growth arrest and DNA damage‐inducible protein, GADD34), which is induced as part of the ISR, or by the constitutively expressed paralogue PPP1R15B (also known as constitutive repressor of eIF2α phosphorylation, CReP) that is responsible for targeting the enzyme to eIF2α (Fig 1) 15, 76. CReP normally operates in a complex with PP1c in unstressed cells to sustain translational homeostasis by maintaining low levels of eIF2α phosphorylation 76. In contrast, GADD34 expression is induced downstream of phosphorylated eIF2α and ATF4 during the later stages of the ISR to significantly increase eIF2α dephosphorylation 77. Thus, the GADD34–PP1 complex acts as an important negative feedback loop to restore protein synthesis once the ER stress has been resolved, and as such aids in cell survival 78, 79. However, eIF2α dephosphorylation may also be important to accommodate the translation of accumulated mRNAs of stress‐responsive genes during the ER stress and oxidative stress 16. It may also facilitate the execution of cell death when cellular homeostasis cannot be restored, by allowing the synthesis of death‐inducing proteins and also other proteins that accumulate in the cell and aggravate the proteotoxicity and oxidative stress 16, 80. The importance of eIF2α dephosphorylation is highlighted by the fact that Ppp1r15a and Ppp1r15b‐double‐knockout mice exhibit early embryonic lethality, which can be rescued by eIF2α S51A mutation that prevents eIF2α phosphorylation 81.
Recently, in addition to PP1c and GADD34, G‐actin was identified as a crucial and conserved component of the eIF2α phosphatase complex, providing an insight into the importance of cytoskeletal dynamics in regulating the ER stress‐induced ISR 82, 83. It is possible that the eIF2α phosphatase complex contains other important regulatory factors that may differ among cell types or species. The mechanism of ISR termination may also involve CReP degradation as indicated by a recent study 84. Interestingly, the role of GADD34 in a cell may not be limited to aiding cellular recovery after protein synthesis shut off, but it may also play an important, independent role in apoptosis induction 77, 85, 86.
Components of the ISR pathway
eIF2α at the core of the ISR
eIF2α, eIF2β, and eIF2γ together form the eIF2 complex and eIF2α is the main regulatory subunit of this complex since it contains both the phosphorylation and RNA binding sites. Under normal conditions, eIF2 plays a key role in the initiation of mRNA translation and recognition of the AUG start codon 87, 88. It forms a ternary complex with GTP and Met‐tRNAi that binds the 40S ribosome subunit, and together with two small initiation factors, eIF1 and eIF1A, it forms the 43S pre‐initiation complex (PIC) 89, 90. The 43S PIC is recruited to the 5′methylguanine Cap of mRNA in a process that is facilitated by the eIF4F complex among others. The eIF4F complex consists of the Cap‐binding protein eIF4E, eIF4G that constitutes as a scaffold protein, and the RNA helicase eIF4A 91. The interaction between eIF4G and eIF3 further stabilizes the PIC, leading to its migration to the AUG start codon 89, 90, 92. Upon binding of the Met‐tRNAi anticodon and the AUG start codon, eIF1 dissociates from the complex and GTP on eIF2 is hydrolyzed with the aid of eIF5. This leads to the dissociation of the eIF2–GDP complex from the 40S ribosomal complex and its recycling for another round of initiation of mRNA translation. Exchange of GDP for GTP is catalyzed by a guanine nucleotide exchange factor eIF2B, and this converts eIF2 back to its active form 87, 88.
In response to ISR activation, phosphorylated eIF2α blocks the eIF2B‐mediated exchange of GDP for GTP, thereby preventing the formation of the 43S PIC. This results in the global attenuation of 5′Cap‐dependent protein synthesis and concomitant translation of selected mRNAs that contain a short upstream open reading frame (uORF) in their 5′ untranslated region (5′UTR). Examples of these preferentially translated mRNAs include ATF4, ATF5, CHOP, and GADD34 (Fig 2) 92, 93, 94. These mRNAs do not require Cap recognition by the eIF4F complex, and their translation relies on a re‐initiation mechanism or the direct recruitment of ribosomes to internal ribosome entry sites (IRES) 6, 95.
Figure 2. Model for eIF2‐mediated translational control of ATF4.
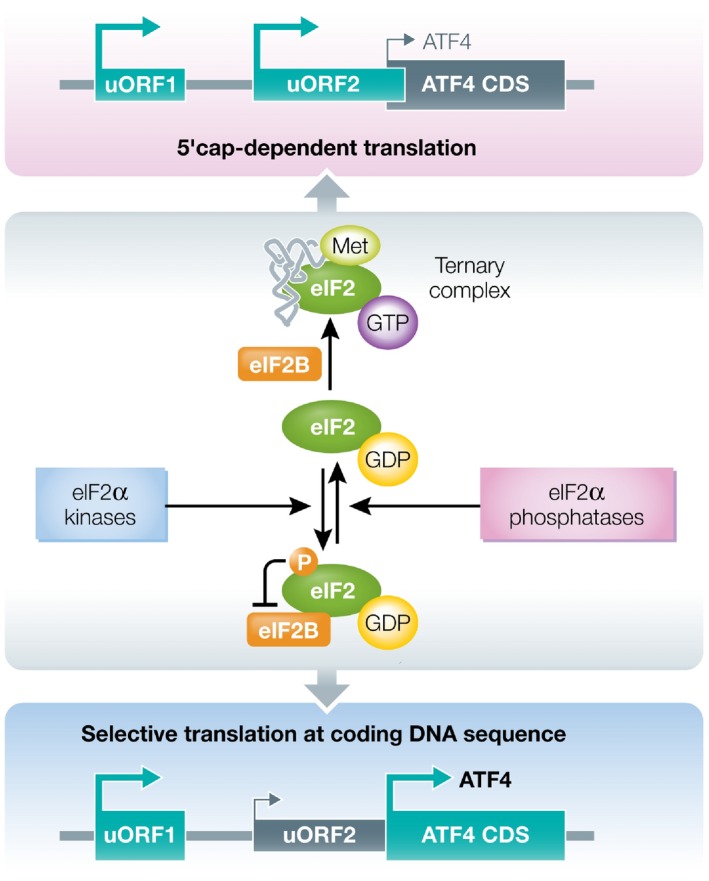
During the ISR, phosphorylation of eIF2α subunit of eIF2 complex inhibits eIF2B‐mediated exchange of eIF2‐GDP to eIF2‐GTP, thus reducing the formation of ternary complex consisting of eIF2‐GTP‐methionyl‐initiator tRNA. Under normal conditions, due to abundance of the ternary complex, ribosomes initiate scanning at upstream open reading frame uORF1 of ATF4 transcript and quickly re‐initiate at uORF2. uORF2 overlaps out‐of‐frame with ATF4 coding DNA sequence (CDS) preventing ATF4 translation. During cell stress, limiting ternary complex availability leads to longer ribosomal scanning along the ATF4 transcript enabling re‐initiation of translation at the ATF4 CDS.
Phosphorylation of eIF2α occurs at S51. Homozygous mutation at this site effectively prevents eIF2α phosphorylation permitting normal mRNA translation even under ER stress or glucose deprivation 96. Early work showed that mice with homozygous mutation of the eIF2α phosphorylation site die shortly after birth due to prolonged hypoglycemia, demonstrating the importance of translation initiation in glucose metabolism 96, 97. These data suggest that eIF2α phosphorylation is crucial for sustaining proper physiological function of the liver and pancreas after birth, as eIF2α phosphorylation is required for the induction of gluconeogenic enzymes and insulin 96. Also, it is noteworthy that eIF2α S51A/S51A MEF cells are hypersensitive to ER stress and require supplementation by nonessential amino acids and a reductive agent, which indicates that eIF2α phosphorylation protects against metabolic and oxidative stress 96. Moreover, cells with homozygous eIF2α S51A mutation are unable to induce autophagy upon viral infection, starvation, and ER stress 98, 99. Thus, the phosphorylation of eIF2α at S51 appears to be crucial for stress‐induced autophagy (this will be discussed in a later section).
ATF4 is the best characterized effector of the ISR
ATF4 is a basic leucine zipper (bZIP) transcription factor that belongs to the activating transcription factor/cyclic AMP response element binding protein (ATF/CREB family) 100, 101, 102. ATF4 has several dimerization partners that influence its regulation of gene transcription and can guide cellular outcome. In fact, ATF4 is a key deciding factor in cellular fate in response to ISR activation. It is regulated at the transcriptional, translational, and post‐translational level, and moreover, its ability to interact with other transcription factors provides a further level of regulation. A consequence of this intricate regulation is that despite the common mediator, the ISR produces distinct tailored responses to different cellular stresses, with the activation of different ATF4 target genes also being highly dependent on stress intensity and the cellular context 2, 101, 103.
ATF4 functions as a transcription factor
ATF4 has an important role in regulating both normal metabolic and redox processes as well as acting as a master transcription factor during the ISR. ATF4 has a vital role in many tissues and plays a central function in the regulation of obesity, glucose homeostasis, energy expenditure, and neural plasticity. Atf4‐knockout mice reveal critical roles for ATF4 in embryonic lens formation 104, 105, fetal liver hematopoiesis 106, and bone development 107. Atf4‐knockout mice are also smaller and leaner 106, suggesting a role for ATF4 in lipid metabolism. They have decreased fat mass caused by a reduction in the volume rather than the number of cells, suggesting that the deletion of ATF4 provides an increased fat mobilization and suppressed the fatty acid synthesis 108. Knockout of Atf4 in mice has also revealed a physiological role in thermoregulation as these mice exhibit higher core body temperatures in response to cold stress 109. ATF4 has also been implicated as a vital mediator of muscle weakness and atrophy in aging muscles as the reduction in its expression improves muscle quality, strength, and mass in mice 110. Although the role of ATF4 in models of learning and memory formation is somewhat controversial, with both positive and negative effects suggested 111, 112, 113, 114, targeted knockdown of Atf4 in the mouse hippocampus indicated that it is necessary for memory formation and neural plasticity 115.
During stressful conditions, elevated translation of ATF4 mRNA facilitates transcriptional upregulation of stress‐responsive genes. ATF4 regulates the transcription of its target genes through binding to C/EBP‐ATF response element (CARE) sequences that can mediate the transcriptional activation in response to various stimuli 116, 117, 118. During amino acid starvation, these sequences function as amino acid response element (AARE) and ATF4 is the only transcription factor known to bind all known AARE sequences 103, 119, 120. ATF4 can form homodimers and heterodimers with several other bZIP transcription factors, including its downstream target CHOP 100, 120. Recent studies have connected ATF4 and CHOP with autophagy induction in mammalian cells 6, 103, 121. ATF4 and/or CHOP can bind to the specific promoter cis element in their target genes, leading to the induction of the AARE‐dependent transcription of genes involved in autophagy activation upon starvation 103. Moreover, the formation of an ATF4–CHOP heterodimer increases its binding affinity for AARE and this is reported as an early event in transcriptional induction of autophagy genes 103. Upon ER stress and amino acid depletion in mouse cells, ATF4 alone, or together with CHOP, preferentially binds to proximal promoter regions of target genes, including Atf3, Ppp1r15a, Trib3, Wars, and Rpl7 122. Although it is well established that ATF4 alone regulates the expression of genes involved in amino acid transport and biosynthesis, Kaufman and colleagues showed that in response to ER stress, ATF4 and CHOP interact to regulate common genes involved in cellular amino acid metabolic processes, mRNA translation, and the unfolded protein response (UPR) 122.
Although ATF4 mainly acts as a transcriptional activator of the cohort of genes involved in cellular stress adaptation, it can also act as a transcriptional repressor 100. Some examples of the repressor activity of ATF4 include repression of CRE‐dependent transcription of genes such as enkephalin and chromogranin B 101, 123. The repressive effect of ATF4 on transcription can be linked to the inhibition of induction and maintenance of long‐term memory by ATF4 124. ATF4 is implicated in amino acid metabolism through the induction of asparagine synthetase (ASNS) during both the amino acid and UPR signaling where it binds promoters that contain nutrient‐sensing response elements 1 and 2 (NSRE‐1 and NSRE‐2) 120. Together, these two elements function as an enhancer and mediate the activation of the ASNS gene in response to ATF4, regardless of whether the initial stimulus was amino acid limitation or ER stress 120, 125. ATF4 was identified as a major regulator of VEGF expression induced by oxidized phospholipids, a common lipid component of atheroma, thus implicating ATF4 as a pathogenic factor in vascular damage 126. Recently, several studies have shown ATF4 to be a link between stresses such as glucose starvation or ER stress and mammalian target of rapamycin (mTOR) inhibition through upregulation of cytoprotective Sestrin2 127, 128, 129.
ATF4 acts in combination with other proteins
Due to the presence of a leucine zipper domain, ATF4 can interact with other proteins forming homodimers and/or heterodimers. When ATF4 is not bound to its DNA target, it exists as a monomer 130. Transcriptional selectivity of ATF4 is modulated by the formation of heterodimers with other bZIP or AP‐1 members. These interactions of ATF4, with other transcription factors or interacting partners, influence the outcome of ISR signaling; for example, interactions with ATF3 enhance cellular efforts to re‐establish homeostasis, while interactions with CHOP promote cell death upon ER stress 131, 132. Consequently, by association with other proteins and transcription factors, ATF4 can modulate the transcriptome of the cell in response to diverse stress stimuli. Some interactors, such as PHD1 133, PHD3 134 during hypoxia, and TRIB3 135 upon ER stress and amino acid starvation, can effectively decrease ATF4 transcriptional activity. Interestingly, TRIB3 is a downstream target of ATF4, and thus, its activation provides a negative feedback loop to control ATF4 activity during ER stress and amino acid deprivation 131, 135. The interacting partners that cooperate with ATF4 are summarized in Table 1. It seems that the interaction between ATF4 and a precise set of bZIP family members and co‐activators regulates specific gene transcription, deciding on the signaling outcome. This interaction network must be therefore highly coordinated and time dependent to induce and modulate gene transcription tailored to the cellular demand.
Table 1.
ATF4 interacting partners
| Interacting partner | Name | Function | References |
|---|---|---|---|
| β‐TrCP | F‐box protein β‐transducin repeat‐containing protein | Interaction of β‐TrCP with ATF4 controls ATF4 stability and inhibits its transcriptional activity | 153 |
| CENP‐F | Centromere protein F | Interacts with ATF4 and negatively regulates ATF4 transcriptional activation in interphase | 256 |
| CEP290 | Centrosomal protein of 290 kDa | Interacts with ATF4 and induces its transcriptional activity | 257 |
| CHOP | C/EBP homologous protein |
Interaction between ATF4 and CHOP results in negative regulation of ATF4‐dependent transcriptional activation of the ASNS gene ATF4 and CHOP interact to induce target genes involved in protein synthesis and the UPR |
122 258 |
| PHD3 | Prolyl hydroxylase 3 | Regulates ATF4 protein stability | 134 |
| FAM175B (Abro1) | BRISC complex subunit Abro1 | Interaction with ATF4 is crucial for the cytoprotective function of Abro1 in response to oxidative stress | 259 |
| FIAT | Factor inhibiting ATF4‐mediated transcription | Modifies early osteoblast activity by interacting with ATF4 | 260 |
| FOS | Proto‐oncogene c‐Fos | Forms heterodimers with ATF4 | 261 |
| Fra1 | Fos‐related antigen 1 | Forms heterodimers with ATF4 | 261 |
| GABA(B) | Gamma‐aminobutyric acid type B | Interacts with ATF4 in mouse brain during the postnatal development | 262 |
| GPE‐1 | GPE1‐binding protein | Forms heterodimers with ATF4 that regulate G‐CSF gene expression | 263 |
| HIF1A | Hypoxia inducible factor 1‐alpha | Interaction with ATF4 initiates bone angiogenesis in hypoxic osteoblasts | 264 |
| Jun | Proto‐oncogene c‐Jun | Forms heterodimers with ATF4 | 261 |
| NIPK | Neuronal cell death‐inducible putative kinase (TRIB3) | Interacts with ATF4 and inhibits ATF4 transcriptional activity | 265 |
| Nrf‐2 | Nuclear factor erythroid 2‐related factor 2 | Interaction between ATF4 and Nrf2 regulates the expression of heme oxygenase‐1 gene | 266 |
| p300 | Histone acetyltransferase p300 | Interaction with p300 stabilizes ATF4 and increases its transcriptional activity | 164 |
| RPB3 | RNA polymerase II subunit 3 | Interaction with RPB3 enhances ATF4 transcriptional potential in muscle differentiation | 267 |
| RSK2 (RPS6KA3) | Ribosomal protein S6 kinase alpha‐2 | ATF4 and RSK2 interact to regulate osteoblast differentiation and function | 107 |
| RUNX2 | Runt‐related transcription factor 2 | Interaction with ATF4 stimulates osteoblast differentiation and bone formation | 268 |
| SATB2 | DNA‐binding protein SATB2 | Regulates skeletal development and osteoblast differentiation through the direct interaction with ATF4 and enhances its activity | 269 |
| SKIP3 | Orthologue of the Drosophila TRIB3 and rat NIPK | Interaction between SKIP3 and ATF4 is associated with ATF4 proteolysis | 161 |
| Tax of HTLV‐1 | Trans‐activating transcriptional regulatory protein of HTLV‐1 | ATF4 interacts with Tax and triggers Tax transactivation of HTLV | 270 |
| TRIB3 | Tribbles homolog 3 | Interacts with ATF4 and negatively regulates the ATF4‐dependent transcription. | 135 197 |
| Zhangfei | HCF‐binding transcription factor Zhangfei | Forms heterodimers with ATF4 and enhances its transcriptional activity | 271 |
| ZIP | Zipper‐interacting protein kinase | Forms heterodimers with ATF4, which inhibits the apoptotic activities mediated by ZIP | 160 |
Translational regulation of ATF4
The primary mechanism by which the levels of ATF4 protein are adjusted in response to different stresses, such as ER stress, hypoxia, or oxidative stress, is through translational regulation 17, 136, 137. The mechanism is highly conserved, with yeast regulating levels of Gcn4p (a functional homologue of ATF4) using a very similar mechanism 138, 139. Since yeast do not have PERK, global inhibition of translation is related to the activation of Gcn2p (the yeast homologue of GCN2) and eIF2α phosphorylation upon amino acid depletion 139. This results in preferential synthesis of Gcn4p, a transcription factor that controls the expression of genes involved in amino acid synthesis and transport 138.
Translational control of yeast GCN4 requires four uORFs 140. When the ternary complex is abundant, ribosomes initiate scanning at uORF1 and re‐initiate at inhibitory downstream uORFs that precludes the translation of GCN4 coding sequence (CDS). Under amino acid depletion, more time is required for the ribosome to re‐initiate translation, due to low levels of ternary complex, so that ribosomes scan through uORF4 and re‐initiate at the GCN4 CDS 141. In mammals, mouse Atf4 has features similar to the GCN4 13. The mouse gene contains two uORFs (uORF1 and uORF2) that are located 5′ to the Atf4 CDS (Fig 2), while human ATF4 mRNA contains three upstream open reading frames (uORF1, uORF2, and uORF3) 142. Under normal cellular conditions, the translation of mouse Atf4 mRNA is initiated at uORF1, which encodes a peptide just three amino acids in length, and re‐initiates at uORF2. This precludes translation of Atf4 mRNA because, unlike GCN4, the uORF2 (uORF3 in human) sequence overlaps with the Atf4 CDS in an out‐of‐frame manner 100, 142, 143, 144. In stressed cells, ribosomes scan through uORF2 and re‐initiate at the Atf4 CDS 5, 143.
Thus, upon eIF2α phosphorylation, re‐initiation of translation at the Atf4 CDS results in approximately fivefold higher ATF4 expression (Fig 2) 13, 87, 143. However, Walter's group recently observed high levels of peptide synthesis from both ATF4 uORFs during ER stress and oxidative stress conditions 145. These authors suggest that this deviation from the current re‐initiation model may indicate that either only a few ribosomes skip uORF2 or that a combination of re‐initiation and scanning past uORF2 (“leaky scanning”) occurs. Interestingly, apart from the ribosome re‐initiation mechanism, in HeLa and THP‐1 cells the translation of an alternatively spliced variant of ATF4 mRNA is reported to be also controlled by an IRES sequence in 5′UTR 95.
Transcriptional regulation of ATF4
Under non‐stressed conditions, the levels of ATF4 mRNA transcripts available are low 87. ATF4 gene transcription is induced in response to different stresses and is mediated by different transcription factors 17. For example, nuclear factor‐like 2 (NRF2) induces ATF4 in response to oxidative stress 126, CLOCK regulates ATF4 expression in response to the chemotherapeutic drugs cisplatin and etoposide 146, TFEB and TFE3 induce ATF4 transcription under ER stress and starvation conditions 147, and a pancreas‐specific transcription factor (PDX1) mediates transcriptional induction of Atf4 during ER stress in mouse islet β cells 148. Interestingly, in a positive feedback loop, ATF4 downstream gene targets, such as NUPR1, can also elevate ATF4 mRNA levels 149.
In contrast, hypoxia does not alter ATF4 mRNA levels, although it can increase ATF4 levels by translational and post‐translational mechanisms 136. Furthermore, there is evidence of transcriptional repression of ATF4 by some cellular stressors. For example, C/EBPβ, a member of the C/EBP family, has been identified as a transcriptional repressor of Atf4 during UV irradiation, revealing a novel role for C/EBPβ in regulating the ISR 150. Thus, despite an increase in eIF2α phosphorylation upon UV irradiation, ATF4 induction is not observed because of diminished levels of available ATF4 transcript 150, 151. Transcriptional repression of ATF4 is also observed in nonalcoholic fatty liver and nonalcoholic steatohepatitis 152.
ATF4 post‐translational modifications and protein stability
ATF4 is an unstable protein with a half‐life of < 1 h under normal conditions 102, 153, 154. Several post‐translational modifications regulate ATF4 stability, including phosphorylation and ubiquitination, which are the two most significant in this regard (Table 2). ATF4 degradation is mediated by the ubiquitin‐proteasome pathway 153. Proteasomal inhibitors such as bortezomib and MG132 cause an increase in ATF4 protein levels as a dual consequence of inhibited proteasomal degradation (which stabilizes the protein), and ISR activation (which is triggered by the depletion of intracellular amino acids as a result of proteasome inhibition) 153, 155, 156, 157. Phosphorylation of ATF4 at S219 (within its degradation motif sequence DSGICMS) is essential for its interaction with the F‐box protein β‐transducin repeat‐containing protein (β‐TrCP), the receptor component of SCF E3 ubiquitin ligase complex (SCFβTrCP) that targets ATF4 for proteasomal degradation (Fig 3) 153. Also, phosphorylation of ATF4 at S224 within the same motif, and proline‐directed phosphorylation at multiple proline residues surrounding β‐TrCP have been implicated in regulating ATF4 interaction with β‐TrCP, indicating the tight control of ATF4 degradation 153, 158.
Table 2.
ATF4 post‐translational modifications and their role
| Modification | Residue | Function | References |
|---|---|---|---|
| Acetylation | K311 | Inhibition of ATF4 ubiquitination | 164 |
| Hydroxylation |
P60 P235 |
Possible decrease in ATF4 transcriptional activity | 162 |
| Phosphorylation |
S219 S224 |
bTrCP‐dependent degradation | 153 272 |
|
S245 S251 S254 |
Upregulation of ATF4 transcriptional activity | 107 159 | |
| S215 | ATF4 stabilization | 273 | |
| Ubiquitination |
S219 D218 |
ATF4 degradation | 153 |
Figure 3. Schematic ATF4 protein structure.

ATF4 protein consists of 351 amino acids organized into several domains and motifs. It contains basic domain for DNA binding and leucine zipper domain involved in protein–protein interactions. ATF4 has an oxygen‐dependent degradation domain, which is recognized by PHD3. ATF4 has β‐TrCP recognition motif, which is a degradation motif. Depending on the phosphorylation events mainly at S219, ATF4 is recognized by β‐TrCP and targeted for proteasomal degradation.
Phosphorylation of ATF4 at S245 by ribosomal protein S6 kinase α‐2 (RSK2) or S254 by protein kinase A (PKA) can enhance its transcriptional activity during osteoblast and osteoclast differentiation 107, 159. In addition, there are several other kinases that have been shown to interact with ATF4, for example, zipper‐interacting protein kinase (ZIPK) 160, casein kinases 1 and 2 (CK1 and CK2) 158, and SKIP3 161, but their full role in regulating ATF4 activity in response to physiological stimuli and hypoxia, respectively, remains to be addressed.
In addition to phosphorylation during ER stress, ATF4 stability and activity can be negatively regulated by prolyl hydroxylase domain protein 3 (PHD3) leading to hydroxylation at proline residues 60 and 235 162. This process is regulated by Siah 1/2 ubiquitin ligases that target PHD1 and PHD3 for degradation and thus increase ATF4 stability 162. These findings demonstrate an important link between phosphorylation and hydroxylation, as P235 is localized close to the β‐TrCP recognition DSGICMS motif 162. Interaction of ATF4 with PHD1, but not with PHD3, is a stabilizing mechanism for ATF4. However, both interactions lead to a decrease in ATF4 transcriptional activity 133, 134, 163. Additionally, ATF4 stability can be enhanced by histone acetyltransferase p300, independently of its catalytic activity, resulting in the inhibition of ATF4 ubiquitination 164.
Cellular outcome of the ISR
Although multiple stresses converge on eIF2α phosphorylation to activate the ISR, the cellular outcome is not always the same. The effect of ISR activation depends not only on the nature of the stress, its duration and severity, but also on the extent of eIF2α phosphorylation and translation of ATF4 mRNA and other bZIP transcription factors 17, 165. It is commonly accepted that a short‐lived ISR is an adaptive, pro‐survival response aiming at resolving stress and restoring homeostasis, while a prolonged ISR can signal toward cell death induction 154. Therefore, this dual effect of eIF2α phosphorylation raises an important question concerning how the switch between pro‐survival and pro‐death signaling by ISR is regulated, as well as whether a threshold of cell stress signals exists that favors the activation of cell death proteins.
Cellular recovery signaling by the ISR
The ISR, together with other cellular adaptation pathways, functions as an important part of the cellular defense strategy in response to stress. It does this mainly through altering global protein synthesis and through the regulation of genes that promote pro‐survival signaling such as through the activation of autophagy, or that counteract pathways that lead to cell death such as apoptosis or proteotoxicity (impairment of cell function due to the effects of misfolded proteins). Notably, there is also a cross‐talk between the ISR and other pro‐survival pathways such as the UPR, phosphatidylinositol‐3 kinase (PI3K) signaling, autophagy, and the ubiquitin‐proteasome system. These are discussed below.
One of the major effects of the ISR is on protein synthesis. The initial repression of global mRNA translation plays a very important role in promoting cell survival in the face of different stresses that activate the ISR. The accumulation of unfolded proteins in the ER induces a condition of ER stress, which is relieved by the reduced level of incoming proteins when global protein synthesis is inhibited 1. PKR activation of the ISR during viral infection helps to reduce the translation of viral mRNAs, thus protecting the cells 8, 42. Under conditions of amino acid depletion, activation of the ISR by GCN2 reduces the need for amino acids for protein synthesis, thus alleviating this stress 34. Activation of the ISR by HRI under conditions of low heme lessens the need for heme by attenuating the translation of globin mRNAs, thus reducing the stress and promoting survival 52. It is important, however, to note that timely termination of the ISR also plays a key role in promoting long‐term cell survival, by re‐starting synthesis of essential proteins. This is achieved through dephosphorylation of eIF2α by the phosphatase GADD34, which is induced by ATF4 and its downstream targets CHOP and ATF3 15, 78, 79, 166.
Through the activation of macroautophagy, commonly known as autophagy, the ISR can regulate cell survival and cell death pathways. Autophagy promotes the bulk removal and degradation of unfolded proteins or aggregates as well as damaged organelles while also serving as a means to replenish depleted amino acids for building proteins and to provide energy to a starved cell. Although the precise mechanisms by which phosphorylated eIF2α leads to autophagy are still poorly understood, it is interesting to note that distinct intrinsic and extrinsic stresses that lead to the phosphorylation of eIF2α have also been shown to induce autophagy. For example, ER stress leads to an increased phosphorylation of eIF2α and the subsequent upregulation of certain autophagy receptors such as SQSTM1, NBR1, and BNIP3L in a PERK‐dependent manner 167. Pharmacological inhibition of PERK suppressed transcriptional upregulation of these autophagy receptors in mammalian cells 167. In addition, PERK‐mediated phosphorylation of eIF2α mediates increased ATG12 and LC3 conversion upon the expression of polyQ72 aggregates in C2C5 cells 98. In fact, the PERK arm of the UPR can regulate all stages of autophagy including induction, vesicle nucleation, phagophore elongation, and maturation 168. Consistent with this, another study showed that ER stress caused by bluetongue virus induces autophagy through a PERK‐eIF2α pathway in infected cells 169. In that study, both pharmacological inhibition of PERK activity and genetic silencing of eIF2α were shown to significantly reduce the level of the autophagy marker LC3 169. Furthermore, the induction of autophagy during hypoxia is also dependent on PERK‐mediated eIF2α phosphorylation, as the disruption of PERK signaling or exposure of non‐phosphorylatable eIF2α S51A mutant to hypoxia decreases the levels of MAP1LC3B and ATG5 transcripts 121. It has also been shown that GCN2‐mediated phosphorylation of eIF2α is essential for the induction of autophagy upon amino acid starvation in tumor cells 7. While GCN2‐knockout cells displayed the reduced level of LC3, the eIF2α S51A‐mutant cells could not induce LC3 processing 7. Similarly, S51 of eIF2α has also been demonstrated to be important for amino acid starvation‐induced autophagy in yeast and MEFs 99. Together, these findings place eIF2α phosphorylation as the central link between different stresses and induction of autophagy.
Downstream of eIF2α phosphorylation, ATF4 has been shown to be required for autophagy induction. However, it has been suggested that mechanisms directed from eIF2α phosphorylation other than selective translation of ATF4 mRNA might also be involved in the activation of autophagy 170. During hypoxia, ER stress, or amino acid deprivation, there is ATF4‐dependent transcriptional upregulation of essential autophagy genes involved in autophagosome biogenesis and function, for example, MAP1LC3B and ATG5 6, 103, 167. ATF4 can also lead to an increased expression of regulated in development and DNA damage response 1 (REDD1; also known as Ddit4), which suppresses mTOR complex 1 (mTORC1) activity leading to autophagy induction upon ER stress and starvation 171, 172. ATF4 activation due to amino acid deprivation stimulates the upregulation of several genes involved in autophagy including Atg3, Atg5, Atg7, Atg10, Atg12, Atg16, Becn1, Gabarap, Gabarapl2, Map1lc3b, and Sqstm1 103. Through the upregulation of key autophagy genes, the ISR contributes to increased autophagic flux that helps the cell to deal with the stress caused by hypoxia and nutrient deprivation through the enhanced recycling of cytoplasmic components and maintenance of cellular biosynthetic capacity and ATP levels, for example, by supplying amino acids for de novo protein synthesis and providing substrates for the tricarboxylic acid cycle, such as amino acids and free fatty acids 7, 173. It is worth noting that different autophagy genes can have different levels of dependence on ATF4 and CHOP signaling and that the activation of such genes is determined by the ratio of ATF4 and CHOP proteins that are bound to specific promoter cis elements, thus allowing subtle tuning of gene expression that can be tailored specifically to meet cellular needs 103. Interestingly, a cytoprotective function of eIF2α phosphorylation against conditions that mimic viral infection or induce ER stress has been reported to occur through the activation of pathways that promote cell survival, such as PI3K and its downstream target Akt/mTOR 174. In this regard, it is also of interest to consider the effect of proteasome inhibition on survival signaling by the ISR. Proteasome inhibition, for example, using bortezomib, leads to the activation of the ISR through GCN2 as a result of depletion of amino acids for protein synthesis 157. In mammalian cells, the depletion of amino acids due to proteasome inhibition also activates autophagy through mTOR in an attempt to restore amino acid levels 157. Supplementation with critical amino acids that are depleted by proteasome inhibition attenuates eIF2α phosphorylation and suppresses autophagy 157. Thus, amino acid depletion constitutes a link between autophagy induction and ISR activation in an attempt to promote the survival of cells.
The UPR that is activated by ER stress is mainly an adaptive response and encompasses the activation of the ISR. In this pathway, PERK, which phosphorylates eIF2α to activate the ISR, functions in concert with the other arms of the UPR, IRE1, and ATF6, to ameliorate proteotoxicity by enhancing the transcription of genes that promote protein folding and degradation of misfolded or aggregated polypeptides 175, 176. The interplay between the different arms of the UPR controls the cellular outcome 177. The survival benefit provided by the ISR during ER stress reveals ATF4 as a focal point linking PERK‐mediated translational control with IRE1‐ and ATF6‐mediated gene expression 1. It has been proposed that the relative duration of PERK and IRE1 signaling dictates the cellular outcome, with the sustained PERK activation promoting apoptosis and the prolonged IRE1 activation leading to cell survival 178, 179.
The ISR also promotes survival signaling by negative regulation of cell death pathways, particularly apoptosis. For example, in response to ER stress, PERK‐mediated activation of the ISR leads to an upregulation of cellular inhibitor of apoptosis proteins (cIAP1 and cIAP2) in both tumor‐derived cell lines and non‐tumor cells 180, 181, 182. ATF4 has also been shown to mediate bortezomib‐induced upregulation of anti‐apoptotic MCL‐1 leading to resistance to the drug 183. Both MCL‐1 and cIAPs can inhibit apoptosis at different points in the pathway that are upstream and downstream of the release of cytochrome c from the mitochondria (a critical step in the intrinsic apoptosis pathway). PERK can also mediate a pro‐survival role by the induction of microRNA 211 (miR‐211), which attenuates a stress‐dependent expression of CHOP and which was suggested by the authors as a mechanism that allows cells to re‐establish homeostasis before the initiation of apoptosis 184. However, despite having a beneficial role in restoring homeostasis, these mechanisms could also contribute to tumor development. Increased expression of miR‐211 was detected in two mouse models of mammary carcinoma as well as in human B‐cell lymphoma and was found to be PERK dependent 184.
Several other genes activated during ISR appear to contribute to pro‐survival signaling, although their importance and function in determining cellular outcome remains to be fully addressed. An example is IBTKα, the α isoform of inhibitor of Bruton's tyrosine kinase, an important substrate adaptor for protein ubiquitination, which has emerged recently as an important pro‐survival factor activated during ER stress 185. Translational repression by eIF2α has also been implicated in nuclear factor kappa light chain enhancer of activated B cells (NF‐κB) pathway induction to protect against ER stress 186. ATF4 also activates the transcription factor NUPR1, which regulates the expression of metabolic stress‐responsive genes, especially those involved in DNA repair and cell cycle regulation, and therefore may be considered as a pro‐survival factor 149, 187.
Cell death signaling by the ISR
Within the ISR are embedded pathways that can lead to the induction of cell death if the adaptive response does not restore homeostasis. These are mainly regulated through the transcriptional activity of ATF4 and some of its downstream targets, particularly CHOP and ATF3. Post‐transcriptional regulation of gene expression through the upregulation of microRNAs also plays a role in cell death signaling by the ISR. These will be discussed below.
One of the best studied mechanisms of ISR‐induced cell death is through ATF4‐mediated activation of CHOP. CHOP is a transcription factor that promotes cell death signaling through multiple mechanisms. In several cellular models, CHOP has been shown to induce cell death by upregulation of the BH3‐only pro‐apoptotic BCL‐2 family members BCL2L11 and BBC3, thus promoting ER stress‐induced apoptosis 188, 189. CHOP can also contribute to cell death by enhancing the expression of one of the death receptors, DR5, that plays a role in the induction of apoptosis under ER stress 190. In addition, CHOP induces the expression of the oxidase ERO1α that destabilizes the homeostasis of the oxidizing environment of ER 78. Additionally, CHOP can further regulate gene expression by binding to other ATF/CREB family members, such as ATF4 or ATF3, thus altering their DNA binding specificity 103, 191. For example, CHOP–ATF4 heterodimers can upregulate ATF5 expression, amplifying cell death signaling by regulating the expression of several pro‐apoptotic genes such as PMAIP1, APAF‐1, and TXNIP 192. CHOP and ATF3 can interact to increase the expression of TNFRSF10B 191, 193. CHOP is also essential for the induction of PPP1R15A, which is important for both adaptation and cell death signaling 78. It is important to note that although there is a well‐established role for CHOP in cell death signaling, CHOP expression alone is not sufficient to induce cell death 194. In fact, CHOP‐deficient cells are only partially resistant to ER stress‐induced cell death, indicating the role of other factors in mediating the cell death 195.
ATF4 itself can promote the cell death. It has been shown to be a direct transcriptional activator of the pro‐apoptotic BCL‐2 family member PMAIP1, which was found to mediate ER stress‐induced cell death in neuroectodermal tumor cells 196. ATF4 can also promote the cell death through the formation of heterodimers with CHOP (as described in the previous paragraph) or with ATF3, both of which are transcriptional targets of ATF4. ATF4–ATF3 heterodimers can increase the transcription of PMAIP1 and thus promote apoptosis through the intrinsic pathway 132. ATF4 and CHOP together can also induce TRIB3, a pseudokinase, which under normal conditions regulates NF‐κB‐induced gene expression, while during the cellular response to ER stress, it negatively regulates ATF4, thus limiting the transcription of other ATF4‐dependent stress‐responsive genes 135, 197. TRIB3 was identified as an ATF4 binding partner in a yeast two‐hybrid assay and was shown to promote ATF4 degradation 161. Moreover, TRIB3 can interact with CHOP and downregulate its own induction through a negative feedback loop whereby it represses ATF4‐CHOP transactivation 131. Notably, TRIB3‐silenced HEK293 and HeLa cells display an increased resistance to ER stress‐induced apoptosis 131. Additionally, TRIB3 has also been shown to inhibit Akt activation in the liver cells, suggesting an alternative mechanism by which TRIB3 could promote the cell death 198. In this regard, it is interesting to note that the loss of TRIB3 was shown to promote Akt‐driven tumorigenesis via forkhead box O (FOXO) inactivation 199.
In addition, the ISR can modulate the cell death through the regulation of miR expression. PERK‐mediated increases in miR30c‐2 negatively regulate the expression of XBP1 (part of the pro‐survival arm of the UPR) in mammalian cells 200. Both ATF4 and NRF2 (which are directly phosphorylated by PERK) repress the miR‐106b‐25 cluster, which functions as an oncogene and whose overexpression has been documented in lung cancer, B‐cell lymphoma, and gastric cancer 201, 202. ATF4‐ or NRF2‐induced reduction in the miR‐106b‐25 cluster expression is accompanied by an increased expression of the pro‐apoptotic BCL‐2 family member BIM and the induction of apoptosis in mammalian cells 201.
Additionally, the ISR can also promote the cell death through translational and post‐translational regulation of the caspase inhibitor X chromosome‐linked inhibitor of apoptosis (XIAP) 203. PERK has been shown to downregulate XIAP translation through the phosphorylation of eIF2α, while ATF4 promotes XIAP protein degradation through the ubiquitin‐proteasome system 203. This reduction in XIAP levels removes the brake on caspases and permits their activation with ensuing apoptosis 204, 205. It is worth noting that other IAPs such as cIAP1, cIAP2, Livin, Survivin, and NAIP can also be downregulated by the ISR in the cases of chronic PERK signaling 203.
During the prolonged ER stress, ATF4 induces an amino acid transporter network, which leads to mTORC1 activation, the inhibition of autophagy and the phosphorylation of multiple factors including eukaryotic elongation factor 2 (eEF2), and as such, positively regulates translation 165. While this series of events could serve to restore cellular homeostasis, it can also contribute to cell death execution during chronic ER stress, as constant synthesis of nascent peptides without the production of mature proteins causes proteotoxic stress.
Although apoptosis is the main cell death pathway regulated by ISR, it can also trigger other forms of cell death. For example, the ISR can initiate ATF4‐dependent necrosis, especially in response to glucose deprivation, as recently reported 206. Intriguingly, ATF4‐dependent apoptosis can be induced by 2‐deoxyglucose in the same cellular model 206. The ISR can also regulate autophagic cell death in human alveolar epithelial A549 cells in response to lipopolysaccharide treatment 207.
In the context of understanding the role of the ISR in cellular survival/death decisions, it is instructive to consider the utility of mathematical models. To gain a deeper understanding as to how UPR signaling produces different cellular outcomes, Erguler and colleagues developed a mathematical model, based on up‐to‐date literature, that integrates the three arms of the UPR and the associated genetic and post‐translational interactions in a coherent mechanistic model 208. This model reveals that the UPR has three distinct activity states depending on the level and duration of stress and on the availability of GRP78. The low, intermediate, and high activity states of the UPR are associated with three cellular outcomes of stress adaptation, tolerance, and initiation of apoptosis, respectively. This model revealed for the first time evidence of UPR‐induced oscillations in translation attenuation and apoptotic signals, which provides valuable new insights into how the UPR switches between survival and death. Interestingly, the Erguler's mathematical model can also be considered in association with another model for mitochondrial BAX/BAK/BH3 apoptosis 209, 210. Importantly, the predictions of the Erguler model are supported by several experimental observations. For example, PERK signaling in that model is constantly elevated, indicating that the duration of PERK signaling is important for the life and death decision of a cell. However, the majority of the parameter values used in the model remain to be measured experimentally. The construction of a similar mathematical model to gain insight into the ISR is likely to prove helpful to understanding its role in cell death and survival decision making. Given the large amount of experimental data currently available on the ISR, it is likely that a robust model could be developed that would yield valuable new observations that may be useful in the development of therapeutic approaches for relevant diseases.
Pharmacological modulation of ISR
Dysregulation of ISR signaling has important pathologic consequences linked to inflammation, diabetes, cancer, and neurodegenerative diseases 211, 212, 213, 214. Thus, pharmacological modulation of the ISR represents a promising therapeutic strategy. In recent years, encouraging progress has been made in this area through the development of several drugs that target ISR components to enhance or inhibit this signaling pathway. Interestingly, strategies aimed both to suppress and to enhance ISR signaling have been reported to inhibit tumor growth in vivo, which reflects ISR dual role in promoting cell survival and cell death 7, 215.
Enhancers of ISR signaling
Pharmacological activation of ISR signaling can be achieved either by activating eIF2α kinases leading to eIF2α phosphorylation in the absence of upstream stress or by inhibiting eIF2α dephosphorylation using phosphatase inhibitors. Some of the compounds that have been shown to promote eIF2α phosphorylation include CCT020312 (a selective PERK activator that does not elicit a general UPR) 216; BTdCPU and other related N,N'‐diarylureas (HRI activators) 215; histidinol, halofuginone, asparaginase, and arginine deiminase (GCN2 activators) 217, 218, 219, 220; and BEPP monohydrochloride (PKR activator) 221. Drug‐induced enhancement of ISR signaling through eIF2α kinases can be exploited in combined therapies against resistant cancers as it can lead to G1/S checkpoint activation through eIF2α‐dependent depletion of cyclin D 216. Halofuginone, for example, is a potent inhibitor of angiogenesis progression 222 and is being evaluated in several clinical trials.
Inhibitors of eIF2α dephosphorylation include drugs such as salubrinal 223 and guanabenz (2,6‐dichlorobenzylidene aminoguanidine acetate) or its derivative Sephin1, which inactivate the binding of GADD34 to PP1c 224, 225, 226. Salubrinal is an inhibitor of GADD34–PP1 and CReP–PP1 complexes that dephosphorylate eIF2α 223, 225. Although it is well established that salubrinal produces high levels of eIF2α phosphorylation and its action is independent of PERK, GCN2, PKR, or HRI, the mechanism by which salubrinal leads to eIF2α phosphorylation is still incompletely understood 225. It has been suggested that salubrinal might inhibit GADD34–PP1 and CReP–PP1 complexes through the direct binding or that an additional indirect signaling may be involved 223. Since protein phosphatases are involved in many pathological processes, salubrinal has been used to block the replication of herpes simplex virus (HSV) in both cultured cells and a mouse model of viral infection 223, 227. Salubrinal also produces beneficial effects in experimental models of Alzheimer's and Huntington's diseases 228, 229.
In contrast, the mechanism of action of guanabenz and its derivative Sephin1 is better understood. Guanabenz acts by inhibiting the stress‐induced phosphatase, GADD34, that causes the dephosphorylation of eIF2α 226. However, it should be noted that guanabenz does not selectively inhibit GADD34 and that it has nanomolar affinity for the α2‐adrenergic receptor and is used in the treatment for hypertension 230. Moreover, in mice, guanabenz exhibited an adverse effect by decreasing rotarod performance, which can be attributed to its adrenergic agonist activity as Sephin1 showed no such effect 224. Similarly, in humans, treatment with guanabenz revealed the adrenergic side effects characterized by drowsiness and coma 230. In contrast, Sephin1 (selective inhibitor of holophosphatase) selectively and safely inhibits stress‐induced GADD34 in vivo 224. Neither Sephin1 nor guanabenz inhibits the constitutive CReP, thus permitting low basal levels of eIF2α dephosphorylation; however, Sephin1 is devoid of α2‐adrenergic activity and therefore has less adverse effects than guanabenz 224.
Some other drugs have also been shown to enhance the ISR. Nelfinavir is a HIV protease inhibitor that was recently shown to induce the ISR in several cell lines as well as in mice 231. Nelfinavir treatment downregulates CReP levels and leads to a decrease in PP1 association with eIF2α. In this way, nelfinavir mediates an increase in eIF2α phosphorylation and thus ISR activation. Although the precise molecular target of nelfinavir is not yet known, it is apparent that nelfinavir itself does not activate any one of the four eIF2α kinases although it is dependent on basal activity of these enzymes. Regardless, nelfinavir seems to be a promising compound to restore homeostasis in pathologies characterized by elevated levels of eIF2α phosphorylation. Another drug, ONC201, is an anticancer drug that triggers cell death in various tumor types, which has recently been reported to trigger the ISR. Using the cells derived from various types of solid tumors, it was shown to activate ATF4 through PKR and HRI eIF2α kinases, leading to an increase in the abundance of TRAIL and its receptor DR5 232. Together, these data provide strong evidence that approaches aimed at enhancing or prolonging phosphorylation of eIF2α represent a promising strategy to treat a number of disorders associated with dysregulated ISR signaling.
Recent studies report the beneficial effects of enhancers of ISR in models of degenerative diseases, particularly where protein misfolding plays a role in disease pathogenesis. For example, guanabenz produces the beneficial effects in a rodent model of traumatic brain injury 233, in mouse models of multiple sclerosis 234, and in amyotrophic lateral sclerosis (ALS) 235. Similarly, Sephin1, a guanabenz derivative, has been shown to prevent several molecular defects in mouse models of Charcot–Marie–Tooth syndrome (CMT) and ALS 224. In diabetes, translational recovery during unresolved ER stress and thus increased protein synthesis promote apoptosis 236. Pharmacological intervention with salubrinal applied during translational recovery upon chronic ER stress results in the attenuation of protein synthesis and protects against cell death 236. Drugs such as salubrinal, guanabenz, and Sephin1 appear to act by slowing down protein synthesis, allowing increased time for protein folding within the ER, and thus protecting the cells from the deleterious effects of proteotoxicity.
The beneficial effects of enhanced ISR activity in some conditions are further supported by genetic manipulation of the ISR pathway in mouse model of diseases. Upregulation of PERK activity in oligodendrocytes attenuates disease severity in a mouse model of multiple sclerosis 237. Enhanced eIF2 phosphorylation by GADD34 manipulation delays disease onset in a mouse model of ALS 238. Furthermore, Wrabetz and colleagues showed that ablation of Gadd34 in P0S63del transgenic mouse model of CMT restores motor function and attenuates demyelination 239.
Inhibition of ISR signaling
Inhibition of eIF2α kinases also represents a strategy to overcome resistant cancers, or to target neurodegenerative diseases and diabetes. Some examples of eIF2α kinase inhibitors include GSK2606414 and GSK2656157 that trap PERK in an inactive state and prevent its autophosphorylation 240, 241. PERK is involved in tumorigenesis and cancer cell proliferation, and both of the GSK inhibitors have been shown to exert antitumor activity against xenografts of human pancreatic cancer in mice 242, 248. Application of GSK2656157 significantly reduced the growth of human xenograft multiple myeloma in mice and also displayed antiangiogenic activity 248. C16 is a small‐molecule inhibitor that acts in an ATP‐competitive manner to block autophosphorylation of PKR 242. Aminopyrazolindane is an ATP‐competitive HRI kinase inhibitor 243. The ATP analogs indirubin‐3′‐monoxime, SP600125, and a SyK all inhibit GCN2; however, they are not GCN2 specific 244. Another small‐molecule inhibitor of the ISR is called integrated stress response inhibitor (ISRIB), which renders cells insensitive to eIF2α phosphorylation and thus inhibits the ISR downstream of eIF2α phosphorylation resulting in the attenuation of ATF4 synthesis (Fig 4) 245. ISRIB restores the translational capacity and thus impairs the adaptation of cells to chronic ER stress. Additionally, ISRIB has been shown to prevent the formation of stress granules caused by eIF2α phosphorylation. ISRIB seems to be a promising tool for the treatment of neurodegenerative diseases that are associated with the formation of RNA–protein aggregates 245. Inhibition of the translational block induced by ISR signaling can significantly enhance cognitive memory and, as such, represents a strategy to combat memory disorders 245.
Figure 4. Pharmacological approaches for targeting the components of ISR .
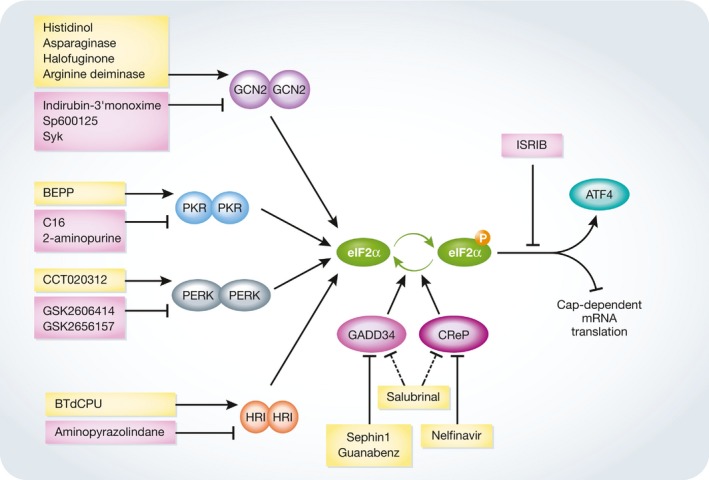
Schematic representation of ISR signaling indicates the effect of eIF2α phosphorylation on translational control. eIF2α phosphorylation can be either stimulated through chemical activators of eIF2α kinases such as histidinol 220, asparaginase 217, halofuginone 253, arginine deiminase 219, BTdCPU 215, BEPP monohydrochloride 221, and CCT020312 216, or prevented using indirubin‐3′‐monoxime, SP600125, and SyK to inhibit GSK2 244, GSK2606414 and GSK2656157 to block PERK activation 240, 254, C16 and 2‐aminopurine to modulate PKR 242, 255, and aminopyrazolindane to inhibit HRI 243. Salubrinal 223 prolongs ISR through an unknown mechanism, while guanabenz 226 and Sephin1 224 block GADD34, and nelfinavir 231 decreases CReP expression and affects CReP–PP1 complex binding to eIF2α. The consequences of eIF2α phosphorylation can be reversed using ISRIB 245. See text for more details. Arrows point to enzyme activation, bar‐headed lines point to enzyme inhibition, dashed lines indicate the lack of known target, yellow boxes indicate the activators of ISR signaling, and pink boxes indicate the negative regulators of ISR signaling.
Another approach to regulate ISR involves the modulation of ATF4 post‐translational modifications. Phosphorylation of ATF4 at S251 can be blocked by SL0101, an RSK2 kinase inhibitor 246. Inhibition of phosphorylation of ATF4 at S254 can be achieved by inhibiting PKA with H‐89 159. Also, nuclear to cytoplasmic shuttling of ATF4 followed by its phosphorylation‐dependent proteasomal degradation can be induced by the synthesized RPL41 peptide, which may have potential in cancer therapy 247.
Conclusions and future perspectives
Since it was first coined in 2002 by David Ron, Heather Harding, and coworkers 1, 2, the term “integrated stress response” has garnered increasing attention as a useful way to organize our understanding of the effect of multiple stresses that converge on the phosphorylation of eIF2α through the activation of eIF2α kinases. There are still gaps in our knowledge regarding many aspects of the ISR. For example, the role of cross‐talk between the eIF2α kinases and its downstream targets in determining death or survival is unclear. It is not yet understood how cellular responses are tailored to address the different stresses that lead to eIF2α phosphorylation downstream of different eIF2α kinases. There is still much to be learned about the mechanisms that direct preferential mRNA translation during the ISR 194. The fact that ATF4 upregulates less than 50% genes in MEF cells in response to ER stress rises the question about the existence of other effectors of ISR 2. Also, yet to be clarified is how the regulation of specific target genes depends on the duration of eIF2α phosphorylation, type of stress, extent of stimuli, and cell type. Walter's group has recently shown that the cells exhibit noncanonical eIF2A‐mediated synthesis of selected proteins during the ISR, thus highlighting divergent mechanisms employed by the ISR to regulate protein synthesis that we still do not fully understand 145. Intriguingly, the authors suggested that the translation products of uORFs during the ISR could serve a variety of biological functions, for example, as antigens that shape an immune response.
The ISR is important in several diseases due to its role in cell death and survival, and thus, the possibility of targeting it to modulate pathological conditions is a strong rationale for understanding this in greater detail. It is evident that for protein folding diseases, such as certain neurodegenerative diseases, there are beneficial effects to slowing down translation, allowing more time for the ER to fold proteins properly. Targeting the ISR through the inhibition of GADD34, which catalyzes dephosphorylation of eIF2α, shows promise in models of several neurodegenerative diseases that are characterized by proteotoxicity 224. In tumors, the activation of ISR has been implicated in several anticancer effects, including promoting cell survival. In fact, the cytoprotective ISR can be induced in response to anticancer therapies, thus promoting the development of chemoresistance, as it is the case in pancreatic ductal carcinoma cells and an orthotopic mouse model subjected to gemcitabine treatment 248. Therefore, cancer treatment with chemotherapeutic drugs in combination with inhibitors to block the cytoprotective ISR may be beneficial. Also, there may be other drugs, already used in the clinic, that could be repurposed for the treatment of diseases where it is desirable to target the ISR.
For therapeutic manipulation of the ISR, it is important to recognize that the consequences of targeting ISR are not the same in all cell types and that there may be undesirable side effects. ATF4 has the potential to regulate more than 400 genes that are important in various, often opposing processes such as cell survival and cell death 115, 122. For example, research on ONC201‐induced toxicity revealed that different stress response pathways are activated by the drug in solid tumors and hematological malignancies 232, 249. Clearly, when targeting the ISR, what needs to be considered is a global organism physiology and benefit for the whole body rather than for individual cells. For example, cells growing in vitro or as xenografts in mice are not subjected to physiological nonautonomous tumor microenvironment conditions. Knockdown of GCN2 in human HT1080 sarcoma cells results in reduced tumor growth in xenograft mice model, while in a genetically engineered mouse model of soft tissue sarcoma, Gcn2 knockdown has no effect on tumor progression due to a compensatory activation of PERK 65. Thus, both PERK and GCN2 might need to be inhibited to reduce the phosphorylation of eIF2α, and indeed, this has been shown to be the case in mouse fibroblasts 64.
Research conducted in vitro using the cultured cells isolated from tissues shows the ability of cells to autonomously respond to stress stimuli. However, recent advances on manipulating stress responses at an organismal level reveal multilayered intracellular cross‐talk between and across the tissues to regulate organism homeostasis 250. During aging in the nematode Caenorhabditis elegans (C. elegans), the accumulation of neurotoxic protein aggregates may influence protein homeostasis (proteostasis) in somatic tissues 251. Furthermore, sensory neurons can downregulate noncanonical UPR activity in non‐neuronal cells of C. elegans to suppress immune responses to pathogen infection 252. This is an exciting, new area and understanding how higher organisms communicate local stress to the entire organism is yet to be clarified. Thus, understanding the consequences of targeting the ISR in different tissues, especially vulnerable cells such as neurons, cardiomyocytes, and pancreatic β cells, and different disease models, and the mechanisms engaged to regulate different responses across the whole organism are essential for the development of effective pharmacological agents. There are several physiological conditions that alter cell homeostasis, for example, high secretory demand on pancreatic β cells, hepatocytes, plasma cells, and barrier epithelial cells. Thus, pharmacological alterations of physiological responses might result in diabetes development, compromised immune response, hepatocarcinoma, and sepsis as unwanted side effects.
To gain an important insight into the physiological and pathological roles of the ISR, a better understanding of the molecular mechanisms linking the ISR to other cellular processes, such as autophagy, immunomodulation, metabolism, secretion, cell cycle regulation, and differentiation, would greatly facilitate approaches to modulate the ISR for therapeutic benefit. Considering autophagy as a key component of the ISR, there are still many unanswered questions concerning this in regard to signaling networks and links to biological processes that await further investigation 170. Although a number of mechanisms underlying the interplay between the ISR and autophagy have been proposed, including upregulation of ATF4‐regulated genes, a more comprehensive knowledge of how mTOR activity is regulated by ISR is required.
In conclusion, the ISR represents an important cell response that controls translation and mounts a cell protective response. It is poised at the balance of cell death and survival, and as such, it is an important target in the treatment for many disorders.
Conflict of interest
A.S. is a cofounder and director of Aquila Bioscience Ltd. A.G., A.S., K.M., M.L., and K.P.Z. are all cofounders of Cell Stress Discoveries Ltd.
Sidebar A: In need of answers.
What are the best therapeutic combinations that should be employed to usefully target the ISR in different disease conditions while minimizing unwanted side effects?
Does the ISR elicit extracellular signaling to regulate organismal homeostasis and could such extracellular signals be used as biomarkers of disease pathologies?
What are the molecular mechanisms that shift the balance between opposing cellular fates due to ISR activation?
Apart from ATF4, which other factors control the ISR?
How do post‐translational modifications of ATF4 govern its interactions with binding partners to regulate gene expression?
How does the ISR regulate autophagy?
Acknowledgements
This work was funded in part by the Health Research Board (Grant Number HRA/2009/59; HRA‐POR‐2014‐643), Belgian Science Policy Office Interuniversity Attraction Poles program (IAP 7/32), and Breast Cancer Campaign grant (2010NovPR13) to A.S. A Science Foundation Ireland (SFI) grant cofunded under the European Regional Development Fund under Grant Number 13/RC/2073. I.K. was funded by an Irish Research Council Scholarship (GOIPG/2014/508) and by the Thomas Crawford Hayes Fund. K.M. is funded by an Irish Research Council Fellowship (GOIPD/2014/53).
EMBO Reports (2016) 17: 1374–1395
See the Glossary for abbreviations used in this article.
References
- 1. Ron D (2002) Translational control in the endoplasmic reticulum stress response. J Clin Invest 110: 1383–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R et al (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11: 619–633 [DOI] [PubMed] [Google Scholar]
- 3. Brostrom CO, Prostko CR, Kaufman RJ, Brostrom MA (1996) Inhibition of translational initiation by activators of the glucose‐regulated stress protein and heat shock protein stress response systems. Role of the interferon‐inducible double‐stranded RNA‐activated eukaryotic initiation factor 2alpha kinase. J Biol Chem 271: 24995–25002 [DOI] [PubMed] [Google Scholar]
- 4. Dever TE, Feng L, Wek RC, Cigan AM, Donahue TF, Hinnebusch AG (1992) Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene‐specific translational control of GCN4 in yeast. Cell 68: 585–596 [DOI] [PubMed] [Google Scholar]
- 5. Wek RC, Jiang HY, Anthony TG (2006) Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans 34: 7–11 [DOI] [PubMed] [Google Scholar]
- 6. Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L, Pires I , Hammond E, Ragoussis I, Harris AL (2010) Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 29: 4424–4435 [DOI] [PubMed] [Google Scholar]
- 7. Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN, Bobrovnikova‐Marjon E, Diehl JA, Ron D, Koumenis C (2010) The GCN2‐ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J 29: 2082–2096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Garcia MA, Meurs EF, Esteban M (2007) The dsRNA protein kinase PKR: virus and cell control. Biochimie 89: 799–811 [DOI] [PubMed] [Google Scholar]
- 9. Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic‐reticulum‐resident kinase. Nature 397: 271–274 [DOI] [PubMed] [Google Scholar]
- 10. Denoyelle C, Abou‐Rjaily G, Bezrookove V, Verhaegen M, Johnson TM, Fullen DR, Pointer JN, Gruber SB, Su LD, Nikiforov MA et al (2006) Anti‐oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat Cell Biol 8: 1053–1063 [DOI] [PubMed] [Google Scholar]
- 11. Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, Qiu B, Zhang H, Cerniglia G, Bi M et al (2012) ER stress‐mediated autophagy promotes Myc‐dependent transformation and tumor growth. J Clin Invest 122: 4621–4634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Donnelly N, Gorman AM, Gupta S, Samali A (2013) The eIF2alpha kinases: their structures and functions. Cell Mol Life Sci 70: 3493–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu PD, Harding HP, Ron D (2004) Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol 167: 27–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nagahara N, Yoshii T, Abe Y, Matsumura T (2007) Thioredoxin‐dependent enzymatic activation of mercaptopyruvate sulfurtransferase. An intersubunit disulfide bond serves as a redox switch for activation. J Biol Chem 282: 1561–1569 [DOI] [PubMed] [Google Scholar]
- 15. Novoa I, Zeng H, Harding HP, Ron D (2001) Feedback inhibition of the unfolded protein response by GADD34‐mediated dephosphorylation of eIF2alpha. J Cell Biol 153: 1011–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Novoa I, Zhang Y, Zeng H, Jungreis R, Harding HP, Ron D (2003) Stress‐induced gene expression requires programmed recovery from translational repression. EMBO J 22: 1180–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dey S, Baird TD, Zhou D, Palam LR, Spandau DF, Wek RC (2010) Both transcriptional regulation and translational control of ATF4 are central to the integrated stress response. J Biol Chem 285: 33165–33174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Scheuner D, Patel R, Wang F, Lee K, Kumar K, Wu J, Nilsson A, Karin M, Kaufman RJ (2006) Double‐stranded RNA‐dependent protein kinase phosphorylation of the alpha‐subunit of eukaryotic translation initiation factor 2 mediates apoptosis. J Biol Chem 281: 21458–21468 [DOI] [PubMed] [Google Scholar]
- 19. Perkins DJ, Barber GN (2004) Defects in translational regulation mediated by the alpha subunit of eukaryotic initiation factor 2 inhibit antiviral activity and facilitate the malignant transformation of human fibroblasts. Mol Cell Biol 24: 2025–2040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berlanga JJ, Herrero S, de Haro C (1998) Characterization of the hemin‐sensitive eukaryotic initiation factor 2alpha kinase from mouse nonerythroid cells. J Biol Chem 273: 32340–32346 [DOI] [PubMed] [Google Scholar]
- 21. Chen JJ, Throop MS, Gehrke L, Kuo I, Pal JK, Brodsky M, London IM (1991) Cloning of the cDNA of the heme‐regulated eukaryotic initiation factor 2 alpha (eIF‐2 alpha) kinase of rabbit reticulocytes: homology to yeast GCN2 protein kinase and human double‐stranded‐RNA‐dependent eIF‐2 alpha kinase. Proc Natl Acad Sci USA 88: 7729–7733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, Wek RC (1998) Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha‐subunit kinase, PEK, involved in translational control. Mol Cell Biol 18: 7499–7509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meurs E, Chong K, Galabru J, Thomas NS, Kerr IM, Williams BR, Hovanessian AG (1990) Molecular cloning and characterization of the human double‐stranded RNA‐activated protein kinase induced by interferon. Cell 62: 379–390 [DOI] [PubMed] [Google Scholar]
- 24. Ramirez M, Wek RC, Hinnebusch AG (1991) Ribosome association of GCN2 protein kinase, a translational activator of the GCN4 gene of Saccharomyces cerevisiae. Mol Cell Biol 11: 3027–3036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lavoie H, Li JJ, Thevakumaran N, Therrien M, Sicheri F (2014) Dimerization‐induced allostery in protein kinase regulation. Trends Biochem Sci 39: 475–486 [DOI] [PubMed] [Google Scholar]
- 26. Korennykh A, Walter P (2012) Structural basis of the unfolded protein response. Annu Rev Cell Dev Biol 28: 251–277 [DOI] [PubMed] [Google Scholar]
- 27. Wang M, Kaufman RJ (2016) Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529: 326–335 [DOI] [PubMed] [Google Scholar]
- 28. Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded‐protein response. Nat Cell Biol 2: 326–332 [DOI] [PubMed] [Google Scholar]
- 29. Carrara M, Prischi F, Nowak PR, Kopp MC, Ali MM (2015) Noncanonical binding of BiP ATPase domain to Ire1 and Perk is dissociated by unfolded protein CH1 to initiate ER stress signaling. eLife 4: e03522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gardner BM, Walter P (2011) Unfolded proteins are Ire1‐activating ligands that directly induce the unfolded protein response. Science 333: 1891–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. de la Cadena SG, Hernandez‐Fonseca K, Camacho‐Arroyo I, Massieu L (2014) Glucose deprivation induces reticulum stress by the PERK pathway and caspase‐7‐ and calpain‐mediated caspase‐12 activation. Apoptosis 19: 414–427 [DOI] [PubMed] [Google Scholar]
- 32. Moore CE, Omikorede O, Gomez E, Willars GB, Herbert TP (2011) PERK activation at low glucose concentration is mediated by SERCA pump inhibition and confers preemptive cytoprotection to pancreatic beta‐cells. Mol Endocrinol 25: 315–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Castilho BA, Shanmugam R, Silva RC, Ramesh R, Himme BM, Sattlegger E (2014) Keeping the eIF2 alpha kinase Gcn2 in check. Biochim Biophys Acta 1843: 1948–1968 [DOI] [PubMed] [Google Scholar]
- 34. Vazquez de Aldana CR, Wek RC, Segundo PS, Truesdell AG, Hinnebusch AG (1994) Multicopy tRNA genes functionally suppress mutations in yeast eIF‐2 alpha kinase GCN2: evidence for separate pathways coupling GCN4 expression to unchanged tRNA. Mol Cell Biol 14: 7920–7932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Deng J, Harding HP, Raught B, Gingras AC, Berlanga JJ, Scheuner D, Kaufman RJ, Ron D, Sonenberg N (2002) Activation of GCN2 in UV‐irradiated cells inhibits translation. Curr Biol 12: 1279–1286 [DOI] [PubMed] [Google Scholar]
- 36. Lu W, Laszlo CF, Miao Z, Chen H, Wu S (2009) The role of nitric‐oxide synthase in the regulation of UVB light‐induced phosphorylation of the alpha subunit of eukaryotic initiation factor 2. J Biol Chem 284: 24281–24288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Clemens MJ, Elia A (1997) The double‐stranded RNA‐dependent protein kinase PKR: structure and function. J Interferon Cytokine Res 17: 503–524 [DOI] [PubMed] [Google Scholar]
- 38. Lemaire PA, Anderson E, Lary J, Cole JL (2008) Mechanism of PKR activation by dsRNA. J Mol Biol 381: 351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dey M, Cao C, Dar AC, Tamura T, Ozato K, Sicheri F, Dever TE (2005) Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell 122: 901–913 [DOI] [PubMed] [Google Scholar]
- 40. Zhang F, Romano PR, Nagamura‐Inoue T, Tian B, Dever TE, Mathews MB, Ozato K, Hinnebusch AG (2001) Binding of double‐stranded RNA to protein kinase PKR is required for dimerization and promotes critical autophosphorylation events in the activation loop. J Biol Chem 276: 24946–24958 [DOI] [PubMed] [Google Scholar]
- 41. Vattem KM, Staschke KA, Wek RC (2001) Mechanism of activation of the double‐stranded‐RNA‐dependent protein kinase, PKR: role of dimerization and cellular localization in the stimulation of PKR phosphorylation of eukaryotic initiation factor‐2 (eIF2). Eur J Biochem 268: 3674–3684 [DOI] [PubMed] [Google Scholar]
- 42. Balachandran S, Roberts PC, Brown LE, Truong H, Pattnaik AK, Archer DR, Barber GN (2000) Essential role for the dsRNA‐dependent protein kinase PKR in innate immunity to viral infection. Immunity 13: 129–141 [DOI] [PubMed] [Google Scholar]
- 43. Lee SB, Esteban M (1993) The interferon‐induced double‐stranded RNA‐activated human p68 protein kinase inhibits the replication of vaccinia virus. Virology 193: 1037–1041 [DOI] [PubMed] [Google Scholar]
- 44. Shimazawa M, Hara H (2006) Inhibitor of double stranded RNA‐dependent protein kinase protects against cell damage induced by ER stress. Neurosci Lett 409: 192–195 [DOI] [PubMed] [Google Scholar]
- 45. Lee ES, Yoon CH, Kim YS, Bae YS (2007) The double‐strand RNA‐dependent protein kinase PKR plays a significant role in a sustained ER stress‐induced apoptosis. FEBS Lett 581: 4325–4332 [DOI] [PubMed] [Google Scholar]
- 46. Garcia MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, Esteban M (2006) Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev 70: 1032–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Williams BR (1999) PKR; a sentinel kinase for cellular stress. Oncogene 18: 6112–6120 [DOI] [PubMed] [Google Scholar]
- 48. Zhou HR, He K, Landgraf J, Pan X, Pestka JJ (2014) Direct activation of ribosome‐associated double‐stranded RNA‐dependent protein kinase (PKR) by deoxynivalenol, anisomycin and ricin: a new model for ribotoxic stress response induction. Toxins 6: 3406–3425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Reineke LC, Lloyd RE (2015) The stress granule protein G3BP1 recruits protein kinase R to promote multiple innate immune antiviral responses. J Virol 89: 2575–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hovanessian AG, Galabru J (1987) The double‐stranded RNA‐dependent protein kinase is also activated by heparin. Eur J Biochem 167: 467–473 [DOI] [PubMed] [Google Scholar]
- 51. Saelens X, Kalai M, Vandenabeele P (2001) Translation inhibition in apoptosis: caspase‐dependent PKR activation and eIF2‐alpha phosphorylation. J Biol Chem 276: 41620–41628 [DOI] [PubMed] [Google Scholar]
- 52. Han AP, Yu C, Lu L, Fujiwara Y, Browne C, Chin G, Fleming M, Leboulch P, Orkin SH, Chen JJ (2001) Heme‐regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J 20: 6909–6918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bruns GP, London IM (1965) The effect of hemin on the synthesis of globin. Biochem Biophys Res Commun 18: 236–242 [DOI] [PubMed] [Google Scholar]
- 54. Chefalo PJ, Oh J, Rafie‐Kolpin M, Kan B, Chen JJ (1998) Heme‐regulated eIF‐2alpha kinase purifies as a hemoprotein. Eur J Biochem 258: 820–830 [DOI] [PubMed] [Google Scholar]
- 55. Suragani RN, Zachariah RS, Velazquez JG, Liu S, Sun CW, Townes TM, Chen JJ (2012) Heme‐regulated eIF2alpha kinase activated Atf4 signaling pathway in oxidative stress and erythropoiesis. Blood 119: 5276–5284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rafie‐Kolpin M, Chefalo PJ, Hussain Z, Hahn J, Uma S, Matts RL, Chen JJ (2000) Two heme‐binding domains of heme‐regulated eukaryotic initiation factor‐2alpha kinase. N terminus and kinase insertion. J Biol Chem 275: 5171–5178 [DOI] [PubMed] [Google Scholar]
- 57. Rafie‐Kolpin M, Han AP, Chen JJ (2003) Autophosphorylation of threonine 485 in the activation loop is essential for attaining eIF2alpha kinase activity of HRI. Biochemistry 42: 6536–6544 [DOI] [PubMed] [Google Scholar]
- 58. Acharya P, Chen JJ, Correia MA (2010) Hepatic heme‐regulated inhibitor (HRI) eukaryotic initiation factor 2alpha kinase: a protagonist of heme‐mediated translational control of CYP2B enzymes and a modulator of basal endoplasmic reticulum stress tone. Mol Pharmacol 77: 575–592 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59. Hirai K, Martinkova M, Igarashi J, Saiful I, Yamauchi S, El‐Mashtoly S, Kitagawa T, Shimizu T (2007) Identification of Cys385 in the isolated kinase insertion domain of heme‐regulated eIF2 alpha kinase (HRI) as the heme axial ligand by site‐directed mutagenesis and spectral characterization. J Inorg Biochem 101: 1172–1179 [DOI] [PubMed] [Google Scholar]
- 60. Ill‐Raga G, Tajes M, Busquets‐Garcia A, Ramos‐Fernandez E, Vargas LM, Bosch‐Morato M, Guivernau B, Valls‐Comamala V, Eraso‐Pichot A, Guix FX et al (2015) Physiological control of nitric oxide in neuronal BACE1 translation by heme‐regulated eIF2alpha kinase HRI induces synaptogenesis. Antioxid Redox Signal 22: 1295–1307 [DOI] [PubMed] [Google Scholar]
- 61. McEwen E, Kedersha N, Song B, Scheuner D, Gilks N, Han A, Chen JJ, Anderson P, Kaufman RJ (2005) Heme‐regulated inhibitor kinase‐mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J Biol Chem 280: 16925–16933 [DOI] [PubMed] [Google Scholar]
- 62. Yerlikaya A, Kimball SR, Stanley BA (2008) Phosphorylation of eIF2alpha in response to 26S proteasome inhibition is mediated by the haem‐regulated inhibitor (HRI) kinase. Biochem J 412: 579–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lu L, Han AP, Chen JJ (2001) Translation initiation control by heme‐regulated eukaryotic initiation factor 2alpha kinase in erythroid cells under cytoplasmic stresses. Mol Cell Biol 21: 7971–7980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hamanaka RB, Bennett BS, Cullinan SB, Diehl JA (2005) PERK and GCN2 contribute to eIF2alpha phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Mol Biol Cell 16: 5493–5501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lehman SL, Ryeom S, Koumenis C (2015) Signaling through alternative Integrated Stress Response pathways compensates for GCN2 loss in a mouse model of soft tissue sarcoma. Sci Rep 5: 11781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Devi L, Ohno M (2013) Deletion of the eIF2alpha kinase GCN2 fails to rescue the memory decline associated with Alzheimer's disease. PLoS ONE 8: e77335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Roobol A, Roobol J, Bastide A, Knight JR, Willis AE, Smales CM (2015) p58IPK is an inhibitor of the eIF2alpha kinase GCN2 and its localization and expression underpin protein synthesis and ER processing capacity. Biochem J 465: 213–225 [DOI] [PubMed] [Google Scholar]
- 68. Anderson BR, Kariko K, Weissman D (2013) Nucleofection induces transient eIF2alpha phosphorylation by GCN2 and PERK. Gene Ther 20: 136–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Berlanga JJ, Ventoso I, Harding HP, Deng J, Ron D, Sonenberg N, Carrasco L, de Haro C (2006) Antiviral effect of the mammalian translation initiation factor 2alpha kinase GCN2 against RNA viruses. EMBO J 25: 1730–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cheng G, Feng Z, He B (2005) Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF‐2alpha dephosphorylation by the gamma(1)34.5 protein. J Virol 79: 1379–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Baker BM, Nargund AM, Sun T, Haynes CM (2012) Protective coupling of mitochondrial function and protein synthesis via the eIF2alpha kinase GCN‐2. PLoS Genet 8: e1002760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pyo CW, Lee SH, Choi SY (2008) Oxidative stress induces PKR‐dependent apoptosis via IFN‐gamma activation signaling in Jurkat T cells. Biochem Biophys Res Commun 377: 1001–1006 [DOI] [PubMed] [Google Scholar]
- 73. Li L, Tan H, Gu Z, Liu Z, Geng Y, Liu Y, Tong H, Tang Y, Qiu J, Su L (2014) Heat stress induces apoptosis through a Ca(2)(+)‐mediated mitochondrial apoptotic pathway in human umbilical vein endothelial cells. PLoS ONE 9: e111083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhao M, Tang D, Lechpammer S, Hoffman A, Asea A, Stevenson MA, Calderwood SK (2002) Double‐stranded RNA‐dependent protein kinase (pkr) is essential for thermotolerance, accumulation of HSP70, and stabilization of ARE‐containing HSP70 mRNA during stress. J Biol Chem 277: 44539–44547 [DOI] [PubMed] [Google Scholar]
- 75. Chen H, Song R, Wang G, Ding Z, Yang C, Zhang J, Zeng Z, Rubio V, Wang L, Zu N et al (2015) OLA1 regulates protein synthesis and integrated stress response by inhibiting eIF2 ternary complex formation. Sci Rep 5: 13241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jousse C, Oyadomari S, Novoa I, Lu P, Zhang Y, Harding HP, Ron D (2003) Inhibition of a constitutive translation initiation factor 2alpha phosphatase, CReP, promotes survival of stressed cells. J Cell Biol 163: 767–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kojima E, Takeuchi A, Haneda M, Yagi A, Hasegawa T, Yamaki K, Takeda K, Akira S, Shimokata K, Isobe K (2003) The function of GADD34 is a recovery from a shutoff of protein synthesis induced by ER stress: elucidation by GADD34‐deficient mice. FASEB J 17: 1573–1575 [DOI] [PubMed] [Google Scholar]
- 78. Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D (2004) CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 18: 3066–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ma Y, Hendershot LM (2003) Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J Biol Chem 278: 34864–34873 [DOI] [PubMed] [Google Scholar]
- 80. Liu L, Ito S, Nishio N, Sun Y, Chen N, Tanaka Y, Isobe K (2015) GADD34 facilitates cell death resulting from proteasome inhibition. Anticancer Res 35: 5317–5324 [PubMed] [Google Scholar]
- 81. Harding HP, Zhang Y, Scheuner D, Chen JJ, Kaufman RJ, Ron D (2009) Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2alpha) dephosphorylation in mammalian development. Proc Natl Acad Sci USA 106: 1832–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chambers JE, Dalton LE, Clarke HJ, Malzer E, Dominicus CS, Patel V, Moorhead G, Ron D, Marciniak SJ (2015) Actin dynamics tune the integrated stress response by regulating eukaryotic initiation factor 2alpha dephosphorylation. eLife 4: e04872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chen R, Rato C, Yan Y, Crespillo‐Casado A, Clarke HJ, Harding HP, Marciniak SJ, Read RJ, Ron D (2015) G‐actin provides substrate‐specificity to eukaryotic initiation factor 2alpha holophosphatases. eLife 4: e04871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Loveless TB, Topacio BR, Vashisht AA, Galaang S, Ulrich KM, Young BD, Wohlschlegel JA, Toczyski DP (2015) DNA damage regulates translation through beta‐TRCP targeting of CReP. PLoS Genet 11: e1005292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Farook JM, Shields J, Tawfik A, Markand S, Sen T, Smith SB, Brann D, Dhandapani KM, Sen N (2013) GADD34 induces cell death through inactivation of Akt following traumatic brain injury. Cell Death Dis 4: e754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Shi W, Sun C, He B, Xiong W, Shi X, Yao D, Cao X (2004) GADD34‐PP1c recruited by Smad7 dephosphorylates TGFbeta type I receptor. J Cell Biol 164: 291–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jackson RJ, Hellen CU, Pestova TV (2010) The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11: 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Pain VM (1996) Initiation of protein synthesis in eukaryotic cells. Eur J Biochem 236: 747–771 [DOI] [PubMed] [Google Scholar]
- 89. Aitken CE, Lorsch JR (2012) A mechanistic overview of translation initiation in eukaryotes. Nat Struct Mol Biol 19: 568–576 [DOI] [PubMed] [Google Scholar]
- 90. Lomakin IB, Steitz TA (2013) The initiation of mammalian protein synthesis and mRNA scanning mechanism. Nature 500: 307–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Hinnebusch AG, Lorsch JR (2012) The mechanism of eukaryotic translation initiation: new insights and challenges. Cold Spring Harb Perspect Biol 4: a011544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hinnebusch AG (2011) Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol Mol Biol Rev 75: 434–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lee YY, Cevallos RC, Jan E (2009) An upstream open reading frame regulates translation of GADD34 during cellular stresses that induce eIF2alpha phosphorylation. J Biol Chem 284: 6661–6673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Palam LR, Baird TD, Wek RC (2011) Phosphorylation of eIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translation. J Biol Chem 286: 10939–10949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Chan CP, Kok KH, Tang HM, Wong CM, Jin DY (2013) Internal ribosome entry site‐mediated translational regulation of ATF4 splice variant in mammalian unfolded protein response. Biochim Biophys Acta 1833: 2165–2175 [DOI] [PubMed] [Google Scholar]
- 96. Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner‐Weir S, Kaufman RJ (2001) Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 7: 1165–1176 [DOI] [PubMed] [Google Scholar]
- 97. Back SH, Scheuner D, Han J, Song B, Ribick M, Wang J, Gildersleeve RD, Pennathur S, Kaufman RJ (2009) Translation attenuation through eIF2α phosphorylation prevents oxidative stress and maintains the differentiated state in β cells. Cell Metab 10: 13–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T (2007) ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine‐induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 14: 230–239 [DOI] [PubMed] [Google Scholar]
- 99. Talloczy Z, Jiang W, Virgin HWT, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B (2002) Regulation of starvation‐ and virus‐induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA 99: 190–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ameri K, Harris AL (2008) Activating transcription factor 4. Int J Biochem Cell Biol 40: 14–21 [DOI] [PubMed] [Google Scholar]
- 101. Karpinski BA, Morle GD, Huggenvik J, Uhler MD, Leiden JM (1992) Molecular cloning of human CREB‐2: an ATF/CREB transcription factor that can negatively regulate transcription from the cAMP response element. Proc Natl Acad Sci USA 89: 4820–4824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Vallejo M, Ron D, Miller CP, Habener JF (1993) C/ATF, a member of the activating transcription factor family of DNA‐binding proteins, dimerizes with CAAT/enhancer‐binding proteins and directs their binding to cAMP response elements. Proc Natl Acad Sci USA 90: 4679–4683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. B'Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A (2013) The eIF2alpha/ATF4 pathway is essential for stress‐induced autophagy gene expression. Nucleic Acids Res 41: 7683–7699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hettmann T, Barton K, Leiden JM (2000) Microphthalmia due to p53‐mediated apoptosis of anterior lens epithelial cells in mice lacking the CREB‐2 transcription factor. Dev Biol 222: 110–123 [DOI] [PubMed] [Google Scholar]
- 105. Tanaka T, Tsujimura T, Takeda K, Sugihara A, Maekawa A, Terada N, Yoshida N, Akira S (1998) Targeted disruption of ATF4 discloses its essential role in the formation of eye lens fibres. Genes Cells 3: 801–810 [DOI] [PubMed] [Google Scholar]
- 106. Masuoka HC, Townes TM (2002) Targeted disruption of the activating transcription factor 4 gene results in severe fetal anemia in mice. Blood 99: 736–745 [DOI] [PubMed] [Google Scholar]
- 107. Yang X, Matsuda K, Bialek P, Jacquot S, Masuoka HC, Schinke T, Li L, Brancorsini S, Sassone‐Corsi P, Townes TM et al (2004) ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin‐Lowry Syndrome. Cell 117: 387–398 [DOI] [PubMed] [Google Scholar]
- 108. Wang C, Huang Z, Du Y, Cheng Y, Chen S, Guo F (2010) ATF4 regulates lipid metabolism and thermogenesis. Cell Res 20: 174–184 [DOI] [PubMed] [Google Scholar]
- 109. Wang C, Xia T, Du Y, Meng Q, Li H, Liu B, Chen S, Guo F (2013) Effects of ATF4 on PGC1alpha expression in brown adipose tissue and metabolic responses to cold stress. Metabolism 62: 282–289 [DOI] [PubMed] [Google Scholar]
- 110. Ebert SM, Dyle MC, Bullard SA, Dierdorff JM, Murry DJ, Fox DK, Bongers KS, Lira VA, Meyerholz DK, Talley JJ et al (2015) Identification and small molecule inhibition of an activating transcription factor 4 (ATF4)‐dependent pathway to age‐related skeletal muscle weakness and atrophy. J Biol Chem 290: 25497–25511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Costa‐Mattioli M, Gobert D, Harding H, Herdy B, Azzi M, Bruno M, Bidinosti M, Ben Mamou C, Marcinkiewicz E, Yoshida M et al (2005) Translational control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature 436: 1166–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Sidrauski C, McGeachy AM, Ingolia NT, Walter P (2015) The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. eLife 4: e05033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Trinh MA, Kaphzan H, Wek RC, Pierre P, Cavener DR, Klann E (2012) Brain‐specific disruption of the eIF2alpha kinase PERK decreases ATF4 expression and impairs behavioral flexibility. Cell Rep 1: 676–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wei W, Coelho CM, Li X, Marek R, Yan S, Anderson S, Meyers D, Mukherjee C, Sbardella G, Castellano S et al (2012) p300/CBP‐associated factor selectively regulates the extinction of conditioned fear. J Neurosci 32: 11930–11941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Pasini S, Corona C, Liu J, Greene LA, Shelanski ML (2015) Specific downregulation of hippocampal ATF4 reveals a necessary role in synaptic plasticity and memory. Cell Rep 11: 183–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Fawcett TW, Martindale JL, Guyton KZ, Hai T, Holbrook NJ (1999) Complexes containing activating transcription factor (ATF)/cAMP‐responsive‐element‐binding protein (CREB) interact with the CCAAT/enhancer‐binding protein (C/EBP)‐ATF composite site to regulate Gadd153 expression during the stress response. Biochem J 339(Pt 1): 135–141 [PMC free article] [PubMed] [Google Scholar]
- 117. Kilberg MS, Balasubramanian M, Fu L, Shan J (2012) The transcription factor network associated with the amino acid response in mammalian cells. Adv Nutr 3: 295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kilberg MS, Shan J, Su N (2009) ATF4‐dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol Metab 20: 436–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Averous J, Bruhat A, Jousse C, Carraro V, Thiel G, Fafournoux P (2004) Induction of CHOP expression by amino acid limitation requires both ATF4 expression and ATF2 phosphorylation. J Biol Chem 279: 5288–5297 [DOI] [PubMed] [Google Scholar]
- 120. Siu F, Bain PJ, LeBlanc‐Chaffin R, Chen H, Kilberg MS (2002) ATF4 is a mediator of the nutrient‐sensing response pathway that activates the human asparagine synthetase gene. J Biol Chem 277: 24120–24127 [DOI] [PubMed] [Google Scholar]
- 121. Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken JW et al (2010) The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest 120: 127–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M et al (2013) ER‐stress‐induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15: 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Bartsch D, Ghirardi M, Skehel PA, Karl KA, Herder SP, Chen M, Bailey CH, Kandel ER (1995) Aplysia CREB2 represses long‐term facilitation: relief of repression converts transient facilitation into long‐term functional and structural change. Cell 83: 979–992 [DOI] [PubMed] [Google Scholar]
- 124. Chen A, Muzzio IA, Malleret G, Bartsch D, Verbitsky M, Pavlidis P, Yonan AL, Vronskaya S, Grody MB, Cepeda I et al (2003) Inducible enhancement of memory storage and synaptic plasticity in transgenic mice expressing an inhibitor of ATF4 (CREB‐2) and C/EBP proteins. Neuron 39: 655–669 [DOI] [PubMed] [Google Scholar]
- 125. Chen H, Pan YX, Dudenhausen EE, Kilberg MS (2004) Amino acid deprivation induces the transcription rate of the human asparagine synthetase gene through a timed program of expression and promoter binding of nutrient‐responsive basic region/leucine zipper transcription factors as well as localized histone acetylation. J Biol Chem 279: 50829–50839 [DOI] [PubMed] [Google Scholar]
- 126. Afonyushkin T, Oskolkova OV, Philippova M, Resink TJ, Erne P, Binder BR, Bochkov VN (2010) Oxidized phospholipids regulate expression of ATF4 and VEGF in endothelial cells via NRF2‐dependent mechanism: novel point of convergence between electrophilic and unfolded protein stress pathways. Arterioscler Thromb Vasc Biol 30: 1007–1013 [DOI] [PubMed] [Google Scholar]
- 127. Bruning A, Rahmeh M, Friese K (2013) Nelfinavir and bortezomib inhibit mTOR activity via ATF4‐mediated sestrin‐2 regulation. Mol Oncol 7: 1012–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Ding B, Parmigiani A, Divakaruni AS, Archer K, Murphy AN, Budanov AV (2016) Sestrin2 is induced by glucose starvation via the unfolded protein response and protects cells from non‐canonical necroptotic cell death. Sci Rep 6: 22538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Saveljeva S, Cleary P, Mnich K, Ayo A, Pakos‐Zebrucka K, Patterson JB, Logue SE, Samali A (2016) Endoplasmic reticulum stress‐mediated induction of SESTRIN 2 potentiates cell survival. Oncotarget 7: 12254–12266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Podust LM, Krezel AM, Kim Y (2001) Crystal structure of the CCAAT box/enhancer‐binding protein beta activating transcription factor‐4 basic leucine zipper heterodimer in the absence of DNA. J Biol Chem 276: 505–513 [DOI] [PubMed] [Google Scholar]
- 131. Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H (2005) TRB3, a novel ER stress‐inducible gene, is induced via ATF4‐CHOP pathway and is involved in cell death. EMBO J 24: 1243–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Wang Q, Mora‐Jensen H, Weniger MA, Perez‐Galan P, Wolford C, Hai T, Ron D, Chen W, Trenkle W, Wiestner A et al (2009) ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3‐only protein NOXA in cancer cells. Proc Natl Acad Sci USA 106: 2200–2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Hiwatashi Y, Kanno K, Takasaki C, Goryo K, Sato T, Torii S, Sogawa K, Yasumoto K (2011) PHD1 interacts with ATF4 and negatively regulates its transcriptional activity without prolyl hydroxylation. Exp Cell Res 317: 2789–2799 [DOI] [PubMed] [Google Scholar]
- 134. Koditz J, Nesper J, Wottawa M, Stiehl DP, Camenisch G, Franke C, Myllyharju J, Wenger RH, Katschinski DM (2007) Oxygen‐dependent ATF‐4 stability is mediated by the PHD3 oxygen sensor. Blood 110: 3610–3617 [DOI] [PubMed] [Google Scholar]
- 135. Jousse C, Deval C, Maurin AC, Parry L, Cherasse Y, Chaveroux C, Lefloch R, Lenormand P, Bruhat A, Fafournoux P (2007) TRB3 inhibits the transcriptional activation of stress‐regulated genes by a negative feedback on the ATF4 pathway. J Biol Chem 282: 15851–15861 [DOI] [PubMed] [Google Scholar]
- 136. Blais JD, Filipenko V, Bi M, Harding HP, Ron D, Koumenis C, Wouters BG, Bell JC (2004) Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol Cell Biol 24: 7469–7482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Roybal CN, Hunsaker LA, Barbash O, Vander Jagt DL, Abcouwer SF (2005) The oxidative stressor arsenite activates vascular endothelial growth factor mRNA transcription by an ATF4‐dependent mechanism. J Biol Chem 280: 20331–20339 [DOI] [PubMed] [Google Scholar]
- 138. Hinnebusch AG (2005) Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol 59: 407–450 [DOI] [PubMed] [Google Scholar]
- 139. Hinnebusch AG, Natarajan K (2002) Gcn4p, a master regulator of gene expression, is controlled at multiple levels by diverse signals of starvation and stress. Eukaryot Cell 1: 22–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Mueller PP, Hinnebusch AG (1986) Multiple upstream AUG codons mediate translational control of GCN4. Cell 45: 201–207 [DOI] [PubMed] [Google Scholar]
- 141. Hinnebusch AG (1997) Translational regulation of yeast GCN4. A window on factors that control initiator‐trna binding to the ribosome. J Biol Chem 272: 21661–21664 [DOI] [PubMed] [Google Scholar]
- 142. Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D (2000) Regulated translation initiation controls stress‐induced gene expression in mammalian cells. Mol Cell 6: 1099–1108 [DOI] [PubMed] [Google Scholar]
- 143. Vattem KM, Wek RC (2004) Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci USA 101: 11269–11274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Huret JL, Ahmad M, Arsaban M, Bernheim A, Cigna J, Desangles F, Guignard JC, Jacquemot‐Perbal MC, Labarussias M, Leberre V et al (2013) Atlas of genetics and cytogenetics in oncology and haematology in 2013. Nucleic Acids Res 41: D920–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Starck SR, Tsai JC, Chen K, Shodiya M, Wang L, Yahiro K, Martins‐Green M, Shastri N, Walter P (2016) Translation from the 5′ untranslated region shapes the integrated stress response. Science 351: aad3867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Igarashi T, Izumi H, Uchiumi T, Nishio K, Arao T, Tanabe M, Uramoto H, Sugio K, Yasumoto K, Sasaguri Y et al (2007) Clock and ATF4 transcription system regulates drug resistance in human cancer cell lines. Oncogene 26: 4749–4760 [DOI] [PubMed] [Google Scholar]
- 147. Martina JA, Diab HI, Brady OA, Puertollano R (2016) TFEB and TFE3 are novel components of the integrated stress response. EMBO J 35: 479–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Sachdeva MM, Claiborn KC, Khoo C, Yang J, Groff DN, Mirmira RG, Stoffers DA (2009) Pdx1 (MODY4) regulates pancreatic beta cell susceptibility to ER stress. Proc Natl Acad Sci USA 106: 19090–19095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Jin HO, Seo SK, Woo SH, Choe TB, Hong SI, Kim JI, Park IC (2009) Nuclear protein 1 induced by ATF4 in response to various stressors acts as a positive regulator on the transcriptional activation of ATF4. IUBMB Life 61: 1153–1158 [DOI] [PubMed] [Google Scholar]
- 150. Dey S, Savant S, Teske BF, Hatzoglou M, Calkhoven CF, Wek RC (2012) Transcriptional repression of ATF4 gene by CCAAT/enhancer‐binding protein beta (C/EBPbeta) differentially regulates integrated stress response. J Biol Chem 287: 21936–21949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Collier AE, Wek RC, Spandau DF (2015) Translational repression protects human keratinocytes from UVB‐induced apoptosis through a discordant eIF2 kinase stress response. J Invest Dermatol 135: 2502–2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Puri P, Mirshahi F, Cheung O, Natarajan R, Maher JW, Kellum JM, Sanyal AJ (2008) Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 134: 568–576 [DOI] [PubMed] [Google Scholar]
- 153. Lassot I, Segeral E, Berlioz‐Torrent C, Durand H, Groussin L, Hai T, Benarous R, Margottin‐Goguet F (2001) ATF4 degradation relies on a phosphorylation‐dependent interaction with the SCF(betaTrCP) ubiquitin ligase. Mol Cell Biol 21: 2192–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, Mori K, Sadighi Akha AA, Raden D, Kaufman RJ (2006) Adaptation to ER stress is mediated by differential stabilities of pro‐survival and pro‐apoptotic mRNAs and proteins. PLoS Biol 4: e374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Jiang HY, Wek RC (2005) Phosphorylation of the alpha‐subunit of the eukaryotic initiation factor‐2 (eIF2alpha) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J Biol Chem 280: 14189–14202 [DOI] [PubMed] [Google Scholar]
- 156. Milani M, Rzymski T, Mellor HR, Pike L, Bottini A, Generali D, Harris AL (2009) The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res 69: 4415–4423 [DOI] [PubMed] [Google Scholar]
- 157. Suraweera A, Munch C, Hanssum A, Bertolotti A (2012) Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol Cell 48: 242–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Frank CL, Ge X, Xie Z, Zhou Y, Tsai LH (2010) Control of activating transcription factor 4 (ATF4) persistence by multisite phosphorylation impacts cell cycle progression and neurogenesis. J Biol Chem 285: 33324–33337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Elefteriou F, Ahn JD, Takeda S, Starbuck M, Yang X, Liu X, Kondo H, Richards WG, Bannon TW, Noda M et al (2005) Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature 434: 514–520 [DOI] [PubMed] [Google Scholar]
- 160. Kawai T, Matsumoto M, Takeda K, Sanjo H, Akira S (1998) ZIP kinase, a novel serine/threonine kinase which mediates apoptosis. Mol Cell Biol 18: 1642–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Bowers AJ, Scully S, Boylan JF (2003) SKIP3, a novel Drosophila tribbles ortholog, is overexpressed in human tumors and is regulated by hypoxia. Oncogene 22: 2823–2835 [DOI] [PubMed] [Google Scholar]
- 162. Scortegagna M, Kim H, Li JL, Yao H, Brill LM, Han J, Lau E, Bowtell D, Haddad G, Kaufman RJ et al (2014) Fine tuning of the UPR by the ubiquitin ligases Siah1/2. PLoS Genet 10: e1004348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Natarajan R, Salloum FN, Fisher BJ, Smithson L, Almenara J, Fowler AA III (2009) Prolyl hydroxylase inhibition attenuates post‐ischemic cardiac injury via induction of endoplasmic reticulum stress genes. Vascul Pharmacol 51: 110–118 [DOI] [PubMed] [Google Scholar]
- 164. Lassot I, Estrabaud E, Emiliani S, Benkirane M, Benarous R, Margottin‐Goguet F (2005) p300 modulates ATF4 stability and transcriptional activity independently of its acetyltransferase domain. J Biol Chem 280: 41537–41545 [DOI] [PubMed] [Google Scholar]
- 165. Guan BJ, Krokowski D, Majumder M, Schmotzer CL, Kimball SR, Merrick WC, Koromilas AE, Hatzoglou M (2014) Translational control during endoplasmic reticulum stress beyond phosphorylation of the translation initiation factor eIF2alpha. J Biol Chem 289: 12593–12611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Jiang HY, Wek SA, McGrath BC, Lu D, Hai T, Harding HP, Wang X, Ron D, Cavener DR, Wek RC (2004) Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol 24: 1365–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Deegan S, Koryga I, Glynn SA, Gupta S, Gorman AM, Samali A (2015) A close connection between the PERK and IRE arms of the UPR and the transcriptional regulation of autophagy. Biochem Biophys Res Commun 456: 305–311 [DOI] [PubMed] [Google Scholar]
- 168. Deegan S, Saveljeva S, Gorman AM, Samali A (2013) Stress‐induced self‐cannibalism: on the regulation of autophagy by endoplasmic reticulum stress. Cell Mol Life Sci 70: 2425–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Lv S, Sun EC, Xu QY, Zhang JK, Wu DL (2015) Endoplasmic reticulum stress‐mediated autophagy contributes to bluetongue virus infection via the PERK‐eIF2alpha pathway. Biochem Biophys Res Commun 466: 406–412 [DOI] [PubMed] [Google Scholar]
- 170. Kroemer G, Marino G, Levine B (2010) Autophagy and the integrated stress response. Mol Cell 40: 280–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Dennis MD, McGhee NK, Jefferson LS, Kimball SR (2013) Regulated in DNA damage and development 1 (REDD1) promotes cell survival during serum deprivation by sustaining repression of signaling through the mechanistic target of rapamycin in complex 1 (mTORC1). Cell Signal 25: 2709–2716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Whitney ML, Jefferson LS, Kimball SR (2009) ATF4 is necessary and sufficient for ER stress‐induced upregulation of REDD1 expression. Biochem Biophys Res Commun 379: 451–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Rzymski T, Milani M, Singleton DC, Harris AL (2009) Role of ATF4 in regulation of autophagy and resistance to drugs and hypoxia. Cell Cycle 8: 3838–3847 [DOI] [PubMed] [Google Scholar]
- 174. Kazemi S, Mounir Z, Baltzis D, Raven JF, Wang S, Krishnamoorthy JL, Pluquet O, Pelletier J, Koromilas AE (2007) A novel function of eIF2alpha kinases as inducers of the phosphoinositide‐3 kinase signaling pathway. Mol Biol Cell 18: 3635–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D (2000) Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell 5: 897–904 [DOI] [PubMed] [Google Scholar]
- 176. Liu CY, Schroder M, Kaufman RJ (2000) Ligand‐independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J Biol Chem 275: 24881–24885 [DOI] [PubMed] [Google Scholar]
- 177. Szegezdi E, Logue SE, Gorman AM, Samali A (2006) Mediators of endoplasmic reticulum stress‐induced apoptosis. EMBO Rep 7: 880–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P (2007) IRE1 signaling affects cell fate during the unfolded protein response. Science 318: 944–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Lin JH, Li H, Zhang Y, Ron D, Walter P (2009) Divergent effects of PERK and IRE1 signaling on cell viability. PLoS ONE 4: e4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Hamanaka RB, Bobrovnikova‐Marjon E, Ji X, Liebhaber SA, Diehl JA (2009) PERK‐dependent regulation of IAP translation during ER stress. Oncogene 28: 910–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Hu P, Han Z, Couvillon AD, Exton JH (2004) Critical role of endogenous Akt/IAPs and MEK1/ERK pathways in counteracting endoplasmic reticulum stress‐induced cell death. J Biol Chem 279: 49420–49429 [DOI] [PubMed] [Google Scholar]
- 182. Warnakulasuriyarachchi D, Cerquozzi S, Cheung HH, Holcik M (2004) Translational induction of the inhibitor of apoptosis protein HIAP2 during endoplasmic reticulum stress attenuates cell death and is mediated via an inducible internal ribosome entry site element. J Biol Chem 279: 17148–17157 [DOI] [PubMed] [Google Scholar]
- 183. Hu J, Dang N, Menu E, De Bruyne E, Xu D, Van Camp B, Van Valckenborgh E, Vanderkerken K (2012) Activation of ATF4 mediates unwanted Mcl‐1 accumulation by proteasome inhibition. Blood 119: 826–837 [DOI] [PubMed] [Google Scholar]
- 184. Chitnis NS, Pytel D, Bobrovnikova‐Marjon E, Pant D, Zheng H, Maas NL, Frederick B, Kushner JA, Chodosh LA, Koumenis C et al (2012) miR‐211 is a prosurvival microRNA that regulates chop expression in a PERK‐dependent manner. Mol Cell 48: 353–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Baird TD, Palam LR, Fusakio ME, Willy JA, Davis CM, McClintick JN, Anthony TG, Wek RC (2014) Selective mRNA translation during eIF2 phosphorylation induces expression of IBTKalpha. Mol Biol Cell 25: 1686–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, Harding HP, Ron D (2004) Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol Cell Biol 24: 10161–10168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Hamidi T, Cano CE, Grasso D, Garcia MN, Sandi MJ, Calvo EL, Dagorn JC, Lomberk G, Urrutia R, Goruppi S et al (2012) Nupr1‐aurora kinase A pathway provides protection against metabolic stress‐mediated autophagic‐associated cell death. Clin Cancer Res 18: 5234–5246 [DOI] [PubMed] [Google Scholar]
- 188. Galehdar Z, Swan P, Fuerth B, Callaghan SM, Park DS, Cregan SP (2010) Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4‐CHOP‐mediated induction of the Bcl‐2 homology 3‐only member PUMA. J Neurosci 30: 16938–16948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm‐Breschkin J, Motoyama N et al (2007) ER stress triggers apoptosis by activating BH3‐only protein Bim. Cell 129: 1337–1349 [DOI] [PubMed] [Google Scholar]
- 190. Zou W, Yue P, Khuri FR, Sun SY (2008) Coupling of endoplasmic reticulum stress to CDDO‐Me‐induced up‐regulation of death receptor 5 via a CHOP‐dependent mechanism involving JNK activation. Cancer Res 68: 7484–7492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Xu L, Su L, Liu X (2012) PKCdelta regulates death receptor 5 expression induced by PS‐341 through ATF4‐ATF3/CHOP axis in human lung cancer cells. Mol Cancer Ther 11: 2174–2182 [DOI] [PubMed] [Google Scholar]
- 192. Teske BF, Fusakio ME, Zhou D, Shan J, McClintick JN, Kilberg MS, Wek RC (2013) CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol Biol Cell 24: 2477–2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Liu GB, Su L, Hao XX, Zhong N, Zhong DS, Singhal S, Liu XG (2012) Salermide up‐regulates death receptor 5 expression through the ATF4‐ATF3‐CHOP axis and leads to apoptosis in human cancer cells. J Cell Mol Med 16: 1618–1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Young SK, Palam LR, Wu C, Sachs MS, Wek RC (2016) Ribosome elongation stall directs gene‐specific translation in the integrated stress response. J Biol Chem 291: 6546–6558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Oyadomari S, Takeda K, Takiguchi M, Gotoh T, Matsumoto M, Wada I, Akira S, Araki E, Mori M (2001) Nitric oxide‐induced apoptosis in pancreatic beta cells is mediated by the endoplasmic reticulum stress pathway. Proc Natl Acad Sci USA 98: 10845–10850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Armstrong JL, Flockhart R, Veal GJ, Lovat PE, Redfern CP (2010) Regulation of endoplasmic reticulum stress‐induced cell death by ATF4 in neuroectodermal tumor cells. J Biol Chem 285: 6091–6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Liew CW, Bochenski J, Kawamori D, Hu J, Leech CA, Wanic K, Malecki M, Warram JH, Qi L, Krolewski AS et al (2010) The pseudokinase tribbles homolog 3 interacts with ATF4 to negatively regulate insulin exocytosis in human and mouse beta cells. J Clin Invest 120: 2876–2888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Du K, Herzig S, Kulkarni RN, Montminy M (2003) TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 300: 1574–1577 [DOI] [PubMed] [Google Scholar]
- 199. Salazar M, Lorente M, Garcia‐Taboada E, Perez Gomez E, Davila D, Zuniga‐Garcia P, Maria Flores J, Rodriguez A, Hegedus Z, Mosen‐Ansorena D et al (2015) Loss of Tribbles pseudokinase‐3 promotes Akt‐driven tumorigenesis via FOXO inactivation. Cell Death Differ 22: 131–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Byrd AE, Aragon IV, Brewer JW (2012) MicroRNA‐30c‐2* limits expression of proadaptive factor XBP1 in the unfolded protein response. J Cell Biol 196: 689–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Gupta S, Read DE, Deepti A, Cawley K, Gupta A, Oommen D, Verfaillie T, Matus S, Smith MA, Mott JL et al (2012) Perk‐dependent repression of miR‐106b‐25 cluster is required for ER stress‐induced apoptosis. Cell Death Dis 3: e333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Mendell JT (2008) miRiad roles for the miR‐17‐92 cluster in development and disease. Cell 133: 217–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Hiramatsu N, Messah C, Han J, LaVail MM, Kaufman RJ, Lin JH (2014) Translational and posttranslational regulation of XIAP by eIF2alpha and ATF4 promotes ER stress‐induced cell death during the unfolded protein response. Mol Biol Cell 25: 1411–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Deveraux QL, Takahashi R, Salvesen GS, Reed JC (1997) X‐linked IAP is a direct inhibitor of cell‐death proteases. Nature 388: 300–304 [DOI] [PubMed] [Google Scholar]
- 205. Salvesen GS, Duckett CS (2002) IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol 3: 401–410 [DOI] [PubMed] [Google Scholar]
- 206. Leon‐Annicchiarico CL, Ramirez‐Peinado S, Dominguez‐Villanueva D, Gonsberg A, Lampidis TJ, Munoz‐Pinedo C (2015) ATF4 mediates necrosis induced by glucose deprivation and apoptosis induced by 2‐deoxyglucose in the same cells. FEBS J 282: 3647–3658 [DOI] [PubMed] [Google Scholar]
- 207. Li S, Guo L, Qian P, Zhao Y, Liu A, Ji F, Chen L, Wu X, Qian G (2015) Lipopolysaccharide induces autophagic cell death through the PERK‐dependent branch of the unfolded protein response in human alveolar epithelial A549 cells. Cell Physiol Biochem 36: 2403–2417 [DOI] [PubMed] [Google Scholar]
- 208. Erguler K, Pieri M, Deltas C (2013) A mathematical model of the unfolded protein stress response reveals the decision mechanism for recovery, adaptation and apoptosis. BMC Syst Biol 7: 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Zhang T, Brazhnik P, Tyson JJ (2009) Computational analysis of dynamical responses to the intrinsic pathway of programmed cell death. Biophys J 97: 415–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Tyson JJ, Baumann WT, Chen C, Verdugo A, Tavassoly I, Wang Y, Weiner LM, Clarke R (2011) Dynamic modelling of oestrogen signalling and cell fate in breast cancer cells. Nat Rev Cancer 11: 523–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, Harding H, Novoa I, Varia M, Raleigh J et al (2005) ER stress‐regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J 24: 3470–3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C (2000) EIF2AK3, encoding translation initiation factor 2‐alpha kinase 3, is mutated in patients with Wolcott‐Rallison syndrome. Nat Genet 25: 406–409 [DOI] [PubMed] [Google Scholar]
- 213. Lin W, Lin Y, Li J, Fenstermaker AG, Way SW, Clayton B, Jamison S, Harding HP, Ron D, Popko B (2013) Oligodendrocyte‐specific activation of PERK signaling protects mice against experimental autoimmune encephalomyelitis. J Neurosci 33: 5980–5991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Mouton‐Liger F, Paquet C, Dumurgier J, Lapalus P, Gray F, Laplanche JL, Hugon J, Groupe d'Investigation du Liquide Cephalorachidien Study N (2012) Increased cerebrospinal fluid levels of double‐stranded RNA‐dependant protein kinase in Alzheimer's disease. Biol Psychiatry 71: 829–835 [DOI] [PubMed] [Google Scholar]
- 215. Chen T, Ozel D, Qiao Y, Harbinski F, Chen L, Denoyelle S, He X, Zvereva N, Supko JG, Chorev M et al (2011) Chemical genetics identify eIF2alpha kinase heme‐regulated inhibitor as an anticancer target. Nat Chem Biol 7: 610–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Stockwell SR, Platt G, Barrie SE, Zoumpoulidou G, Te Poele RH, Aherne GW, Wilson SC, Sheldrake P, McDonald E, Venet M et al (2012) Mechanism‐based screen for G1/S checkpoint activators identifies a selective activator of EIF2AK3/PERK signalling. PLoS ONE 7: e28568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Bunpo P, Dudley A, Cundiff JK, Cavener DR, Wek RC, Anthony TG (2009) GCN2 protein kinase is required to activate amino acid deprivation responses in mice treated with the anti‐cancer agent L‐asparaginase. J Biol Chem 284: 32742–32749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Keller TL, Zocco D, Sundrud MS, Hendrick M, Edenius M, Yum J, Kim YJ, Lee HK, Cortese JF, Wirth DF et al (2012) Halofuginone and other febrifugine derivatives inhibit prolyl‐tRNA synthetase. Nat Chem Biol 8: 311–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Long Y, Tsai WB, Wangpaichitr M, Tsukamoto T, Savaraj N, Feun LG, Kuo MT (2013) Arginine deiminase resistance in melanoma cells is associated with metabolic reprogramming, glucose dependence, and glutamine addiction. Mol Cancer Ther 12: 2581–2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Zhang P, McGrath BC, Reinert J, Olsen DS, Lei L, Gill S, Wek SA, Vattem KM, Wek RC, Kimball SR et al (2002) The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Mol Cell Biol 22: 6681–6688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Hu W, Hofstetter W, Wei X, Guo W, Zhou Y, Pataer A, Li H, Fang B, Swisher SG (2009) Double‐stranded RNA‐dependent protein kinase‐dependent apoptosis induction by a novel small compound. J Pharmacol Exp Ther 328: 866–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Elkin M, Miao HQ, Nagler A, Aingorn E, Reich R, Hemo I, Dou HL, Pines M, Vlodavsky I (2000) Halofuginone: a potent inhibitor of critical steps in angiogenesis progression. FASEB J 14: 2477–2485 [DOI] [PubMed] [Google Scholar]
- 223. Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D et al (2005) A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 307: 935–939 [DOI] [PubMed] [Google Scholar]
- 224. Das I, Krzyzosiak A, Schneider K, Wrabetz L, D'Antonio M, Barry N, Sigurdardottir A, Bertolotti A (2015) Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 348: 239–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Tsaytler P, Bertolotti A (2013) Exploiting the selectivity of protein phosphatase 1 for pharmacological intervention. FEBS J 280: 766–770 [DOI] [PubMed] [Google Scholar]
- 226. Tsaytler P, Harding HP, Ron D, Bertolotti A (2011) Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 332: 91–94 [DOI] [PubMed] [Google Scholar]
- 227. Bryant KF, Macari ER, Malik N, Boyce M, Yuan J, Coen DM (2008) ICP34.5‐dependent and ‐independent activities of salubrinal in herpes simplex virus‐1 infected cells. Virology 379: 197–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Lee d, Lee KS, Lee HJ, Kim d, Noh YH, Yu K, Jung HY, Lee SH, Lee JY, Youn YC et al (2010) Activation of PERK signaling attenuates Abeta‐mediated ER stress. PLoS ONE 5: e10489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Reijonen S, Putkonen N, Norremolle A, Lindholm D, Korhonen L (2008) Inhibition of endoplasmic reticulum stress counteracts neuronal cell death and protein aggregation caused by N‐terminal mutant huntingtin proteins. Exp Cell Res 314: 950–960 [DOI] [PubMed] [Google Scholar]
- 230. Holmes B, Brogden RN, Heel RC, Speight TM, Avery GS (1983) Guanabenz. A review of its pharmacodynamic properties and therapeutic efficacy in hypertension. Drugs 26: 212–229 [DOI] [PubMed] [Google Scholar]
- 231. De Gassart A, Bujisic B, Zaffalon L, Decosterd LA, Di Micco A, Frera G, Tallant R, Martinon F (2016) An inhibitor of HIV‐1 protease modulates constitutive eIF2alpha dephosphorylation to trigger a specific integrated stress response. Proc Natl Acad Sci USA 113: E117–E126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Kline CL, Van den Heuvel AP, Allen JE, Prabhu VV, Dicker DT, El‐Deiry WS (2016) ONC201 kills solid tumor cells by triggering an integrated stress response dependent on ATF4 activation by specific eIF2alpha kinases. Sci Signal 9: ra18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Dash PK, Hylin MJ, Hood KN, Orsi SA, Zhao J, Redell JB, Tsvetkov AS, Moore AN (2015) Inhibition of eukaryotic initiation factor 2 alpha phosphatase reduces tissue damage and improves learning and memory after experimental traumatic brain injury. J Neurotrauma 32: 1608–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Way SW, Podojil JR, Clayton BL, Zaremba A, Collins TL, Kunjamma RB, Robinson AP, Brugarolas P, Miller RH, Miller SD et al (2015) Pharmaceutical integrated stress response enhancement protects oligodendrocytes and provides a potential multiple sclerosis therapeutic. Nat Commun 6: 6532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Jiang HQ, Ren M, Jiang HZ, Wang J, Zhang J, Yin X, Wang SY, Qi Y, Wang XD, Feng HL (2014) Guanabenz delays the onset of disease symptoms, extends lifespan, improves motor performance and attenuates motor neuron loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neuroscience 277: 132–138 [DOI] [PubMed] [Google Scholar]
- 236. Krokowski D, Han J, Saikia M, Majumder M, Yuan CL, Guan BJ, Bevilacqua E, Bussolati O, Broer S, Arvan P et al (2013) A self‐defeating anabolic program leads to beta‐cell apoptosis in endoplasmic reticulum stress‐induced diabetes via regulation of amino acid flux. J Biol Chem 288: 17202–17213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Lin W, Bailey SL, Ho H, Harding HP, Ron D, Miller SD, Popko B (2007) The integrated stress response prevents demyelination by protecting oligodendrocytes against immune‐mediated damage. J Clin Invest 117: 448–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Wang L, Popko B, Roos RP (2014) An enhanced integrated stress response ameliorates mutant SOD1‐induced ALS. Hum Mol Genet 23: 2629–2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. D'Antonio M, Musner N, Scapin C, Ungaro D, Del Carro U, Ron D, Feltri ML, Wrabetz L (2013) Resetting translational homeostasis restores myelination in Charcot‐Marie‐Tooth disease type 1B mice. J Exp Med 210: 821–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, Li WH, Heerding DA, Minthorn E, Mencken T et al (2012) Discovery of 7‐methyl‐5‐(1‐{[3‐(trifluoromethyl)phenyl]acetyl}‐2,3‐dihydro‐1H‐indol‐5‐yl)‐7H‐p yrrolo[2,3‐d]pyrimidin‐4‐amine (GSK2606414), a potent and selective first‐in‐class inhibitor of protein kinase R (PKR)‐like endoplasmic reticulum kinase (PERK). J Med Chem 55: 7193–7207 [DOI] [PubMed] [Google Scholar]
- 241. Axten JM, Romeril SP, Shu A, Ralph J, Medina JR, Feng Y, Li WH, Grant SW, Heerding DA, Minthorn E et al (2013) Discovery of GSK2656157: an optimized PERK inhibitor selected for preclinical development. ACS Med Chem Lett 4: 964–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Jammi NV, Whitby LR, Beal PA (2003) Small molecule inhibitors of the RNA‐dependent protein kinase. Biochem Biophys Res Commun 308: 50–57 [DOI] [PubMed] [Google Scholar]
- 243. Rosen MD, Woods CR, Goldberg SD, Hack MD, Bounds AD, Yang Y, Wagaman PC, Phuong VK, Ameriks AP, Barrett TD et al (2009) Discovery of the first known small‐molecule inhibitors of heme‐regulated eukaryotic initiation factor 2alpha (HRI) kinase. Bioorg Med Chem Lett 19: 6548–6551 [DOI] [PubMed] [Google Scholar]
- 244. Robert F, Williams C, Yan Y, Donohue E, Cencic R, Burley SK, Pelletier J (2009) Blocking UV‐induced eIF2alpha phosphorylation with small molecule inhibitors of GCN2. Chem Biol Drug Des 74: 57–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Sidrauski C, Acosta‐Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, Gamache K, Gallagher CM, Ang KK, Wilson C et al (2013) Pharmacological brake‐release of mRNA translation enhances cognitive memory. eLife 2: e00498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Masuda M, Miyazaki‐Anzai S, Levi M, Ting TC, Miyazaki M (2013) PERK‐eIF2alpha‐ATF4‐CHOP signaling contributes to TNFalpha‐induced vascular calcification. J Am Heart Assoc 2: e000238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Wang A, Xu S, Zhang X, He J, Yan D, Yang Z, Xiao S (2011) Ribosomal protein RPL41 induces rapid degradation of ATF4, a transcription factor critical for tumour cell survival in stress. J Pathol 225: 285–292 [DOI] [PubMed] [Google Scholar]
- 248. Palam LR, Gore J, Craven KE, Wilson JL, Korc M (2015) Integrated stress response is critical for gemcitabine resistance in pancreatic ductal adenocarcinoma. Cell Death Dis 6: e1913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Ishizawa J, Kojima K, Chachad D, Ruvolo P, Ruvolo V, Jacamo RO, Borthakur G, Mu H, Zeng Z, Tabe Y et al (2016) ATF4 induction through an atypical integrated stress response to ONC201 triggers p53‐independent apoptosis in hematological malignancies. Sci Signal 9: ra17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. van Oosten‐Hawle P, Morimoto RI (2014) Transcellular chaperone signaling: an organismal strategy for integrated cell stress responses. J Exp Biol 217: 129–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Labbadia J, Morimoto RI (2015) The biology of proteostasis in aging and disease. Annu Rev Biochem 84: 435–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Sun J, Singh V, Kajino‐Sakamoto R, Aballay A (2011) Neuronal GPCR controls innate immunity by regulating noncanonical unfolded protein response genes. Science 332: 729–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Sundrud MS, Koralov SB, Feuerer M, Calado DP, Kozhaya AE, Rhule‐Smith A, Lefebvre RE, Unutmaz D, Mazitschek R, Waldner H et al (2009) Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science 324: 1334–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Atkins C, Liu Q, Minthorn E, Zhang SY, Figueroa DJ, Moss K, Stanley TB, Sanders B, Goetz A, Gaul N et al (2013) Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res 73: 1993–2002 [DOI] [PubMed] [Google Scholar]
- 255. Hu Y, Conway TW (1993) 2‐Aminopurine inhibits the double‐stranded RNA‐dependent protein kinase both in vitro and in vivo . J Interferon Res 13: 323–328 [DOI] [PubMed] [Google Scholar]
- 256. Zhou X, Wang R, Fan L, Li Y, Ma L, Yang Z, Yu W, Jing N, Zhu X (2005) Mitosin/CENP‐F as a negative regulator of activating transcription factor‐4. J Biol Chem 280: 13973–13977 [DOI] [PubMed] [Google Scholar]
- 257. Sayer JA, Otto EA, O'Toole JF, Nurnberg G, Kennedy MA, Becker C, Hennies HC, Helou J, Attanasio M, Fausett BV et al (2006) The centrosomal protein nephrocystin‐6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet 38: 674–681 [DOI] [PubMed] [Google Scholar]
- 258. Su N, Kilberg MS (2008) C/EBP homology protein (CHOP) interacts with activating transcription factor 4 (ATF4) and negatively regulates the stress‐dependent induction of the asparagine synthetase gene. J Biol Chem 283: 35106–35117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Ambivero CT, Cilenti L, Zervos AS (2012) ATF4 interacts with Abro1/KIAA0157 scaffold protein and participates in a cytoprotective pathway. Biochim Biophys Acta 1823: 2149–2156 [DOI] [PubMed] [Google Scholar]
- 260. St‐Arnaud R, Mandic V, Elchaarani B (2010) FIAT, the factor‐inhibiting ATF4‐mediated transcription, also represses the transcriptional activity of the bZIP factor FRA‐1. Ann N Y Acad Sci 1192: 338–343 [DOI] [PubMed] [Google Scholar]
- 261. Hai T, Curran T (1991) Cross‐family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc Natl Acad Sci USA 88: 3720–3724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Ritter B, Zschuntsch J, Kvachnina E, Zhang W, Ponimaskin EG (2004) The GABA(B) receptor subunits R1 and R2 interact differentially with the activation transcription factor ATF4 in mouse brain during the postnatal development. Brain Res Dev Brain Res 149: 73–77 [DOI] [PubMed] [Google Scholar]
- 263. Nishizawa M, Nagata S (1992) cDNA clones encoding leucine‐zipper proteins which interact with G‐CSF gene promoter element 1‐binding protein. FEBS Lett 299: 36–38 [DOI] [PubMed] [Google Scholar]
- 264. Zhu K, Jiao H, Li S, Cao H, Galson DL, Zhao Z, Zhao X, Lai Y, Fan J, Im HJ et al (2013) ATF4 promotes bone angiogenesis by increasing VEGF expression and release in the bone environment. J Bone Miner Res 28: 1870–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Ord D, Ord T (2003) Mouse NIPK interacts with ATF4 and affects its transcriptional activity. Exp Cell Res 286: 308–320 [DOI] [PubMed] [Google Scholar]
- 266. He CH, Gong P, Hu B, Stewart D, Choi ME, Choi AM, Alam J (2001) Identification of activating transcription factor 4 (ATF4) as an Nrf2‐interacting protein. Implication for heme oxygenase‐1 gene regulation. J Biol Chem 276: 20858–20865 [DOI] [PubMed] [Google Scholar]
- 267. De Angelis R, Iezzi S, Bruno T, Corbi N, Di Padova M, Floridi A, Fanciulli M, Passananti C (2003) Functional interaction of the subunit 3 of RNA polymerase II (RPB3) with transcription factor‐4 (ATF4). FEBS Lett 547: 15–19 [DOI] [PubMed] [Google Scholar]
- 268. Xiao G, Jiang D, Ge C, Zhao Z, Lai Y, Boules H, Phimphilai M, Yang X, Karsenty G, Franceschi RT (2005) Cooperative interactions between activating transcription factor 4 and Runx2/Cbfa1 stimulate osteoblast‐specific osteocalcin gene expression. J Biol Chem 280: 30689–30696 [DOI] [PubMed] [Google Scholar]
- 269. Dobreva G, Chahrour M, Dautzenberg M, Chirivella L, Kanzler B, Farinas I, Karsenty G, Grosschedl R (2006) SATB2 is a multifunctional determinant of craniofacial patterning and osteoblast differentiation. Cell 125: 971–986 [DOI] [PubMed] [Google Scholar]
- 270. Reddy TR, Tang H, Li X, Wong‐Staal F (1997) Functional interaction of the HTLV‐1 transactivator Tax with activating transcription factor‐4 (ATF4). Oncogene 14: 2785–2792 [DOI] [PubMed] [Google Scholar]
- 271. Hogan MR, Cockram GP, Lu R (2006) Cooperative interaction of Zhangfei and ATF4 in transactivation of the cyclic AMP response element. FEBS Lett 580: 58–62 [DOI] [PubMed] [Google Scholar]
- 272. Pons J, Evrard‐Todeschi N, Bertho G, Gharbi‐Benarous J, Benarous R, Girault JP (2007) Phosphorylation‐dependent structure of ATF4 peptides derived from a human ATF4 protein, a member of the family of transcription factors. Peptides 28: 2253–2267 [DOI] [PubMed] [Google Scholar]
- 273. Ampofo E, Sokolowsky T, Gotz C, Montenarh M (2013) Functional interaction of protein kinase CK2 and activating transcription factor 4 (ATF4), a key player in the cellular stress response. Biochim Biophys Acta 1833: 439–451 [DOI] [PubMed] [Google Scholar]