

 Longitudinal proteomic analysis of bovine milk shows consistent changes over time across cows after intramammary challenge with Streptococcus uberis.
Longitudinal proteomic analysis of bovine milk shows consistent changes over time across cows after intramammary challenge with Streptococcus uberis.
Abstract
Mastitis, inflammation of the mammary gland, is the most common and costly disease of dairy cattle in the western world. It is primarily caused by bacteria, with Streptococcus uberis as one of the most prevalent causative agents. To characterize the proteome during Streptococcus uberis mastitis, an experimentally induced model of intramammary infection was used. Milk whey samples obtained from 6 cows at 6 time points were processed using label-free relative quantitative proteomics. This proteomic analysis complements clinical, bacteriological and immunological studies as well as peptidomic and metabolomic analysis of the same challenge model. A total of 2552 non-redundant bovine peptides were identified, and from these, 570 bovine proteins were quantified. Hierarchical cluster analysis and principal component analysis showed clear clustering of results by stage of infection, with similarities between pre-infection and resolution stages (0 and 312 h post challenge), early infection stages (36 and 42 h post challenge) and late infection stages (57 and 81 h post challenge). Ingenuity pathway analysis identified upregulation of acute phase protein pathways over the course of infection, with dominance of different acute phase proteins at different time points based on differential expression analysis. Antimicrobial peptides, notably cathelicidins and peptidoglycan recognition protein, were upregulated at all time points post challenge and peaked at 57 h, which coincided with 10 000-fold decrease in average bacterial counts. The integration of clinical, bacteriological, immunological and quantitative proteomics and other-omic data provides a more detailed systems level view of the host response to mastitis than has been achieved previously.
1. Introduction
Bovine milk is a complex physiological secretion and contains protein at an average concentration of 32 g L–1. Caseins form 80% of the total milk protein while whey proteins constitute about 16% of the total milk protein.1 Whey comprises several hundred heterogeneous, mostly water-soluble proteins including beta-lactoglobulin, alpha-lactalbumin, blood serum albumin and immunoglobulins (IgG, IgA, IgM and IgE). These proteins have a number of functions such as ion binding, protein binding, carbohydrate binding, pattern binding, cell surface binding, lipid binding, enzyme regulating, cell-to-cell signalling and cell cycle regulating activities.1,2 There are substantial changes in the whey proteome (the set of proteins present in whey) during mastitis, inflammation of the mammary gland. The pathogenesis of mastitis, which is largely due to intra-mammary infections (IMI), includes an inflammatory reaction involving the release of cytokines and acute-phase proteins (APP).3,4 Several studies have shown changes in the milk or whey proteome due to mastitis.5–7
This is the second of three studies integrating omic approaches to the investigation of experimentally induced mastitis with Streptococcus uberis, a major cause of the disease in the UK and many other parts of the world.8 Using the same milk samples, temporal changes in the milk peptidome,4 proteome (this paper) and metabolome9 were determined during the acute phase of infection and its resolution. The peptidome was considered to consist of peptides, polypeptides and short protein sequences, usually degradation-derived protein fragments, with masses between ∼400 Da and ∼20 000 Da.4 The proteome includes whole proteins with masses ranging up to 3 MDa,10 which can be experimentally broken down into peptide pools using proteolytic enzymes, usually trypsin, and identified by comparing the mass spectra from experiments with the theoretical enzyme-specific fragmentation patterns derived from protein sequences. Finally, the metabolome consists of the entirety of molecules, small and large, that undergo metabolism, most of which have a mass less than 1500 Da, with the exception of lipids, which have masses up to 5000 Da.10
In addition to identification, quantitation of proteins in complex biological samples is possible.10,11 The classical method used for quantitative analysis of complex mixtures of proteins such as milk is by protein separation and comparison by two-dimensional polyacrylamide gel electrophoresis (2D-PAGE), followed by mass spectrometry (MS) analysis.11,12 However, the gel-based quantitative proteomics techniques are laborious and suffer poor representation of hydrophobic, very high or low molecular weight proteins.11 To overcome the shortcomings of the gel-based methods and to increase the dynamic range and quantitative accuracy, non-gel-based quantitative proteomics methods have been developed.11,13 Non-gel-based quantitative proteomics approaches can be divided into methods using metabolic or chemical labelling and label-free approaches.13 Some of the labelling approaches utilize isotope-labelled compounds (such as isotope labelled amino acids) that are functionally and chemically identical to the properties of their unlabelled equivalent except in mass, which allows for their discrimination in mass spectrometry. Stable labelling approaches include stable isotope labelling by amino acids in cell culture (SILAC), isotope-coded affinity tag (ICAT), isobaric tags for relative and absolute quantification (iTRAQ), dimethyl labelling and tandem mass tags.13–15 Label-free relative quantification is an alternative method that can be applied to clinical samples, and offers better dynamic range than some labelling approaches15–17 and requires minimal manipulation of samples, which reduces the possibility of introducing artefactual changes.
In this study, we describe the application of a label-free relative quantification method to analyse the quantitative changes in the proteome of bovine milk whey in the experimental model of S. uberis mastitis and compare those to data obtained from clinical, immunological, and peptidomic studies.
2. Materials and methods
Cows (n = 6) were challenged with Streptococcus uberis strain FSL Z1-048 in a single bacteriologically negative udder quarter per cow as previously described.3 Aliquots of milk samples collected from six time points (0, 36, 42, 57, 81 and 312 hours post-challenge (PC)) were used to generate quantitative label-free proteomics data. These were the same samples as used in the associated peptidomic4 and metabolomic9 studies and were selected on the basis of clinical manifestation, bacterial load and SCC.3 Body temperature of the cows and bacterial concentrations in milk from challenged quarters peaked from 24 h (bacteria) or 30 h (temperature) PC up to 57 h PC and had decreased to a plateau by 81 h PC, whereby body temperature had returned to normal and bacterial concentrations in culture positive quarters stayed constant until the end of the study at 312 h PC. The challenge study was conducted at the Moredun Research Institute (Penicuik, UK) and had received ethical approval from the Institute's Experiments and Ethical Review Committee in accordance with the UK's Animals (Scientific Procedures) Act 1986.3
2.1. Label-free quantitative proteomic data generation
2.1.1. Separation of whey
The aliquots of frozen skimmed milk samples,3 ranging between 0.5 mL and 1.5 mL in volume per sample, were thawed at 4 °C. The volume of every sample was brought to 1.5 mL using phosphate buffered saline (PBS). To remove the residual milk fat globules and cell pellets, the samples were centrifuged at 13 000 × g for 30 min at 4 °C in an Eppendorf centrifuge (model 5804R) with a fixed-angle rotor (FA-45-30-11). Using a pipette, the middle clear portion (1 mL) was carefully drawn from each sample and transferred into an ultracentrifuge tube (Beckman Coulter Thickwall polycarbonate, part no. 343778) and centrifuged in a Beckman Coulter benchtop ultracentrifuge (model TL-100) with a fixed-angle rotor (TLA-100.2) at 150 000 × g (59 000 rpm) for 60 minutes at 4 °C. Most of the caseins in the samples sedimented to the bottom of the ultracentrifuge tubes, and above them exosomes formed a loose pellet layer with crude whey forming the supernatant. This crude whey was transferred to a clean ultracentrifuge tube and centrifuged again at 150 000 × g for 60 minutes at 4 °C to remove the residual caseins.7
2.1.2. Whey protein extraction, purification and normalization
Total protein quantity in the whey was measured by Bradford protein assay in 250 μL microplate assay format using Bio-Rad protein assay dye reagent concentrate (product no. 500-0006) and bovine serum albumin (BSA) fraction V (Roche, product no. 10735086001) as the standard. To remove substances that might interfere with downstream proteomic analysis, proteins from whey were purified by precipitating them with absolute acetone.7,18 Using the measured total protein concentration in each sample, whey was diluted with HPLC grade water to 2 mg mL–1 total protein. For every diluted whey sample, an aliquot of 100 μL was transferred into a 1.5 mL micro test tube and six volumes (600 μL) of ice-cold 100% acetone (VWR International, product no. 20066.330) was added and kept at –80 °C for 12 hours. Precipitated proteins were centrifuged at 20 000 × g for 40 minutes at –4 °C in an Eppendorf centrifuge (model 5804R). The supernatant was discarded, and the pellets (precipitated proteins) were washed three times with 400 μL of 80% (v/v) acetone to remove salts, and then dried under a fume hood for 10 minutes. The dried pelleted proteins from each sample were re-suspended in 50 μL of 50 mM ammonium bicarbonate (Sigma-Aldrich, product no. A6141) buffer (NH4CO3 buffer) and the extracted protein quantity was measured by Bradford protein assay as described before. The re-suspended proteins in each sample were normalized by diluting them with the required volume of NH4CO3 buffer to arrive at 2.5 mg mL–1 total protein concentration.
2.1.3. Preparation of trypsin digests
For every sample, an aliquot of 40 μL of the normalized re-suspended proteins, containing 100 μg of total proteins in buffer was transferred into a 1.5 mL micro test tube. For each aliquot, 12 μL of 10% (w/v) sodium deoxycholate (SDC) solution in buffer (Sigma-Aldrich, product no. D6750), 8 μL of 80% (v/v) acetonitrile (Fisher Scientific, product no. 10660131) in buffer and 50 μL of 10% (w/v) modified trypsin (Promega, product no. V5111) in trypsin re-suspension buffer were added. The digest was incubated for 18 hours at 37 °C in a heating block. Then, 12 μL of 1% (v/v) formic acid (Sigma-Aldrich, product no. 94318) was added to the digest (final formic acid concentration 0.1%) to precipitate SDC, and samples were centrifuged at 16 000 × g for 10 minutes at 4 °C. For every sample, supernatant containing 2 μg (calculated) of digested protein was transferred into a well of a conical bottom microplate and dried in a SpeedVac (Thermo Fisher Scientific, model no. SPD1010).
2.1.4. On-line liquid chromatography and tandem mass spectrometry
For on-line reversed-phase liquid chromatography and mass spectrometry (LC-MS), a Dionex UltiMate 3000 RSLCnano (liquid chromatography) system coupled to a Thermo Scientific Orbitrap Elite mass spectrometer was used. A stainless steel Nano-Trap column with 300 μm inside diameter, 5 mm length, particle size 5 μm and pore size 10 nm, packed with stationary phase Acclaim PepMap C18 (Thermo Scientific, part no. 160454) and a resolving Nano LC column with 75 μm inside diameter, 15 cm length, particle size 2 μm and pore diameter 10 nm with stationary phase Acclaim PepMap RSLC C18 (Thermo Fisher Scientific, part no. 164534) were used in the HPLC. The dried protein digests in the microplate were loaded on the Rapid Separation LC (RSLC) Autosampler connected to the C18 trap column equilibrated in 96% solution A (0.1% formic acid in HPLC grade water (v/v)) and 4% solution B (80% acetonitrile and 0.08% formic acid in HPLC grade water (v/v)) with a flow rate of 25 μL min–1. The trap column was washed for 12 minutes at the same flow rate and then switched to the in-line resolving C18 column. A constant flow rate of 300 nL min–1 was maintained with a linear gradient from 4% solution B to 40% solution B in 108 minutes, then to 100% solution B by the 124th minute. Then the column was washed with 100% solution B for 5 minutes followed by recalibration with 96% solution A for 6 minutes. In the mass spectrometer, one scan cycle comprised MS1 scan (m/z range from 400–2000) in the Orbitrap Elite followed by up to 20 data-dependent MS2 scans (threshold value 1000 and maximum injection time 200 ms) in the Velos LTQ in collision-induced dissociation (CID) mode. To account for any retention time drift, carryover or other errors that might occur during the run, the sample loading order was randomized using Microsoft Excel. After every six samples, a blank was analysed to monitor carryover. All samples were run consecutively without breaks, which took about 4 days of mass spectrometer time.
2.2. Label-free quantitative proteomic data analysis
2.2.1. Exploration of the raw data
The raw MS/MS data obtained from each sample were visually examined by generating a number of plots using MZmine (version 2.10) software.19 To examine sample loading and peak resolution, total ion current (TIC) chromatograms and base peak chromatograms were generated from the MS1 data obtained from each sample. To detect chromatographic shifts in retention time, MS1 spectra were visualized by generating 2D and 3D plots using the 2D and 3D plot functions in MZmine software. In addition, 2D plots and TIC chromatograms of the MS1 spectra were generated using the integrated viewer in the MaxQuant software (version 1.5.2.8)20 and examined for overall consistency and retention time shifts between the samples.
2.2.2. Peptide identification and protein quantification
After initial quality control, the raw MS/MS data from all samples and blanks were imported into MaxQuant software (version 1.5.2.8) for label-free relative quantification analysis.21 Feature detection and mass recalibration were automatically performed in MaxQuant, and peptides were identified using its integrated Andromeda search engine.22 Reporter quantification, retention time alignment, protein assembly, label-free quantification and MaxLFQ normalization were also performed in MaxQuant.23 For identification and quantification, N-terminal acetylation, oxidation of methionine and deamidation of asparagine or glutamine were set as variable modifications, and carbamidomethylation of cysteines was set as a fixed modification. For in silico digestion, Trypsin/P was used and a maximum of 2 missed cleavages were allowed. Up to 6 ppm peptide mass tolerance was allowed during the main search. A false discovery rate (FDR) up to 1% was allowed for peptide spectrum match and protein assembly, and the FDR was estimated using the reversed peptide sequences. At least one unique or “razor” peptide was required for identification. For label-free quantification, the ‘Fast LFQ’ option was turned off and a minimum of one quantified peptide pair was required for pair-wise comparisons of a protein between two samples. The ‘match-between-runs’ option with a match time window of 2 minutes was used to transfer identifications across the replicate experiments, whereby the 6 individual cows were treated as biological replicates for each time point. In addition, absolute protein quantitation was performed using the intensity based absolute quantification (iBAQ) method.
Proteins from the Bos taurus proteome were identified using the 23 868 protein reference proteome (UniProt Proteome ID: UP000009136; last modified 10 May 2015), which was downloaded from the UniProt Knowledgebase and imported into the Andromeda search engine. Conflicts of multiple protein assignments were manually resolved taking into account the peptide counts, the razor and/or unique peptide counts, and the evidence status of the protein annotation (annotation score) in the UniProt database. Where a protein was identified based on comparison with both the Bos taurus reference proteome and the MaxQuant contaminant list, they were assigned to Bos Taurus, because many proteins on this list, e.g. keratin or bovine serum proteins, are of bovine.24
2.2.3. Statistical analysis
Statistical analysis was performed using Perseus (version 1.5.2.6), the Partek Genomics Suite (version 6.6, Partek Inc., St. Louis) and R (version 3.1.2) software. The normalized protein intensities from the MaxQuant analysis were imported into Perseus software. Protein intensities (abundances) in the linear scale were transformed into logarithmic scale with base two. The missing values were replaced with a constant value of 10 to simulate signals from low abundant proteins. For exploratory analysis, histograms were generated to examine the dataset. Hierarchical clustering analysis and principal component analysis (PCA) were performed using Perseus and Partek Genomics Suite software. To identify differentially expressed proteins one-way analysis of variance (ANOVA) was performed with time as factor. From the ANOVA results, protein lists were created by comparing each time-point PC to the pre-challenge results (0 h PC). Proteins with an absolute fold change >2 and FDR-adjusted p-value < 0.05 were considered differentially expressed and included in the protein lists.
2.2.4. Pathway analysis
The differentially expressed proteins were analysed for enrichment of canonical pathways using ingenuity pathway analysis (IPA) software (QIAGEN, Redwood City, CA) and the STRING database (version 10.0) and search tool.25 IPA computes an enrichment score for the overlap between the observed and the predicted regulated gene sets using a Fisher's exact test and p-value >0.05. The direction of regulation, i.e. up- or downregulation, was inferred from the activation z-score in the IPA.26 In addition, the ratio of identified (i.e. present in the sample) to potentially identifiable proteins (i.e. present in the pathway) is calculated for each pathway.
3. Results
3.1. Quantification and analysis of bovine proteins
TIC chromatograms, base peak chromatograms and 2D plots were generated for each sample and showed overall consistency with a retention time drift of about 2 minutes, demonstrating high quality of the raw data (Fig. S1, ESI†). A total of 2552 non-redundant bovine peptides were quantified, and 570 proteins were assembled from the quantified peptides (Table S1, ESI†). Exploratory data analysis such as histograms, hierarchical clustering analysis (HCA) and principal components analysis (PCA) were performed on the quantified protein data.
3.1.1. Hierarchical clustering analysis
To explore the dataset, HCA using Euclidean distance and average linkage was performed on the 570 proteins that were quantified from the cow reference proteome. The analysis (Fig. 1) shows three major clusters in the column dendrogram, corresponding to different phases of the infection process. Cluster C includes samples from before challenge (0 h PC) and at late resolution stage (312 h PC), by which time 5 of 6 cows had cleared the infection.3 It also includes 36 h and 42 h PC samples from cow 5, which was previously identified as a late responder based on clinical signs and cytokine profiling.3 Cluster B includes samples from 36 and 42 h PC, corresponding to the early stage of infection, which is characterized by bacterial growth and neutrophil influx.3 Cluster A predominantly contains samples from 57 h and 81 h PC, during which time bacterial numbers had started to decrease.3
Fig. 1. Heat map of the bovine proteome after intramammary challenge with Streptococcus uberis with hierarchical clustering analysis of cow and time. This heat map is based on 570 proteins. Hierarchical clustering analysis was performed using Euclidean distance as distance metric and average linkage as agglomeration method. Clusters are identified by letters (C = pre-challenge and resolution stage; B = early to peak infection based on bacterial numbers; A = post peak infection), time points by colours (see inset), and individual cows by numbers. Scale bar indicates intensity of upregulation on a log 2 scale.

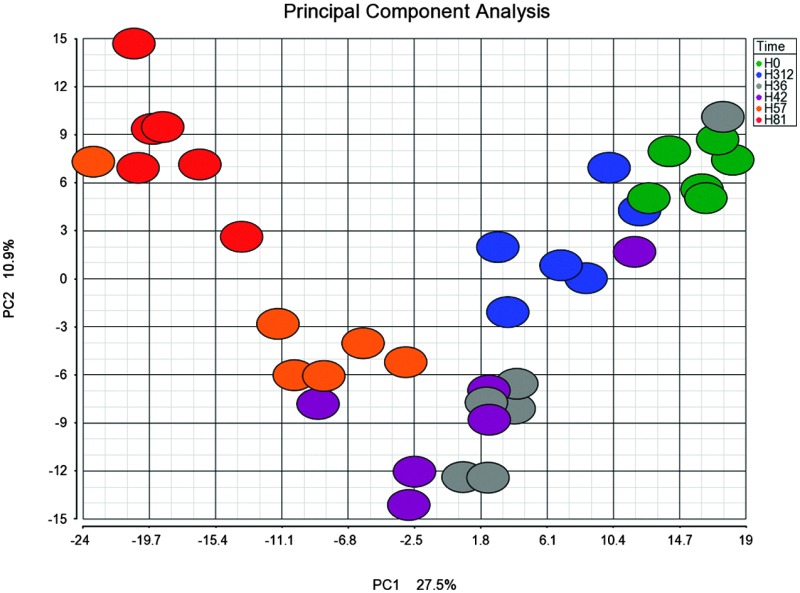
3.1.2. Principal component analysis
To further examine the set of 570 bovine proteins, PCA was performed (Fig. 2). The PCA shows clustering of samples by time-point with a few exceptions. As in HCA, results are similar for the pre-challenge (0 h) and resolution time points (312 h). Samples collected at 81 h PC were most divergent. Outliers at 36 and 42 h PC, which cluster with samples from 0 h, correspond to the slow responder (cow 5) that is also visible in Fig. 1 and in clinical, bacteriologic and inflammatory parameters.3
Fig. 2. Principal component analysis (PCA) of the bovine proteome after intramammary challenge with Streptococcus uberis. The PCA was based on 570 proteins and the plot was generated using the Partek Genomic suite. The data points refer to milk samples obtained from 6 cows at 6 time points post challenge (PC). Cows are identified by number and time points by colour, with hours PC shown in the legend.

3.1.3. Differential expression analysis
One-way ANOVA was performed with time as factor to identify proteins that were differentially expressed between pre- and post-challenge time points. No distinction was made between proteins that were detected in all samples and those that were detected in a subset of samples only, but this information is reflected in the LFQ intensities (quantities) listed in ESI,† Table S1 where intensity is shown as 0 if a protein is not detected. Differentially expressed protein lists were created for each time point, and proteins with an absolute fold change more than 2 and FDR-adjusted p-value less than 0.05 were included in the protein lists. For time points 36, 42, 57, 81 and 312 h PC, there were 76 (54 upregulated, 22 downregulated), 126 (96 upregulated, 30 downregulated), 237 (186 upregulated, 51 downregulated), 292 (248 upregulated, 44 downregulated) and 56 (49 upregulated, 7 downregulated) differentially expressed proteins, respectively (Table S2, ESI†). The top-15 upregulated and downregulated bovine proteins for each time point, as compared to 0 h PC, are given in Tables 1–5. Patterns of up- and down regulation differed both qualitatively (proteins) and quantitatively (fold change) between time points, with strongest up- and down-regulation observed at 57 and 81 h PC. Upregulated proteins include APP, e.g. haptoglobin and serum amyloid A (SAA); antimicrobial proteins, e.g. the cathelicidin family and peptidoglycan recognition protein; and proteins with dual APP and antimicrobial function, e.g. histidine-rich glycoprotein (HRG) and lipopolysaccharide-binding protein (LBP). Down-regulated proteins included cystatin-B, dystroglycan, and mucin-1 in the early stage of infection (36 and 42 h PC; Tables 1 and 2), and myozenin-1 and alpha-lactalbumin at the subsequent stage (57 and 81 h PC; Tables 3 and 4). During the resolution phase (312 h PC), both the number of differentially expressed proteins and the fold change were smaller than at earlier infection stages, with only 7 proteins still significantly downregulated (Table 5), and in agreement with results from HCA and PCA, which also showed a return to normal at 312 h PC.
Table 1. Top-15 most up-and down-regulated bovine proteins at 36 hours after intramammary challenge with Streptococcus uberis .
| Up/down-regulated | UniProt ID | Protein name | Fold change | P-Value a |
| Up | Q8SPP7 | Peptidoglycan recognition protein 1 | 3305 | 4.5 × 10–10 |
| Up | P54229 | Cathelicidin-5 | 1444 | 1.9 × 10–8 |
| Up | P56425 | Cathelicidin-7 | 1217 | 1.6 × 10–6 |
| Up | P22226 | Cathelicidin-1 | 1026 | 2.8 × 10–8 |
| Up | Q2TBU0 | Haptoglobin | 997 | 3.8 × 10–8 |
| Up | F1N465 | Uncharacterized protein GN = KBTBD8 | 527 | 1.5 × 10–3 |
| Up | E1BCU6 | Uncharacterized protein GN = TCN1 | 401 | 1.5 × 10–6 |
| Up | Q9TU03 | Rho GDP-dissociation inhibitor 2 | 313 | 1.6 × 10–4 |
| Up | P52176 | Matrix metalloproteinase-9 | 219 | 1.1 × 10–4 |
| Up | P33046 | Cathelicidin-4 | 208 | 2.7 × 10–4 |
| Up | Q0VCG9 | Pentraxin-related protein PTX3 | 194 | 1.5 × 10–8 |
| Up | Q58CQ9 | Pantetheinase | 189 | 8.5 × 10–4 |
| Up | G3MXK8 | Uncharacterized protein (Fragment) GN = PRTN3 | 167 | 1.2 × 10–3 |
| Up | Q28085 | Complement factor H | 134 | 1.6 × 10–3 |
| Up | Q3SZV7 | Hemopexin | 131 | 4.9 × 10–6 |
| Down | P81265 | Polymeric immunoglobulin receptor | –6 | 2.0 × 10–4 |
| Down | Q3MHX6 | Protein OS-9 | –6 | 4.9 × 10–3 |
| Down | P10790 | Fatty acid-binding protein, heart | –7 | 3.2 × 10–4 |
| Down | Q8WML4 | Mucin-1 | –38 | 2.7 × 10–3 |
| Down | P13696 | Phosphatidylethanolamine-binding protein 1 | –39 | 1.8 × 10–3 |
| Down | Q9XSG3 | Isocitrate dehydrogenase [NADP] cytoplasmic | –50 | 5.0 × 10–5 |
| Down | Q9TUM6 | Perilipin-2 | –61 | 2.0 × 10–3 |
| Down | E1BLC6 | Uncharacterized protein GN = TTC17 | –67 | 4.3 × 10–3 |
| Down | F1N1D2 | Uncharacterized protein GN = DMC1 | –77 | 4.6 × 10–3 |
| Down | O18738 | Dystroglycan | –77 | 1.2 × 10–3 |
| Down | P26201 | Platelet glycoprotein 4 | –87 | 1.0 × 10–4 |
| Down | E1B9W6 | Uncharacterized protein GN = ADCY10 | –145 | 2.5 × 10–3 |
| Down | F6PZ29 | Uncharacterized protein GN = MCFD2 | –191 | 3.1 × 10–3 |
| Down | F6QEL0 | Cystatin-B | –204 | 1.8 × 10–4 |
| Down | E1BN90 | Uncharacterized protein GN = ZKSCAN2 | –214 | 4.6 × 10–3 |
aFalse discovery rate adjusted.
Table 2. Top-15 most up- or down-regulated bovine proteins at 42 hours after intramammary challenge with Streptococcus uberis .
| Up/down-regulated | UniProt ID | Protein name | Fold change | P-value a |
| Up | P54229 | Cathelicidin-5 | 9209 | 1.5 × 10–10 |
| Up | P56425 | Cathelicidin-7 | 8922 | 1.7 × 10–8 |
| Up | Q8SPP7 | Peptidoglycan recognition protein 1 | 8453 | 3.7 × 10–11 |
| Up | Q2TBU0 | Haptoglobin | 4794 | 5.2 × 10–10 |
| Up | P22226 | Cathelicidin-1 | 3812 | 7.6 × 10–10 |
| Up | P33046 | Cathelicidin-4 | 2619 | 1.1 × 10–6 |
| Up | E1BCU6 | Uncharacterized protein GN = TCN1 | 1292 | 6.1 × 10–8 |
| Up | P19660 | Cathelicidin-2 | 1159 | 3.9 × 10–5 |
| Up | F1MCC8 | Uncharacterized protein GN = NWD1 | 1144 | 5.3 × 10–4 |
| Up | Q0VCG9 | Pentraxin-related protein PTX3 | 963 | 4.7 × 10–11 |
| Up | F1N465 | Uncharacterized protein GN = KBTBD8 | 961 | 6.0 × 10–4 |
| Up | F1MKS5 | Histidine-rich glycoprotein | 775 | 6.3 × 10–6 |
| Up | P52176 | Matrix metalloproteinase-9 | 708 | 7.1 × 10–6 |
| Up | F1N1F8 | Uncharacterized protein GN = CENPF | 661 | 5.7 × 10–3 |
| Up | Q9TU03 | Rho GDP-dissociation inhibitor 2 | 614 | 3.8 × 10–5 |
| Down | P80457 | Xanthine dehydrogenase/oxidase | –15 | 1.1 × 10–2 |
| Down | P02702 | Folate receptor alpha | –35 | 5.6 × 10–3 |
| Down | P29392 | Spermadhesin-1 | –42 | 8.1 × 10–3 |
| Down | Q8WML4 | Mucin-1 | –44 | 1.8 × 10–3 |
| Down | P08037 | Beta-1,4-galactosyltransferase 1 | –51 | 1.9 × 10–3 |
| Down | F1MNS0 | Uncharacterized protein GN = HERC1 | –58 | 2.6 × 10–3 |
| Down | P63048 | Ubiquitin-60S ribosomal protein L40 | –70 | 3.2 × 10–3 |
| Down | Q0VCX2 | 78 kDa glucose-regulated protein | –73 | 2.1 × 10–3 |
| Down | F1N1D2 | Uncharacterized protein GN = DMC1 | –77 | 4.6 × 10–3 |
| Down | O18738 | Dystroglycan | –78 | 1.2 × 10–3 |
| Down | P13696 | Phosphatidylethanolamine-binding protein 1 | –87 | 2.3 × 10–4 |
| Down | P26201 | Platelet glycoprotein 4 | –87 | 1.0 × 10–4 |
| Down | F6QEL0 | Cystatin-B | –97 | 9.3 × 10–4 |
| Down | F6PZ29 | Uncharacterized protein GN = MCFD2 | –201 | 2.8 × 10–3 |
| Down | E1BN90 | Uncharacterized protein GN = ZKSCAN2 | –230 | 4.1 × 10–3 |
aFalse discovery rate adjusted.
Table 3. Top-15 most up- or down-regulated bovine proteins at 57 hours after intramammary challenge with Streptococcus uberis .
| Up/down-regulated | UniProt ID | Protein name | Fold change | P-value a |
| Up | Q8SPP7 | Peptidoglycan recognition protein 1 | 27 479 | 2.0 × 10–12 |
| Up | P54229 | Cathelicidin-5 | 16 618 | 3.4 × 10–11 |
| Up | Q2TBU0 | Haptoglobin | 14 937 | 3.0 × 10–11 |
| Up | P56425 | Cathelicidin-7 | 11 877 | 9.1 × 10–9 |
| Up | P22226 | Cathelicidin-1 | 7281 | 1.4 × 10–10 |
| Up | P33046 | Cathelicidin-4 | 4753 | 3.0 × 10–7 |
| Up | Q9TU03 | Rho GDP-dissociation inhibitor 2 | 4748 | 5.0 × 10–7 |
| Up | F1N1F8 | Uncharacterized protein GN = CENPF | 4312 | 5.9 × 10–4 |
| Up | F1MYX5 | Uncharacterized protein GN = LCP1 | 2578 | 3.9 × 10–7 |
| Up | Q3ZCJ8 | Dipeptidyl peptidase 1 | 2530 | 7.0 × 10–6 |
| Up | P02584 | Profilin-1 | 2404 | 1.0 × 10–6 |
| Up | P48616 | Vimentin | 2155 | 8.2 × 10–11 |
| Up | P19660 | Cathelicidin-2 | 2104 | 1.2 × 10–5 |
| Up | E1BI67 | Uncharacterized protein GN = IL18BP | 2095 | 9.9 × 10–7 |
| Up | A5PJH7 | LOC788112 protein GN = LOC788112 | 1967 | 1.9 × 10–7 |
| Down | P80457 | Xanthine dehydrogenase/oxidase | –172 | 1.4 × 10–5 |
| Down | P79345 | Epididymal secretory protein E1 | –215 | 4.8 × 10–3 |
| Down | O18738 | Dystroglycan | –222 | 1.1 × 10–4 |
| Down | Q32KV6 | Nucleotide exchange factor SIL1 | –294 | 8.8 × 10–4 |
| Down | P29392 | Spermadhesin-1 | –327 | 1.3 × 10–4 |
| Down | E1BGZ9 | PHD finger protein 20-like protein 1 | –337 | 2.8 × 10–3 |
| Down | P41541 | General vesicular transport factor p115 | –472 | 1.2 × 10–3 |
| Down | E1BN90 | Uncharacterized protein GN = ZKSCAN2 | –585 | 1.0 × 10–3 |
| Down | F6PZ29 | Uncharacterized protein GN = MCFD2 | –675 | 3.9 × 10–4 |
| Down | Q58DJ3 | KIAA1984 | –824 | 2.1 × 10–3 |
| Down | P00711 | Alpha-lactalbumin | –1022 | 4.7 × 10–6 |
| Down | F1MV51 | Uncharacterized protein GN = APC | –1217 | 1.0 × 10–3 |
| Down | Q8SQ24 | Myozenin-1 | –3030 | 7.2 × 10–4 |
| Down | E1BNS8 | Uncharacterized protein GN = SIK1 | –4741 | 3.0 × 10–3 |
| Down | Q3ZC66 | Cysteine-rich PDZ-binding protein | –6094 | 1.5 × 10–3 |
aFalse discovery rate adjusted.
Table 4. Top-15 most up-and down-regulated bovine proteins at 81 hours after intramammary challenge with Streptococcus uberis .
| Up/down-regulated | UniProt ID | Protein name | Fold change | P-value a |
| Up | Q2TBU0 | Haptoglobin | 28 858 | 6.1 × 10–12 |
| Up | Q8SPP7 | Peptidoglycan recognition protein 1 | 17 090 | 6.3 × 10–12 |
| Up | P54229 | Cathelicidin-5 | 11 722 | 8.0 × 10–11 |
| Up | Q9TU03 | Rho GDP-dissociation inhibitor 2 | 7794 | 1.8 × 10–7 |
| Up | P48616 | Vimentin | 7549 | 2.2 × 10–12 |
| Up | P56425 | Cathelicidin-7 | 7316 | 2.6 × 10–8 |
| Up | F1MYX5 | Uncharacterized protein GN = LCP1 | 5417 | 7.3 × 10–8 |
| Up | A6QLL8 | Fructose-bisphosphate aldolase GN = ALDOA | 4918 | 8.9 × 10–10 |
| Up | E1BLI9 | Protein S100-A9 | 4847 | 7.6 × 10–13 |
| Up | P22226 | Cathelicidin-1 | 4743 | 4.3 × 10–10 |
| Up | Q5E9F7 | Cofilin-1 | 4636 | 8.6 × 10–8 |
| Up | Q9XSJ4 | Alpha-enolase | 4619 | 3.9 × 10–11 |
| Up | Q3ZBD7 | Glucose-6-phosphate isomerase | 4533 | 5.7 × 10–8 |
| Up | Q3ZCJ8 | Dipeptidyl peptidase 1 | 3839 | 3.1 × 10–6 |
| Up | P02584 | Profilin-1 | 3799 | 3.7 × 10–7 |
| Down | Q8WML4 | Mucin-1 | –102 | 2.3 × 10–4 |
| Down | F1MIR2 | Uncharacterized protein GN = EXOC6B | –119 | 7.5 × 10–4 |
| Down | A8YXY3 | 15 kDa selenoprotein GN = SEP15 | –123 | 1.4 × 10–3 |
| Down | Q9TUM6 | Perilipin-2 GN = PLIN2 | –166 | 2.2 × 10–4 |
| Down | E1BN90 | Uncharacterized protein GN = ZKSCAN2 | –221 | 4.3 × 10–3 |
| Down | P29392 | Spermadhesin-1 | –327 | 1.3 × 10–4 |
| Down | E1BGZ9 | PHD finger protein 20-like protein 1 | –337 | 2.8 × 10–3 |
| Down | F1MMF2 | Uncharacterized protein (Fragment) | –359 | 4.1 × 10–3 |
| Down | Q3ZC66 | Cysteine-rich PDZ-binding protein | –475 | 1.9 × 10–2 |
| Down | F6PZ29 | Uncharacterized protein GN = MCFD2 | –799 | 2.9 × 10–4 |
| Down | Q58DJ3 | KIAA1984 OS = Bos taurus | –824 | 2.1 × 10–3 |
| Down | E1B9W6 | Uncharacterized protein GN = ADCY10 | –2764 | 1.2 × 10–5 |
| Down | Q8SQ24 | Myozenin-1 | –3030 | 7.2 × 10–4 |
| Down | F1MV51 | Uncharacterized protein GN = APC | –3282 | 2.5 × 10–4 |
| Down | P00711 | Alpha-lactalbumin | –7360 | 5.8 × 10–8 |
aFalse discovery rate adjusted.
Table 5. Differentially expressed proteins at 312 hours after intramammary challenge with Streptococcus uberis .
| Up/down-regulated | UniProt ID | Protein name | Fold change | P-value a |
| Up | Q2TBU0 | Haptoglobin | 4191 | 7.4 × 10–10 |
| Up | G3MZ19 | HRPE773-like | 1254 | 2.6 × 10–6 |
| Up | P48616 | Vimentin | 672 | 3.1 × 10–9 |
| Up | P30922 | Chitinase-3-like protein 1 | 444 | 2.3 × 10–7 |
| Up | E1BKS1 | Syndecan | 403 | 8.7 × 10–6 |
| Up | P54229 | Cathelicidin-5 | 387 | 7.8 × 10–7 |
| Up | F1N1Z8 | Uncharacterized protein (Fragment) | 348 | 2.6 × 10–5 |
| Up | Q8SPP7 | Peptidoglycan recognition protein 1 | 291 | 5.5 × 10–7 |
| Up | F1MYX5 | Uncharacterized protein GN = LCP1 | 246 | 8.7 × 10–5 |
| Up | P22226 | Cathelicidin-1 | 226 | 2.4 × 10–6 |
| Up | Q8SQ28 | Serum amyloid A protein | 220 | 2.6 × 10–6 |
| Up | Q2HJF0 | Similar to Serotransferrin | 210 | 3.1 × 10–5 |
| Up | Q9XSJ4 | Alpha-enolase | 190 | 6.7 × 10–7 |
| Up | G3X746 | Uncharacterized protein (Fragment) GN = CABIN1 | 183 | 4.6 × 10–3 |
| Up | P33046 | Cathelicidin-4 | 175 | 3.9 × 10–4 |
| Down | E1BAU6 | Uncharacterized protein GN = INPP5E | –2 | 2.1 × 10–3 |
| Down | P02192 | Myoglobin | –2 | 6.3 × 10–4 |
| Down | P80195 | Glycosylation-dependent cell adhesion molecule 1 | –3 | 3.8 × 10–3 |
| Down | Q0IIH5 | Nucleobindin 2 | –4 | 3.9 × 10–5 |
| Down | E1BLC6 | Uncharacterized protein GN = TTC17 | –67 | 4.3 × 10–3 |
| Down | P13696 | Phosphatidylethanolamine-binding protein 1 | –87 | 2.3 × 10–4 |
| Down | Q8SQ24 | Myozenin-1 | –642 | 4.9 × 10–3 |
aFalse discovery rate adjusted.
3.1.4. Pathway analysis
To detect enriched canonical pathways and to construct functional networks in the differentially expressed bovine proteins, IPA was used (Fig. 3 and ESI,† Fig. S2–S5). The acute-phase response signalling pathway was the most enriched pathway at each time point, with a positive z-score indicating upregulation. The liver X receptor (LXR), retinoid X receptor (LXR) and Farnesoid X receptor (FXR) activation pathways were also enriched following intramammary challenge. The complement system pathway showed a change from downregulation at 36 h PC to upregulation at 81 h PC. Interleukin (IL) 6 signalling is upregulated at 57 and 81 h PC only. Other pathways are also up-regulated at those time points, including Rho signalling, integrin signalling and leucocyte extravasation signalling, whilst an additional pathway is up-regulated at 81 h PC only, i.e. Cdc42 signalling (Fig. 3).
Fig. 3. Canonical pathways enriched in the differentially expressed bovine proteins (n = 292) at 81 hours after intramammary challenge with Streptococcus uberis. The length of the bar against each pathway shows the negative log of the p-value obtained by a Fisher's exact test (the significance of enrichment; the longer the better), and the colour of the bar indicates the direction and strength of regulation inferred from the activation z-score (orange: upregulation, blue: downregulation, grey: no activity pattern available; white: z-score = 0, indicating upregulation of some proteins and downregulation of others), with colour intensity indicating the strength of the effect. The ratio is the proportion of proteins out of the entire pathway that were identified in the dataset, e.g. for ratio = 0.10, 10% of proteins from the pathway were identified in the dataset. LXR = liver X receptor, RXR = retinoid X receptor, FXR = Farnesoid X receptor, IL = interleukin.

The expression of 38 proteins in the acute-phase response signalling pathway changed over the course of the infection (Table 6), with maximum upregulation observed from as early as 42 h, e.g. for HRG and alpha-2-macroglobulin, to as late as 312 h for complement C1 subcomponent and retinol-binding protein. Less than half of the proteins (n = 16) were significantly upregulated at all time points PC. Of proteins with more than 10-fold upregulation, 5 were most strongly upregulated at 42 h, 6 at 57 h, 11 at 81 h, and 2 at 312 h. Haptoglobin was the most strongly upregulated protein at all time points PC. SAA was also strongly upregulated but differences were observed between different isoforms, whereby SAA4 showed a modest peak at 42 h PC whilst SAA1 and SAA3 showed much stronger and later peaks in upregluation, i.e. over a 1000-fold at 81 h PC. Interleukin-1 receptor agonist was the only protein that was upregulated at 36 through 81 h PC and hard returned to the pre-challenge value during the resolution phase at 312 h. Unlike APP, the antimicrobial proteins showed strong upregulation at all time points and all reached peak expression increases of several 1000 or 10 000 fold at 57 h PC. By 312 h PC, their upregulation levels had decreased to several 100 fold or less.
Table 6. Temporal changes in acute phase proteins and antimicrobial proteins in the bovine whey proteome after intramammary challenge with Streptococcus uberis. Acute phase proteins were identified using the ingenuity pathway analysis database with fold-change (compared to 0 h post challenge, PC) and p-values based on one-way ANOVA (show in italics if not <0.05). Antimicrobial proteins were added for comparison. For proteins with a fold change >10, the time point with strongest up- or down regulation is in bold. Values >10 are rounded to the nearest integer.
| UniProt accession | Protein name | Fold change at specified time PC (h) |
False discovery rate adjusted p-value at specified time PC (h) |
||||||||
| 36 | 42 | 57 | 81 | 312 | 36 | 42 | 57 | 81 | 312 | ||
| Acute phase proteins | |||||||||||
| Q3SZR3 | Alpha-1-acid glycoprotein | 1.6 | 1.8 | 1.8 | 1.8 | 1.2 | 1 × 10 –1 | 6 × 10 –2 | 5 × 10 –2 | 5 × 10 –2 | 5 × 10 –1 |
| P28800 | Alpha-2-antiplasmin | 4.9 | 5.9 | 4.6 | 3.1 | 1.4 | 4 × 10–5 | 8 × 10–6 | 7 × 10–5 | 2 × 10–3 | 4 × 10 –1 |
| P12763 | Alpha-2-HS-glycoprotein | 1.4 | 1.8 | 1.7 | 1.2 | –1.4 | 6 × 10 –2 | 3 × 10–3 | 6 × 10–3 | 4 × 10 –1 | 6 × 10 –2 |
| Q7SIH1 | Alpha-2-macroglobulin | 68 | 170 | 128 | 102 | 33 | 2 × 10–4 | 2 × 10–5 | 4 × 10–5 | 7 × 10–5 | 2 × 10–3 |
| P15497 | Apolipoprotein A-I | 6.3 | 8.0 | 6.8 | 4.1 | 1.5 | 3 × 10–5 | 5 × 10–6 | 2 × 10–5 | 7 × 10–4 | 3 × 10 –1 |
| P81644 | Apolipoprotein A-II | 11 | 22 | 14 | 5.1 | –1.4 | 4 × 10–2 | 1 × 10–2 | 3 × 10–2 | 2 × 10 –1 | 8 × 10 –1 |
| Q0VCX1 | Complement C1s subcomponent | 1.0 | 1.0 | 2.2 | 20 | 31 | 1 × 10 +0 | 1 × 10 +0 | 4 × 10 –1 | 4 × 10–3 | 1 × 10–3 |
| Q3SYW2 | Complement C2 | 11 | 8.7 | 19 | 84 | 81 | 2 × 10–2 | 4 × 10–2 | 6 × 10–3 | 1 × 10–4 | 1 × 10–4 |
| Q2UVX4 | Complement C3 | 1.3 | 1.3 | 1.3 | 1.4 | 2.0 | 1 × 10 –1 | 1 × 10 –1 | 1 × 10 –1 | 6 × 10 –2 | 4 × 10–4 |
| F1MY85 | Complement C5a anaphylatoxin | 32 | 32 | 210 | 129 | 21 | 2 × 10–2 | 2 × 10–2 | 4 × 10–4 | 1 × 10–3 | 3 × 10–2 |
| P81187 | Complement factor B | 3.2 | 4.1 | 7.4 | 8.2 | 2.8 | 1 × 10–4 | 6 × 10–6 | 1 × 10–8 | 4 × 10–9 | 4 × 10–4 |
| F1N076 | CP Protein | 3.5 | 4.2 | 4.4 | 3.7 | 2.9 | 3 × 10–5 | 4 × 10–6 | 3 × 10–6 | 2 × 10–5 | 3 × 10–4 |
| P50448 | Factor XIIa inhibitor | –2.5 | –2.4 | –3.0 | –3.2 | –1.2 | 6 × 10–3 | 7 × 10–3 | 1 × 10–3 | 6 × 10–4 | 6 × 10 –1 |
| P02676 | Fibrinogen beta chain | 1.2 | 1.9 | 13 | 9.9 | 7.5 | 8 × 10 –1 | 2 × 10 –1 | 2 × 10 –5 | 1 × 10–6 | 5 × 10–4 |
| F1MGU7 | Fibrinogen gamma-B chain | –1.7 | 1.1 | 3.4 | 2.9 | 3.1 | 2 × 10 –1 | 9 × 10 –1 | 3 × 10–3 | 7 × 10–3 | 5 × 10–3 |
| Q2TBU0 | Haptoglobin | 997 | 4794 | 14 937 | 28 858 | 4191 | 4 × 10–8 | 5 × 10–10 | 3 × 10–11 | 6 × 10–12 | 7 × 10–10 |
| Q3SZV7 | Hemopexin | 131 | 153 | 170 | 158 | 73 | 5 × 10–6 | 3 × 10–6 | 2 × 10–6 | 3 × 10–6 | 3 × 10–5 |
| Q3T0D0 | Heterogeneous nuclear ribonucleoprotein K | 1.0 | 4.7 | 2.5 | 66 | 1.0 | 1 × 10 +0 | 1 × 10 –1 | 3 × 10 –1 | 8 × 10 –5 | 1 × 10 +0 |
| F1MKS5 | Histidine-rich glycoprotein | 106 | 775 | 760 | 451 | 30 | 6 × 10–4 | 6 × 10–6 | 7 × 10–6 | 2 × 10–5 | 9 × 10–3 |
| F1MNW4 | Inter-alpha-trypsin inhibitor heavy chain H2 | 51 | 143 | 78 | 52 | 38 | 3 × 10–3 | 3 × 10–4 | 1 × 10–3 | 3 × 10–3 | 5 × 10–3 |
| Q3T052 | Inter-alpha-trypsin inhibitor heavy chain H4 | 14 | 21 | 34 | 38 | 16 | 5 × 10–3 | 1 × 10–3 | 3 × 10–4 | 2 × 10–4 | 3 × 10–3 |
| Q0VC51 | Interleukin 1 receptor accessory | 2.4 | 2.4 | 213 | 267 | 1.0 | 3 × 10 –1 | 2 × 10 –1 | 5 × 10–8 | 2 × 10–8 | 1 × 10 +0 |
| O77482 | Interleukin-1 receptor antagonist | 30 | 80 | 325 | 176 | 1.0 | 2 × 10–4 | 8 × 10–6 | 7 × 10–8 | 5 × 10–7 | 1 × 10 +0 |
| Q2TBI0 | Lipopolysaccharide-binding protein | 28 | 84 | 395 | 693 | 113 | 2 × 10–4 | 5 × 10–6 | 2 × 10–8 | 4 × 10–9 | 2 × 10–6 |
| C4T8B4 | Pentaxin | 13 | 7.2 | 45 | 82 | 1.0 | 6 × 10 –2 | 2 × 10 –1 | 8 × 10–3 | 3 × 10–3 | 1 × 10 +0 |
| P06868 | Plasminogen | 31 | 33 | 76 | 71 | 13 | 2 × 10–2 | 2 × 10–2 | 4 × 10–3 | 4 × 10–3 | 7 × 10 –2 |
| P00978 | Protein AMBP | 16 | 5.1 | 26 | 16 | 1.2 | 4 × 10–2 | 2 × 10 –1 | 2 × 10–2 | 4 × 10–2 | 9 × 10 –1 |
| P18902 | Retinol-binding protein 4 | 2.3 | 2.2 | –1.4 | 2.4 | 23 | 4 × 10 –1 | 4 × 10 –1 | 7 × 10 –1 | 4 × 10 –1 | 2 × 10–3 |
| Q29443 | Serotransferrin | 4.3 | 5.4 | 5.1 | 4.0 | 2.2 | 2 × 10–4 | 3 × 10–5 | 5 × 10–5 | 4 × 10–4 | 3 × 10–2 |
| A6QPQ2 | Serpin A3-8 | 20 | 158 | 246 | 283 | 37 | 3 × 10–2 | 5 × 10–4 | 2 × 10–4 | 2 × 10–4 | 10 × 10–3 |
| G8JKW7 | SERPINA3 Protein | 2.7 | 3.0 | 2.9 | 4.0 | 2.8 | 2 × 10–3 | 1 × 10–3 | 1 × 10–3 | 8 × 10–5 | 2 × 10–3 |
| P02769 | Serum albumin | 1.9 | 2.2 | 2.1 | 1.4 | –1.4 | 6 × 10–3 | 1 × 10–3 | 2 × 10–3 | 2 × 10 –1 | 1 × 10 –1 |
| F1MMW8 | Serum amyloid A protein – M-SAA3.2 | 20 | 58 | 107 | 358 | 73 | 5 × 10–4 | 1 × 10–7 | 1 × 10–6 | 1 × 10–8 | 4 × 10–6 |
| P35541 | Serum amyloid A protein – SAA1 | 5 | 49 | 1178 | 1926 | 6.5 | 1 × 10 –1 | 2 × 10–3 | 6 × 10–7 | 2 × 10–7 | 1 × 10 –1 |
| Q8SQ28 | Serum amyloid A protein – SAA3 | 93 | 201 | 556 | 1585 | 220 | 4 × 10–5 | 3 × 10–6 | 2 × 10–7 | 8 × 10–9 | 3 × 10–6 |
| Q32L76 | Serum amyloid A protein – SAA4 | 17 | 66 | 27 | 10 | 2.0 | 4 × 10–2 | 3 × 10–3 | 2 × 10–2 | 9 × 10 –2 | 6 × 10 –1 |
| O46375 | Transthyretin | 2.4 | 2.2 | 1.9 | 1.3 | –1.2 | 3 × 10–3 | 7 × 10–3 | 3 × 10–2 | 3 × 10 –1 | 5 × 10 –1 |
| Antimicrobial proteins | |||||||||||
| P22226 | Cathelicidin-1 | 1026 | 3812 | 7281 | 4743 | 226 | 3 × 10–8 | 8 × 10–10 | 1 × 10–10 | 4 × 10–10 | 2 × 10–6 |
| P19660 | Cathelicidin-2 | 78 | 1159 | 2104 | 1683 | 38 | 6 × 10–3 | 4 × 10–5 | 1 × 10–5 | 2 × 10–5 | 2 × 10–2 |
| P33046 | Cathelicidin-4 | 208 | 2619 | 4753 | 2963 | 175 | 3 × 10–4 | 1 × 10–6 | 3 × 10–7 | 8 × 10–7 | 4 × 10–4 |
| P54229 | Cathelicidin-5 | 1444 | 9209 | 16 618 | 11 722 | 387 | 2 × 10–8 | 2 × 10–10 | 3 × 10–11 | 8 × 10–11 | 8 × 10–7 |
| P56425 | Cathelicidin-7 | 1217 | 8922 | 11 877 | 7316 | 178 | 2 × 10–6 | 2 × 10–8 | 9 × 10–9 | 3 × 10–8 | 3 × 10–2 |
| Q8SPP7 | Peptidoglycan recognition protein 1 | 3305 | 8453 | 27 479 | 17 090 | 291 | 5 × 10–10 | 4 × 10–11 | 2 × 10–12 | 6 × 10–12 | 6 × 10–7 |
4. Discussion
In the present study, a label-free quantitative proteomics approach was used for profiling the bovine whey proteome during experimentally induced S. uberis mastitis. This enabled the dynamic change in 570 proteins of the whey proteome to be studied in synchronisation with the clinical and bacteriological manifestations of infection,3 the peptidome4 and the metabolome,9 and allowed quantification of the relative change in multiple proteins in milk samples from the S. uberis infected quarters. Furthermore, by examining sequential time points following bacterial challenge, the temporal changes in important host response pathways were revealed. Thus at 36 h post challenge, the first time point examined, peptidoglycan recognition protein 1 and the cathelicidins, which are antimicrobial proteins (AMP) derived from phagocytic polymorphonuclear leucocytes (PMNL) cells that cross from the blood into the mammary gland, show the highest fold increase, reaching a peak at 57 h PC. In contrast, APP such as haptoglobin, LBP and SAA, derived from local synthesis in mammary epithelia increase at a slower rate but showed their maximal levels by 81 h PC. The concentrations in the milk samples of haptoglobin and serum amyloid A were also measured by immunoassay,4 giving similar increases at 57 h and 81 h and thus serving as validation of the results obtained by the quantitative proteomics approach. It would be of great interest to similarly validate by immunoassay or Western blot the results obtained for other potential biomarkers over the course of the S. uberis IMI, such as the cathelicidins and histidine rich glycoprotein, but this was beyond the scope of the current investigation.
Changes in the milk proteome during mastitis due to infection with S. uberis, S. aureus or E. coli have been studied previously using mass spectrometry techniques.27 Many of these studies used gel-based techniques, which are semi-quantitative, although recently quantification using labeling such as iTRAQ or calibration standards have been described.7,28–31 The method used here was able to yield relative quantification of 570 proteins, which is among the highest number that have been determined, being exceeded only in the study of Reinhardt and coworkers7 who examined subsets of milk proteins and also depleted both caseins and lactoglobins in order to enhance detection of low abundance protein. In the current study only caseins were depleted by ultracentrifugation. Method refinements introduced here that may have enhanced protein recovery included total protein concentration being normalized after acetone precipitation and the preparation of trypsin digests using SDC as well as acetonitrile to improve complete digestion of proteins.32,33
In a systems biology approach it is appropriate to consider the time course of the changes in the multiple components of milk during IMI caused by S. uberis to put the data generated into context (Fig. 4). Many but not all of the proteome responses to IMI found by quantitative proteomics occurred at the same time as the maximal change in peptidomic and metabolomic responses4,9 with the maximal change in analytes occurring at 81 h PC, at 45 h after the peak in bacterial count (36 h PC) and, with the exception of one cow (cow 5), after rectal temperature had returned to normal levels. This course of events, combined with the cytokine profiles,3 supports the interpretation that the response to bacterial challenge first leads to cytokine release which subsequently causes the resultant change in peptide, protein and metabolite profiles. At 36 h PC, bacterial counts peak, clinical signs are detectable, cytokines IL-1, IL-6, IL-8, IL-10, and IL-12p40 have been released as well as TNF-α and there are detectable changes in the proteins and metabolites. This coincides with massive influx of polymorphonuclear leucocytes (PMNL) into the mammary gland,3 which accounts for the increased milk somatic cell count (SCC). The PMNL influx may be a causative event in both the reduction in bacterial numbers and the change in peptidomic and proteomic profiles.
Fig. 4. The relative responses of analytes following experimental induced S. uberis mastitis combining proteomic results with data from Tassi et al. 3 and Thomas et al. 4 The shading represents strength of the response relative to the peak response Responses were increases compared to day 0 levels except for casein levels (indicated by *), which decreased after challenge. cfu = colony forming unit; SCC = somatic cell count; TNF = tumor necrosis factor; 1DGE = 1 dimensional gel electrophoresis; (m)SAA3 = (milk) derived serum amyloid A; IMI77 = peptidomic profile based on 77 peptides.

The bioinformatics tools used here to examine the overall changes taking place in the whey proteome demonstrate that maximal responses occurred at 57 and 81 h PC, time points that clustered by HCA. PCA demonstrated that milk samples from 81 h PC were the most divergent from the pre-challenge samples while samples from 312 h PC, i.e. the resolution phase, were being restored towards, but were still distinct from the pre-challenge clusters, even though 5 cows had cleared the infection at that point.3 Pathway analysis using IPA identified the APP pathway as having the largest change of any pathway at all time points, supporting the evidence from investigations using immunoassay and transcript analysis that these proteins are among those most affected by IMI.4,34 The second and third most affected pathways were the LXR/RXR activation and FXR/RXR activation pathways, incorporating liver (LXR), retinoid (RXR) and farnesoid (FXR) receptor related proteins. However, a number of APP are also components of these pathways and lead to identified up-regulation by IPA due to this cross-recognition. The IPA also showed that although the PMNL influx increases rapidly between 24 and 42 h post-challenge,3 the leucocyte extravasation signalling pathway was only enriched at 57 and 81 h PC, indicating that there may be a lag between initial influx and detectable levels of protein upregulation in this pathway. Similarly, IL-6 levels were significantly elevated at 36 and 42 h PC based on ELISA assays,3 but enrichment of the IL-6 pathway was not detected until 57 h PC by proteomic analysis and IPA.
Like IPA, analysis of differential protein expression profiles identified APP as being central to the pathophysiological changes following S. uberis challenge. In addition, several AMP featured in the lists of proteins with the highest fold increase in expression. The AMP are a diverse group of proteins that show antimicrobial activity. They are secreted by PMNL and function as primary effectors of innate immunity in the mammary gland.35 Among the AMP, cathelicidins and peptidoglycan recognition protein 1 were strongly upregulated from 36 h PC onwards, with expression levels 1000s of times higher than before challenge. Indeed, cathelecidin-5 and peptidoglycan recognition protein showed the largest fold increase of any of the proteins quantified by LC-MS/MS up to and including 57 h PC. Previous studies also reported up-regulation of AMPs, particularly cathelicidins, in mastitic milk.6,7 Other AMPs, e.g. lactoperoxidase and mucin, which is thought to be an inducible innate immune effector,36 were detected at lower level after challenge, which could indicate decreased expression, or increased use without replenishment. Interestingly, the highest levels of cathelicidins were detected from 42 to 81 h, a period that coincides with a massive decrease in bacterial numbers3 from an average of 108 cfu ml–1 down to 104 cfu ml–1, and cathelicidin expression decreased after this reduction in cfu count. Unlike some other mastitis pathogens, S. uberis is resistant to phagocytosis and killing by neutrophils.37 The massive increase in cathelicidin levels, which followed PMNL influx and preceded or coincided with bacterial clearance, may provide an alternative mechanism by which PMNL contribute to resolution of IMI caused by S. uberis.
As the acute-phase response is a swift systemic inflammatory reaction in response to infections and is already implicated in responses to IMI38,39 it is no surprise that changes were found among the APP in this investigation. However, the profile of changes in multiple APP, in response to the S. uberis challenge, was shown here in much more detail than has been previously possible and within the APP, differing profiles were found. A number of the APP showed their highest fold increase at 42 h PC. Thus, alpha-2-macroglobulin and HRG had fold changes of 170× and 775× respectively at this time point. In contrast, a number of APP showed continuing elevation in their fold increase up to 81 h PC with haptoglobin, SAA1 and LBP having fold increases of 28 858×, 1926× and 693× respectively. The differences found in the profile of responses of the APP are likely to be due to cellular mechanisms in the control of their synthesis and release, dependent on the cytokine cocktail developed in response to infection.40,41 Cytokine profiles differ between bacterial species42 and hence differing profiles of both the APP and AMP responses can be expected for different mastitis pathogens. Further investigation of these profiles and of interaction with the peptide and metabolomics changes4,9 may lead to multiplexed biomarker analysis capable of providing pathogen specific diagnosis which would be of great value in mastitis diagnosis and therapy.
Examining the expression of individual APP, increased expression of haptoglobin is known to occur during mastitis38,43 and has been quantified previously in proteomic investigation.44,45 It was apparent that Hp detection by quantitative proteomic analysis was more sensitive than detection by ELISA, as substantial increases in Hp levels were detected at 36 h PC in by the proteomic approach, but not by Thomas and colleagues where ELISA was used.4 The high fold increase of Hp which was still present at 312 h PC at 4191× indicates that Hp may be useful as an indicator of high SCC, which was still high at that time, but may have limited value as indicator of the IMI, which had been resolved in 5 of 6 animals.3 SAA, in the isoforms found here, also reached a maximum at 81 h PC. These are a family of apolipoproteins that are associated with high density lipoprotein when in serum.38 The mammary associated SAA3 isoform is also one of the first APP reported to increase during mastitis and previous proteomics studies have shown up-regulation of isoforms of SAA in milk in response to gram-negative and gram-positive pathogens.6,7,38 As for Hp, proteomic analysis identified the increase in SAA levels earlier than ELISA-based analysis4 demonstrating further that quantitative proteomics may be more sensitive than the forms of ELISA used previously. However the use of relative quantitation may give a misleading impression of the change taking place when the level of the protein in the control (0 h PC) is very low or not detectable in the LC-MS/MS analysis. Absolute quantification by calibrated standard in quantitative proteomics or in immunoassay is needed to determine the change in the absolute concentration of the milk proteins in IMI.
Among the APP with an early maximum fold increase at 42 h PC, alpha-2-macroglobulin is a protease inhibitor that can inhibit all four classes of proteases (serine, cysteine, aspartyl and metalloproteases). It is present in milk in its native, active state and its concentration is known to increase during mastitis.7,46 HRG was also identified as an early elevated APP and is a major plasma protein in a range of mammals, including cattle.47,48 It plays a role in blood coagulation, fibrinolysis, and innate immune systems and is also thought to have antibacterial properties.48 As HRG was upregulated as early as 36 h PC and was returned to normal levels towards the end of the experiment, it may have a role as a diagnostic marker in detecting the occurrence and resolution of IMI. However, the only protein with significantly increased expression at 36 h PC which had returned to pre-challenge levels in the resolution phase was interleukin-1 receptor agonist. During the resolution phase of IMI (57 to 312 h PC), increased levels of vimentin were detected. Vimentin is a fibroblast marker, whilst there are conflicting reports on its presence in myoepithelial cells.49,50 Its elevated expression in milk would appear to indicate damage or repair of the subalveolar tissue of the mammary gland.
In addition to quantifying host proteins in whey, we attempted to quantify bacterial peptides and identify bacterial proteins using the S. uberis reference proteome (data not shown). Despite massive increase in bacterial numbers over the course of infection with peak concentrations around 108 colony forming units per ml of milk,3 differential expression analysis showed much lower fold increases than for bovine proteins (maximum of 706 fold increase for a bacterial putative lipoprotein versus maximum of 28 858 fold change for haptoglobin). Separation of bacteria from whey or other modifications to the sample processing methods may be needed for better characterisation of the bacterial proteome during IMI.
5. Conclusion and outlook
Using a label-free relative quantification method, changes in protein expression in bovine whey in an experimental model of S. uberis mastitis have been determined. In particular, the dynamic changes in the proteome during establishment and resolution of infection, with emphasis on APP and AMP has been determined. Our results were in agreement with previous proteomic studies but provide a time-course rather than a snapshot of protein profiles. Proteins that have not been previously associated with mastitis, including HRG, an acute-phase and antimicrobial protein, have been quantified. In addition, the time course of events observed in our linked studies provides a potential explanation for the clearance of S. uberis after influx of PMNL, whereby cathelicidins produced by the PMNL rather than neutrophil phagocytosis and killing may be the main effector mechanism. Quantitative proteomics has provided an additional layer of analysis to the milk whey samples obtained during the experimental model of bovine mastitis caused by S. uberis and by integration with the pathophysiological, molecular, peptidomic and metabolomics analyses performed on the same sample set has enabled a more comprehensive, systems level view of the host responses to bovine mastitis than has been achieved previously.
Acknowledgments
The work was carried out in Glasgow Polyomics, which is supported by the Wellcome Trust (grant no. 097821/Z/11/Z). It was also financially supported by the Moredun Innovation Fund (RT) and the Scottish Government's Rural and Environment Science and Analytical Services Division (TM and RZ).
Footnotes
References
- D'Alessandro A., Zolla L., Scaloni A. Mol. BioSyst. 2011;7:579–597. doi: 10.1039/c0mb00027b. [DOI] [PubMed] [Google Scholar]
- Yang Y., Bu D., Zhao X., Sun P., Wang J., Zhou L. J. Proteome Res. 2013;12:1660–1667. doi: 10.1021/pr301001m. [DOI] [PubMed] [Google Scholar]
- Tassi R., McNeilly T. N., Fitzpatrick J. L., Fontaine M. C., Reddick D., Ramage C., Lutton M., Schukken Y. H., Zadoks R. N. J. Dairy Sci. 2013;96:5129–5145. doi: 10.3168/jds.2013-6741. [DOI] [PubMed] [Google Scholar]
- Thomas F. C., Mullen W., Tassi R., Ramirez-Torres A., Mudaliar M., McNeilly T. N., Zadoks R. N., Burchmore R., Eckersall P. D. Mol. BioSyst. 2016 doi: 10.1039/c6mb00239k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerstedt M., Wredle E., Lam V., Johansson M. Journal of Dairy Research. 2012;79:297–303. doi: 10.1017/S0022029912000301. [DOI] [PubMed] [Google Scholar]
- Boehmer J. L. J. Mammary Gland Biol. Neoplasia. 2011;16:323–338. doi: 10.1007/s10911-011-9229-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt T. A., Sacco R. E., Nonnecke B. J., Lippolis J. D. J. Proteomics. 2013;82:141–154. doi: 10.1016/j.jprot.2013.02.013. [DOI] [PubMed] [Google Scholar]
- Zadoks R. N. CAB Rev. 2007;30:1–15. [Google Scholar]
- Thomas F. C., Mudaliar M., Tassi R., McNeilly T. N., Burchmore R., Burgess K., Herzyk P., Zadoks R. N., Eckersall P. D. Mol. BioSyst. 2016 doi: 10.1039/c6mb00289g. [DOI] [PubMed] [Google Scholar]
- Cox J., Mann M. Annu. Rev. Biochem. 2011;80:273–299. doi: 10.1146/annurev-biochem-061308-093216. [DOI] [PubMed] [Google Scholar]
- Abdallah C., Dumas-Gaudot E., Renaut J., Sergeant K. Int. J. Plant Genomics. 2012;2012:494572. doi: 10.1155/2012/494572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M., Murakami K., Wallingford J. C., Yuki Y. Electrophoresis. 2002;23:1153–1160. doi: 10.1002/1522-2683(200204)23:7/8<1153::AID-ELPS1153>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Bantscheff M., Schirle M., Sweetman G., Rick J., Kuster B. Anal. Bioanal. Chem. 2007;389:1017–1031. doi: 10.1007/s00216-007-1486-6. [DOI] [PubMed] [Google Scholar]
- Aebersold R. J. Infect. Dis. 2003;187(suppl 2):S315–320. doi: 10.1086/374756. [DOI] [PubMed] [Google Scholar]
- Megger D. A., Bracht T., Meyer H. E., Sitek B. Biochim. Biophys. Acta. 2013;1834:1581–1590. doi: 10.1016/j.bbapap.2013.04.001. [DOI] [PubMed] [Google Scholar]
- Latosinksa A., Vougas K., Makridakis M., Klein J., Mullen W., Abbaas M., Stavodimos K., Katafigiotis I., Merseburger A. S., Zoidakis J., Mischak H., Vlahou A., Jankowski V. PLoS One. 2015;10:e0137048. doi: 10.1371/journal.pone.0137048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W., Smith J. W., Huang C. M. J. Biomed. Biotechnol. 2010;2010:840518. doi: 10.1155/2010/840518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowell A. M., Wall M. J., Doucette A. A. Anal. Chim. Acta. 2013;796:48–54. doi: 10.1016/j.aca.2013.08.005. [DOI] [PubMed] [Google Scholar]
- Pluskal T., Castillo S., Villar-Briones A., Orešič M. BMC Bioinf. 2010;11:395. doi: 10.1186/1471-2105-11-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S., Temu T., Carlson A., Sinitcyn P., Mann M., Cox J. Proteomics. 2015;15:1453–1456. doi: 10.1002/pmic.201400449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Mann M. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. J. Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- Cox J., Hein M. Y., Luber C. A., Paron I., Nagaraj N., Mann M. Mol. Cell. Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Matic I., Hilger M., Nagary N., Selbach M., Olsen J. V., Mann M. Nat. Protoc. 2009;4:698–705. doi: 10.1038/nprot.2009.36. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K. P., Kuhn M., Bork P., Jensen L. J., Von Mering C. Nucleic Acids Res. 2015;43:D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer A., Green J., Pollard J., Tugendreich S. Bioinformatics. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira A. M., Bislev S. L., Bendixen E., Almeida A. M. J. Proteomics. 2013;94:110–123. doi: 10.1016/j.jprot.2013.09.012. [DOI] [PubMed] [Google Scholar]
- Hettinga K., van Valenberg H., de Vries S., Boeren S., van Hooijdonk T., van Arendonk J., Vervoort J. PLoS One. 2011;6:e19433. doi: 10.1371/journal.pone.0019433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese S., Reidgeld K. A., Meyer H. E., Warscheid B. Proteomics. 2007;7:1004. doi: 10.1002/pmic.200600422. [DOI] [PubMed] [Google Scholar]
- Bisleve S. L., Kusebauch U., Codrea M. C., Beynon R. J., Harman V. M., Rontved C., Aebersold R., Moritz R. L., Bendixen E. J. Proteome Res. 2012;11:1832–1843. doi: 10.1021/pr201064g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Boeren S., Hageman J. A., van Hooijdonk T., Vervoort J., Hettinga K. PLoS One. 2015;10:e0116710. doi: 10.1371/journal.pone.0116710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon I. R., Schwammle V., Jensen O. N., Sprenger R. R. Mol. Cell. Proteomics. 2013;12:2992–3005. doi: 10.1074/mcp.M112.025585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y., Zhou J., Bi D., Chen P., Wang X., Liang S. Anal. Biochem. 2008;377:259–266. doi: 10.1016/j.ab.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Whelehan C. J., Meade K. G., Eckersall P. D., Young F. J., O'Farrelly C. Vet. Immunol. Immunopathol. 2011;140:181–189. doi: 10.1016/j.vetimm.2010.11.013. [DOI] [PubMed] [Google Scholar]
- Stelwagen K., Carpenter E., Haigh B., Hodgkinson A., Wheeler T. T. J. Anim. Sci. 2009;87(suppl 13):3–9. doi: 10.2527/jas.2008-1377. [DOI] [PubMed] [Google Scholar]
- Sando L., Pearson R., Gray C., Parker P., Hawken R., Thomson P. C., Meadows J. R., Kongsuwan K., Smith S., Tellam R. L. J. Dairy Sci. 2009;92:5276–5291. doi: 10.3168/jds.2009-2216. [DOI] [PubMed] [Google Scholar]
- Leigh J. A. Vet. J. 1999;157:225–238. doi: 10.1053/tvjl.1998.0298. [DOI] [PubMed] [Google Scholar]
- Ceciliani F., Ceron J. J., Eckersall P. D., Sauerwein H. J. Proteomics. 2012;75:4207–4231. doi: 10.1016/j.jprot.2012.04.004. [DOI] [PubMed] [Google Scholar]
- Tothova C., Nagy O., Kovac G. Vet. Med. 2014;59:163–180. [Google Scholar]
- Bode J. G., Albrecht U., Haeussinger D., Heinrich P. C., Schaper F. Eur. J. Cell Biol. 2012;91:496–505. doi: 10.1016/j.ejcb.2011.09.008. [DOI] [PubMed] [Google Scholar]
- Moshage H. J. Pathol. 1997;181:257–266. doi: 10.1002/(SICI)1096-9896(199703)181:3<257::AID-PATH756>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Bannerman D. D. J. Anim. Sci. 2009;87:10–25. doi: 10.2527/jas.2008-1187. [DOI] [PubMed] [Google Scholar]
- Eckersall P. D., Young F. J., McComb C., Hogarth C. J., Safi S., Fitzpatrick J. L., Nolan A. M., Weber A., McDonald T. Vet. Rec. 2001;148:35–41. doi: 10.1136/vr.148.2.35. [DOI] [PubMed] [Google Scholar]
- Ibeagha-Awemu E. M., Ibeagha A. E., Messier S., Zhao X. J. Proteome Res. 2010;9:4604–4619. doi: 10.1021/pr100336e. [DOI] [PubMed] [Google Scholar]
- Smolenski G. A., Broadhurst M. K., Stelwagen K., Haigh B. J., Wheeler T. T. Proteome Sci. 2014;12:19. doi: 10.1186/1477-5956-12-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rantamäki L. K., P Müller H. J. Dairy Res. 1992;59:273–285. doi: 10.1017/s0022029900030557. [DOI] [PubMed] [Google Scholar]
- Turk R., Piras C., Kovacic M., Samardzija M., Ahmed H., De Canio M., Urbani A., Mestric Z. F., Soggiu A., Bonizzi L., Roncada P. J. Proteomics. 2012;75:4412–4428. doi: 10.1016/j.jprot.2012.05.021. [DOI] [PubMed] [Google Scholar]
- Wakabayashi S. Int. Rev. Cell Mol. Biol. 2013;304:467–493. doi: 10.1016/B978-0-12-407696-9.00009-9. [DOI] [PubMed] [Google Scholar]
- Cravero D., Marignani E., Miretti S., Macchi E., Accornero P., Baratta M. Res. Vet. Sci. 2014;97:367–375. doi: 10.1016/j.rvsc.2014.07.022. [DOI] [PubMed] [Google Scholar]
- Zavizion B., Politis I., Gorewit R. C. J. Dairy Sci. 1992;75:3367–3380. doi: 10.3168/jds.S0022-0302(92)78113-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.