

Abstract
We have developed a novel real‐time quaking‐induced conversion RT‐QuIC‐based assay to detect alpha‐synuclein aggregation in brain and cerebrospinal fluid from dementia with Lewy bodies and Parkinson's disease patients. This assay can detect alpha‐synuclein aggregation in Dementia with Lewy bodies and Parkinson's disease cerebrospinal fluid with sensitivities of 92% and 95%, respectively, and with an overall specificity of 100% when compared to Alzheimer and control cerebrospinal fluid. Patients with neuropathologically confirmed tauopathies (progressive supranuclear palsy; corticobasal degeneration) gave negative results. These results suggest that RT‐QuiC analysis of cerebrospinal fluid is potentially useful for the early clinical assessment of patients with alpha‐synucleinopathies.
Introduction
Alpha‐synuclein (a‐syn) is well conserved, small acidic protein of 140 amino acids, and a molecular weight of 19 kDa that is encoded by the SNCA gene located on chromosome 4.1 It is located in high concentration in the presynaptic nerve terminals within the central nervous system where it plays a role in synaptic vesicle biology.2 The synucleinopathies are a set of neurodegenerative disorders associated with the deposition of fibrillary aggregates of a‐syn within selective populations of neurons and glia. These deposits can be found within neuronal soma as Lewy bodies (LB) or in dystrophic neurites in diseases such as Parkinson's disease (PD) or dementia with Lewy bodies (DLB), or in glial cytoplasmic inclusions in multiple system atrophy (MSA).3
The presence of a‐syn has been detected in biological fluids such as cerebrospinal fluid (CSF)4 and serum.5 The measurement of a‐syn concentrations in CSF by ELISAs has been proposed as a biomarker for a‐syn‐related disorders. However, despite many studies showing a reduction in CSF a‐syn levels in PD and in DLB,4, 6 overall results are not consistent.7 Even in those studies where a reduction in CSF a‐syn has been demonstrated, the differences are small8 and there is considerable overlap within patient groups and between patient and control groups. In addition the standardization of CSF a‐syn measurement between laboratories has proven difficult.9
The aggregation properties of a‐syn have recently been compared to prion protein, the aggregation of which causes transmissible spongiform encephalopathies (TSEs).10 Indeed, there is much discussion in current literature as to whether the alpha‐synucleinopathies are actually prion‐like diseases.11 A recently described technique called real‐time quaking‐induced conversion (RT‐QuIC), which exploits the ability of prion protein to induce self‐aggregation, has been used to develop a diagnostic CSF test for sporadic Creutzfeldt–Jakob disease (sCJD), the most common human form of TSE.12 Using our expertize in RT‐QuIC,12 we have developed and tested a similar assay for the detection of a‐syn in CSF.
Materials and Methods
Real‐time quaking induced aggregation for alpha‐synuclein
The RT‐QuiC reaction buffer (RB) was composed of 100 mmol/L phosphate buffer (pH 8.2), 10 μmol/L thioflavin T (ThT), and 0.1 mg/mL human recombinant full‐length (1–140 aa) alpha‐synuclein (Stratech, Cambridge, UK). Each well of a black 96‐well plate with a clear bottom (Nalgene Nunc International, Fisher Scientific Ltd, Loughborough, UK) contained 98 μL, 90 μL, or 85 μL RB (depending on volume of seed added) and 37 ± 3 mg of 0.5 mm zirconium/silica beads (Thistle Scientific Ltd, Glasgow, UK). Reactions were seeded with 2 μL of working strength brain homogenate, 5 μL, 10 μL or 15 μL, of undiluted CSF to a final reaction volume of 100 μL. The plates were sealed with a plate sealer film (Fisher Scientific Ltd) and incubated in a BMG OPTIMA FluoSTAR plate reader at 30°C for 120 h with intermittent shaking cycles: double orbital with 1 min shake (200 rpm), 14 min rest. ThT fluorescence measurements (450 nm excitation and 480 nm emission) were taken every 15 min. Each sample was run in duplicate, allowing two negative control samples (reactions seeded with SD and AD brain homogenate), one positive control (reaction seeded with DLB brain homogenate), an unseeded reaction, and 44 CSF samples to be tested on one plate.
Patient groups
Initial phase of CSF RT‐QuIC development was carried out on 99 CSF samples obtained from the OPTIMA cohort (Oxford Project to Investigate Memory and Ageing) with clinically and neuropathologically confirmed diagnosis of pure DLB (n = 12), PD (n = 2), progressive supranuclear palsy (PSP) (n = 2), corticobasal degeneration (n = 3), DLB with AD pathology (n = 17), AD with incidental LBs (n = 13), pure AD (n = 30), and controls (n = 20). OPTIMA initiated in 1988, is a prospective, longitudinal clinico‐pathological study of dementia and aging including CSF collection at multiple time points during clinical follow‐up. All clinical and pathological protocols have been described in detail13 and were approved by the local ethics committee and participants provided informed consent prior to enrollment.
The validation phase of RT‐QuIC was carried out on CSF samples (20 PD, 15 controls, and 3 at‐risk) obtained from the Oxford Discovery cohort (http://opdc.medsci.ox.ac.uk) which is one of the largest, clinically best‐characterized longitudinal PD cohorts to date. Full clinical details of this cohort have been described previously.14 In brief, patients with idiopathic PD diagnosed within 3.5 years according to UK PD Society Brain Bank diagnostic criteria15 were recruited between September 2010 and September 2014 from a 2.9 million population (ethics study 10/H0505/71). Mean disease duration among 20 PD patients was 1.6 ± 1.1 years (range 0.1–3.2 years) and Hoehn and Yahr stage 1.9 ± 0.4 (range: 1–3, maximum possible score 5). The control population was recruited from spouses and friends of patients taking part in the study, as well as the general public. The at‐risk group comprised patients with REM sleep behavior confirmed on overnight polysomnography,16 80% of which have shown to develop Lewy body disorder over time.17 Demographic information is given in Table 1.
Table 1.
Patient demographic information for the Optima and Discovery patients investigated
| Age at death Mean ± SD (range) | F/M | |
|---|---|---|
| OPTIMA patients (n) | ||
| Pure LBD (12) | 80.8 ± 6.5 (71–92) | 4/8 |
| Parkinson's disease (2) | 77.5 ± 7.8 (72–83) | 0/2 |
| Mixed LBD/AD (17) | 80.1 ± 6.4 (69–90) | 10/7 |
| AD with incidental LB (13) | 79.8 ± 7.8 (67–91) | 9/4 |
| Pure AD (30) | 77.7 ± 8.6 (61–93) | 17/13 |
| Progressive supranuclear palsy (PSP) (2) | 69.5 ± 3.5 (67–72) | 2/0 |
| Corticobasal degeneration (CBD) (3) | 64.0 ± 10.6 (52–72) | 1/2 |
| Controls (20) | 82.9 ± 6.9 (68–93) | 10/10 |
| Discovery patients (n) | ||
| Parkinson's disease (20) | 65.1 ± 9.1 (42–80) | 6/14 |
| At‐risk RBD patients (3) | 67.6 ± 7.7 (59–74) | 0/3 |
| Controls (15) | 65.8 ± 7.4 (55–83) | 8/7 |
Brain homogenates
Brain tissue was provided by the MRC Brain Bank in the NCJDRSU (ethical license 11/ES/0022). All tissue was frozen at −80°C within 2 h of being sampled and stored at −80°C prior to analysis. Brain tissues had been stored between 2 and 18 years prior to use.
Frontal cortex tissue was taken from patients with Alzheimer's disease (AD), sporadic Creutzfeldt–Jakob disease (sCJD), and diffuse Lewy body dementia (DLB). In addition, frontal cortex was obtained from individuals without neurodegenerative disease from the MRC Sudden Death Brain and Tissue Bank (Sudden Death [SD] controls). Both frontal cortex and substantia nigra tissue was obtained from patients with mixed AD/DLB, mixed sCJD/DLB, and mixed AD/PD. All cases used had been examined histologically and the diagnosis reached using internationally accepted criteria.18 Initial 10% w/v brain homogenates were prepared using phosphate‐buffered saline (PBS) containing 1 mmol/L EDTA, 150 mmol/L NaCl, 0.5% Triton X, and complete protease inhibitor cocktail from Roche. Subsequent working strength BHs were prepared by diluting the above 1:20,000 with PBS.
Cerebrospinal fluid samples
CSF was drawn from patients by lumbar puncture and samples with visible red color were excluded to avoid blood contamination. A total quantity of 15 mL of CSF was collected in 20 mL polypropylene tubes and samples were stored on ice until centrifuged for 5 min at 1300g at 4°C to remove cellular components. The supernatant was then divided into 1 mL aliquots stored in polypropylene cryotubes and frozen at −80°C for long‐term storage, prior to analysis. Although all CSF samples with visible red coloration were excluded from RT‐QuIC analysis in this study, experiments were undertaken to show that a‐syn RT‐QuIC analysis is not inhibited or falsely elevated in samples containing red cell concentrations of up to 1250 × 106/μL.
Ninety‐nine CSF samples from the OPTIMA cohort and 38 CSF samples from the Oxford Parkinson's Disease Centre (OPDC) Discovery study were received from the Nuffield Department of Clinical Neurosciences, University of Oxford. All CSF samples were transported from Oxford to Edinburgh on dry‐ice and stored at −80°C on arrival. In addition, CSF samples from patients with neuropathologically confirmed sCJD or DLB from the NCJDRSU CSF Bank were analyzed. Ethical approval for the use of CSF samples from the NCJDRSU CSF Bank was covered by Multi‐centre Research Ethics Committee for Scotland 05/MRE00/67. All CSF were spun and stored at −80°C prior to analysis.
Results
The development of RT‐QuIC was undertaken using frontal cortex BH from patients with a clinico‐pathological diagnosis of DLB, Alzheimer's disease (AD), and sCJD. Patients with no neuropathological evidence of neurological disease who died suddenly and were part of the MRC Sudden Death brain bank were used as controls (SD) (Fig. 1A). The RT‐QuIC reactions seeded with BH from DLB had a lag phase of 50 h, after which an increasing thioflavin T fluorescent signal was seen that became maximal at 70 h. None of the reactions seeded with brain homogenates from patients with other protein misfolding disorders (AD or sCJD) or the SD controls, gave a positive response even after 120 h.
Figure 1.
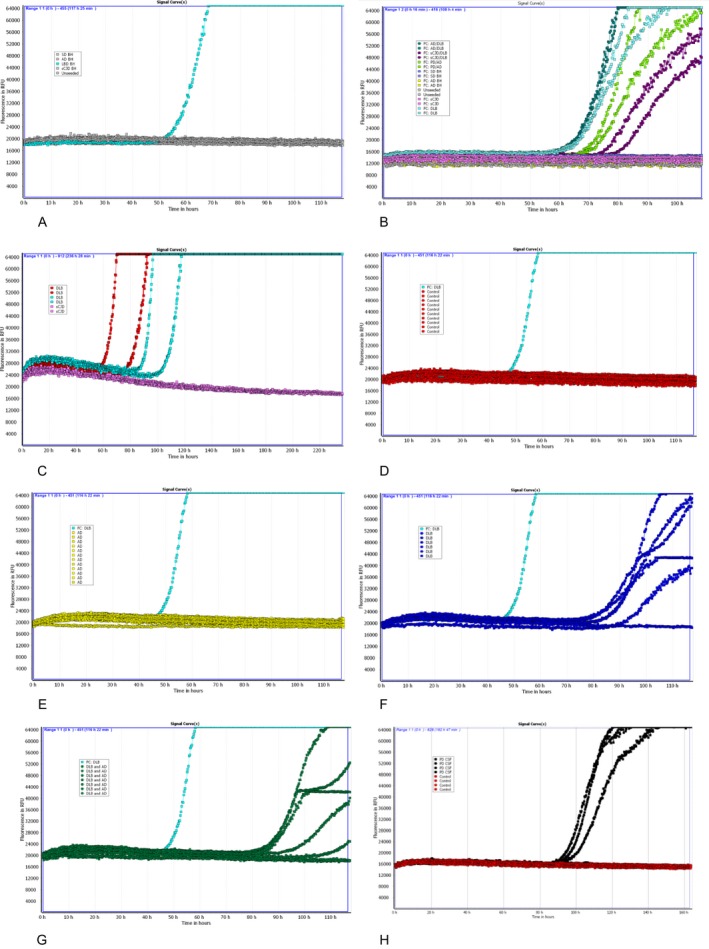
RT‐QuIC responses observed with reactions seeded with brain homogenates A, B, and CSF samples C–H. (A) Frontal cortex: DLB (light blue); AD, sCJD, SD, and unseeded reactions (gray); (B) Frontal cortex: DLB (light blue); mixed AD/DLB (dark green), mixed sCJD/DLB (purple), mixed AD/PD (light green), AD (yellow), sCJD (pink), SD (dark blue), unseeded reaction (gray); (C) CSF from two DLB patients (red, light blue) and one sCJD patient (pink); (D) CSF from 10 control patients (red); (E) CSF from 12 AD patients (yellow); (F) CSF from six DLB patients (dark blue); (G) CSF from seven mixed DLB/AD patients (dark green), frontal cortex DLB (e–h) (light blue); (H) CSF from four PD patients (one negative for RT‐QuIC) (light green) and four controls (red). A, D–H: reactions performed in duplicate and mean of duplicates illustrated; B, C: reactions performed in duplicate and both duplicates illustrated. CJD, Creutzfeldt–Jakob disease; CSF, cerebrospinal fluid; DLB, Lewy bodies; SD, sudden death.
Many disease pathologies commonly coexist, especially AD‐related and a‐syn pathology.19 To investigate whether the presence of an alternative protein misfolding disorder can interfere with the a‐syn aggregation induced by either DLB or PD, brain homogenates from the frontal cortex of patients with mixed pathologies were examined (Fig. 1B). The presence of second protein misfolding disorders such as AD or sCJD does not inhibit the RT‐QuIC reaction induced by DLB or PD brain homogenates (Fig. 1A and B). To investigate whether the RT‐QuIC method developed was sensitive enough to detect a‐syn in CSF, two CSF samples from patients with neuropathologically confirmed DLB and one CSF from a neuropathologically confirmed case of sCJD were analyzed (Fig. 1C). Both CSF samples from the DLB patients gave positive responses with a lag phase between 60 and 100 h.
An exploratory set of 99 in vivo CSF samples obtained as part of the OPTIMA study from patients with subsequent neuropathologically confirmed disease were analyzed at three different volumes (i.e., 5, 10, and 15 μL) to investigate the sensitivity and specificity of the RT‐QuIC and to calculate the optimal CSF volume for the analysis (Table 2). Using a volume of 15 μL, a sensitivity of 92% was obtained for CSF samples from DLB (Fig. 1F and G) and a sensitivity of 65% was obtained for CSF samples from patients with mixed DLB/AD pathology. None of the CSF samples from the control subjects (Fig. 1D) or patients with pure AD (Fig. 1E), corticobasal degeneration (CBD), or PSP were positive. Using this exploratory set of CSF samples, a positive response was defined as a relative fluorescence unit (rfu) value of >2SD above the mean of the negative controls at 120 h of at least one of the CSF duplicates.
Table 2.
Positive RT‐QuIC reactions seeded with CSF samples from patients with neuropathologically confirmed DLB, mixed DLB/AD, AD with incidental LB, AD, PD, and healthy controls (Exploratory group) and patients with clinically diagnosed PD, at‐risk PD, neuropathologically confirmed corticobasal degeneration and supranuclear palsy and PD controls (Confirmatory group)
| Number of positive RT‐QuIC (%) using 5 μL | Number of positive RT‐QuIC (%) using 10 μL | Number of positive RT‐QuIC (%) using 15 μL | |
|---|---|---|---|
| Exploratory patient group (n) | |||
| AD with incidental LB (13) | 2 (15%) | 4 (31%) | 2 (15%) |
| Healthy Controls (20) | 0 (0%) | 0 (0%) | 0 (0%) |
| Mixed DLB/AD (17) | 9 (53%) | 11 (65%) | 11 (65%) |
| Parkinson's disease (2) | 2 (100%) | 2 (100%) | 2 (100%) |
| Progressive supranuclear palsy (2) | 0 (0%) | 0 (0%) | 0 (0%) |
| Corticobasal degeneration (3) | 0 (0%) | 0 (0%) | 0 (0%) |
| Pure AD (30) | 2 (7%) | 1 (3%) | 0 (0%) |
| Pure DLB (12) | 10 (83%) | 11 (92%) | 11 (92%) |
| Sensitivity (DLB) | 83% | 92% | 92% |
| Specificity (vs. controls) | 100% | 100% | 100% |
| Specificity (vs. AD) | 93% | 97% | 100% |
| Specificity (vs. controls + AD) | 96% | 98% | 100% |
| Confirmatory patient group (n) | |||
| Parkinson disease (20) | – | – | 19 (95%) |
| At‐risk PD patients (3) | – | – | 3 (100%) |
| Parkinson's disease controls (15) | – | – | 0 (0%) |
| Sensitivity (PD) | – | – | 95% |
| Specificity | – | – | 100% |
A positive RT‐ASA response was classified as a relative fluorescence unit (rfu) value of >2SD above the mean of the negative controls at 120 h of at least one of the CSF duplicates.
The second phase of the study was to apply these analytical conditions and cutoff criteria to a set of confirmatory in vivo CSF samples from 20 patients with clinically diagnosed PD, 15 control patients and three patients with REM sleep behavior disorder (RBD) recognized to be at high risk of developing future alpha‐synucleinopathies,17 obtained from the large prospective, OPDC Discovery cohort.17 These CSF samples were coded, analyzed, and reported without prior knowledge of the final diagnosis. After the samples were decoded, the results showed that 19 of the 20 PD patients had a positive RT‐QuIC response (Fig. 1H) and none of the 15 controls were found to be positive. This resulted in a RT‐QuIC sensitivity and specificity for PD of 95% and 100%, respectively (Table 2). Interestingly, all three patients at‐risk of developing PD had a positive RT‐QuIC response. These patients had RBD, of whom 80% have been shown to progress to develop a Lewy body disorder.17
Discussion
The early diagnosis of both DLB and PD is hampered by the lack of sensitive and reliable clinical diagnostic tests. Both conditions are underpinned by the neuropathological deposition of an aggregated form of a‐syn which is released into the CSF. We have exploited the ability of the aggregated a‐syn to induce further aggregation of nonaggregated a‐syn in a cyclic manner to develop a technique that can detect abnormal CSF a‐syn in DLB and PD with a sensitivity of 92% and 95%, respectively, with 100% specificity. Uniquely, we found that three RBD patients at high future risk of developing a Lewy body disorder gave a positive RT‐QuIC response, suggesting that this test could be used as an early diagnostic test for prodromal PD. Future work focusing on test validation in a larger cohort of RBD patients, followed by ongoing longitudinal assessment, will test the assay's utility in risk stratifying those prodromal individuals most at risk of early PD conversion in whom neuroprotective therapies might be trialed. We also found that CSF samples from CBD and PSP patients do not give positive RT‐QuIC responses. These are movement disorders associated with abnormalities in tau protein rather than a‐syn which may be mistaken for PD in the early stages. Therefore, RT‐QuIC offers a new approach for the detection of abnormal a‐syn and one which has the potential to improve the early clinical diagnosis of PD and DLB in addition to other alpha‐synucleinopathies such as MSA.
Author Contributions
A.G. directed and designed the study; G.F., L.McG., S. P., M.E, R.W‐M, M.H, and L.P. contributed to the design of the study; G.F. performed all the experiments; A.G. and G.F. interpreted the results; JI providing brain tissue and neuropathological expertize; L.P. provided samples and their clinico‐pathological description; M.H. is the director of the Discovery cohort and identified and diagnosed patients; R.W‐M is the director of the OPDC; S.E collected and collated Discovery samples; M.R, F.B, and C.R performed in vivo lumbar punctures, sample analysis, and provided clinical information; M.E. is a founder of OPTIMA and provided samples and their clinico‐pathological description; S.C is the OPTIMA project manager and provided clinical information and C.J and J.N. provided pathological and clinical description of the OPTIMA cohort, respectively; A.G., and G.F. wrote the manuscript; L.P. contributed to the manuscript. All authors reviewed and commented on the manuscript.
Conflict of Interest
MR reports grants from NIHR Oxford BRC during the conduct of the study; MTMH reports grants from Parkinson's UK, grants and nonfinancial support from NIHR Oxford Biomedical Research Centre, grants from Michael J Fox Foundation during the conduct of the study, and grants from UCB Pharmaceutical company, outside the submitted work; AJEG has a patent GB1611840.8 pending.
Acknowledgments
This study has been funded by the Scottish Government's Chief Scientists Office, Government of Scotland (ETM/315, RA2647: GF; LMcG), and the Michael J Fox foundation (R43828: GF). The National CJD Research & Surveillance Unit is funded by the Department of Health and the Scottish Home Office Department of Health (R43401: AG). The Edinburgh Brain Bank is supported by the Medical Research Council (G1000681). We are indebted to Professor Colin Smith (University of Edinburgh) for providing brain tissue and neuropathological expertize. The views expressed in this publication are those of the authors and not necessarily those of the Department of Health or the Scottish Government. We acknowledge the Oxford Brain Bank, supported by the Medical Research Council (MRC), Brains for Dementia Research (BDR) (Alzheimer Society and Alzheimer Research UK), Autistica UK and the NIHR Oxford Biomedical Research Centre. The OPDC Discovery cohort is funded by the Monument Trust Discovery Award from Parkinson's UK and supported by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre based at Oxford University Hospitals NHS Trust and University of Oxford, and the Dementias and Neurodegenerative Diseases Research Network (DeNDRoN).
References
- 1. Spillantini MG, Divane A, Goedert M. Assignment of human alpha‐synuclein (SNCA) and beta‐synuclein (SNCB) geners to chromosome 4q21 and 5q35. Genomics 1995;27:379–381. [DOI] [PubMed] [Google Scholar]
- 2. Goedert M, Spillantini MG, Del Tredici K, et al. 100 years of lewy pathology. Nat Rev Neurol 2013;9:13–24. [DOI] [PubMed] [Google Scholar]
- 3. Martf MJ, Tolsa E, Campdelacreu J. Clinical overview of the synucleinopathies. Mov Disord 2003;18:S21–S27. [DOI] [PubMed] [Google Scholar]
- 4. Mollenhauer M, Cullen V, Kahn I, et al. Direct quantification of CSF alpha‐synuclein by ELISA and first cross‐sectional study in patients with neurodegeneration. Exp Neurol 2008;213:315–325. [DOI] [PubMed] [Google Scholar]
- 5. Williams SM, Schult P, Sierks MR. Oligomeric a‐synuclein an B‐amyloid variants as potential biomarkers for Parkinson's and Alzheimer's diseases. Eur J Neurosci 2016;43(1):3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shi M, Bradner J, Hancock AM. Cerebrospinal fluid biomarkers for Parkinson's disease diagnosis and progression. Ann Neurol 2011;69:570–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ohrfelt A, Grognet P, Andreasen N, et al. Cerebrospinal fluid alpha‐synuclein in neurodegenerative disorders‐ a marker of synapse loss? Neurosci Lett 2009;450:332–335. [DOI] [PubMed] [Google Scholar]
- 8. Hong Z, Shi M, Chung KA, et al. DJ‐1 and alpha‐synuclein in human cerebrospinal fluid as biomarkers of Parkinson's disease. Brain 2010;133:713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kruse N, Persson S, Alcolea D, et al. Validation of a quantitative cerebrospinal fluid alpha‐synuclein assay in a European‐wide interlaboratory study. Neurobiol Aging 2015;36:2587–2596. [DOI] [PubMed] [Google Scholar]
- 10. Bernis ME, Babila JT, Breid S, et al. Prion‐lie propagation of human brain‐derived alpha‐synuclein in transgenic mice expressing human wild‐type alpha‐synuclein. Acta Neuropathol Commun 2015;3:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brandel JP, Corbille AG, Derkinderen P, et al. Is Parkinson's disease a prion disease? Rev Neurol (Paris) 2015;171:812–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McGuire L, Peden A, Orru C, et al. Prion seeding activity in cerebrospinal fluid from patients with sporadic Creutzfeldt‐Jakob disease patients using real‐time QuIC analysis: a potential new clinical diagnostic test with high sensitivity and specificity. Ann Neurol 2012;72:278–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clarke R, Smith AD, Jobst KA, et al. Folate, vitamin B12 and serum total homocysteine levels n confirmed Alzheimer's disease. Arch Neurol 1998;55:1449–1455. [DOI] [PubMed] [Google Scholar]
- 14. Szewwczyk‐Krolikowski K, Tomlinson P, Nithi K, et al. The influence of age and gender on motor and non‐motor features of early Parkinson's disease: initial findings from the Oxford Parkinson Disease Center (OPDC) discovery cohort. Parkinsonism Relat Disord 2014;20:99–105. [DOI] [PubMed] [Google Scholar]
- 15. Hughes AJ, Daniel SE, Kilford L, et al. Accuracy of clinical diagnosis of idiopathic Parkinson's disease ‐ a clinicopathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rolinski M, Zokaei N, Baig F, et al. Visual short‐term memory deficits in REM sleep behaviour disorder mirror those in Parkinson's disease. Brain 2016;139:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iranzo A, Tolosa E, Gelpi E, et al. Neurodegenerative disease status and post‐mortem pathology in idiopathic rapid‐eye‐movement sleep behaviour disorder: an observational cohort study. Lancet 2013;12:443–453. [DOI] [PubMed] [Google Scholar]
- 18. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathological assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 2012;123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kovacs GG, Alafuzoff I, AI‐Sarraj S, et al. Mixed brain pathologies in dementia: the BrainNet Europe consortium experience. Dement Geriatr Cogn Disord 2008;26:343–350. [DOI] [PubMed] [Google Scholar]