

Abstract
The 7SK snRNA sequesters P‐TEFb, a general transcription elongation factor and human co‐factor for HIV‐1 Tat protein, into the catalytically inactive 7SK snRNP. Little is known about how 7SK RNA is regulated to perform this function. Here, we show that most of 7SK is pseudouridylated at position U250 by the predominant cellular pseudouridine synthase machinery, the DKC1–box H/ACA RNP. Pseudouridylation is critical to stabilize 7SK snRNP, as its abolishment by either mutation at or around U250 or depletion of DKC1, the catalytic component of the box H/ACA RNP, disrupts 7SK snRNP and releases P‐TEFb to form the super elongation complex (SEC) and the Brd4–P‐TEFb complex. The SEC is then recruited by Tat to the HIV‐1 promoter to stimulate viral transcription and escape from latency. Thus, although 7SK RNA levels remain mostly unchanged, its function is modulated by pseudouridylation, which in turn controls transcription of both HIV‐1 and cellular genes.
Keywords: 7SK snRNA, HIV‐1 transcription, latency, pseudouridylation, P‐TEFb
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; RNA Biology
Introduction
The 7SK RNA is an abundant non‐coding nuclear RNA highly conserved in vertebrates 1, 2. Despite its first detection in human cells in the 1970s 3, little was known about its function for the next quarter of century. In 2001, two independent studies 4, 5 identified 7SK as a critical regulator of the homeostasis and activity of P‐TEFb, a key human transcription factor and a specific host cofactor for HIV‐1 transcription.
7SK was identified during the isolation of nuclear factors that can bind and control the activity of P‐TEFb. Composed of cyclin‐dependent kinase‐9 (CDK9) and its partner cyclin T (CycT1 or the minor T2a and T2b) 2, P‐TEFb phosphorylates a pair of negative elongation factors (DSIF and NELF) and the C‐terminal domain of RNA polymerase (Pol) II. These events antagonize the inhibitory actions of the negative factors and stimulate Pol II elongation and co‐transcriptional RNA processing 2.
P‐TEFb is an integral component of the multi‐subunit super elongation complex (SEC) that is recruited by the HIV‐1‐encoded Tat protein to the viral long terminal repeat (LTR) to stimulate viral transcription elongation 6, 7. It cooperates with another elongation stimulatory factor ELL1 or ELL2 that resides in the same SEC to synergistically produce full‐length HIV‐1 transcripts essential for productive viral replication. As such, P‐TEFb and the encompassing SEC are cellular co‐factors required for maximal Tat‐transactivation and efficient escape from viral latency 6, 8, 9.
Under normal growth conditions and depending on the cell type, up to 90% of cellular P‐TEFb is sequestered in another multi‐subunit complex called the 7SK snRNP 2. This RNP also contains HEXIM1 or HEXIM2, which inhibit CDK9, and MePCE and LARP7, which protect 7SK integrity by binding to the 5′ and 3′ end of the RNA, respectively. The 7SK RNP functions as a cellular reservoir for excess P‐TEFb to keep it in an inactive state 2. Under conditions that globally affect cell growth or induce stress response, P‐TEFb is released from 7SK RNP to form the SEC and the Brd4–P‐TEFb complex to stimulate transcription and regulate growth. The bromodomain protein Brd4 present in the latter complex recruits P‐TEFb to many primary response genes to promote their expression 10, 11, 12. Notably, P‐TEFb can also be extracted from 7SK RNP by Tat to form Tat–SEC for activation of the HIV‐1 LTR 2.
The 7SK RNA is constitutively produced by RNA Pol III from a multi‐gene family 1, 4. Although its levels remain relatively constant, a recent genomewide study 13 suggests that it may undergo a type of post‐transcriptional modification termed pseudouridylation, which could potentially alter its structure and function. Pseudouridylation is one of the most frequent internal modifications found in stable non‐coding RNA and produces the C5‐glycosidic isomer of uridine (Ψ) 14. While the pseudouridylation of 7SK has been suggested in this high‐throughput study, biochemical and functional validation of this modification has yet to be performed. Here, we provide direct biochemical proof that the vast majority of cellular 7SK RNA is pseudouridylated at residue U250. Our data demonstrate a key role for Ψ250 in controlling the formation of 7SK RNP and show that the DKC1–box H/ACA RNP is mostly responsible for this modification. Impairment of the modification through DKC1 depletion preferentially activates Tat‐dependent HIV‐1 transcription and promotes reversal of viral latency by enhancing SEC's binding to Tat and the HIV‐1 LTR.
Results and Discussion
7SK RNA is pseudouridylated exclusively on U250 by an enzyme in HeLa nuclear extract (NE)
We first sought to biochemically confirm results from a recent genomewide study suggesting that 7SK RNA is pseudouridylated at residue U250 13. To avoid possible false‐positive signals that can arise in this type of study that employed reverse transcription and primer extension of structured RNAs, we instead evaluated 7SK pseudouridylation directly using an in vitro modification assay and thin‐layer chromatography (TLC). 7SK was divided into six fragments (Fig 1A) and in vitro transcribed in the presence of 32P‐UTP. Following incubation with HeLa NE and digestion by nuclease P1, products were resolved by one‐dimensional TLC (1D‐TLC) along with pU and pΨ as controls. Only fragment 5 containing U250 yielded pΨ (Fig 1B). To confirm that the modification indeed selectively occurred at U250, the same analysis was performed with mutant 7SK containing U250 replaced with G (U250G). This change completely abolished the production of pΨ (Fig 1C). Furthermore, using a modified SCARLET (Site‐specific Cleavage And Radioactive‐labeling followed by Ligation‐assisted Extraction and Thin‐layer chromatography) method 15 (Fig 1D) that enables site‐specific labeling of U250 or the control U203 in full‐length 7SK produced by in vitro transcription (IVT) or isolated from HeLa cells, only the latter sample contained pΨ at position 250 in 94% of the population (Fig 1E). These data indicate that most 7SK RNA are pseudouridylated at U250 in vivo. Notably, U250 is present in all identified vertebrate 7SK RNA 16, suggesting a critical role of this residue in 7SK function. It remains to be tested whether this residue is also pseudouridylated in other vertebrates besides humans.
Figure 1. Most cellular 7SK RNA including those bound to P‐TEFb are pseudouridylated at U250.
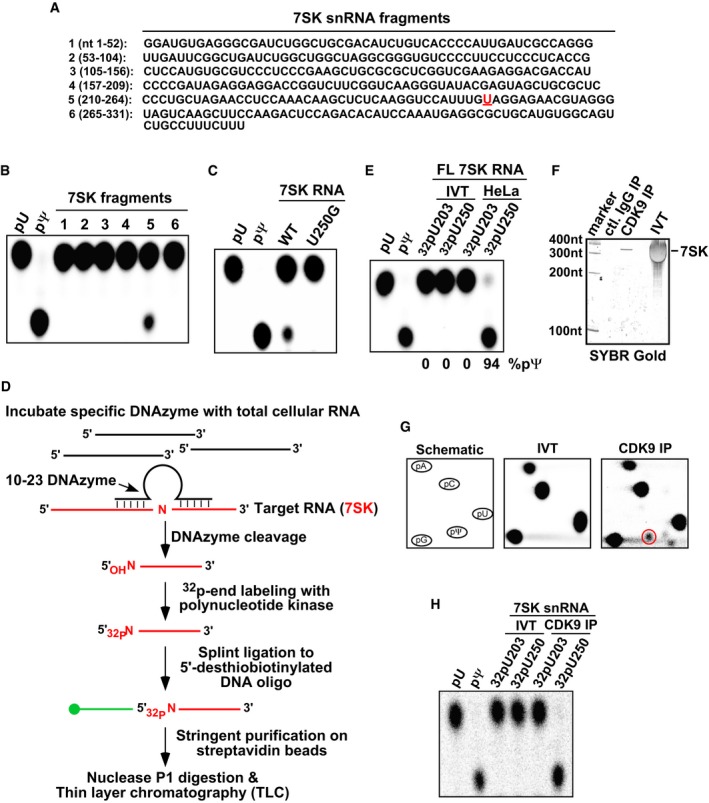
- 7SK was divided into six non‐overlapping fragments and in vitro transcribed in the presence of 32P‐UTP for use in an in vitro modification assay. U250 in fragment 5 is highlighted in red.
- In vitro modification reactions containing the six fragments and HeLa NE were performed. After nuclease P1 digestion, the products were analyzed by 1D‐TLC with pU and pΨ as controls.
- WT and mutant 7SK fragment 5 containing U250G were analyzed as in (B).
- Outline of the biotin‐facilitated extraction and analysis of the modification status of site‐specifically labeled nucleotides in 7SK by 1D‐TLC.
- 7SK RNA, either generated by in vitro transcription (IVT) or purified from HeLa total RNA and then site‐specifically labeled at the indicated positions through the procedure described in (D), was analyzed by 1D‐TLC.
- SYBR Gold staining of 7SK obtained by either IVT or immunoprecipitation (IP) with the indicated antibodies.
- 2D‐TLC analysis of 32P‐labeled mononucleotides derived from IVT or CDK9‐bound 7SK RNA, with the first panel depicting a schematic migration pattern of the indicated 5′‐phosphorylated mononucleotides. The red circle indicates the spot corresponding to pΨ.
- 7SK RNA, obtained by either IVT or anti‐CDK9 IP and then site‐specifically labeled at the indicated positions through the procedure described in (D), was analyzed by 1D‐TLC.
P‐TEFb‐bound 7SK RNA is pseudouridylated on U250
To analyze the modification status of 7SK RNA bound to P‐TEFb, we purified 7SK by anti‐Flag immunoprecipitation from NE of F1C2 cells, a HeLa‐based cell line stably expressing CDK9‐Flag (Fig 1F). The purified 7SK was compared with the IVT 7SK by nuclease digestion and two‐dimensional (2D)‐TLC. Only the CDK9‐bound but not the IVT 7SK produced a spot corresponding to pΨ (Fig 1G, right panel, red circle). This level of modification is not low considering that U250 is only one of 75 U residues in 7SK RNA. Finally, when the CDK9‐bound 7SK was site‐specifically labeled at either U250 or U203 and subjected to 1D‐TLC analysis, pseudouridylation at U250 but not U203 was confirmed (Fig 1H).
Pseudouridylation of U250 is required for efficient formation of 7SK RNP
To determine the impact of Ψ250 on formation of 7SK RNP, U250 was mutated to G to block the modification. The mutation was introduced into a version of 7SK RNA that carries a sequence tag (nt 216–221, Fig 2A) to allow discrimination from endogenous 7SK by primer extension. Compared to the tagged 7SK containing WT sequence at and around U250 (Fig 2A), the U250G mutant (mut1, Fig 2B) showed significantly reduced binding to the 7SK RNP components CDK9 and HEXIM1 but not LARP7 when expressed in HeLa cells (Fig 2G–I). As a La‐related protein, LARP7 binds to the 3′‐poly(U) end of 7SK 17, which may explain why its binding to 7SK was unaffected by the mutation. Notably, the U250G change also decreased the binding to hnRNP R (Fig 2J), a protein known to interact with 7SK RNA upon its release from 7SK RNP 18. To rule out the possibility that the compromised 7SK RNP formation was due to disruption of the hairpin structure by U250G, we restored base‐pairing of the upper stem by mutating A228 to C at the complementary site on the opposite strand (mut2, Fig 2C). Like mut1, mut2 showed decreased bindings to CDK9, HEXIM1, and hnRNP R, but displayed the same binding capacity as WT and mut1 toward LARP7 (Figs 2K–N and EV1).
Figure 2. Ψ250 in 7SK RNA is critical for 7SK RNP formation.
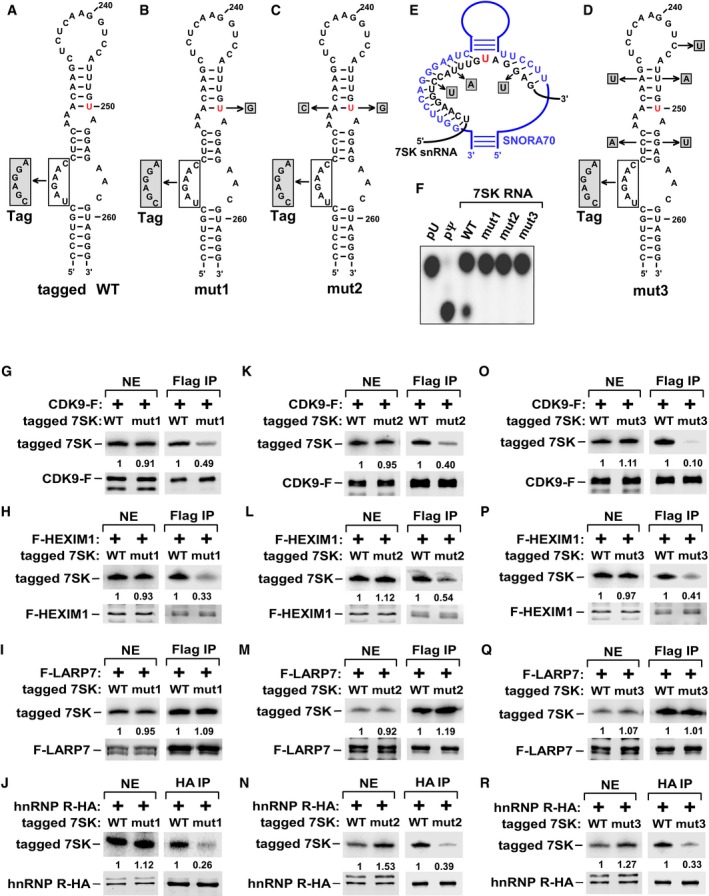
-
A–DSequence alternations, in WT and 7SK RNA mutants mut1, mut2, and mut3, are shown in shaded boxes. U250 is marked in red. The tag refers to a 6‐nt sequence replacement to enable discrimination from endogenous 7SK by primer extension.
-
EAlignment between the 7SK sequences (black) flanking U250 (red) and a portion of the box H/ACA snoRNA (blue) showing base‐pairing between the two. The altered nucleotides in shaded boxes were introduced into 7SK to disrupt the base‐pairing.
-
FThe indicated 7SK RNA were in vitro transcribed in the presence of 32P‐UTP and subjected to in vitro modification reactions. The nuclease P1‐digested products were analyzed by 1D‐TLC with pU and pΨ as controls.
-
G–RTagged WT or mutant 7SK RNA were co‐transfected with the indicated Flag‐ or HA‐tagged proteins into HeLa cells. Anti‐Flag or anti‐HA immunoprecipitates (IP) were analyzed by immunoblotting for the indicated proteins and primer extension for the bound 7SK RNA.
Figure EV1. Mutations at or around U250 in 7SK RNA prevent endogenous CDK9 from binding.
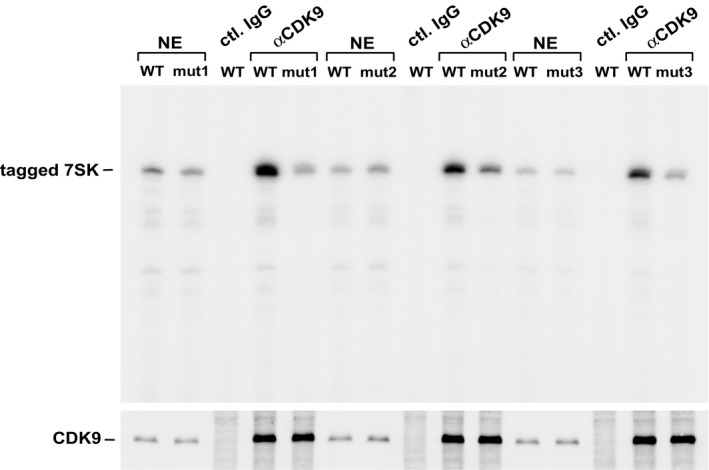
Tagged WT or mutant 7SK (mut1, mut2, and mut3) were transfected separately into HeLa cells. Anti‐CDK9 IPs were analyzed by immunoblotting to detect CDK9 and primer extension for the bound 7SK RNA.
Pseudouridylation of U250 requires base‐pairing with H/ACA snoRNA
In eukaryotes, pseudouridylation of non‐coding RNAs is catalyzed primarily by the box H/ACA ribonucleoprotein (RNP) complex, in which dyskerin (DKC1) serves as the catalytic subunit and the box H/ACA snoRNA guides the RNP to the targeted uridine by base‐pairing with the flanking sequences 14 (Fig 2E). Importantly, the bioinformatics pipeline RNAsnoop 19 reveals extensive base‐pairing between the 7SK sequences flanking U250 and the second pseudouridylation pocket of snoRNA U70.
As a first step toward determining a possible role for the box H/ACA RNP in 7SK pseudouridylation, we disrupted the predicated snoRNA‐7SK RNA interactions by mutating three nucleotides (C243U, U247A, and G253U) surrounding U250 in 7SK to disrupt the base‐pairing with the snoRNA (Fig 2E). Two additional changes (C224A and A231U) were also made in 7SK in order to restore base‐pairing to the stems both above and below U250 (Fig 2D). Just like mut1 and mut2, the new mut3 (Fig 2D), which still maintains the intact U250, also displayed defective interactions with CDK9, HEXIM1, and hnRNP R but not LARP7 (Fig 2O–R). An in vitro modification assay confirmed that all three mutants failed to undergo pseudouridylation at U250 (Fig 2F). Finally, ruling out a potential artifact caused by transient expression of tagged proteins, the ability of endogenous CDK9 to discriminate between WT and the three 7SK mutants was found to be the same as that of transfected CDK9‐F (Fig EV1).
Isomerization of uridine to Ψ results in the formation of an extra hydrogen bond donor capable of coordinating a water molecule 20. The resulting structural changes are largely stabilizing as they promote the RNA structural and thermal stability, and can also influence RNA–protein and RNA–RNA interactions 14. Consistent with this general view of the function of pseudouridylation, our results above demonstrate that Ψ250 in 7SK RNA promotes 7SK RNP formation by stabilizing the interactions with HEXIM1 and P‐TEFb. Moreover, the box H/ACA RNP is likely responsible for the modification.
DKC1 is involved in 7SK pseudouridylation
As DKC1 is the catalytic subunit of a box H/ACA RNP, we examined whether it interacted with 7SK RNA. Indeed, ectopically expressed V5‐DKC1 co‐immunoprecipitated with endogenous 7SK RNA (Fig 3A). Moreover, immunoprecipitated CDK9‐F also pulled down DKC1 in addition to the 7SK RNP components HEXIM1 and LARP7 (Fig 3B). The physical interaction between DKC1 and 7SK snRNP supports the notion that the box H/ACA RNP directs 7SK RNA pseudouridylation.
Figure 3. DKC1–H/ACA snoRNP is responsible for 7SK pseudouridylation and DKC1 KD releases P‐TEFb from 7SK RNP to form Brd4‐P–TEFb and Tat–SEC complexes.
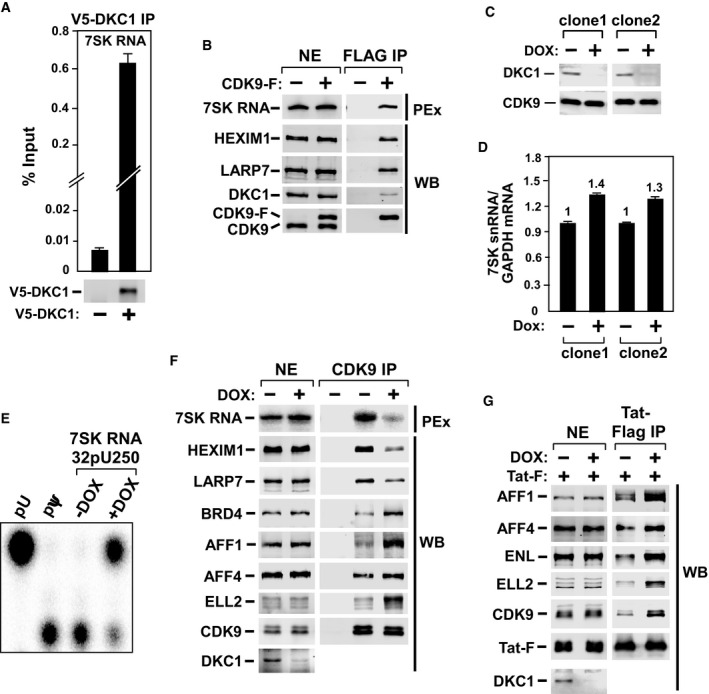
- Anti‐V5 immunoprecipitates (IP) from HeLa cells expressing no V5–DKC1 (−) or V5‐DKC1 (+) were analyzed by immunoblotting for V5‐DKC1 and qRT–PCR for the bound 7SK RNA. The bars represent mean ± SD from three independent experiments.
- Anti‐Flag IP from NEs of HeLa or the HeLa‐derived F1C2 cells expressing CDK9‐F were analyzed by Western blotting (WB) for the indicated proteins and primer extension (PEx) for 7SK RNA.
- WB analysis of DKC1 and CDK9 levels in two HeLa clones inducibly expressing shDKC1 upon doxycycline (DOX) treatment for 5 days.
- The 7SK RNA levels in the two clones from (C) were detected by qRT–PCR before and after DOX treatment and normalized to the GAPDH mRNA levels. The bars represent mean ± SD from three independent experiments.
- 1D‐TLC analysis of Ψ250 in 7SK RNA from inducible DKC1 KD clone 1 before and after DOX treatment.
- NEs and anti‐CDK9 IP from NEs of inducible DKC1 KD clone 1, either untreated or treated with DOX, were analyzed by WB for the indicated proteins and PEx for 7SK RNA.
- Clone 1 was treated with DOX for 3 days and then transfected with the Tat‐Flag cDNA (2 μg/150‐mm culture dish). Two days later, NEs and anti‐Flag IP from NEs were analyzed as in (F).
To further test this idea, we constructed HeLa‐based cell lines inducibly expressing a short hairpin RNA (shRNA) that targets DKC1 (shDKC1). The DKC1 protein level was markedly decreased upon doxycycline (DOX) treatment in two independent knockdown (KD) clones (Fig 3C). Although the 7SK RNA level remained largely unaffected by the KD (even slightly increased, Fig 3D), the Ψ250 level was drastically decreased (Fig 3E). Given that the KD efficiency was ~80%, these results indicate the box H/ACA RNP as the predominant enzyme catalyzing the pseudouridylation of 7SK in vivo, although the possible involvement of other minor modifiers cannot be completely ruled out at this stage.
DKC1 KD releases P‐TEFb from 7SK RNP to form Brd4‐P–TEFb and Tat–SEC complexes
To determine whether the impairment of 7SK RNA pseudouridylation by DKC1 KD affects 7SK RNP formation, we performed anti‐CDK9 immunoprecipitations in NE of DKC1 KD cells that were either untreated or treated with DOX to induce shDKC1 expression. The KD significantly decreased the levels of 7SK RNA, HEXIM1, and LARP7 bound to the immunoprecipitated CDK9 (Fig 3F), indicating disruption of 7SK RNP and release of P‐TEFb. The same conclusion was also reached when the effect of DKC1 KD on endogenous 7SK RNP was analyzed in a glycerol gradient (Fig EV2). The KD caused HEXIM1, and to a lesser degree, CDK9 to move out of fractions 9–15 (dashed box) corresponding to the large‐size 7SK snRNP to the top of the gradient that contained smaller size complexes and free proteins. Notably, although 7SK RNP can also be disrupted by certain drugs such as DRB and the CDK9 inhibitor i‐CDK9 21 (Fig EV3A), the drug‐induced disruption did not change the modification state of U250 (Fig EV3B).
Figure EV2. Glycerol gradient analysis of DKC1 KD‐induced 7SK RNP disruption.
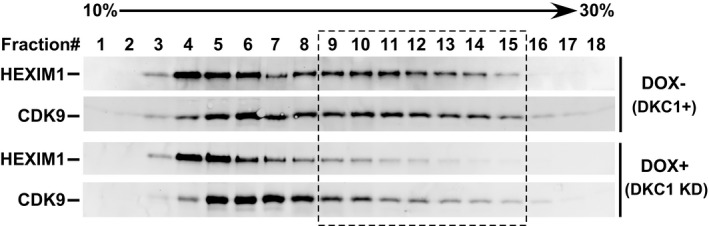
NEs from inducible DKC1 KD clone 1 before and after the DOX‐induced shDKC1 expression were analyzed by sedimentation through a 10–30% glycerol gradient. HEXIM1 and CDK9 in collected fractions were detected by immunoblotting. The dashed box highlights the fractions in which the levels of the two proteins drop upon KD.
Figure EV3. Drug‐induced 7SK RNP destruction does not change Ψ250 level in 7SK RNA.
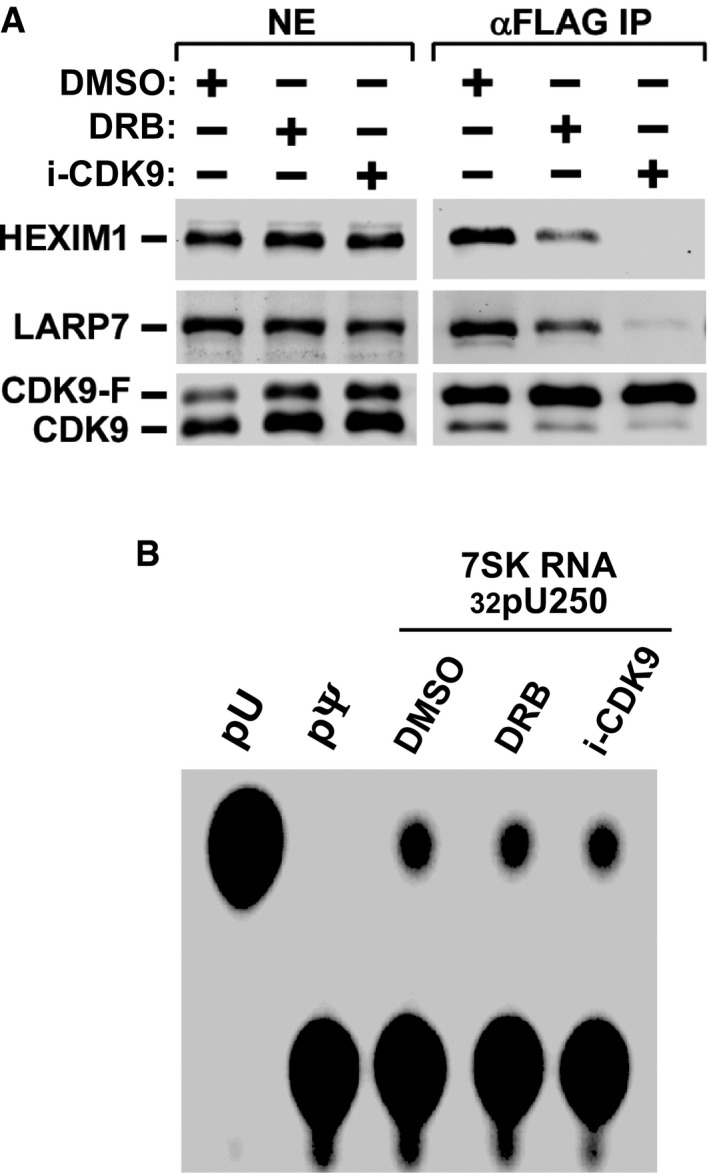
- F1C2 cells expressing CDK9‐F were treated with the indicated drugs or DMSO. Anti‐Flag IP from NEs were analyzed by immunoblotting.
- Total RNA was purified from the treated cells, subjected to the procedure described in Fig 1D, and analyzed by 1D‐TLC.
In addition to disrupting 7SK RNP, the KD also markedly increased the interactions of CDK9 with Brd4 and the SEC subunits especially AFF1 and ELL2, revealing a transfer of P‐TEFb from the inactive 7SK RNP to the active Brd4‐P–TEFb complex and SEC under the KD conditions. The SEC family of complexes, particularly the ones containing AFF1 and ELL2, play an important role in mediating Tat‐transactivation 6, 8, 9, 22. In addition to the above demonstration that DKC1 KD promoted the SEC formation, examination of the Tat‐associated proteins under the KD conditions also revealed a significant increase in the interaction of the AFF1/ELL2‐SEC with Tat (Fig 3G). Together, these data support a model in which impairment of 7SK pseudouridylation by DKC1 KD shifts the P‐TEFb functional equilibrium toward the active state by promoting the formation of both the Tat–SEC and Brd4‐P–TEFb complexes.
DKC1 KD preferentially activates Tat‐dependent HIV‐1 transcription by increasing SEC binding to viral promoter
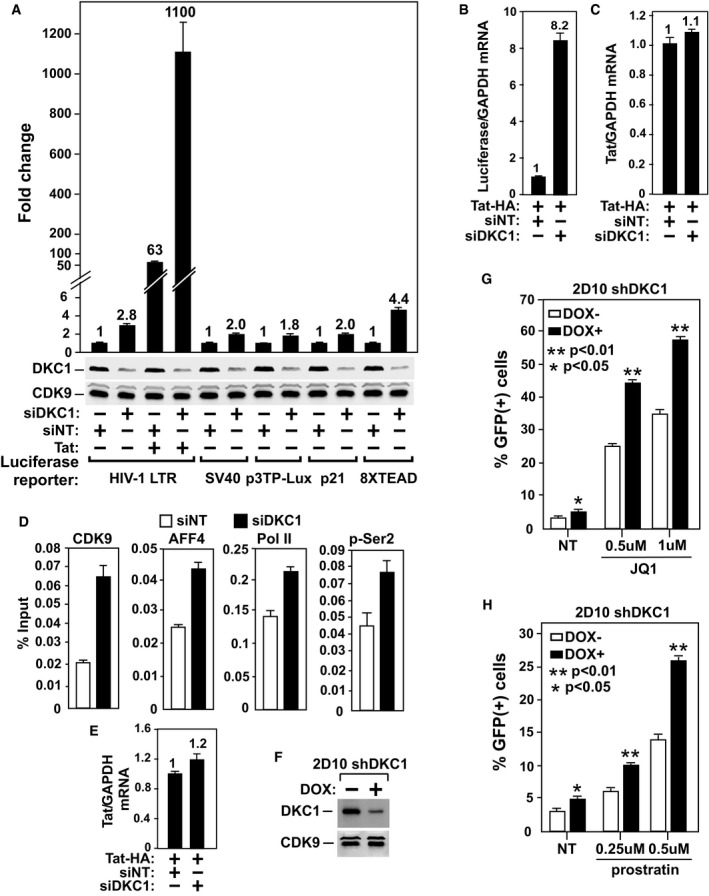
To investigate the functional significance of the DKC1 KD‐induced release of P‐TEFb from 7SK snRNP, we first examined the effect of the KD on expression of a luciferase reporter gene placed under the control of various viral and cellular gene promoters. Consistent with the observation above that the KD shifted the P‐TEFb equilibrium toward the active Brd4‐P–TEFb and SEC complexes, the introduction of the DKC1‐specific siRNA (siDKC1) into HeLa cells produced a small but consistent increase ranging from 1.8‐ to 4.4‐fold in luciferase expression from all the promoters tested (Fig 4A). Interestingly, although the basal, Tat‐independent HIV‐1 promoter activity was induced by only 2.8‐fold, and the Tat‐dependent activity was elevated 17.5‐fold. Notably, Tat failed to activate any of the non‐HIV‐1 promoters used in this assay (Fig EV4).
Figure 4. DKC1 KD preferentially activates Tat‐dependent HIV‐1 transcription and latency reversal by increasing SEC binding to HIV‐1 promoter.
-
ALuciferase activities were measured in HeLa cells transfected with the indicated reporter constructs, an empty vector (−) or the Tat cDNA, and either siDKC1 or a non‐targeting siRNA (siNT). For each reporter, the activity in cells expressing siNT was set to 1.
-
B, CThe mRNA levels of luciferase (B) or Tat (C) in cells expressing the indicated siRNAs were determined by qRT–PCR and normalized to the GAPDH mRNA and plotted.
-
DNH1 cells containing the integrated HIV‐1 LTR‐luciferase reporter and expressing Tat were transfected with siDKC1 or siNT. ChIP–qPCR analysis was performed to determine the levels of the indicated factors bound to the HIV‐1 promoter. The signals were normalized to those of input and plotted.
-
EThe mRNA levels of Tat in cells expressing the indicated siRNAs were determined by qRT–PCR and normalized to the GAPDH mRNA and plotted.
-
FWestern blot analysis of DKC1 and CDK9 levels in a Jurkat 2D10‐derived clone inducibly expressing shDKC1 upon exposure to DOX.
-
G, HThe inducible DKC1 KD clone, either untreated or treated with DOX for 4 days, was incubated with the indicated concentrations of JQ1 (G) or prostratin (H) for 24 h. GFP expression was measured by flow cytometry and expressed as percentage of GFP(+) cells of the entire population.
Figure EV4. Tat‐transactivation is specific for HIV‐1.
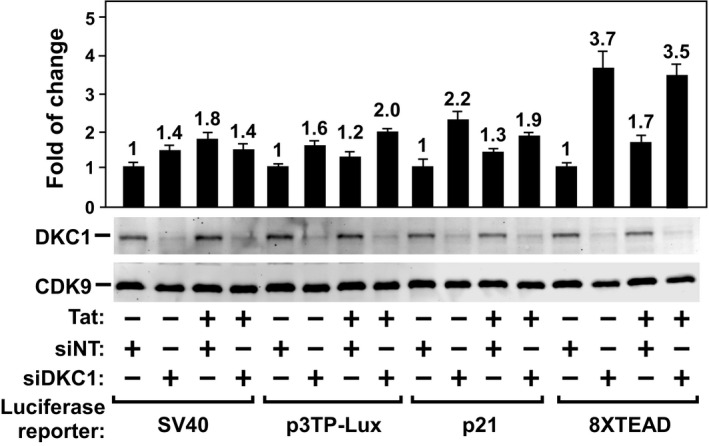
Luciferase activities were measured in HeLa cells transfected with the indicated reporter constructs and siRNA, and an empty vector (−) or the Tat cDNA. For each reporter, the activity in cells expressing siNT was set to 1. The bars represent mean ± SD from three independent experiments.
The preferential effect of the DKC1 KD on Tat‐transactivation was not due to any major increase in Tat expression but instead reflects elevated HIV‐1 transcription, as the qRT–PCR analyses showed an increase in the mRNA levels of luciferase but not Tat upon the KD (Fig 4B and C). It is highly likely that the enhanced Tat–SEC interaction (Fig 3G) that is known to support optimal Tat function 6, 22, 23 was responsible for this preferential effect in the KD cells. Consistently, our ChIP–qPCR analysis showed that the KD increased occupancy of SEC at the HIV‐1 promoter, which in turn resulted in higher levels of both total Pol II and Ser2‐phosphorylated Pol II detected at this region (Fig 4D and E).
Impairment of 7SK pseudouridylation promotes reversal of HIV‐1 latency
The suppression of P‐TEFb activity in resting T cells through sequestration in 7SK RNP contributes to establishment of HIV‐1 latency 24, 25, raising the possibility that the decreased 7SK pseudouridylation upon DKC1 KD may facilitate the reversal of viral latency by releasing P‐TEFb from the RNP. To test this idea, we created inducible DKC1 KD cells in the Jurkat 2D10 system, a widely used post‐integrative latency model 26. The DOX‐induced depletion of ~50% of cellular DKC1 (Fig 4F) did not cause obvious cell death, but activated the LTR‐driven GFP expression by ~85% (Fig 4G and H). Moreover, the KD enhanced the ability of two well‐established latency‐reversing agents (LRAs), JQ1 and prostratin, to induce the GFP production by ~80–100% depending on the concentrations of the drugs (Fig 4G and H). The lack of a more robust activation was likely caused by the inefficient DKC1 KD. Nevertheless, these results suggest that although the KD‐induced release of P‐TEFb from 7SK RNP alone may not be enough to fully reverse HIV‐1 latency, it can be used in conjunction with other LRAs to further enhance their effectiveness.
The data presented in this study indicate that 7SK is pseudouridylated predominately by the DKC1–box H/ACA RNPs. Although these RNPs also modify other RNA including spliceosomal snRNA and ribosomal rRNA, which could broadly influence gene expression, we believe this is unlikely to account for the preferential activation of HIV‐1 Tat function observed here. Both the DKC1 KD and mutagenesis that disrupt the pseudouridylation target sequence in 7SK resulted in a redistribution of P‐TEFb from the inactive 7SK RNP to the active Tat–SEC that is required for optimal Tat‐transactivation.
According to the structural model proposed by Wassarman and Steitz 1, 7SK RNA contains four hairpins and U250 resides in the third one. Previously, HEXIM1 and P‐TEFb have been shown to bind to two distinct elements located in the 5′‐ and 3′‐terminal hairpins of 7SK 27. Surprisingly, the deletion of the middle portion of 7SK including the third hairpin did not disrupt these interactions. There are two possible explanations for the apparent discrepancy between these results and our current findings. The first is that the lack of pseudouridylation at U250 may change the overall folding and structure of the entire 7SK molecule including the terminal 5′ and 3′ hairpins, which in turn decreases binding by HEXIM1 and P‐TEFb. In contrast, the simple deletion of the 3rd hairpin does not produce such a global effect and may still allow the two terminal hairpins to form independently and bind HEXIM1 and P‐TEFb. The second possibility is that the loss of pseudouridylation at U250 may enable the recruitment of an unknown factor that interferes with 7SK RNP formation. In this scenario, the deletion of the 7SK middle section causes loss of the binding site for this factor, and thus, no disruption of 7SK RNP occurs. Future studies are necessary to test these two possibilities and directly measure the impact of Ψ250 on 7SK's global structure and folding.
Our findings that targeting DKC1 inhibits 7SK pseudouridylation and affects P‐TEFb transcriptional activity also have important clinical implications, as mutations in DKC1 are associated with a number of human diseases, including X‐linked dyskeratosis congenita (X‐DC) and the Hoyeraal–Hreidarsson syndrome 28, 29. Furthermore, X‐DC mutations in DKC1 are known to impair pseudouridylation and stability of various box H/ACA snoRNAs 30. In light of these, future studies are needed to determine whether defects in 7SK RNA pseudouridylation and P‐TEFb regulation contribute to the pathology of the DKC1 diseases.
Efforts are currently underway to develop effective strategies to “shock” the latent HIV reservoirs for their subsequent “kill” by anti‐retroviral therapy 24, 31. Several LRAs have been used in the “shock” phase of the therapy but are highly toxic or ineffective, and thus, new and improved LRAs are needed 32, 33. Our present data support the idea that the 7SK RNP plays an inhibitory role in HIV's transcription and escape from latency. The demonstration that this RNP can be destabilized by reducing 7SK RNA pseudouridylation, which activates HIV transcription and promotes latency reversal, implicates the DKC1–box H/ACA RNPs as a promising new target for developing novel LRAs to eradicate latent viral reservoirs.
Materials and Methods
Antibodies
Antibodies against DKC1 (Bethyl, Cat. A302‐591A), AFF1 (Bethyl, Cat. A302‐344A; A302‐345A), ELL2 (Bethyl, Cat. A302‐505A‐1), ENL (Bethyl, Cat. A302‐267A), AFF4 (Abcam, Cat. ab57077; Bethyl, Cat. A302‐539A‐1), Flag (M2) (Sigma‐Aldrich, Cat. F1804), HA (3F10) (Roche, Cat. 11867423001), pSer2 Pol II (Abcam, Cat. ab5095), and total Pol II (Santa Cruz, Cat. sc‐899X) were purchased commercially. The antibodies against CDK9, LARP7, HEXIM1 were generated in our own laboratory and have been described previously 5, 17, 34.
Transfection, co‐IP, and primer extension
HeLa cells (60–80% confluent) were transfected with the constructs expressing the various tagged protein and tagged 7SK snRNA and harvested 48 h post‐transfection. The co‐IP assay was performed as described 6 with minor modifications. Briefly, the preparation of nuclear extracts (NE) followed the classic procedure by Dignam et al 35 without any further dialysis. For anti‐Flag or anti‐HA immunoprecipitations (IP), NE prepared from the transfected HeLa cells were incubated with anti‐Flag (Sigma‐Aldrich) or anti‐HA agarose beads (Roche) for 2 h at 4°C. After incubation, the immunoprecipitates were washed extensively with buffer D [20 mM Hepes‐KOH (pH 7.9), 15% (vol/vol) glycerol, 0.2 mM EDTA, 0.2% Nonidet P‐40, 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride, and 0.3 M KCl] before elution with Flag or HA peptides (0.5 mg/ml) dissolved in this buffer. The eluted materials were analyzed by Western blotting or primer extension. For anti‐CDK9 IP, NE were incubated with anti‐CDK9 antibodies or total rabbit IgG overnight and then with protein A beads (Invitrogen) for 1 h. After extensive wash in buffer D, one‐third of the beads were saved for RNA extraction by TRIzol reagent and then subjected to analyses by primer extension as described 36. The remaining beads were eluted by a low pH solution (200 mM glycine, pH 2.5). The neutralized eluate (with 1/20 volume of 2 M Tris–HCl, pH 8.8) was subjected to analysis by Western blotting. The primer extension results were quantified with GelQuant.NET software provided by biochemlabsolutions.com.
Glycerol gradient analysis
The assay was performed essentially as described 22 with minor modifications. Briefly, glycerol gradients (10–30%) were established in buffer D in 13.5‐ml Beckman centrifugation tubes. The nuclear pellets prepared from inducible DKC1 KD clone 1 before (−) and after (+) the DOX‐induced shDKC1 expression for 5 days were lysed in 0.5 ml of buffer D for 30 min at 4°C. The lysates were carefully loaded over the top of the glycerol gradients after centrifugation at 17,900 g for 10 min. NE were fractioned by centrifugation in an SW 41 Ti rotor (Beckman) at 38,000 rpm for 21 h. Fractions were analyzed by immunoblotting with the appropriate antibodies after precipitation with trichloroacetic acid.
Pseudouridylation assays
The in vitro assay was performed as previously described 37 with some modifications. Briefly, one T75 flask (80–100% confluent) of HeLa cells was collected through microfugation. Nuclear fractionation was performed using the REAP method as described previously 38, except that the nuclear pellets were re‐suspended in 200 μl of extraction buffer containing 20 mM HEPES at pH 7.9, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 0.5 mM PMSF, and 25% glycerol. Following incubation at 4°C, extracts were microfuged for 10 min at 14,000 g, the supernatant was recovered, and used for the pseudouridylation assay. About 1,000 cpm 32P‐labeled substrate was mixed with 5 μl of nuclear extract, in a final volume of 10 μl containing 100 mM Tris–HCl at pH 8.0, 100 mM ammonium acetate, 5 mM MgCl2, 2 mM DTT, 0.1 mM EDTA. Reactions were conducted at 37°C for < 1 h. RNA was then recovered through phenol–chloroform extraction and ethanol precipitation and subjected to nuclease P1 digestion and TLC analysis as described previously 39. For 2D‐TLC, 7SK RNA isolated from CDK9‐Flag immunoprecipitates or in vitro transcribed was digested to completion with 0.5–1 U of RNase T2 (Worthington) in 50 mM ammonium acetate, 0.05% SDS, and 1 mM EDTA. The samples were then 5′ 32P‐labeled with T4 polynucleotide kinase and resolved by 2D‐TLC as previously described 40.
The validation of Ψ sites by SCARLET was performed as previously described 15, with a few modifications. Briefly, 7SK RNA was cleaved immediately 5′ of the nucleotides of interest using 10 pM of 10–23 DNAzymes in T4 PNK buffer as previously described 15. DNAzyme cleavage generates a 5′‐OH, which was directly 32P‐labeled with T4 PNK. The mixture was then annealed with 10 pmol corresponding splint oligos and 10 pmol desthiolbiotinylated DNA oligos by heating at 75°C for 5 min followed by addition of 2.5 μl ligation buffer (1.4× PNK buffer, 0.3 mM ATP, 57% DMSO, 5 U/μl T4 DNA ligase) and incubation for 4 h at 37°C and overnight at room temperature. The reaction was brought up to 500 μl with 150 mM NaCl, 10 mM Tris, pH 7.5, captured by Streptavidin‐magnetic C1 beads (Invitrogen), and digested for 1D‐TLC as described above.
DKC1 knockdown (KD)
To generate the HeLa‐ or Jurkat 2D10‐based inducible DKC1 KD cells, a specific shRNA (shDKC1) sequence 5′‐CCGGGGACAGGTTTCATTAATCTTTCAAGAGAAGATTAATGAAACCTGTCCTTTTTTG‐3′ was inserted into the Tet‐on‐pLKO lentiviral vector 41, 42. shRNA targeting GFP was used as a non‐targeting control. Lentivirus production and infection were conducted as described 43. The KD was induced by DOX for 5 days. To reduce DKC1 expression in the HeLa‐based NH1 cells 44, siDKC1 SMARTpool (L‐013639‐00‐0005, Dharmacon) or the non‐targeting pool (D‐001810‐10, Dharmacon) was transfected using INTERFERin siRNA transfection reagents (Polyplus‐transfection, Cat. 409‐10).
ChIP–qPCR
The assay was performed as described 44 with some modifications. Briefly, NH1 cells containing the integrated HIV‐1 LTR‐luciferase reporter construct and transfected with the Tat expression constructs were cross‐linked with 1% formaldehyde for 10 min and then quenched by 0.125 M glycine for 5 min. Fixed cells were collected and lysed in SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris, pH 8.1). Chromatin DNA was fragmented to ~200–1,000 bp in length with the Covaris‐S2 sonicator (Covaris). One‐tenth of the sonicated lysates were incubated overnight with 3 μg of the indicated antibodies per reaction and then with Protein A Dynabeads (Life Technologies) for 1 h. After extensively wash, immunoprecipitated DNA was purified by PCR Purification kit (QIAGEN) and analyzed by qPCR. All qPCR signals were normalized to the input, and signals generated by immunoprecipitations with rabbit total IgG were subtracted from the signals obtained by the specific antibodies. The sequences of the PCR primers used for amplification of the HIV‐1 LTR are: Forward, 5′‐GTTAGACCAGATCTGAGCCCT‐3′; Reverse, 5′‐GTGGGTTCCCTAGTTAGCCA‐3′.
Author contributions
YZ and JK performed experiments and analyzed data. YZ, JK, and QZ conceived/designed experiments. YZ and QZ wrote the manuscript. JK and BG reviewed and edited the manuscript. QZ and BG provided support and supervised the project.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank T. Kiss (Université Paul Sabatier, France) and N. J. Krogan (University of California, San Francisco) for HA‐tagged hnRNP R and V5‐tagged DKC1 plasmids, respectively. This work was supported by Public Health Service grants from the National Institutes of Health (R01AI041757 and R01AI095057) to Q.Z., grants from the California HIV/AIDS Research Program (ID13‐B‐529) and Howard Hughes Medical Institute to B.G., and a Damon Runyon Cancer Research Foundation fellowship (DRG# 2121‐12) to J.K.
EMBO Reports (2016) 17: 1441–1451
References
- 1. Wassarman DA, Steitz JA (1991) Structural analyses of the 7SK ribonucleoprotein (RNP), the most abundant human small RNP of unknown function. Mol Cell Biol 11: 3432–3445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhou Q, Li T, Price DH (2012) RNA polymerase II elongation control. Annu Rev Biochem 81: 119–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zieve G, Benecke BJ, Penman S (1977) Synthesis of two classes of small RNA species in vivo and in vitro . Biochemistry 16: 4520–4525 [DOI] [PubMed] [Google Scholar]
- 4. Nguyen VT, Kiss T, Michels AA, Bensaude O (2001) 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature 414: 322–325 [DOI] [PubMed] [Google Scholar]
- 5. Yang Z, Zhu Q, Luo K, Zhou Q (2001) The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 414: 317–322 [DOI] [PubMed] [Google Scholar]
- 6. He N, Liu M, Hsu J, Xue Y, Chou S, Burlingame A, Krogan NJ, Alber T, Zhou Q (2010) HIV‐1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV‐1 transcription. Mol Cell 38: 428–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sobhian B, Laguette N, Yatim A, Nakamura M, Levy Y, Kiernan R, Benkirane M (2010) HIV‐1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol Cell 38: 439–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li Z, Lu H, Zhou Q (2016) A minor subset of Super Elongation Complexes plays a predominant role in reversing HIV‐1 latency. Mol Cell Biol 36: 1194–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lu H, Li Z, Zhang W, Schulze‐Gahmen U, Xue Y, Zhou Q (2015) Gene target specificity of the Super Elongation Complex (SEC) family: how HIV‐1 Tat employs selected SEC members to activate viral transcription. Nucleic Acids Res 43: 5868–5879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q (2005) Recruitment of P‐TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell 19: 535–545 [DOI] [PubMed] [Google Scholar]
- 11. Wu SY, Chiang CM (2007) The double bromodomain‐containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem 282: 13141–13145 [DOI] [PubMed] [Google Scholar]
- 12. Hargreaves DC, Horng T, Medzhitov R (2009) Control of inducible gene expression by signal‐dependent transcriptional elongation. Cell 138: 129–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carlile TM, Rojas‐Duran MF, Zinshteyn B, Shin H, Bartoli KM, Gilbert WV (2014) Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515: 143–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Karijolich J, Yi C, Yu YT (2015) Transcriptome‐wide dynamics of RNA pseudouridylation. Nat Rev Mol Cell Biol 16: 581–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu N, Parisien M, Dai Q, Zheng G, He C, Pan T (2013) Probing N6‐methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 19: 1848–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gruber AR, Koper‐Emde D, Marz M, Tafer H, Bernhart S, Obernosterer G, Mosig A, Hofacker IL, Stadler PF, Benecke BJ (2008) Invertebrate 7SK snRNAs. J Mol Evol 66: 107–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. He N, Jahchan NS, Hong E, Li Q, Bayfield MA, Maraia RJ, Luo K, Zhou Q (2008) A La‐related protein modulates 7SK snRNP integrity to suppress P‐TEFb‐dependent transcriptional elongation and tumorigenesis. Mol Cell 29: 588–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Van Herreweghe E, Egloff S, Goiffon I, Jady BE, Froment C, Monsarrat B, Kiss T (2007) Dynamic remodelling of human 7SK snRNP controls the nuclear level of active P‐TEFb. EMBO J 26: 3570–3580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tafer H, Kehr S, Hertel J, Hofacker IL, Stadler PF (2010) RNAsnoop: efficient target prediction for H/ACA snoRNAs. Bioinformatics 26: 610–616 [DOI] [PubMed] [Google Scholar]
- 20. Charette M, Gray MW (2000) Pseudouridine in RNA: what, where, how, and why. IUBMB Life 49: 341–351 [DOI] [PubMed] [Google Scholar]
- 21. Lu H, Xue Y, Yu GK, Arias C, Lin J, Fong S, Faure M, Weisburd B, Ji X, Mercier A et al (2015) Compensatory induction of MYC expression by sustained CDK9 inhibition via a BRD4‐dependent mechanism. Elife 4: e06535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lu H, Li Z, Xue Y, Schulze‐Gahmen U, Johnson JR, Krogan NJ, Alber T, Zhou Q (2014) AFF1 is a ubiquitous P‐TEFb partner to enable Tat extraction of P‐TEFb from 7SK snRNP and formation of SECs for HIV transactivation. Proc Natl Acad Sci USA 111: E15–E24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schulze‐Gahmen U, Upton H, Birnberg A, Bao K, Chou S, Krogan NJ, Zhou Q, Alber T (2013) The AFF4 scaffold binds human P‐TEFb adjacent to HIV Tat. Elife 2: e00327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Richman DD, Margolis DM, Delaney M, Greene WC, Hazuda D, Pomerantz RJ (2009) The challenge of finding a cure for HIV infection. Science 323: 1304–1307 [DOI] [PubMed] [Google Scholar]
- 25. Karn J (2011) The molecular biology of HIV latency: breaking and restoring the Tat‐dependent transcriptional circuit. Curr Opin HIV AIDS 6: 4–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pearson R, Kim YK, Hokello J, Lassen K, Friedman J, Tyagi M, Karn J (2008) Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J Virol 82: 12291–12303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Egloff S, Van Herreweghe E, Kiss T (2006) Regulation of polymerase II transcription by 7SK snRNA: two distinct RNA elements direct P‐TEFb and HEXIM1 binding. Mol Cell Biol 26: 630–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Knight SW, Heiss NS, Vulliamy TJ, Aalfs CM, McMahon C, Richmond P, Jones A, Hennekam RC, Poustka A, Mason PJ et al (1999) Unexplained aplastic anaemia, immunodeficiency, and cerebellar hypoplasia (Hoyeraal‐Hreidarsson syndrome) due to mutations in the dyskeratosis congenita gene, DKC1. Br J Haematol 107: 335–339 [DOI] [PubMed] [Google Scholar]
- 29. Heiss NS, Knight SW, Vulliamy TJ, Klauck SM, Wiemann S, Mason PJ, Poustka A, Dokal I (1998) X‐linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet 19: 32–38 [DOI] [PubMed] [Google Scholar]
- 30. Bellodi C, McMahon M, Contreras A, Juliano D, Kopmar N, Nakamura T, Maltby D, Burlingame A, Savage SA, Shimamura A et al (2013) H/ACA small RNA dysfunctions in disease reveal key roles for noncoding RNA modifications in hematopoietic stem cell differentiation. Cell Rep 3: 1493–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deeks SG (2012) HIV: shock and kill. Nature 487: 439–440 [DOI] [PubMed] [Google Scholar]
- 32. Rasmussen TA, Tolstrup M, Sogaard OS (2016) Reversal of latency as part of a cure for HIV‐1. Trends Microbiol 24: 90–97 [DOI] [PubMed] [Google Scholar]
- 33. Dahabieh MS, Battivelli E, Verdin E (2015) Understanding HIV latency: the road to an HIV cure. Annu Rev Med 66: 407–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yik JH, Chen R, Nishimura R, Jennings JL, Link AJ, Zhou Q (2003) Inhibition of P‐TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol Cell 12: 971–982 [DOI] [PubMed] [Google Scholar]
- 35. Dignam JD, Lebovitz RM, Roeder RG (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res 11: 1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Karijolich J, Abernathy E, Glaunsinger BA (2015) Infection‐induced retrotransposon‐derived noncoding RNAs enhance herpesviral gene expression via the NF‐kappaB pathway. PLoS Pathog 11: e1005260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Karijolich J, Yu YT (2011) Converting nonsense codons into sense codons by targeted pseudouridylation. Nature 474: 395–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Suzuki K, Bose P, Leong‐Quong RY, Fujita DJ, Riabowol K (2010) REAP: a two minute cell fractionation method. BMC Res Notes 3: 294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhao X, Yu YT (2004) Detection and quantitation of RNA base modifications. RNA 10: 996–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zebarjadian Y, King T, Fournier MJ, Clarke L, Carbon J (1999) Point mutations in yeast CBF5 can abolish in vivo pseudouridylation of rRNA. Mol Cell Biol 19: 7461–7472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wiederschain D, Wee S, Chen L, Loo A, Yang G, Huang A, Chen Y, Caponigro G, Yao YM, Lengauer C et al (2009) Single‐vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle 8: 498–504 [DOI] [PubMed] [Google Scholar]
- 42. Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R, Stegmeier F, Yao YM, Lengauer C (2008) PTEN‐deficient cancers depend on PIK3CB. Proc Natl Acad Sci USA 105: 13057–13062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK et al (2006) A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high‐content screen. Cell 124: 1283–1298 [DOI] [PubMed] [Google Scholar]
- 44. Li Z, Guo J, Wu Y, Zhou Q (2013) The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat‐transactivation. Nucleic Acids Res 41: 277–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File