

Abstract
Correct orientation of cell division is considered an important factor for the achievement of normal brain size, as mutations in genes that affect this process are among the leading causes of microcephaly. Abnormal spindle orientation is associated with reduction of the neuronal progenitor symmetric divisions, premature cell cycle exit, and reduced neurogenesis. This mechanism has been involved in microcephaly resulting from mutation of ASPM, the most frequently affected gene in autosomal recessive human primary microcephaly (MCPH), but it is presently unknown how ASPM regulates spindle orientation. In this report, we show that ASPM may control spindle positioning by interacting with citron kinase (CITK), a protein whose loss is also responsible for severe microcephaly in mammals. We show that the absence of CITK leads to abnormal spindle orientation in mammals and insects. In mouse cortical development, this phenotype correlates with increased production of basal progenitors. ASPM is required to recruit CITK at the spindle, and CITK overexpression rescues ASPM phenotype. ASPM and CITK affect the organization of astral microtubules (MT), and low doses of MT‐stabilizing drug revert the spindle orientation phenotype produced by their knockdown. Finally, CITK regulates both astral‐MT nucleation and stability. Our results provide a functional link between two established microcephaly proteins.
Keywords: astral microtubules, asymmetric division, microcephaly, mitotic spindle orientation, neurogenesis
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Neuroscience; Cell Cycle
Introduction
The orientation of cell division is carefully controlled in both embryonic and adult tissues, in which it may regulate cell fate, generate tissue shape, and maintain normal histological architecture 1, 2, 3. Studies conducted over the last two decades have established that this process may play an important role to regulate the delicate balance between proliferation and differentiation that underlies normal brain development, with particular regard to the generation of a normal number of cortical neurons 2, 4, 5. During embryogenesis, the cerebral cortex is first composed of a single layer of neuroepithelial (NE) cells, which initially expand through symmetric divisions 6, 7, 8, 9. As development progresses, NE progenitors give rise to radial glia (RG) cells that may further expand by dividing symmetrically or may switch to asymmetric division, producing a self‐renewing progenitor and a daughter cell committed to differentiation 5, 10, 11, 12. A characteristic feature of proliferative divisions of NE and RG cells is that cleavage occurs perpendicular to the ventricular surface of the neuroepithelium, while the switch from symmetric to asymmetric divisions is accompanied by a deviation of the cleavage plane 13, 14, 15. Oriented cell division is achieved through the proper positioning of the mitotic spindle 16, 17, 18, which depends on the formation of molecular links between actin‐rich cell cortex and astral microtubules (MT) emanating from centrosome‐derived spindle poles 15, 19, 20. Indeed, either loss of doublecortin (DCX), which destabilizes MT 21, or mutations of LIS1, NDE1, and NDEL1, which disrupt dynein–dynactin function at the cell cortex 22, 23, 24, randomize the mitotic spindle in NE progenitors and lead to their early exhaustion. A similar depletion of progenitor cells, correlated with randomized spindle orientation, can be produced by disruption of proteins involved in centrosome function 25, 26, 27, 28. Whether the relationship between spindle orientation and cell fate choice in cortical development is causal or only correlative is currently debated, on the basis of recent studies suggesting that spindle orientation may not be essential for cortical neurogenesis 26, 29, 30. Mutation of many proteins contributing to centriole biogenesis, centrosome maturation, and spindle organization 31, 32 has been associated with human primary microcephaly (MCPH). MCPH is characterized by reduced head circumference, accompanied by relatively preserved brain architecture, resulting in mild‐to‐moderate intellectual disability and few associated symptoms 33, 34. At least thirteen MCPH loci (MCPH1–MCPH13) have been mapped to date 35, 36, 37, and it has been established that most of the encoded proteins are capable of localizing at the centrosome, at the spindle poles, or at the spindle. Nevertheless, our understanding of how these proteins may affect spindle orientation and cell fate determination is still very limited. ASPM (abnormal spindle‐like microcephaly‐associated, MCPH5) is the most frequently mutated gene in MCPH 28, 38. ASPM is a conserved protein that associates with the MT minus ends, is recruited at the spindle poles during mitosis, and controls spindle MT organization, spindle function, and cytokinesis from insects to mammals 39, 40, 41. In mammals, NE cells with reduced ASPM expression fail to orient the mitotic spindle perpendicular to the ventricular surface of the neuroepithelium and show increased frequency of asymmetric divisions, with a reduction of the pool of neuronal precursors 25, 42. Recent evidence indicates that ASPM may control cell fate choice by regulating the activity of Cdk2/cyclin E complex 43. However, it is presently unknown how ASPM regulates spindle orientation. Previous studies 44 showed that ASPM physically interacts with the cytokinesis regulator citron kinase (CITK) 45, 46. CITK is a conserved protein prominently localized at the cleavage furrow and at the midbody of mitotic cells 46, 47 and is involved in abscission control at the end of cytokinesis 48, 49. In rodents, CITK inactivation leads to severe microcephaly and lethal epilepsy 45, 50. This phenotype is believed to result from apoptosis due to cytokinesis failure. Delayed metaphase–anaphase transition in CITK‐deficient neuronal progenitors has also been described 51. Although ASPM and CITK are both associated with microcephaly and may form a complex in HeLa cells and in developing neural tissue 44, it remained unclear whether their interaction could have functional relevance.
In this report, we show that in addition to its role in cytokinesis, CITK plays a phylogenetically conserved role in the control of mitotic spindle orientation, by promoting the nucleation and stabilization of astral MT. Moreover, we show that CITK is recruited to the spindle by ASPM and that CITK overexpression rescues the spindle orientation defect elicited by ASPM knockdown. These results provide new insight into the mechanisms by which ASPM loss may cause microcephaly and suggest that a spindle orientation defect may contribute to the CITK microcephaly phenotype.
Results and Discussion
CITK is a phylogenetically conserved determinant of spindle orientation
In light of the previously reported association between CITK and ASPM 44 and of the role played by ASPM in regulating spindle orientation 25, 39, we asked whether CITK may also play a role in controlling spindle orientation, besides promoting abscission 49. To address this question, we first examined whether CITK contributes to maintaining mitotic spindle perpendicular to the apical–basal axis of mouse NE progenitors. To do so, we immunostained fixed cryosections of E14.5 mice neocortices for spindle‐pole protein γ‐tubulin and DNA, and we measured the angle formed by the cleavage plan of apical progenitors (AP) with the apical surface of cortex 13, 14, 25, 30 (Fig 1A). This angle was defined as vertical when it was comprised between 76° and 90° and oblique when it was in the range from 0 to 75°. Most AP (75%) divide vertically in CitK+/+ cortices, while vertical divisions occurred in only 24% of AP in CitK−/− mice (Fig 1B), indicating that spindle orientation is perturbed by CITK loss.
Figure 1. CITK controls mitotic spindle orientation in developing mouse and fly brains.
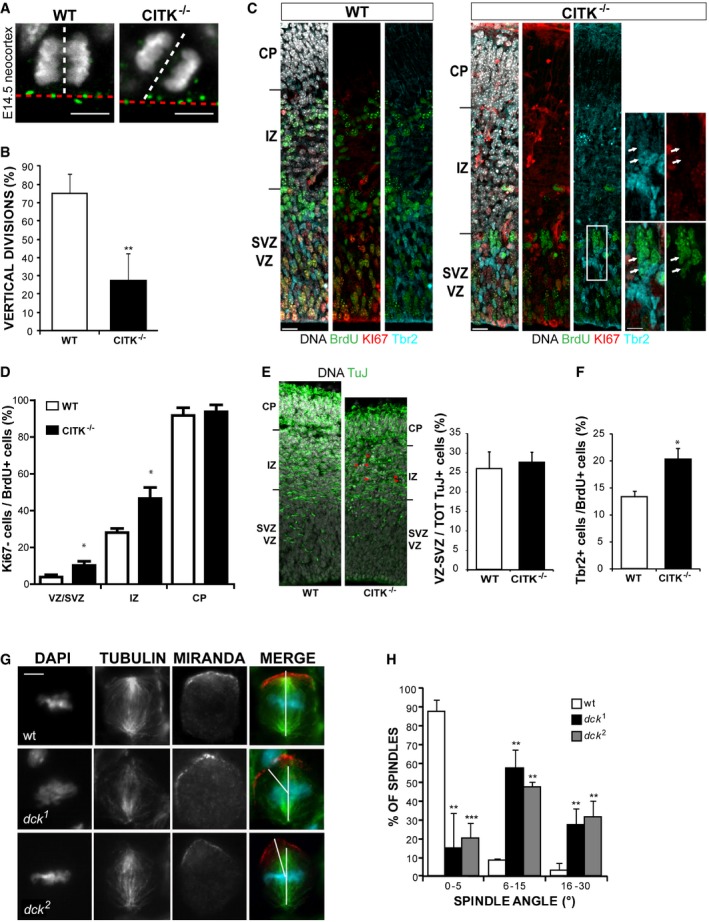
- WT and CITK−/− embryonic (E14.5) mouse cerebral cortex was stained for γ‐tubulin (green) and DNA (gray). The ventricular plane is marked by a red dashed line, and the spindle axis of apical progenitors is indicated by a white dashed line. The angle between these two lines represents the mitotic angle. Scale bars, 5 μm.
- Quantification of vertical divisions of apical progenitors in WT and CITK−/− mice. n = 3 per each genotype.
- Pregnant CITK+/− females, crossed with CITK+/− males, were injected with BrdU at E13.5; 24 h later, WT and CITK−/− embryonic (E14.5) mouse cerebral cortex was fixed in 4% PFA, cryosectioned, and stained for Ki67 (red), BrdU (green), Tbr2 (blue), and DAPI (gray). The different cortical regions are indicated: CP, cortical plate; IZ, intermediate zone; SVZ, subventricular zone; VZ, ventricular zone. Scale bars, 10 μm. White arrows in the inset indicate two Ki67− cells positive for BrdU and Tbr2.
- Quantification of cell cycle exit (ratio of cells BrdU+/Ki67−) in the proliferative regions (VZ/SVZ) and in the neuronal layers (IZ and CP) of sections prepared as in panel (C) (n = 4 per each genotype).
- Sections prepared as in (C) were stained for TUBB3 (TuJ) to reveal post‐mitotic neurons and with DAPI. Red arrowheads indicate apoptotic neurons (defined by pyknotic nuclei). Quantification of neuronal distribution in VZ/SVZ versus total neurons population is shown on the right.
- Quantification of the percentage of Tbr2‐positive cells in the BrdU‐positive population of the same sections analyzed for panel (D) (n = 4 per each genotype).
- Neuroblasts of wild‐type and dck alleles immunostained for α‐tubulin and Miranda. Note that whereas in wild‐type cells there is a tight coupling of the mitotic spindle with the polarity axis, in dck alleles the spindle shows a more oblique orientation with respect to Mira crescent. Scale bar, 5 μm.
- Distribution of spindle angle amplitude (°) in neuroblasts of wild‐type and ck alleles (n = 58).
To evaluate whether this phenotype correlates with increased commitment to differentiation, we pulse‐labeled E13.5 embryos with BrdU and analyzed how many cells had left the cell cycle 24 h later, measuring the percentage of cells positive for BrdU, but negative for the cell cycle marker Ki67 52. A significant increase in BrdU+/Ki67− cells was observed in the proliferative regions and in the intermediate zone (Fig 1C and D), suggesting a shift from proliferative to differentiative divisions. Indeed, the increase in cells exited from cell cycle in these regions could not be explained by neuron migration defects, because the percentage of neurons in VZ/SVZ relative to the total neuron population is the same in WT and CITK−/− mice (Fig 1E). Interestingly, the percentage of BrdU+/Tbr2+ cells in the proliferative regions was significantly increased (Fig 1F). In contrast, the total number of Tbr2+ cells was not different in control and knockout cortices (55.4 ± 4.6 and 52.2 ± 4.1 per 100‐μm‐wide cortical column, respectively). It must be noted that the number of neurons produced in CITK−/− may be strongly underestimated, because of their apoptotic death (Fig 1E). Indeed, we observed a strong absolute reduction of BrdU‐positive cells in the neuronal layers of CitK−/− mice (Fig 1C), consistent with the previous finding that in this model apoptosis occurs especially in post‐mitotic neuroblasts and neurons 53. Altogether, these results indicate that CitK−/− developing cortex is characterized by increased cell cycle exit and increased generation of basal progenitors.
Since the sequence of CITK is well conserved between Drosophila and mammals, and since the loss of CITK in Drosophila produces a cytokinesis‐failure phenotype remarkably similar to the phenotype detected in mammalian cells 47, 54, 55, 56, we asked whether the role of CITK in spindle orientation is conserved as well. To address this question, we analyzed neuroblast (NB) divisions in larval brains from individuals homozygous for either dck 1 or dck 2, two presumptive null alleles at the locus encoding the Drosophila orthologue of CITK 47. Drosophila NBs are stem cells that divide asymmetrically, to give rise to another NB and to a smaller ganglion mother cell (GMC) committed to differentiation. To ensure a correct asymmetric division, NB spindle must be aligned to the cell polarity axis determined by the differential apico‐basal concentration of several proteins. The basal cortex is enriched in proteins that are preferentially segregated into the GMC at the end of division and whose localization is in turn mediated by a large multiprotein complex that concentrates at the apical cortex 57. We immunostained wild‐type and dck mutant larval brains for tubulin and for the basal marker Miranda (Mira) and measured the angle between a line connecting the two spindle poles and a line bisecting the crescent formed by Mira in metaphase NBs (Fig 1G). The angle ranged between 0° and 5° in 88% of control NBs (Fig 1H), indicating a tight coupling of the mitotic spindle with the polarity axis. Conversely, although both dck 1 and dck 2 mutant NBs consistently displayed well‐formed Mira crescents, the majority of the spindles showed more oblique orientations, ranging from 6° to 45° (n = 33 for dck1, and 45 for dck2, respectively; Fig 1H).
These results indicate that besides its role in abscission control, CITK plays a phylogenetically conserved role earlier in mitosis, to ensure correct positioning of the mitotic spindle.
To better characterize this function, we resorted to HeLa cells, which are sensitive to CITK depletion 49, 58 and have extensively been used to study spindle positioning mechanisms 59. We depleted CITK in these cells by RNAi using validated sequences 49 (Fig EV1A) and analyzed cell division through phase‐contrast time‐lapse microscopy. Interestingly, divisions of cells treated with CITK‐specific siRNAs were misoriented, with the two forming daughters partially overlapping rather than being adjacent and with one daughter cell dividing outside of the focal plan, thus delaying adherence to the substrate (Fig 2A–C and Movies EV1 and EV2). We then analyzed, in fixed samples, the angles formed by metaphase spindles with the culture dish. CITK‐depleted cells displayed angles distribution skewed toward high values and significant increase in angles average (Fig 2D and E), whereas spindle length was not affected by CITK depletion (Fig EV4A). A similar phenotype was observed by inducing CITK depletion with a second, independent siRNA sequence (Fig EV1B and C). The phenotype was rescued by restoring CITK levels through the expression of an RNAi‐resistant construct (Fig EV1C), further confirming that it is due to CITK depletion rather than to off‐target effects of the siRNA sequences used. The phenotype observed in CITK‐depleted cells was similar to the phenotype produced by ASPM depletion 39 (Fig 2F). Moreover, the defect on mitotic spindle orientation caused by CITK and ASPM simultaneous knockdown was the same as single knockdown (Fig 2F), indicating that these two proteins are on the same pathway.
Figure EV1. Specificity of the CITK‐RNAi experiments.
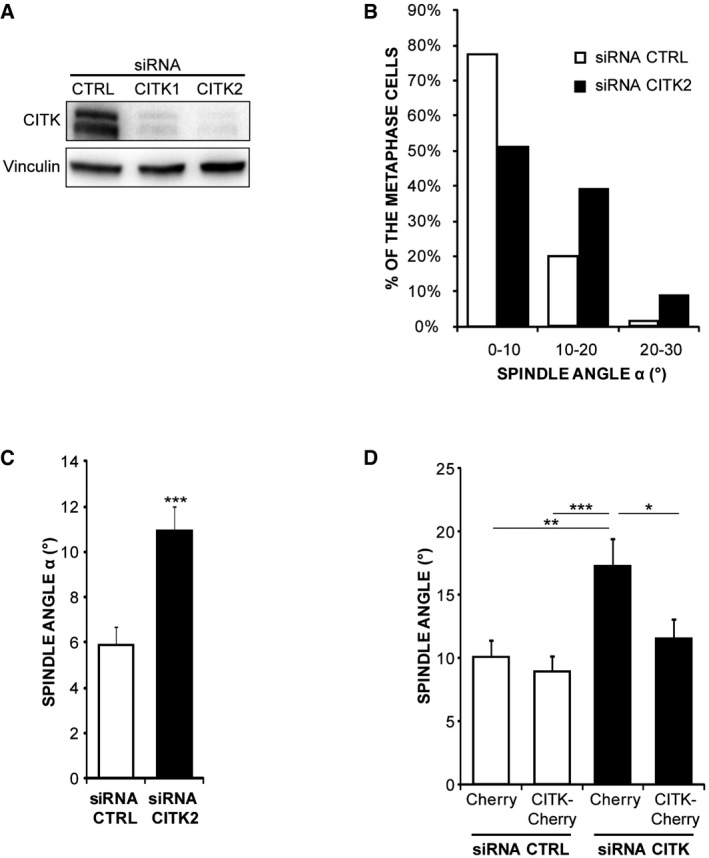
- Western blot analysis performed with the indicated antibodies on lysates of HeLa cells transfected with control or with two different CITK‐specific siRNAs (CITK1 and CITK2). The results of experiments shown in the main figures were obtained with the CITK1 sequence.
- Distribution of spindle angles (°) in HeLa cells depleted of CITK using the CITK2 sequence or transfected with control sequence. The values represent the angles between the axis crossing the two poles of metaphase spindles and the coverslip (n ≥ 150 cells in three independent experiments).
- Quantification of average and distribution of spindle angles (°) in HeLa cells depleted of CITK using the CITK2 (n ≥ 50 cells in three independent experiments).
- Quantification of angles average in HeLa cells transfected with either control or CITK1 siRNAs and cotransfected with expression plasmids expressing mCherry alone or fused to RNAi‐resistant (mouse) CITK wild‐type sequence (n ≥ 50 cells in three independent experiments).
Figure 2. CITK controls mitotic spindle orientation in HeLa cells.
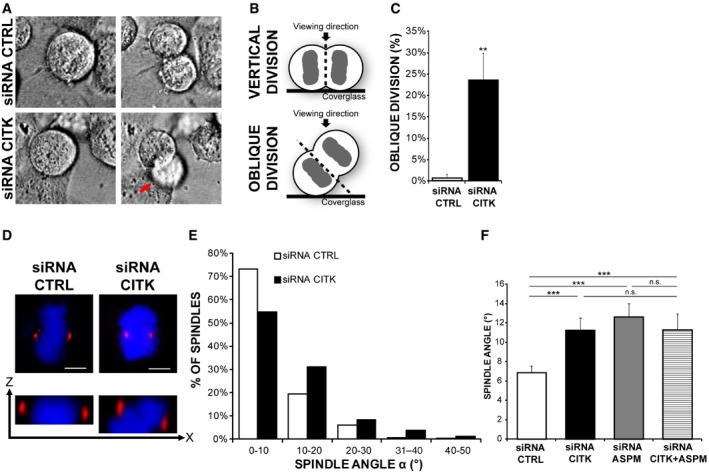
- Selected frames from time‐lapse imaging experiments (see Movies EV1 and EV2) showing two dividing cells transfected with either control (CTRL) or CITK‐specific siRNAs, respectively. Note one of the two daughter cells in the lower panel (red arrow) going out of focus as a consequence of oblique division.
- Graphical representation of vertical and oblique divisions. The cleavage plane is indicated by a black dashed line.
- Quantification of divisions showing uneven timing of daughter cell flattening onto the substrate after mitosis (oblique division) in CITK‐siRNA‐treated HeLa cells compared to control (n > 50 cells, three independent experiments).
- Control or CITK‐depleted cells were immunostained for γ‐tubulin (red) and DNA (blue) and imaged in z (0.3‐μm‐thick sections). Upper panel: maximum‐intensity projections of confocal z‐stacks are shown. Lower panel: cross section (XZ) through the two poles of the same cell. Scale bars, 5 μm.
- Distribution of spindle angles (°) in control and in CITK‐depleted cells. The values represent the angles between the axis crossing the two poles of metaphase spindles and the coverslip (n ≥ 150 cells, six independent experiments).
- Quantification of spindle angles average in control cells and in cells depleted of CITK or ASPM or codepleted of the two proteins (n ≥ 150 cells, in at least three independent experiments).
Figure EV4. Loss of CITK does not alter spindle length or symmetry.
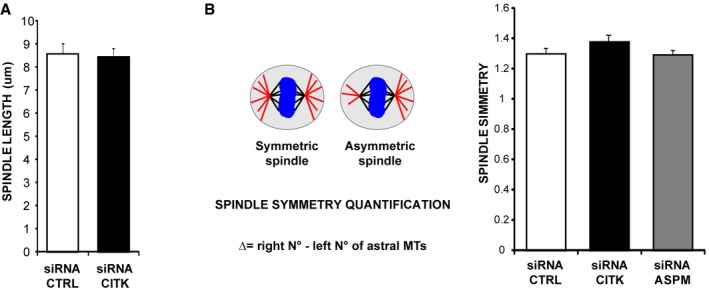
- Quantification of spindle length in HeLa cells transfected with control sequence or with CITK‐specific siRNA.
- Quantification of spindle symmetry in HeLa cells transfected with control sequence or with CITK‐specific siRNA.
Altogether, these results show that CITK is a conserved determinant of spindle orientation and demonstrate that it acts in mitosis even before becoming enriched at the cleavage furrow and midbody during cytokinesis.
CITK is associated with mitotic spindle through ASPM
Several proteomic studies reported that CITK is associated with the mitotic spindle 60, 61, 62, but this localization was not confirmed by other techniques. Previous immunolocalization studies have shown that CITK is localized to the nucleus in interphase cells, accumulates in the cytoplasm before anaphase, and becomes enriched at the cleavage furrow and at the midbody during cytokinesis 46, 63. A similar pattern was detected by live cell imaging in HeLa cells expressing physiological levels of GFP‐tagged CITK from a stably integrated BAC transgene 64, 65, 66 (Movie EV3). As expected, during mitosis the protein appears to be evenly distributed in the cytoplasm and no enrichment is visible at the spindle or at the cell cortex (Movie EV3).
Although these data confirmed the previous reports 46, 63, they did not exclude the possibility that a pool of the protein may associate with mitotic spindle. To address this issue, we performed mild detergent extraction before fixation, which removes cytosolic proteins and facilitates the visualization of cytoskeleton‐associated proteins. Under these conditions, immunofluorescence with anti‐CITK antibodies detected a clear enrichment of the protein on both the spindle and the spindle poles, as it partially overlapped with the γ‐tubulin signal (Fig 3A). This signal disappears in cells treated with CITK siRNA (Fig 3A). Similar results were obtained using anti‐GFP antibodies in HeLa cells expressing GFP‐tagged CITK from a BAC transgene 66 (Fig 3B). Specific association of CITK with spindle poles was further validated through biochemistry, as we found that CITK is enriched in centrosomal preparations obtained from HeLa cells synchronized in metaphase (Fig 3C). In addition, we observed that the signals of both CITK endogenous protein and CITK‐GFP extensively overlap with ASPM immunoreactivity at the spindle poles (Fig 3D and E).
Figure 3. CITK is associated with mitotic spindle poles through ASPM .
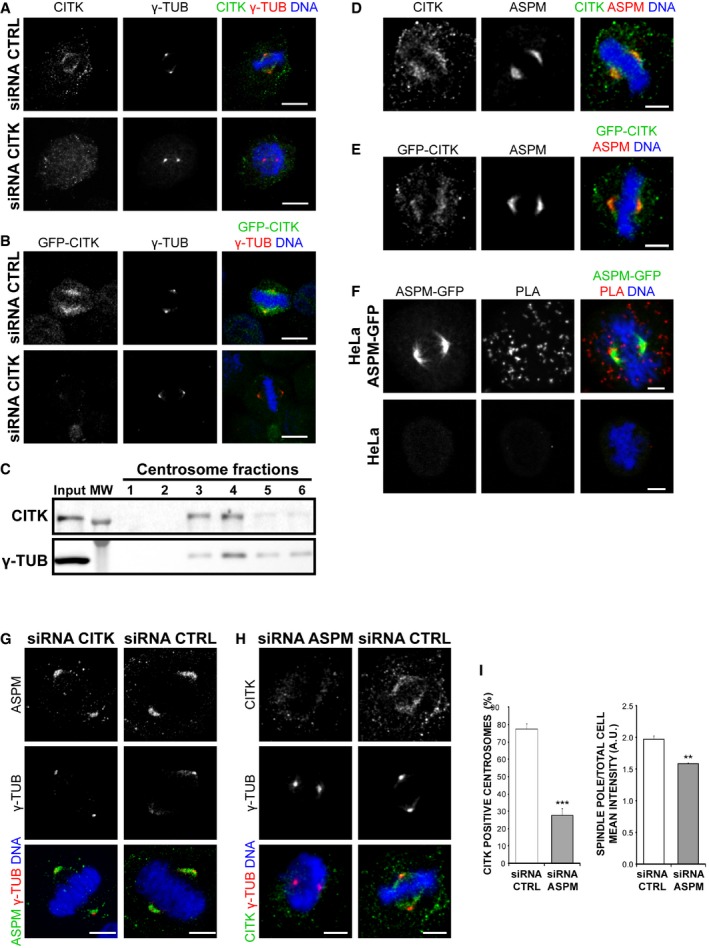
- HeLa cells pre‐extracted for 1 min with 0.5% Triton X‐100 in PHEM buffer and immunostained for CITK (green), γ‐tubulin (red), and DNA (blue).
- HeLa cells expressing CITK‐GFP (green) from a BAC transgene treated as in (A) and immunostained for γ‐tubulin (red) and DNA (blue).
- Western blot of centrosome‐containing fractions from HeLa cells, showing that CITK copurifies with γ‐tubulin.
- HeLa cells pre‐extracted 1 min with 0.5% Triton X‐100 in PHEM buffer and immunostained for CITK (green), ASPM (red), and DNA (blue).
- HeLa cells expressing CITK‐GFP treated as in (A) and immunostained for GFP (green), ASPM (red), and DNA (blue).
- Close physical proximity between CITK and ASPM, revealed by PLA on ASPM‐GFP‐expressing HeLa cells, incubated with GFP and CITK antibodies and developed with PLA‐specific secondary reagents.
- Control or CITK‐depleted cells immunostained for ASPM (green), γ‐tubulin (red), and DNA (blue).
- Control or ASPM‐depleted cells treated as in (A) and immunostained for CITK (green), γ‐tubulin (red), and DNA (blue).
- Quantification of CITK‐positive centrosomes and of the ratio of CITK spindle pole intensity versus total cell mean intensity, in control and ASPM‐depleted cells (n ≥ 75 cells, four independent experiments). Data presented are means ± SEM. ***P < 0.001; **P < 0.01. Statistical significance was assessed using a two‐tailed Student's t‐test.
To confirm the physical interaction between CITK and ASPM, we performed in situ proximity ligation assay (PLA) which can reveal whether two proteins are at a distance < 40 nm 67. In mitotic cells expressing physiological levels of ASPM‐GFP 66 and immunostained for GFP and CITK, we observed a strong signal, which was completely absent in normal HeLa cells, not expressing tagged ASPM (Fig 3F). Fluorescence dots were observed throughout the cytoplasm and in proximity of spindle and spindle poles (Fig 3F). Since these cells were not pre‐extracted, the result indicates that CITK interacts with both the cytoplasmic ASPM pool and spindle‐associated ASPM.
We then examined which CITK regions are involved in the interaction with ASPM. Based on the previously shown interaction between CITK and ASPM C‐terminal region (CTR) 44, we tested the ability of ASPM‐CTR to coimmunoprecipitate with different myc‐CITK fusion proteins (Fig EV2A), including kinase‐active (FL) or kinase‐dead (KD) full‐length CITK, citron‐N (CN), which is a brain‐specific isoform of CITK completely lacking the kinase domain 68, and the N‐terminal and the C‐terminal halves of CITK (CITK‐Nt and CITK‐Ct, respectively). We found that ASPM‐CTR can bind all these proteins (Fig EV2B), suggesting that it may interact, directly or indirectly, with different regions of CITK, independently from its kinase activity.
Figure EV2. Analysis of CITK regions involved in the interaction with ASPM.
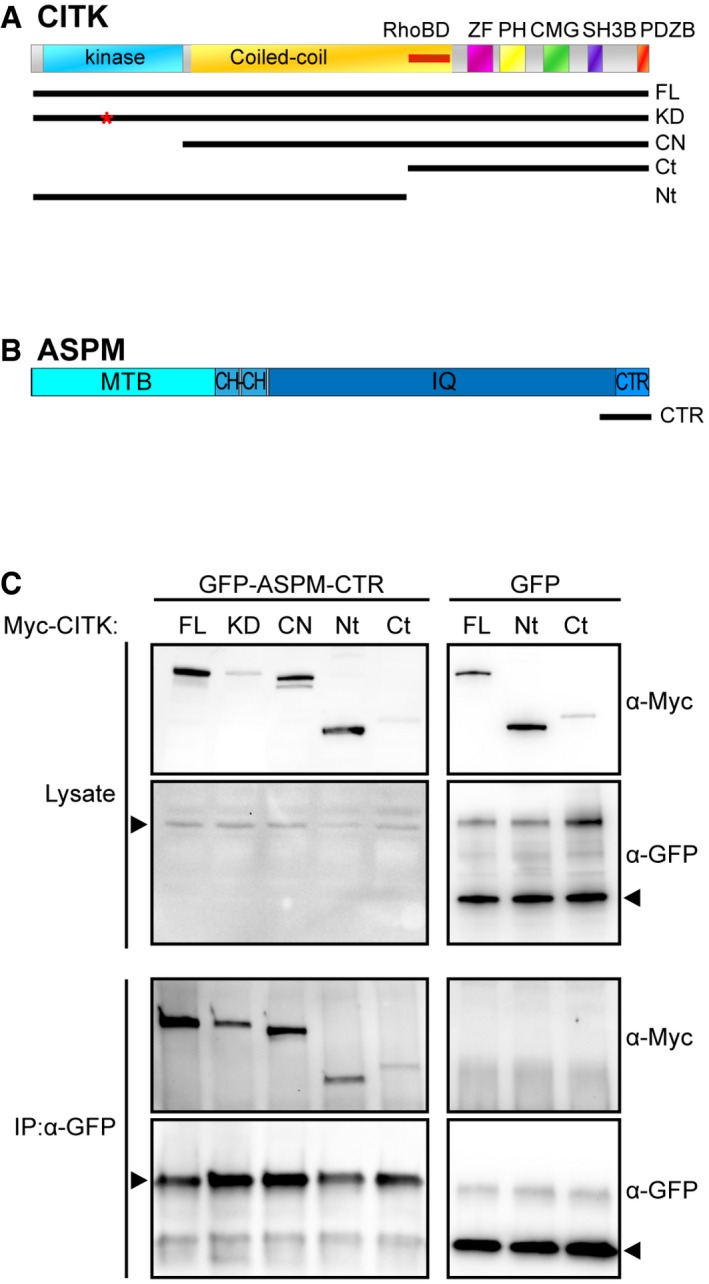
- Representation of domain structure of CITK and its fragments: Ser/Thr kinase domain (kinase), coiled‐coil region (Coiled‐coil), Rho‐binding domain (RhoBD), type‐2 zinc finger (ZF), pleckstrin homology domain (PH), CMG domain (CMG), proline‐rich, putative SH3 binding domain (SH3B), PDZ binding domain (PDZB); full‐length CITK (FL), kinase‐dead full‐length CITK (KD), citron‐N (CN), and the N‐terminal (Nt) and the C‐terminal halves of CITK (Ct).
- Representation of domain structure of ASPM and CTR fragment: putative microtubule binding domain (MTB), calponin homology domain (CH), calmodulin binding domains (IQ), and the C‐terminal region (CTR).
- The indicated Myc‐CITK fusion constructs were cotransfected with ASPM‐CTR‐GFP in HEK293T. After 48 h, immunoprecipitations were performed from total cell lysates using anti‐GFP antibody and the immunoprecipitated proteins were revealed by Western blotting for the GFP and Myc epitopes. The results shown are representative examples of at least three different experiments.
We next analyzed the localization of CITK in ASPM‐depleted cells and vice versa. While the localization pattern of ASPM was not affected by CITK‐RNAi (Fig 3G), the percentage of cells with CITK‐positive spindle poles and the mean intensity of the CITK at the spindle poles of positive cells were significantly decreased by ASPM depletion (Fig 3H and I). These results confirm the interaction between CITK and ASPM in mitotic cells and indicate that a pool of CITK is recruited through ASPM at the spindle and spindle poles, suggesting that CITK may act downstream of ASPM.
CITK regulates astral‐MT organization downstream of ASPM
Proper orientation of mitotic spindle during mitosis requires a dynamic connection of astral MT to the cortical cytoskeleton through the MT motor complex dynein/dynactin, which is localized at a cortical crescent by cortical adaptor proteins such as NuMA 19, 69, 70, 71. We therefore analyzed by immunofluorescence whether CITK absence alters the localization of these proteins at metaphase. No significant changes were observed in the localization pattern of dynein or NuMA (Fig EV3A–C), while the percentage of cells with a dynactin (p150) crescent was slightly increased (Fig EV3A–C), indicating that the phenotype induced by CITK depletion cannot be explained by defective localization of these proteins. Moreover, no differences were observed in the fluorescence intensity of these proteins at spindle poles (Fig EV3A–C).
Figure EV3. Loss of CITK does not impair the localization of the dynein/dynactin motor complex and NuMA.
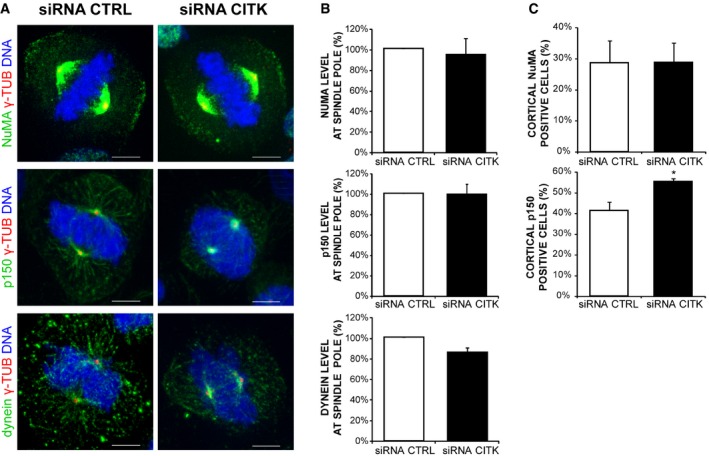
- Control and CITK‐depleted cells were immunostained for γ‐tubulin (red), DNA (blue), and NuMA, dynactin (p150), or dynein (green). Images represent maximum‐intensity projections of confocal z‐stacks. Scale bars, 5 μm.
- Quantification of fluorescence intensity at spindle poles of NuMA, dynactin (p150), or dynein in HeLa cells transfected with either CTRL or CITK siRNA (n > 150 cells, five independent experiments).
- Percentage of mitosis with cortical localization of NuMA or dynactin (p150) in HeLa cells transfected with either CTRL or CITK siRNA. The results shown are representative examples of at least three different experiments.
In contrast, if compared to controls, CITK‐depleted cells displayed a significant reduction in the number and length of astral MT (Fig 4A and B). A similar phenotype was observed in the case of ASPM depletion (Fig 4A and B). It has been reported that asymmetric neurogenic division is associated with spindle‐size asymmetry, negatively controlled by the Wnt pathway 72. Since ASPM has been shown to act upstream of the Wnt pathway to trigger symmetric divisions of cortical progenitors 73, we assessed whether CITK loss of function impacts spindle symmetry. We found that the difference (Δ) in the number of astral MT on the two centrosomes of a mitotic cell did not change when either CITK or ASPM was knocked down (Fig EV4B). These data further indicate that CITK and ASPM may act through similar mechanisms and reveal that they influence astral‐MT organization. To functionally test the possibility that CITK may work downstream of ASPM, we transfected ASPM‐depleted cells with CITK overexpression constructs. Remarkably, the overexpression of CITK reverted the reduction of astral MT (Fig 4C) and the average increase in mitotic angle (Fig 4D) elicited by ASPM depletion back to control values. No rescue was observed when ASPM‐depleted cells were cotransfected with a kinase‐dead mutant of CITK (CKD) or with CITN (Fig 4D), indicating that CITK catalytic activity may be required for spindle orientation. These results suggest that when CITK is expressed at physiological levels, the presence of ASPM is a limiting factor for its recruitment to spindle poles and for its activity on spindle orientation, while CITK overexpression overcomes the requirement for ASPM. A possible explanation for the latter phenomenon could be provided by the finding that CITK can bind to microtubules 48 through many microtubule‐binding proteins, including KIF14, MKLP1, PRC1 58, KIF4A, and KIF10 65. Since the localization of at least some of these proteins during metaphase may partially overlap with ASPM localization 74, it is conceivable that at high CITK levels the binding with these proteins, or other unknown partners, may compensate for ASPM loss.
Figure 4. CITK regulates astral‐MT and spindle orientation downstream of ASPM and through a kinase‐dependent mechanism.
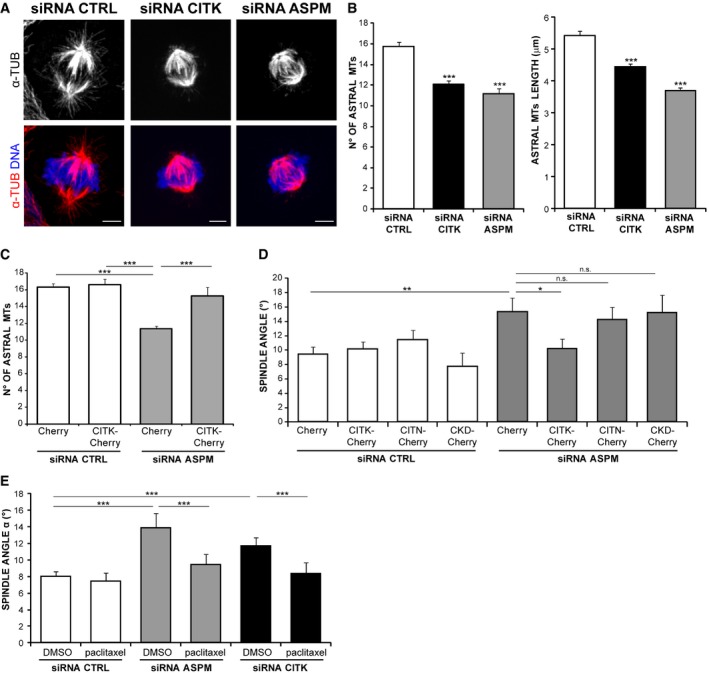
- Control, CITK‐depleted, or ASPM‐depleted HeLa cells immunostained for α‐tubulin (red) and DNA (blue). Maximum‐intensity projections of confocal z‐stacks are shown. Scale bars, 5 μm.
- Quantification of astral‐MT number and maximal length in HeLa cells transfected with CTRL, CITK, or ASPM siRNA (n > 130 cells in three independent experiments).
- Quantification of the rescue of astral‐MT number after CITK overexpression in ASPM‐depleted HeLa cells (n > 50 cells in three independent experiments).
- Quantification of spindle orientation rescues after overexpression of CITK, CITN (lacking the kinase domain), or CKD (citron kinase‐dead) mutants in ASPM‐depleted HeLa cells (n > 50 cells in three independent experiments).
- Synchronized HeLa cells transfected with control, ASPM‐specific, or CITK‐specific siRNAs treated with DMSO or with 250 pM paclitaxel (Taxol) 30 min before immunostaining for α‐tubulin. Spindle angles were measured as above (n > 100 cells in six independent experiments).
Next, we treated HeLa cells, depleted for either protein, with low doses of the MT‐stabilizing drug paclitaxel, which are known to specifically affect the most dynamic pool of MT 75. In both cases, the average of mitotic spindle angles was restored to control values (Fig 4E). Altogether, these results indicate that ASPM and CITK regulate spindle orientation through astral‐MT organization and that CITK is a downstream player of ASPM.
CITK regulates astral‐MT length, stability, and nucleation
To investigate which aspects of the dynamic behavior of MT are affected by CITK, we performed RNAi in HeLa cells stably expressing the MT + TIP protein EB3, fused with the fluorescent Tomato tag, to track the plus ends of growing MT by time‐lapse fluorescence microscopy 20 (Fig 5A). Quantitative analysis of the corresponding movies allows estimation of the different MT dynamic parameters, as it has previously been described 20, 76 (Fig 5B). This technique revealed that CITK loss leads to decreased MT stability and maximal distance traveled from the spindle pole toward the cell cortex, while no differences were observed in MT growth speed (Fig 5C). Moreover, we observed a decrease in nucleation frequency (Fig 5C).
Figure 5. Loss of CITK decreases astral‐MT length, stability, and nucleation.
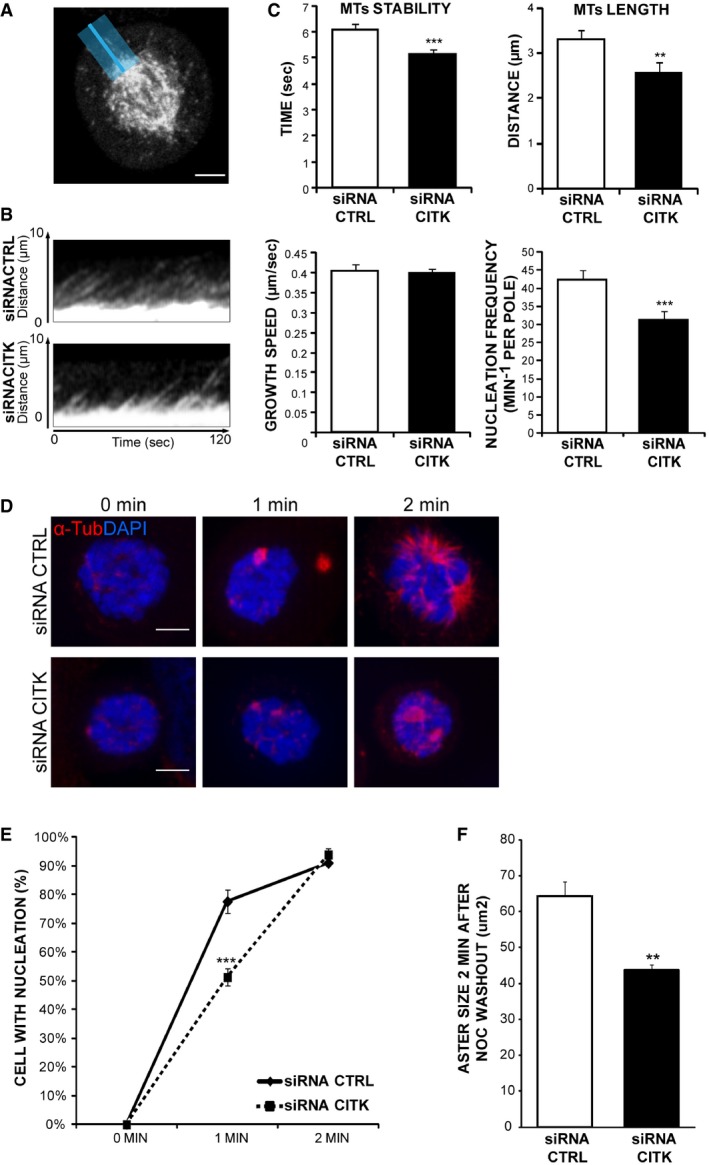
- Exemplar image of EB3–tdTomato HeLa cell used for kymograph analyses. To assess astral‐MT number and dynamics, a line (blue) was added to images from the pole to the cortex.
- Kymographs of pole (bottom)‐to‐cortex (top) line scans of control and CITK‐depleted EB3–tdTomato HeLa cells. The maximum projection of a 20‐pixel‐wide line (exemplified above) was plotted at 500‐ms time intervals from left to right. MT growth results in diagonal lines.
- Quantification of astral‐MT dynamics. MT stability, length, growth speed (n > 250 MT, 30 cells), and nucleation frequency were calculated (n > 30 cells, three independent experiments).
- Immunofluorescence microscopy images showing MT growth at mitotic spindle poles 0, 1, and 2 min after nocodazole washout. Control and CITK‐depleted cells were immunostained for α‐tubulin (red) and DNA (blue).
- Percentage of cells showing detectable MT nucleation (aster size > 1 μm) 0, 1, and 2 min after nocodazole washout (n > 100 cells, four independent experiments).
- Quantification of aster size 2 min after nocodazole washout (n > 100 cells, four independent experiments).
To better address the latter phenotype, we resorted to MT growth assay after cold‐induced depolymerization, by incubating cells at 4°C for 30 min. One minute after switching from 4°C to 37°C, the number of cells showing detectable MT growth (aster size > 1 μm) was significantly decreased in CITK‐depleted cells (Fig 5D and E). Moreover, although at 2 min the value of this parameter was comparable to the control (Fig 5E), the aster size was still significantly smaller in CITK‐depleted cells (Fig 5F). Altogether, these results indicate that CITK loss decreases MT nucleation and MT stability in mitotic cells.
In summary, in this study we have provided evidence that ASPM controls spindle orientation by regulating the dynamics of astral MT and that CITK is a critical downstream partner of ASPM for this activity. Moreover, although the analysis of CITK−/− mice is complicated by the high prevalence of cytokinesis failure and apoptosis, our in vivo data suggest that the two proteins may also cooperate in determining cell fate of dividing NP, a possibility consistent with the reduction of postnatal neural stem cells described in CITK−/− rats 77. Thus, our study suggests that besides apoptosis, disturbed cell fate contributes to microcephalic phenotype of CITK−/− mice. Therefore, it will be interesting to test whether CITK may cooperate with ASPM in controlling the activity of Cdk2/cyclin E complex 43. It also remains to be established how CITK may modulate astral‐MT dynamics. We have shown that CITK affects both MT nucleation and stability. The function of CITK in cytokinesis has already been linked to MT organization. Indeed, CITK promotes midbody maturation by recruiting at the midbody the kinesins KIF14 48, 78 and MLKP1 58, which in turn recruit the MT‐cross‐linking protein PRC1. CITK stabilizes midbody microtubules by indirectly stimulating TUBB3 phosphorylation 53. Finally, systematic proteomic studies have found that CIT‐K may interact with the kinesins KIF4A and KIF10 65. However, the roles of these proteins in astral‐MT organization and in spindle orientation have not been established. Addressing the functional significance of CITK interaction with these proteins and identifying CITK substrates will be an interesting subject for future studies. In addition, it will be very interesting to address whether other MCPH proteins associated with MT function, such as WDR62, CDK5RAP2, CENPJ, and STIL 33, may regulate astral MT functionally cooperating with ASPM and CITK.
Materials and Methods
Cell culture, synchronization, and drug treatments
Unmodified HeLa cells were originally obtained from ATCC, and a batch was frozen after five passages. All cells are routinely screened for mycoplasma contamination.
HeLa cells were cultured in RPMI medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. HeLa cells expressing GFP‐tagged CITK and ASPM from a BAC transgene were obtained by courtesy of Hyman Lab (Max‐Planck Institute of Molecular Cell Biology and Genetics, Dresden) cultured in DMEM–10% heat‐inactivated FBS, 1% penicillin/streptomycin, and 0.4 mg/ml G418 (Geneticin®; Life Technologies). The HeLa cell line expressing EB3–tdTomato 79, obtained by courtesy of Dr. Ann Straube (University of Warwick, Coventry, United Kingdom), was maintained in DMEM‐GlutaMAX (Invitrogen) supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, 200 μg/ml Geneticin (Sigma), and 0.5 μg/ml puromycin 20. All cells were cultured in a humidified 5% CO2 incubator at 37°C.
Cells were synchronized by a double‐ or single‐thymidine block. For the double‐thymidine block, asynchronous cultures were treated with 2 mM thymidine (Sigma) for 16 h, then released for 4–6 h in fresh complete medium, and again blocked for 16 h. Finally, cells were washed twice with fresh medium and allowed to progress through mitosis.
For drug treatments, synchronized HeLa cells were treated with 250 pM of paclitaxel (Sigma) for 30 min before fixation for immunofluorescence.
Transfection, RNAi, and rescue
In this study, we used a previously validated (CK1 = AUGGAAGGCACU AUUUCUCAA) (Gai et al 49) and a second CITK‐siRNA sequence (CK2 = CCAUUCCUUGGCACCUGCUU). The knockdown of ASPM was obtained using a previously validated sequence 39 (GUGGUGAAGGUGACCUUUC). All siRNAs were obtained from GE Healthcare (Dharmacon, Lafayette, CO). The ON‐TARGETplus non‐targeting siRNA #1 (GE Healthcare, Dharmacon) was used as a negative control for potential off‐target effects. The expression construct coding for CITK‐Cherry was generated by inserting the Myc‐CITK cDNA into the pm‐Cherry‐N1 plasmid (Clontech, Mountain View, CA) 49. The same method was used to generate constructs coding for CKD‐Cherry and for CITN‐Cherry. HeLa cells plated on a six‐well plate were transfected using 1 μg of the required plasmid DNA and 3 μl of Trans‐IT‐LT1 transfection reagent (Mirus Bio, Madison, WI), or 6.25 μl of the required siRNA (20 μM) and 2.5 μl Lipofectamine 2000 (Invitrogen, Carlsbad, CA), according to the manufacturers' instructions. For rescue experiments, cells were transiently transfected with siRNAs; 24 h later, cells were transfected with DNA constructs and analyzed 24 h later.
Cell staining
Fixation methods are as follows: for α‐tubulin staining, 4% paraformaldehyde at room temperature for 10 min or with methanol at −20°C for 10 min; for γ‐tubulin, ASPM, and NuMA staining, methanol at −20°C for 10 min; for dynactin and dynein staining, cells were pre‐extracted in 0.5% Triton X‐100 in PHEM buffer (60 mM PIPES, 25 mM HEPES, 10 mM EGTA, and 4 mM MgSO4) for 1 min and then fixed in methanol at −20°C for 10 min; for CITK staining, cells were pre‐extracted in 0.5% Triton X‐100 in PHEM buffer for 1 min and fixed with 4% paraformaldehyde at room temperature for 10 min. In all cases, cells were permeabilized in 0.1% Triton X‐100 in PBS for 10 min, saturated in 5% BSA in PBS for 30 min, and incubated with primary antibody for 45 min at room temperature. Primary antibodies were detected with anti‐rabbit Alexa Fluor 488 or 568 (Molecular Probes, Invitrogen) used at 1:1,000 dilution for 30 min. Counterstaining was performed with the DNA dye DAPI (Sigma) at 0.5 μg/ml for 1 min.
Antibodies
The following primary antibodies were used: mouse anti‐alpha‐tubulin, 1:1,000 (Sigma, cat. no. T5168, clone B‐5‐1‐2); rabbit anti‐alpha‐tubulin, 1:200 (Abcam, cat. no. ab15246); mouse anti‐gamma‐tubulin, 1:200 (Abcam, clone TU‐30, cat. no. ab27074); rabbit anti‐gamma‐tubulin, 1:1,000 (Sigma, cat. no. T5192); mouse anti‐CITK, 1:100 (BD Transduction Laboratories, clone 6/CRIK, cat. no. 611376); rabbit anti‐NuMA, 1:2,000 (Abcam, cat. no. ab36999); rabbit anti‐dynein heavy chain, 1:300 (Santa Cruz, cat. no. sc‐9115); rabbit anti‐ASPM, 1:2,000 39; mouse anti‐p150, 1:1,000 (BD Biosciences, cat. no. 610474); rabbit anti‐GFP, 1:1,000 (Abcam, cat. no. ab290); mouse anti‐Ki67, 1:1,000 (BD Biosciences, clone B56, cat. no. 550609); mouse anti‐BrdU, 1:200 (Chemicon, cat. no. MAB‐1467); mouse anti‐TUBB3, 1:1,000 (clone TuJ1, Covance, cat. no. MMS‐435P); and rabbit anti‐Tbr2, 1:200 (Millipore, cat. no. AB2283).
Mice
CITK−/− E14.5 embryos and matched controls were obtained by crossing CITK+/− mice (mixed genetic background C57/Bl6 x Sv129). Animals were housed in the Animal Facility of Molecular Biotechnology Centre, University of Torino, under license no. 343/2015 PR from Italian Ministry of Health. For estimate of sample size, CITK phenotypes are usually 100% penetrant, with minimal variability due to genetic background. Under these conditions, analysis of three biological replicates is usually sufficient to reveal the existence of significant differences.
Mouse embryonic cortex staining
Embryonic brains were dissected at E14.5 and fixed for 12–16 h at 4°C in 4% PFA. For cell cycle exit analysis, pregnant females were previously injected at E13.5 with 75 mg/kg BrdU. Fixed brains were equilibrated in 30% sucrose in PBS for 12–24 h at 4°C. Brains were embedded with Tissue‐TEK (O.C.T., Sakura Finetek), snap‐frozen in liquid nitrogen, and stored at −20°C. Sectioning was performed at 20 μm with a cryostat. Cryosections were rehydrated five times in PBS before further processing. For measurement of spindle angle, cryosections were subjected to antigen retrieval by heating in 0.01 M citrate buffer pH 6.0 at 70°C for 1 h. Sections were then permeabilized using 0.3% Triton X‐100 in PBS for 30 min and quenched with 0.1 M glycine for 30 min. Sections were then incubated with primary antibodies overnight at 4°C, followed by secondary antibody for 1 h at room temperature in a solution of 0.2% gelatin, 300 mM NaCl, and 0.3% Triton X‐100 in PBS. DNA was stained in the last wash using DAPI. For cell cycle exit experiments, BrdU antigen was retrieved using 2 N HCl for 10 min in a steamer, followed by neutralization with 0.1 M borate buffer pH 8.5. Slides were then processed for IF as above. We usually exclude samples that are not judged of sufficient technical quality by microscopic analysis. In this study, no samples were excluded.
Drosophila strains
The Drosophila citron kinase 1 (ck 1) mutant allele, also called l(3)7 m‐62 or sticky 1 (sti 1), is described by 80 and by FlyBase (http://flybase.bio.indiana.edu/).
The dck 2 allele was isolated from a collection of 1,600 ethylmethanesulfonate (EMS)‐induced third‐chromosome late lethals, generated in Charles Zuker's laboratory (University of California, San Diego, CA) 47. ck 1 and ck 2 mutant alleles were kept in stocks over the TM6 C, a chromosome balancer which carries the dominant larval marker Tubby (http://flybase.bio.indiana.edu/); homozygous mutant larvae were recognized for their non‐Tubby phenotype. All stocks were maintained on standard Drosophila medium at 25°C.
Immunostaining of Drosophila neuroblast cells
Brains from third‐instar larvae were dissected and fixed according to 81. After two rinses in phosphate‐buffered saline (PBS), brain preparations were incubated overnight at 4°C with a monoclonal anti‐alpha‐tubulin antibody (Sigma) diluted 1:1,000 in PBS and rabbit anti‐Miranda antibody (a gift of Y. N. Jan) diluted 1:100 in PBS. After two rinses in PBS, primary antibodies were detected by 1‐h incubation at room temperature with FITC‐conjugated anti‐mouse IgG + IgM (1:20; Jackson Laboratories) and Alexa Fluor 555‐conjugated anti‐rabbit IgG (1:300; Molecular Probes), diluted in PBS. Immunostained preparations were mounted in Vectashield H‐1200 (Vector Laboratories) containing DAPI (4′,6‐diamidino‐2‐phenylindole). The angle between a line connecting the two spindle poles and a line bisecting the crescent formed by Mira in metaphase NBs was measured, by hand with a goniometer, on images captured by a Zeiss Axioplan fluorescence microscope, equipped with an HBO100W mercury lamp and a cooled charge‐coupled device (CCD camera; Photometrics CoolSnap HQ).
Microscopy
Imaging was performed using a Leica TCS SP5‐AOBS 5‐channel confocal system (Leica Microsystems) equipped with a 405‐nm diode, an argon ion, and a 561‐nm DPSS laser. Fixed cells were imaged using a HCX PL APO 63x/1.4 NA oil immersion objective at a pixel resolution of 0.108 × 0.108 μm.
Time lapses were recorded overnight with an interval of 5 min using a 40× PlanApo N.A. 1.4 oil immersion objective on the cells kept in the microscope incubator at 37°C and 5% CO2.
Image analysis
Spindle angle measurements were obtained using a trigonometric formula, once we measured the three‐dimensional (3D) distance (across the x, y, and z planes) and the 2D distance (across the z planes) between the two centrosomes of the spindle.
To quantify NuMA, dynein, and p150Glued signals at the centrosome, we used Fiji (fiji.sc) 82. After obtaining the sum of intensity of all the z‐stacks from multistack images, we built a ROI surrounding the signal at the centrosome and a ROI around the cells. Once the ROIs were established, they were used for all the images analyzed. We evaluated the corrected integrated density (CTCF), for both the centrosomes, calculating as Integrated Density (Area of selection × Mean fluorescence of the background). Analysis requiring manual counting was performed on blinded samples.
Measurement of astral‐MT dynamics
Astral‐MT nucleation frequencies were quantified as described 20 by counting the number of EB3 comets in the focal plane that originated from each spindle pole during the observation time. Cytoplasmic growth speeds for individual comets were measured as the total distance traveled from the spindle pole toward the cell cortex divided by the time. Kymographs of astral‐MT and spindle‐pole movements were obtained using Fiji.
Proximity ligation assay
PLA analysis 67 was performed using the kit provided by Olink Bioscience (Uppsala Science Park, Uppsala, Sweden). Metaphase HeLa cells and ASPM‐GFP HeLa stable cell lines were incubated with the previously described primary antibodies, and reactions were performed following the manufacturer's instructions.
MT regrowth assay
Forty‐eight hours after siRNA transfection, MT were depolymerized at 4°C for 30 min. Cells were then washed and incubated in culture medium at 37°C to allow regrowth. Cells were fixed at different time intervals in methanol at −20°C for 10 min and processed for immunofluorescence microscopy to examine MT regrowth from spindle poles in metaphase cells. The MT signal was quantified using Fiji.
Centrosomal fractionation
Centrosome fractionation was performed as described previously 83. In brief, exponentially growing cells were incubated with 10 μg/ml nocodazole (Sigma‐Aldrich) and 5 μg/ml cytochalasin B (Sigma‐Aldrich) for 90 min and then lysed by hypotonic treatment. Centrosomes were harvested by centrifugation onto a 20% Ficoll cushion and further purified by centrifugation through a discontinuous (70, 50, and 40%) sucrose gradient. Fractions of 0.3 ml were collected and analyzed.
Immunoprecipitation and Western blotting
For all immunoprecipitations, cells were extracted with lysis buffer containing 1% Triton X‐100, 120 mM NaCl, 50 mM Tris–HCl (pH 7.5), protease inhibitors (Roche, Mannheim, Germany), and 1 mM phenylmethylsulfonyl fluoride (PMSF). The extracts were then clarified by centrifugation. Antibodies and protein‐G–Sepharose beads (Amersham Biosciences, Uppsala, Sweden) were added to lysates and incubated overnight at 4°C. Pellets were washed four times with lysis buffer and analyzed by SDS–PAGE.
For immunoblots, immunoprecipitates or equal amounts of proteins from whole‐cell lysates were resolved by reduction with SDS–PAGE and blotted to nitrocellulose filters, which were then incubated with the indicated antibodies and developed using the ECL System (Amersham Biosciences).
Statistics
For the experiments in which we can show statistically significant differences, sample size was determined on the basis of pilot studies estimating the order of magnitude of differences. For the experiments showing non‐significant differences, we used the same sample size of those revealing differences. Normal distribution of data is evaluated by graphical analysis of data plots. All the numbers represented by histograms were obtained by averaging all the data points contained in the replicas (with a comparable number of points per each replica). Statistical significance of the differences was assessed using a two‐tailed Student's t‐test.
Author contributions
All experiments were designed and conceived by MG and FDC. The mouse in vivo experiments were performed by FTB with the support and the advice of WBH. The Drosophila experiments were performed by FV and SB The PLA was performed by LA and AS The analysis of centrosome fractions was performed by AB and NL. All the other experiments were performed by MG, with the support of CV, SP, GEB, FS, and AMAC. ASPM reagents were provided by JB. The manuscript was written by MG and FDC.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Review Process File
Acknowledgements
We are grateful to Anne Straube (University of Warwick, Coventry, UK) for providing the EB3–tdTomato HeLa cell line and to Ina Poser and Antony Hyman (MPI, Dresden, Germany) for providing the GFP‐BAC transgenic ASPM and CITK HeLa cell lines. We thank Gianmarco Pallavicini for help with mouse genotyping and Laura Tasca and Francesca Macaluso for help with image analysis. This work was primarily supported by the Telethon Foundation through grant no. GGP12095 to F.D.C. The laboratory of FDC is also supported by Telethon Foundation grant no. GGP13081, by Associazione Italiana per la Ricerca sul Cancro (AIRC) grant IG17527, and by Consiglio Nazionale delle Ricerche (CNR) through the EPIGEN program.
EMBO Reports (2016) 17: 1396–1409
Contributor Information
Marta Gai, Email: marta.gai@unito.it.
Ferdinando Di Cunto, Email: ferdinando.dicunto@unito.it.
References
- 1. Bergstralh DT, St Johnston D (2014) Spindle orientation: what if it goes wrong? Semin Cell Dev Biol 34: 140–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Noatynska A, Gotta M, Meraldi P (2012) Mitotic spindle (DIS)orientation and DISease: cause or consequence? J Cell Biol 199: 1025–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Panousopoulou E, Green JB (2014) Spindle orientation processes in epithelial growth and organisation. Semin Cell Dev Biol 34: 124–132 [DOI] [PubMed] [Google Scholar]
- 4. Peyre E, Morin X (2012) An oblique view on the role of spindle orientation in vertebrate neurogenesis. Dev Growth Differ 54: 287–305 [DOI] [PubMed] [Google Scholar]
- 5. Taverna E, Gotz M, Huttner WB (2014) The cell biology of neurogenesis: toward an understanding of the development and evolution of the neocortex. Annu Rev Cell Dev Biol 30: 465–502 [DOI] [PubMed] [Google Scholar]
- 6. Arai Y, Pulvers JN, Haffner C, Schilling B, Nusslein I, Calegari F, Huttner WB (2011) Neural stem and progenitor cells shorten S‐phase on commitment to neuron production. Nat Commun 2: 154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fietz SA, Huttner WB (2011) Cortical progenitor expansion, self‐renewal and neurogenesis‐a polarized perspective. Curr Opin Neurobiol 21: 23–35 [DOI] [PubMed] [Google Scholar]
- 8. Morin X, Bellaiche Y (2011) Mitotic spindle orientation in asymmetric and symmetric cell divisions during animal development. Dev Cell 21: 102–119 [DOI] [PubMed] [Google Scholar]
- 9. Paridaen JT, Huttner WB (2014) Neurogenesis during development of the vertebrate central nervous system. EMBO Rep 15: 351–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Calegari F, Huttner WB (2003) An inhibition of cyclin‐dependent kinases that lengthens, but does not arrest, neuroepithelial cell cycle induces premature neurogenesis. J Cell Sci 116: 4947–4955 [DOI] [PubMed] [Google Scholar]
- 11. Florio M, Huttner WB (2014) Neural progenitors, neurogenesis and the evolution of the neocortex. Development 141: 2182–2194 [DOI] [PubMed] [Google Scholar]
- 12. Noctor SC, Martinez‐Cerdeno V, Ivic L, Kriegstein AR (2004) Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci 7: 136–144 [DOI] [PubMed] [Google Scholar]
- 13. Chenn A, McConnell SK (1995) Cleavage orientation and the asymmetric inheritance of Notch1 immunoreactivity in mammalian neurogenesis. Cell 82: 631–641 [DOI] [PubMed] [Google Scholar]
- 14. Kosodo Y, Roper K, Haubensak W, Marzesco AM, Corbeil D, Huttner WB (2004) Asymmetric distribution of the apical plasma membrane during neurogenic divisions of mammalian neuroepithelial cells. EMBO J 23: 2314–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lancaster MA, Knoblich JA (2012) Spindle orientation in mammalian cerebral cortical development. Curr Opin Neurobiol 22: 737–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Castanon I, Gonzalez‐Gaitan M (2011) Oriented cell division in vertebrate embryogenesis. Curr Opin Cell Biol 23: 697–704 [DOI] [PubMed] [Google Scholar]
- 17. Gillies TE, Cabernard C (2011) Cell division orientation in animals. Curr Biol 21: R599–R609 [DOI] [PubMed] [Google Scholar]
- 18. Williams SE, Fuchs E (2013) Oriented divisions, fate decisions. Curr Opin Cell Biol 25: 749–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Busson S, Dujardin D, Moreau A, Dompierre J, De Mey JR (1998) Dynein and dynactin are localized to astral microtubules and at cortical sites in mitotic epithelial cells. Curr Biol 8: 541–544 [DOI] [PubMed] [Google Scholar]
- 20. Samora CP, Mogessie B, Conway L, Ross JL, Straube A, McAinsh AD (2011) MAP4 and CLASP1 operate as a safety mechanism to maintain a stable spindle position in mitosis. Nat Cell Biol 13: 1040–1050 [DOI] [PubMed] [Google Scholar]
- 21. Pramparo T, Youn YH, Yingling J, Hirotsune S, Wynshaw‐Boris A (2010) Novel embryonic neuronal migration and proliferation defects in Dcx mutant mice are exacerbated by Lis1 reduction. J Neurosci 30: 3002–3012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Feng Y, Walsh CA (2004) Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron 44: 279–293 [DOI] [PubMed] [Google Scholar]
- 23. Schwamborn JC, Knoblich JA (2008) LIS1 and spindle orientation in neuroepithelial cells. Cell Stem Cell 2: 193–194 [DOI] [PubMed] [Google Scholar]
- 24. Yingling J, Youn YH, Darling D, Toyo‐Oka K, Pramparo T, Hirotsune S, Wynshaw‐Boris A (2008) Neuroepithelial stem cell proliferation requires LIS1 for precise spindle orientation and symmetric division. Cell 132: 474–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fish JL, Kosodo Y, Enard W, Paabo S, Huttner WB (2006) Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc Natl Acad Sci USA 103: 10438–10443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Insolera R, Bazzi H, Shao W, Anderson KV, Shi SH (2014) Cortical neurogenesis in the absence of centrioles. Nat Neurosci 17: 1528–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Megraw TL, Sharkey JT, Nowakowski RS (2011) Cdk5rap2 exposes the centrosomal root of microcephaly syndromes. Trends Cell Biol 21: 470–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thornton GK, Woods CG (2009) Primary microcephaly: do all roads lead to Rome? Trends Genet 25: 501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Homem CC, Repic M, Knoblich JA (2015) Proliferation control in neural stem and progenitor cells. Nat Rev Neurosci 16: 647–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Konno D, Shioi G, Shitamukai A, Mori A, Kiyonari H, Miyata T, Matsuzaki F (2008) Neuroepithelial progenitors undergo LGN‐dependent planar divisions to maintain self‐renewability during mammalian neurogenesis. Nat Cell Biol 10: 93–101 [DOI] [PubMed] [Google Scholar]
- 31. Bettencourt‐Dias M, Hildebrandt F, Pellman D, Woods G, Godinho SA (2011) Centrosomes and cilia in human disease. Trends Genet 27: 307–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Loffler H, Fechter A, Matuszewska M, Saffrich R, Mistrik M, Marhold J, Hornung C, Westermann F, Bartek J, Kramer A (2011) Cep63 recruits Cdk1 to the centrosome: implications for regulation of mitotic entry, centrosome amplification, and genome maintenance. Cancer Res 71: 2129–2139 [DOI] [PubMed] [Google Scholar]
- 33. Kaindl AM, Passemard S, Kumar P, Kraemer N, Issa L, Zwirner A, Gerard B, Verloes A, Mani S, Gressens P (2010) Many roads lead to primary autosomal recessive microcephaly. Prog Neurobiol 90: 363–383 [DOI] [PubMed] [Google Scholar]
- 34. Mochida GH (2009) Genetics and biology of microcephaly and lissencephaly. Semin Pediatr Neurol 16: 120–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Faheem M, Naseer MI, Rasool M, Chaudhary AG, Kumosani TA, Ilyas AM, Pushparaj P, Ahmed F, Algahtani HA, Al‐Qahtani MH et al (2015) Molecular genetics of human primary microcephaly: an overview. BMC Med Genomics 8(Suppl 1): S4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morris‐Rosendahl DJ, Kaindl AM (2015) What next‐generation sequencing (NGS) technology has enabled us to learn about primary autosomal recessive microcephaly (MCPH). Mol Cell Probes 29: 271–281 [DOI] [PubMed] [Google Scholar]
- 37. Sir JH, Barr AR, Nicholas AK, Carvalho OP, Khurshid M, Sossick A, Reichelt S, D'Santos C, Woods CG, Gergely F (2011) A primary microcephaly protein complex forms a ring around parental centrioles. Nat Genet 43: 1147–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bond J, Roberts E, Mochida GH, Hampshire DJ, Scott S, Askham JM, Springell K, Mahadevan M, Crow YJ, Markham AF et al (2002) ASPM is a major determinant of cerebral cortical size. Nat Genet 32: 316–320 [DOI] [PubMed] [Google Scholar]
- 39. Higgins J, Midgley C, Bergh AM, Bell SM, Askham JM, Roberts E, Binns RK, Sharif SM, Bennett C, Glover DM et al (2010) Human ASPM participates in spindle organisation, spindle orientation and cytokinesis. BMC Cell Biol 11: 85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Saunders RD, Avides MC, Howard T, Gonzalez C, Glover DM (1997) The Drosophila gene abnormal spindle encodes a novel microtubule‐associated protein that associates with the polar regions of the mitotic spindle. J Cell Biol 137: 881–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wakefield JG, Bonaccorsi S, Gatti M (2001) The Drosophila protein asp is involved in microtubule organization during spindle formation and cytokinesis. J Cell Biol 153: 637–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pulvers JN, Bryk J, Fish JL, Wilsch‐Brauninger M, Arai Y, Schreier D, Naumann R, Helppi J, Habermann B, Vogt J et al (2010) Mutations in mouse Aspm (abnormal spindle‐like microcephaly associated) cause not only microcephaly but also major defects in the germline. Proc Natl Acad Sci USA 107: 16595–16600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Capecchi MR, Pozner A (2015) ASPM regulates symmetric stem cell division by tuning Cyclin E ubiquitination. Nat Commun 6: 8763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Paramasivam M, Chang YJ, LoTurco JJ (2007) ASPM and citron kinase co‐localize to the midbody ring during cytokinesis. Cell Cycle 6: 1605–1612 [DOI] [PubMed] [Google Scholar]
- 45. Di Cunto F, Imarisio S, Hirsch E, Broccoli V, Bulfone A, Migheli A, Atzori C, Turco E, Triolo R, Dotto GP et al (2000) Defective neurogenesis in citron kinase knockout mice by altered cytokinesis and massive apoptosis. Neuron 28: 115–127 [DOI] [PubMed] [Google Scholar]
- 46. Madaule P, Eda M, Watanabe N, Fujisawa K, Matsuoka T, Bito H, Ishizaki T, Narumiya S (1998) Role of citron kinase as a target of the small GTPase Rho in cytokinesis. Nature 394: 491–494 [DOI] [PubMed] [Google Scholar]
- 47. Naim V, Imarisio S, Di Cunto F, Gatti M, Bonaccorsi S (2004) Drosophila citron kinase is required for the final steps of cytokinesis. Mol Biol Cell 15: 5053–5063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bassi ZI, Verbrugghe KJ, Capalbo L, Gregory S, Montembault E, Glover DM, D'Avino PP (2011) Sticky/Citron kinase maintains proper RhoA localization at the cleavage site during cytokinesis. J Cell Biol 195: 595–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gai M, Camera P, Dema A, Bianchi F, Berto G, Scarpa E, Germena G, Di Cunto F (2011) Citron kinase controls abscission through RhoA and anillin. Mol Biol Cell 22: 3768–3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sarkisian MR, Li W, Di Cunto F, D'Mello SR, LoTurco JJ (2002) Citron‐kinase, a protein essential to cytokinesis in neuronal progenitors, is deleted in the flathead mutant rat. J Neurosci 22:RC217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. LoTurco JJ, Sarkisian MR, Cosker L, Bai J (2003) Citron kinase is a regulator of mitosis and neurogenic cytokinesis in the neocortical ventricular zone. Cereb Cortex 13: 588–591 [DOI] [PubMed] [Google Scholar]
- 52. Calegari F, Haubensak W, Haffner C, Huttner WB (2005) Selective lengthening of the cell cycle in the neurogenic subpopulation of neural progenitor cells during mouse brain development. J Neurosci 25: 6533–6538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sgro F, Bianchi FT, Falcone M, Pallavicini G, Gai M, Chiotto AM, Berto GE, Turco E, Chang YJ, Huttner WB et al (2016) Tissue‐specific control of midbody microtubule stability by Citron kinase through modulation of TUBB3 phosphorylation. Cell Death Differ 23: 801–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. D'Avino PP, Savoian MS, Glover DM (2004) Mutations in sticky lead to defective organization of the contractile ring during cytokinesis and are enhanced by Rho and suppressed by Rac. J Cell Biol 166: 61–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Echard A, Hickson GR, Foley E, O'Farrell PH (2004) Terminal cytokinesis events uncovered after an RNAi screen. Curr Biol 14: 1685–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shandala T, Gregory SL, Dalton HE, Smallhorn M, Saint R (2004) Citron kinase is an essential effector of the Pbl‐activated Rho signalling pathway in Drosophila melanogaster. Development 131: 5053–5063 [DOI] [PubMed] [Google Scholar]
- 57. Homem CC, Knoblich JA (2012) Drosophila neuroblasts: a model for stem cell biology. Development 139: 4297–4310 [DOI] [PubMed] [Google Scholar]
- 58. Bassi ZI, Audusseau M, Riparbelli MG, Callaini G, D'Avino PP (2013) Citron kinase controls a molecular network required for midbody formation in cytokinesis. Proc Natl Acad Sci USA 110: 9782–9787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Matsumura S, Hamasaki M, Yamamoto T, Ebisuya M, Sato M, Nishida E, Toyoshima F (2012) ABL1 regulates spindle orientation in adherent cells and mammalian skin. Nat Commun 3: 626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Malik R, Lenobel R, Santamaria A, Ries A, Nigg EA, Korner R (2009) Quantitative analysis of the human spindle phosphoproteome at distinct mitotic stages. J Proteome Res 8: 4553–4563 [DOI] [PubMed] [Google Scholar]
- 61. Nousiainen M, Sillje HH, Sauer G, Nigg EA, Korner R (2006) Phosphoproteome analysis of the human mitotic spindle. Proc Natl Acad Sci USA 103: 5391–5396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sauer G, Korner R, Hanisch A, Ries A, Nigg EA, Sillje HH (2005) Proteome analysis of the human mitotic spindle. Mol Cell Proteomics 4: 35–43 [DOI] [PubMed] [Google Scholar]
- 63. Liu H, Di Cunto F, Imarisio S, Reid LM (2003) Citron kinase is a cell cycle‐dependent, nuclear protein required for G2/M transition of hepatocytes. J Biol Chem 278: 2541–2548 [DOI] [PubMed] [Google Scholar]
- 64. Hutchins JR, Toyoda Y, Hegemann B, Poser I, Heriche JK, Sykora MM, Augsburg M, Hudecz O, Buschhorn BA, Bulkescher J et al (2010) Systematic analysis of human protein complexes identifies chromosome segregation proteins. Science 328: 593–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Maliga Z, Junqueira M, Toyoda Y, Ettinger A, Mora‐Bermudez F, Klemm RW, Vasilj A, Guhr E, Ibarlucea‐Benitez I, Poser I, et al (2013) A genomic toolkit to investigate kinesin and myosin motor function in cells. Nat Cell Biol 15: 325–334 [DOI] [PubMed] [Google Scholar]
- 66. Neumann B, Walter T, Heriche JK, Bulkescher J, Erfle H, Conrad C, Rogers P, Poser I, Held M, Liebel U et al (2010) Phenotypic profiling of the human genome by time‐lapse microscopy reveals cell division genes. Nature 464: 721–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG et al (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 3: 995–1000 [DOI] [PubMed] [Google Scholar]
- 68. Furuyashiki T, Fujisawa K, Fujita A, Madaule P, Uchino S, Mishina M, Bito H, Narumiya S (1999) Citron, a Rho‐target, interacts with PSD‐95/SAP‐90 at glutamatergic synapses in the thalamus. J Neurosci 19: 109–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Carminati JL, Stearns T (1997) Microtubules orient the mitotic spindle in yeast through dynein‐dependent interactions with the cell cortex. J Cell Biol 138: 629–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nguyen‐Ngoc T, Afshar K, Gonczy P (2007) Coupling of cortical dynein and G alpha proteins mediates spindle positioning in Caenorhabditis elegans. Nat Cell Biol 9: 1294–1302 [DOI] [PubMed] [Google Scholar]
- 71. Yang Y, Liu M, Li D, Ran J, Gao J, Suo S, Sun SC, Zhou J (2014) CYLD regulates spindle orientation by stabilizing astral microtubules and promoting dishevelled‐NuMA‐dynein/dynactin complex formation. Proc Natl Acad Sci USA 111: 2158–2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Delaunay D, Cortay V, Patti D, Knoblauch K, Dehay C (2014) Mitotic spindle asymmetry: a Wnt/PCP‐regulated mechanism generating asymmetrical division in cortical precursors. Cell Rep 6: 400–414 [DOI] [PubMed] [Google Scholar]
- 73. Buchman JJ, Durak O, Tsai LH (2011) ASPM regulates Wnt signaling pathway activity in the developing brain. Genes Dev 25: 1909–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mollinari C, Kleman JP, Jiang W, Schoehn G, Hunter T, Margolis RL (2002) PRC1 is a microtubule binding and bundling protein essential to maintain the mitotic spindle midzone. J Cell Biol 157: 1175–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mora‐Bermudez F, Matsuzaki F, Huttner WB (2014) Specific polar subpopulations of astral microtubules control spindle orientation and symmetric neural stem cell division. Elife 3: e02875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wandke C, Barisic M, Sigl R, Rauch V, Wolf F, Amaro AC, Tan CH, Pereira AJ, Kutay U, Maiato H et al (2012) Human chromokinesins promote chromosome congression and spindle microtubule dynamics during mitosis. J Cell Biol 198: 847–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ackman JB, Ramos RL, Sarkisian MR, Loturco JJ (2007) Citron kinase is required for postnatal neurogenesis in the hippocampus. Dev Neurosci 29: 113–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gruneberg U, Neef R, Li X, Chan EH, Chalamalasetty RB, Nigg EA, Barr FA (2006) KIF14 and citron kinase act together to promote efficient cytokinesis. J Cell Biol 172: 363–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Straube A, Merdes A (2007) EB3 regulates microtubule dynamics at the cell cortex and is required for myoblast elongation and fusion. Curr Biol 17: 1318–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gatti M, Baker BS (1989) Genes controlling essential cell‐cycle functions in Drosophila melanogaster. Genes Dev 3: 438–453 [DOI] [PubMed] [Google Scholar]
- 81. Bonaccorsi S, Gianti MG, Gatti M (2000) Spindle assembly in Drosophila neuroblasts and ganglion mother cells. Nat Cell Biol 2: 54–56 [DOI] [PubMed] [Google Scholar]
- 82. Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Reber S (2011) Isolation of centrosomes from cultured cells. Methods Mol Biol 777: 107–116 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Review Process File