

Abstract
Hepatocellular carcinoma (HCC) is one of the most common malignant cancers. To elucidate new regulatory mechanisms for heptocarcinogenesis, we investigated the regulation of p21, a cyclin-dependent kinase (CDK) inhibitor encoded by CDKN1A, in HCC. The expression level of p21 is decreased with the progression of HCC. Luciferase assays with a luciferase-p21-3′ UTR reporter and its serial deletions identified a 15-bp repressor element at the 3′-UTR of CDKN1A, which contains a binding site for miR-95-3p. Mutation of the binding site eliminated the regulatory effect of miR-95-3p on p21 expression. Posttranscriptional regulation of p21 expression by miR-95-3p is mainly on the protein level (suppression of translation). Overexpression of miR-95-3p in two different HCC cell lines, HepG2 and SMMC7721, significantly promoted cell proliferation, cell cycle progression and cell migration, whereas a miR-95-3p specific inhibitor decreased cell proliferation, cell cycle progression and cell migration. The effects of miR-95-3p on cellular functions were rescued by overexpression of p21. Overexpression of miR-95-3p promoted cell proliferation and tumor growth in HCC xenograft mouse models. Expression of miR-95-3p was significantly higher in HCC samples than in adjacent non-cancerous samples. These results demonstrate that miR-95-3p is a potential new marker for HCC and regulates hepatocarcinogenesis by directly targeting CDKN1A/p21 expression.
Hepatocellular carcinoma (HCC) is one of the most malignant cancers and causes 662,000 deaths each year worldwide, with more than 50% of deaths occurring in China1−4. Despite achievements in improved diagnosis and surgical therapy in the past few decades, the prognosis of HCC is poor, and the 5 year survival rate of HCC patients is low5. This may be due to incomplete understanding of the molecular pathogenic mechanisms of HCC. Dysregulation of cell cycle progression is a major characteristic of hepatocarcinogenesis, which leads to excessive HCC cell proliferation. Cell cycle is regulated by cyclin-dependent kinases (CDKs), whereas CDK activity is partly inhibited by CDK inhibitors including p21Waf/Cip1 6. The p21 is a well-known CDK inhibitor which belongs to the Cip/Kip family of CDK inhibitors and encoded by the CDKN1A gene7. The p21 protein inhibits the activity of cyclin-CDK2 or -CDK4 complexes and negatively modulates cell cycle progression at phase G1 8. It can also interact with PCNA (proliferating cell nuclear antigen) and is involved in regulation of S phase DNA replication and DNA damage repair9,10,11. As an inhibitor of cell cycle and cell proliferation, p21 plays an important role in tumorigenesis12,13,14.
The expression of CDKN1A/p21 is tightly controlled at the transcriptional level primarily by the tumor suppressor protein p53 in response to DNA damage13. In addition, the stability of p21 was regulated by multiple mechanisms, including interaction with and phosphorylation by extracellular signal-regulated kinase 2 (ERK2), phosphorylation by c-Jun N-terminal kinase, glycogen synthase kinase (GSK) 3 and AKT15,16,17,18. Because of the critical role of p21 in cell cycle and tumor cell proliferation, identification of new regulatory mechanisms for regulation of p21 is important for further understanding of HCC progression and for exploring new treatment and prevention for HCC.
MicroRNAs (miRNAs) are a cluster of noncoding RNA molecules that are approximately 18–25 nucleotides in length and negatively regulate the expression of downstream target genes mainly through direct interaction with the 3′-untranslated regions (3′-UTR) of their corresponding mRNA targets19. MiRNAs generally decrease target gene expression through mRNA degradation and/or translational suppression20. Increasing evidence in the recent years indicates that many microRNAs play critical roles in tumorigenesis and progression21,22. In this study, we found that CDKN1A/p21 was regulated by miR-95-3p by targeting the 3′-UTR. We further showed that expression of miR-95-3p was up-regulated in HCC, whereas expression of p21 was decreased with progression of HCC. Overexpression of miR-95-3p led to increased tumor cell proliferation and growth in mice. Our study defines miR-95-3p as a new oncogenic miRNA involved in hepatocarcinogenesis.
Results
Expression of p21 is decreased with progression of HCC
The p21 protein is a tumor suppressor which has been reported to participate in tumor progression and proliferation6. In this study, we characterized the expression level of p21 in a total of 60 HCC samples and 3 adjacent non-cancerous tissue samples (NCT) using immunohistochemistry. As shown in Fig. 1, the expression level of p21 was high in the 3 adjacent NCT samples. As HCC progresses from stage II to a higher grade of stage IIIC, the expression levels of p21 were decreased (Fig. 1).
Figure 1. The expression level of p21 was decreased with progression of HCC.

(a) Representative images of HCC tissue sections at different stages and immunostained with an anti-p21 antibody. NCT: control adjacent non-cancerous tissue; Scale bar = 50 μm. (b) Correlation between the intensity of immunostaining signals for p21 and the stage of HCC. Negative, no expression or little expression of p21; weak+, weak expression of p21; strong+, strong expression of p21.
Identification of a repressor element regulating p21 expression at the 3′-UTR of CDKN1A
One potential mechanism for down-regulation of p21 in HCC samples is posttranscriptional regulation by microRNA at 3′-UTR of the CDKN1A gene (encoding p21). To identify such a posttranscriptional mechanism for p21 regulation, we constructed a luciferase reporter with the 1,539 bp 3′-UTR of CDKN1A sub-cloned downstream of the luciferase gene in the pMIR-REPORTER vector, resulting in the pMIR-p21-wt reporter (reporter pMIR-1 in Fig. 2a,b). Serial deletions were then made in pMIR-p21-wt, resulting in construction of 8 different deletion mutant reporters (reporter pMIR-2-9 in Fig. 2b). Luciferase assays in HepG2 cells showed that compared to the vector pMIR-REPORTER, luciferase activity of reporter pMIR-1 was reduced, suggesting that there is a repressor element in the 3′-UTR of CDKN1A for regulation of CDKN1A/p21 expression (Fig. 2c). When the region from +1,500 to +1,515 at the 3′-UTR of CDKN1A was deleted, the luciferase activity was significantly increased to the level of pMIR-REPORTER (compare reporter pMIR-9 to others, Fig. 2c). The data suggest that the 3′-UTR repressor element regulating p21 expression is located at the 15-bp region between 1,500 bp and 1,515 bp from the stop codon TAA at the 3′-UTR of CDKN1A.
Figure 2. Identification of a critical regulatory element at the 3′-UTR of the CDKN1A gene (encoding p21) for regulation of p21 expression.
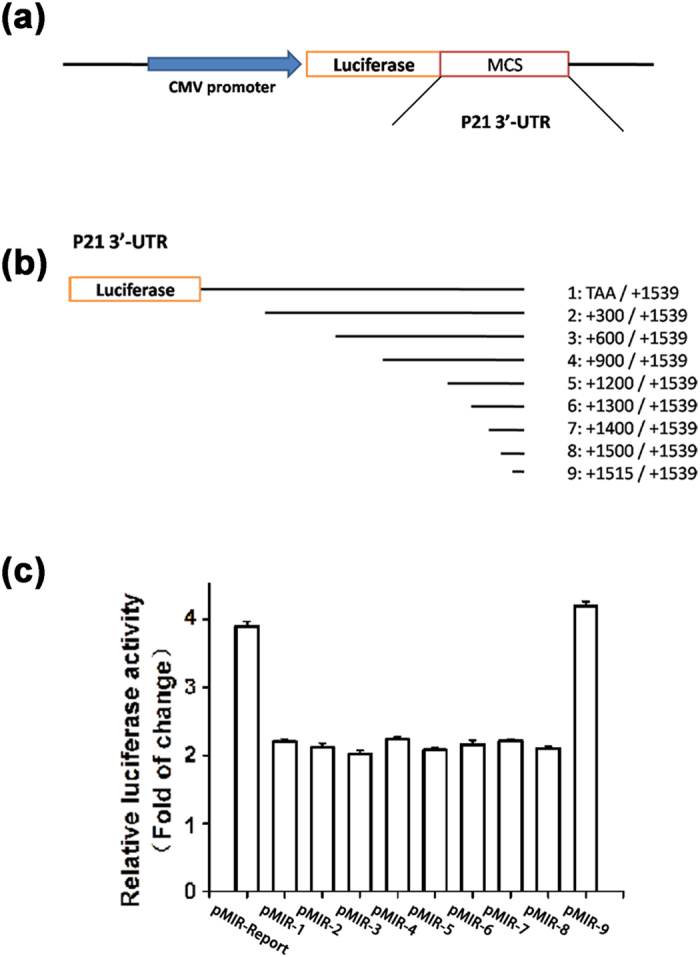
(a) Schematic diagram showing a luciferase reporter with the 3′-UTR of the CDKN1A gene (a 1,539 bp fragment from the stop codon) sub-cloned after the luciferase gene (pMIR-p21-wt or pMIR-1). (b) Serial deletions of the 3′-UTR were created in pMIR-1, resulting in pMIR-2 to pMIR-9. (c) Luciferase activity for different luciferase reporters. Note that a repressor element was present at the region from +1,500 to +1,515 of the 3′-UTR of the CDKN1A gene.
MiR-95-3p regulates expression of p21 by targeting the 3′-UTR of CDKN1A
We analyzed the 3′-UTR repressor element regulating p21 expression for a potential microRNA binding site using miRBase (http://www.mirbase.org/). A potential binding site for Hsa-miR-95-3p was found at the 3′-UTR repressor element (Fig. 3a).
Figure 3. MiR-95-3p inhibits p21 expression by directly targeting the 3′-UTR of the CDKN1A gene.
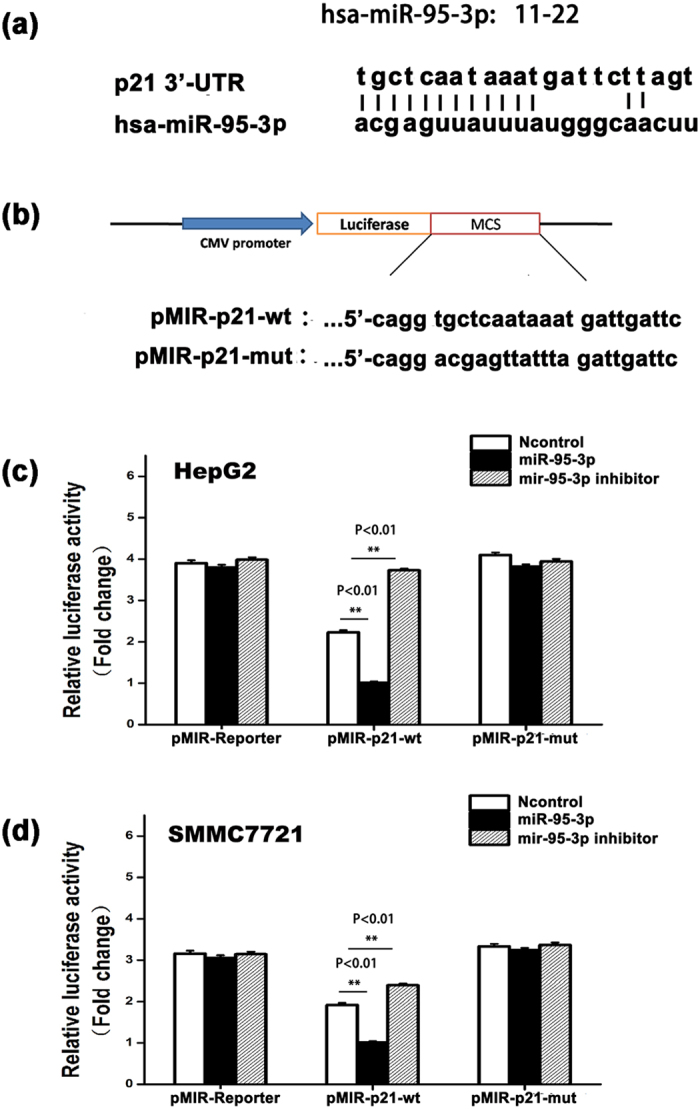
(a) Bioinformatic analysis of the repressor element for the stability of CDKN1A mRNA at the region from +1,500 to +1,515 of the 3′-UTR identified a binding site for miR-95-3p. (b) Schematic diagram showing the wild type (wt) pMIR-p21-wt or mutant pMIR-p21-mut reporter with the miR-95-3p binding site mutated. (c) Luciferase activity of pMIR-p21-wt or mutant pMIR-p21-mut reporters in the presence of miR-95-3p mimics, a negative control miRNA mimics (Ncontrol), and a miR-95-3p specific inhibitor in HepG2 cells. (d) Similar luciferase assays as in (c) but in SMMC7721 cells.
To determine whether miR-95-3p regulates expression of p21, we mutated the miR-95-3p binding site in the luciferase reporter pMIR-p21-wt (wild type reporter), resulting in a mutant pMIR-p21-mut reporter (Fig. 3b). The pMIR-p21-wt or pMIR-P21-mut reporter was co-transfected into two different HCC cell lines, HepG2 and SMMC7721 together with miR-95-3p mimics, a negative control miRNA mimics (Ncontrol), or a miR-95-3p inhibitor and luciferase assays were performed. Compared with empty pMIR-REPORTER vector, the luciferase activity of pMIR-P21-wt was decreased due to endogenous miR-95-3p. For wild type reporter pMIR-P21-wt, transfection of miR-95-3p mimics significantly reduced the reporter luciferase activity compared to Ncontrol, whereas a miR-95-3p specific inhibitor significantly increased the reporter luciferase activity (Fig. 3c,d). However, miR-95-3p mimics and the miR-95-3p inhibitor did not affect the luciferase activity of the mutant pMIR-p21-mut reporter in which the miR-95-3p binding site was mutated (Fig. 3c,d). Together, these data suggest that miR-95-3p down-regulates the expression of p21 by directly targeting the 3′-UTR of CDKN1A.
To further validate the finding that miR-95-3p regulates the expression of p21, we examined the protein expression level of p21 using Western blot analysis with HepG2 and SMMC7721 cells transfected with miR-95-3p mimics vs. Ncontrol, a miR-95-3p specific inhibitor, and a negative control miRNA inhibitor (NC inhibitor). As expected, compared to Ncontrol mimics, miR-95-3p mimics significantly reduced the protein expression level of p21 (Figs 4a and 5a). On the contrary, the miR-95-3p inhibitor significantly increased the protein expression level of p21 in both HepG2 and SMMC7721 cells compared to the NC inhibitor (Figs 4b and 5b).
Figure 4. MiR-95-3p inhibits expression of p21 at the translational level and promotes HepG2 tumor cell proliferation and migration.

(a) Western blot analysis for the expression level of the p21 protein in HepG2 cells transfected with miR-95-3p mimics compared with negative control miRNA mimics (Ncontrol). The Western blot images are shown at the left, quantified and graphed at the right. β-tubulin was used as a loading control. (b) Similar Western blot analysis as in (a) but with a miR-95-3p inhibitor vs. a negative control miRNA inhibitor (NC inhibitor). (c) Proliferation of HepG2 cells transfected with miR-95-3p mimics alone or with a p21 expression plasmid vs. Ncontrol mimics, and miR-95-3p inhibitor vs. NC inhibitor. (d) Percentage of cells at the S phase during cell cycle in HepG2 cells transfected with miR-95-3p mimics alone or with a p21 expression plasmid vs. Ncontrol mimics, and miR-95-3p inhibitor vs. NC inhibitor. (e) Migration of HepG2 cells transfected with miR-95-3p mimics alone or with a p21 expression plasmid, Ncontrol mimics, and miR-95-3p inhibitor using a wound assay.
Figure 5. MiR-95-3p inhibits expression of p21 at the translational level and promotes SMMC7721 tumor cell proliferation and migration.

(a) Western blot analysis for the expression level of the p21 protein in SMMC7721 cells transfected with miR-95-3p mimics compared with negative control miRNA mimics (Ncontrol). The Western blot images are shown at the left, quantified and graphed at the right. β-tubulin was used as a loading control. (b) Similar Western blot analysis as in (a) but with a miR-95-3p inhibitor vs. a negative control miRNA inhibitor (NC inhibitor). (c) Proliferation of SMMC7721 cells transfected with miR-95-3p mimics alone or with a p21 expression plasmid vs. Ncontrol mimics, and miR-95-3p inhibitor vs. NC inhibitor. (d) Percentage of cells at the S phase during cell cycle in SMMC7721 cells transfected with miR-95-3p mimics alone or with a p21 expression plasmid vs. Ncontrol mimics, and miR-95-3p inhibitor vs. NC inhibitor. (e) Migration of SMMC7721 cells transfected with miR-95-3p mimics alone or with a p21 expression plasmid, Ncontrol mimics, and miR-95-3p inhibitor using a wound assay.
The expression level of the CDKN1A mRNA was also assessed using real time RT-PCR analysis. The real-time RT-PCR analysis showed that the CDKN1A mRNA expression level was slightly decreased in HepG2 and SMMC7721 cells transfected with miR-95-3p mimics compared to Ncontrol mimics (Supplementary Fig. S1a and Supplementary Fig. S2a), and slightly increased by the miR-95-3p inhibitor compared to the NC inhibitor (Supplementary Fig. S1b and Supplementary Fig. S2b). However, the differences were not statistically significant (P > 0.05). Taken together, these data suggest that miR-95-3p down-regulates p21 expression mainly at the protein or translational level by directly targeting the 3′-UTR.
MiR-95-3p promotes tumor cell proliferation and migration
Considering the finding that miR-95-3p negatively regulates expression of tumor suppressor p21, we hypothesized that miR-95-3p could promote tumor cell proliferation and migration as well as tumor growth. To determine the impact of miR-95-3p on HCC cell proliferation, we transfected HepG2 and SMMC7721 cells with miR-95-3p mimics vs. Ncontrol, and the miR-95-3p specific inhibitor vs. the NC inhibitor. Cell proliferation assays with the CCK-8 kit revealed that overexpression of miR-95-3p significantly promoted cell proliferation of HepG2 and SMMC7721 cells (Figs 4c and 5c). The miR-95-3p inhibitor significantly reduced cell proliferation of HepG2 and SMMC7721 cells (Figs 4c and 5c). Furthermore, overexpression of miR-95-3p led to an increase of S-phase cells during cell division (Figs 4d and 5d), whereas the miR-95-3p inhibitor significantly decreased the number of S-phase cells (Figs 4d and 5d). The wound healing assays showed that overexpression of miR-95-3p significantly increased migration of HepG2 and SMMC7721 cells (Figs 4e and 5e), whereas the miR-95-3p inhibitor decreased migration of HepG2 and SMMC7721 cells (Figs 4e and 5e).
To examine whether the observed effects of miR-95-3p are due to down-regulation of p21, we co-transfected HepG2 and SMMC7721 cells with a p21 expression plasmid together with miR-95-3p mimics. Overexpression of p21 significantly abrogated the effects of miR-95-3p mimics on cell proliferation, cell division and migration in both HepG2 and SMMC7721 cells (Figs 4c,d,e and 5c,d,e). Taken together, these results suggest that miR-95-3p promotes HCC tumor cell proliferation, cell cycle progression and cell migration by directly targeting p21.
MiR-95-3p promotes tumor growth in mice
We used mouse Hepa1-6 cells to create a xenograft tumor model in mice for HCC. AgomiR-95-3P or AgomiR-NC was transfected into Hepa1-6 cells. Transfected cells were injected subcutaneously into the back of the C57BL/6 mice and tumor growth was monitored. At the end of the study, tumors were excised, weighed and photographed (Fig. 6a). The tumor size from the AgomiR-95-3p group was larger than those from the AgomiR-NC group (Fig. 6a,b). The tumor weight from the AgomiR-95-3p group was significantly heavier than that from the AgomiR-NC group (Fig. 6c). The tumor growth curves showed that the tumors from the AgomiR-95-3p group grew faster than those from the AgomiR-NC control group (Fig. 6d). Real time qPCR analysis showed that the expression level of miR-95-3p was significantly higher in tumors from the AgomiR-95-3p group than those from the AgomiR-NC group (Fig. 6e). Together, these data suggest that overexpression of miR-95-3p promoted tumor growth in vivo.
Figure 6. Overexpression of miR-95-3p promotes tumor growth in mice.

An in vivo hepatoma xenograft model was created by subcutaneous injection of 5 × 105 murine hepatoma Hepa1-6 cells transfected with AgomiR-95-3p or AgomiR-NC in mice. (a) Tumor growth for the AgomiR-95-3p group and the AgomiR-NC group. (b) Photos of tumors excised from mice on experimental day 22. (c) Tumor weight for the AgomiR-95-3p group and the AgomiR-NC group. (d) Tumor growth curves for the AgomiR-95-3p group and the AgomiR-NC group. The tumor volume (V) was computed using a formula of V = (L × W2) × 0.5 (n = 7/group). (e) Quantitative qPCR analysis showing that the expression level of miR-95-3p was significantly increased in tumors isolated from the AgomiR-95-3p group compared with the AgomiR-NC group.
We used immunohistochemical staining with an anti-Ki67 antibody to examine the density of positive Ki67 proliferating cells. An anti-p21 antibody was also used to examine the expression level of p21 in mouse tumor sections. The density of positive Ki67 cells was much higher in tumors from the AgomiR-95-3p group than those from the AgomiR-NC group (Fig. 7a). Immunohistochemical analysis showed that the expression level of p21 was much lower in tumors from the AgomiR-95-3p group than those from the AgomiR-NC group (Fig. 7a). Western blot analysis also showed that p21 expression was lower in the AgomiR-95-3p group than in the AgomiR-NC group (Fig. 7b,c). These data provide in vivo evidence that miR-95-3p down-regulates the expression of p21, which leads to increased cell proliferation and tumor growth.
Figure 7. Overexpression of miR-95-3p decreases p21 expression and promotes tumor cell proliferation in mice.

An in vivo hepatoma xenograft model was established as in Fig. 6. (a) Representative images of tumor sections from the AgomiR-95-3p group and the AgomiR-NC group. H&E, haematoxylin and eosin stain; p21, immunostaining with an anti-p21 antibody; Ki67, immunostaining with an anti-Ki67 antibody; Scale bar = 100 μm. (b) Western blot analysis for p21. β-tubulin was used as an internal control. (c) The p21/tubulin ratio in (b) was quantified.
MiR-95-3p expression was increased in HCC tissues
We used semi-quantitative RT-PCR analysis to measure the relative expression level of miR-95-3p in 10 pairs of HCC samples and their respective adjacent non-cancerous samples. In each pair, the expression level of miR-95-3p was consistently higher in HCC samples than in adjacent non-cancerous samples (Fig. 8a,b,d). Together, the expression level of miR-95-3p was significantly higher in HCC samples than in adjacent non-cancerous samples (P < 0.01; Fig. 8c,e).
Figure 8. The expression level of miR-95-3p is higher in HCC tissues than in normal adjacent non-cancerous tissues.

(a) Semi-quantitative RT-PCR analysis showed a higher expression level of miR-95-3p in HCC tissue samples than in normal adjacent tissue samples. (b) PCR bands in (a) were quantified and plotted. The ratio of the expression level of miR-95-3p over U6 RNA in HCC tissue sample was higher than that in the normal adjacent non-cancerous tissue sample in all 10 groups. (c) The relative miR-95-3p expression level over U6 RNA in human HCC tissues was significantly higher than that in normal adjacent non-cancerous tissues. (d) PCR bands in (a) were quantified and plotted. The ratio of the expression level of miR-95-3p over 5S RNA in HCC tissue sample was higher than that in the normal adjacent non-cancerous tissue sample in all 10 groups. (e) The relative miR-95-3p expression level over 5S RNA in human HCC tissues was significantly higher than that in normal adjacent non-cancerous tissues.
Discussion
In this study, we demonstrated that the expression level of miR-95-3p was consistently up-regulated in HCC tissues compared with adjacent non-cancerous tissues in all 10 groups of samples examined (Fig. 8). Moreover, the expression levels of miR-95-3p in HCC tissues were higher than that from all non-tumorous tissues (Fig. 8). Because the sample size is small, future studies with large sample sizes are needed to further validate this interesting finding. If confirmed, miR-95-3p may serve as a potential marker for diagnosis of HCC.
We found that overexpression of miR-95-3p significantly promoted HepG2 and SMMC7721 cell proliferation and migration in cultured cells (Figs 4 and 5) and Hepa1-6 tumor cell proliferation and tumor growth in mice (Figs 6 and 7). Therefore, it is highly likely that up-regulation of miR-95-3p in HCC tissues is causative to hepatocarcinogenesis and tumor growth. Mechanistically, we showed that miR-95-3p causes hepatocarcinogenesis by posttranscriptional suppression of p21 expression by binding to the 3′-UTR. Several pieces of evidence strongly supports the conclusion. First, we found that the expression level of p21 is reduced with the progression of HCC. When HCC reached stageIIIC, there is little expression of p21 (Fig. 1). Second, we constructed several luciferase reporters which contain different regions of 3′-UTR of CDKN1A cloned downstream of the luciferase gene. Luciferase assays showed that there is a potential repressor element in the 3′-UTR of CDKN1A, which contains a miRNA binding site for miR-95-3p (Fig. 2). Overexpression of miR-95-3p mimics decreased the expression of p21, and the effect was eliminated by mutation of the binding site for miR-95-3p (Figs 3, 4 and 5). Moreover, a miR-95-3p specific inhibitor increased the expression of p21 (Figs 4 and 5). Third, overexpression of miR-95-3p in a mouse hepatoma xenograft model decreased expression of p21 (Fig. 7). Together, we conclude that CDKN1A encoding p21 is a downstream gene regulated by miR-95-3p.
Similar to our finding of up-regulation of miR-95-3p in HCC tissues and promotion of tumorigenesis by overexpression of miR-95-3p, three other reports revealed involvement of miR-95-3p in other types of tumors. The expression level of miR-95-3p was reported to be up-regulated in glioma tissues and down-regulation of miR-95-3p inhibited proliferation and invasion and promoted apoptosis of glioma cells by targeting CELF2 encoding CUGBP- and ETR-3-like family 2 23. The expression level of miR-95-3p was also up-regulated in human prostate and breast cancer tissues or after ionizing radiation. Overexpression of miR-95-3p promoted radiation resistance and cell proliferation following ionizing radiation and increased tumor cell invasiveness as well as tumor growth by targeting the sphingolipid phosphatase SGPP124. The expression level of miR-95-3p was up-regulated in human non-small cell lung cancer tissues and overexpression of miR-95-3p increased tumor growth in xenograft mouse models by targeting SNX1 encoding sorting nexin125. Our study identified the CDKN1A gene encoding p21 as a new target gene for miR-95-3p. In contrast, overexpression of miR-95-3p was found to inhibit brain metastasis of lung adenocarcinoma by suppressing expression of cyclin D126. Together, these studies indicate that miR-95-3p plays important roles in tumorigenesis in different types of cancer by targeting different downstream genes.
There are several limitations with the present study. (1) We have shown that down-regulation of p21 appears to be responsible for the effect of miR-95-3p on cell proliferation, cell cycle progression and cell migration in two independent HCC cell lines because co-expression of p21 rescued the effects of miR-95-3p (Figs 4 and 5). However, as discussed above, miR-95-3p also regulates other target genes such as CELF2, SGPP1, SNX1 and many other unidentified target genes, future studies are needed to investigate the roles of other miR-95-3p target genes in the pathogenesis of HCC. (2) There are other microRNAs that can also regulate the expression of p21. It should be interesting to investigate the roles of other p21-regulating miRNAs in the pathogenesis of HCC. (3) Overexpression of microRNAs may have off-target effects, and caution should be used in interpreting the data, although complementary studies of both miR-95-3p mimics and a miR-95-3p specific inhibitor strengthened the conclusions. (4) The 15-bp 3′-UTR region missing in pMIR-9 (Fig. 2) also contains a less-well matched seed sequence for miR-545-5p (6-nucleotide match). It may be interesting to examine whether miR-545-5p also regulates expression of CDKN1A in the future. (5) The sample sizes of three normal healthy liver specimens for the immunohistochemistry of p21 (Fig. 1) and 10 pairs of HCC tissue samples and adjacent non-cancerous samples for semi-quantitative RT-PCR analysis of miR-95-3p (Fig. 8) are small. Future studies with large sample sizes are needed to further validate the findings of decreased p21 expression and increased miR-95-3p expression in HCC.
In summary, we have found that miR-95-3p is up-regulated in HCC tissues compared with adjacent non-cancerous tissues. Overexpression of miR-95-3p promoted tumor cell proliferation and migration in cultured cells and tumor growth in xenograft mouse models through negative posttranscriptional regulation of p21 by directly targeting the 3′-UTR. This study establishes miR-95-3p as a potential biomarker for diagnosis of HCC and as a new therapeutic target for treatment and prevention of HCC.
Methods
Cell lines and human tumor samples
Three HCC cell lines, including HepG2, SMMC7721, and Hepa1-6, were cultured in DMEM media supplemented with 10% fetal bovine serum (FBS) (Gibco Life Technologies, Gaitherburg, MD, USA) under 5% CO2 and at 37 °C. We screened HepG2 and SMMC7721 cells for mutations in genes encoding p53, p21 and MDM2, but no mutation was found.
HCC tissues and matched human adjacent non-cancerous liver tissue samples (NCT) were collected from patients undergoing surgical resection of tumors in Affiliated Hospitals of Huazhong University of Science and Technology. Clinical stages were classified according to the International Union against Cancer TNM classification system27. The demographical features of age and sex and clinical stages of HCC patients were listed in Supplementary Tables S1 and S2. This study was approved by the Ethics Committees on human subject research of Huazhong University of Science and Technology and local institutions and written informed consent was obtained from all study subjects. This study conformed to guidelines set forth by the Declaration of Helsinki.
Construction of plasmids
The whole 3′-UTR of the CDKN1A gene encoding p21 (1,539 bp) was amplified by polymerase chain reaction (PCR) analysis with human genomic DNA as the template. The PCR product was cut with Spe I and Hind III and cloned into the pMIR-REPORT luciferase vector (Promega, Madison, WI, USA), resulting in a luciferase reporter referred to as pMIR-p21-wt. Serial deletions were created in pMIR-p21-wt (pMIR-1), resulting in different deletion mutants: pMIR-p21-f-300bp (pMIR-2), pMIR-p21-f-600bp (pMIR-3), pMIR-p21-f-900bp (pMIR-4), pMIR-p21-f-1200bp (pMIR-5), pMIR-p21-f-1300bp (pMIR-6), pMIR-p21-f-1400bp (pMIR-7), pMIR-p21-f-1500bp (pMIR-8), and pMIR-p21-f-1515bp (pMIR-9). In all reporters, the 3′-UTR fragment of CDKN1A was cloned downstream of the firefly luciferase coding region, which is under the control of the CMV promoter.
The miR-95-3p binding site at the 3′-UTR of CDKN1A was mutated in the pMIR-p21-wt reporter using site-directed mutagenesis by PCR as previously described28,29,30,31, resulting in reporter pMIR-p21-mut.
Primers used in this study for plasmid construction and mutagenesis were listed in Supplementary Table S3.
MicroRNA reagents
We purchased miR-95-3p mimics, negative control miRNA mimics (Ncontrol), a miR-95-3p inhibitor, a negative control miR-95-3p inhibitor (NC inhibitor), AgomiR-95-3p and control AgomiR-NC from Guangzhou RioboBio (Guangzhou, Guangdong, China).
Dual luciferase assays
Luciferase assays were performed as described previously by us32,33,34. HepG2 and SMMC7721 cells were plated in a 24-well plate and co-transfected with 200 ng of either pMIR-p21-wt, pMIR-p21-mut, or a pMIR-p21-deletion mutants together with 100 nM of miR-95-3p mimics, negative control mimics (NControl) or a miR-95-3p specific inhibitor as well as 10 ng of the pRL-TK vector containing the renilla luciferase gene (Promega, Madison, WI, USA) using Lipofectamine 2000 (Gibco Life Technologies, Gaithersburg, MD, USA). Luciferase assays were carried out using the Dual-Glo luciferase assay kit according to the manufacturer’s instruction (Promega, Madison, WI, USA). The final luciferase activity was expressed as the firefly activity over the renilla luciferase activity.
Measurement of miRNA expression using quantitative RT-PCR (qRT-PCR) analysis
We quantified the relative expression level of miR-95-3p using stem-loop real time PCR as previously described35,36.
Real-time RT-PCR analysis
Total cell or tissue RNA samples were extracted and reverse-transcribed to cDNA by the RevertAid First Strand cDNA synthesis kit (Fermentas) using random primers (Promega, Madison, WI, USA). The expression level of CDKN1A mRNA was quantified using a FastStart Universal SYBR Green Master kit (Roche Applied Science, Mannheim, Germany) as described by us previously31,32,33,37,38. The endogenous control was ACTB (encoding β-actin). Primers for real-time RT- PCR analysis were listed in Supplementary Table S3. Data were analyzed using the 2−△△Ct method as described36.
Western blot analysis
Western blot analysis was carried out as described by us previously31,33. The antibodies used in this study include an anti-p21 antibody (1:500 dilution, Proteintech, Wuhan, China) and an anti-β-tubulin antibody (1:2000 dilution, Proteintech, Wuhan, China).
In vivo tumor growth assays
Both animal care and experimental procedures were approved by the Ethics Committee on Animal Research of College of Life Science and Technology of Huazhong University of Science and Technology and performed according to the Guidelines for the Care and Use of Animals for Research by the Ministry of Science and Technology of the P. R. China. C57BL/6 mice (males, 7-week-old) were purchased from Center for Medical Experimental Animals of Wuhan University (Wuhan, Hubei, China).
In vivo tumor growth assays were performed as previously described39,40. In brief, 150 nM of AgomiR-95-3p or AgomiR-NC was transfected into mouse Hepa1-6 cells using Lipofectamin RNAiMAX (Gibco Life Technologies, Gaithersburg, MD, USA). After 24 h of transfection, cells were harvested, washed with cold PBS and suspended at a concentration of 5 × 106 cells/ml in PBS. C57BL/6 mice were divided into 2 groups (n = 7). These mice were subcutaneously injected with 5 × 105 Hepa1-6 cells (100 μl) on the back. Tumor growth was examined every 2 days beginning at day 4. Tumor length (L) and width (W) were measured and the formula of V = (L × W2) × 0.5 was used to calculate the tumor volume (V).
At the end of the experiment at day 22, the tumors were excised from mice and weighed.
Immunohistochemical staining
Tumors excised from mice were fixed in 4% paraformaldehyde and used for immunohistochemical staining. Immunohistochemical staining was performed as described31,33,41. Briefly, 4.5 μm-tumor sections were immunostained with an antibody against p21 (1:100 dilution, Proteintech, Wuhan, China) and an antibody against Ki-67 (1:100 dilution, Proteintech, Wuhan, China). DAB PI was used for staining of nuclei. Hematoxylin-eosin staining was used to evaluate the morphology of the tumor sections as described by us31. Images were captured under a microscope.
Cell cycle assay
Cell cycle analysis was performed as described31 with cells transfected with miR-95-3p mimics, negative control miRNA mimics (Ncontrol), a miR-95-3p inhibitor or a negative control inhibitor (NC inhibitor). Cells were stained with propidium iodide (PI). Cell cycle analysis was performed using a Beckman Coulter Cytomics FC 500 flow cytometry and CXP software (Beckman Coulter).
Cell proliferation and migration assays
Cells were seeded onto 96-well plates, and assayed for proliferation at 48 h using a CCK-8 kit (Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s instruction. Cell proliferation was analyzed by measurement of absorbance at 450 nm using a microplate reader as described42.
For cell migration, we used a wound assay as described34. Cells (5 × 105) were seeded onto six-well plates and cultured under standard conditions. When the cell density reached confluence, a wound was made by scraping the cell monolayer with a 200 μl pipette tip. Cell migration was determined by measuring the movement of cells into the scraped area. The process of wound closure was monitored and photographed 12 hours after wounding under a microscope.
Bioinformatic and statistical analyses
We used the miRBase (http://www.mirbase.org/) database to predict the putative binding sites of miRNAs.
All quantitative data for statistical analyses were from at least three independent experiments. The data were presented as means ± SD (standard deviation). Statistical analysis was performed using a Student’s t test. A P value of <0.05 was considered to be statistically significant.
Additional Information
How to cite this article: Ye, J. et al. Up-regulation of miR-95-3p in hepatocellular carcinoma promotes tumorigenesis by targeting p21 expression. Sci. Rep. 6, 34034; doi: 10.1038/srep34034 (2016).
Supplementary Material
Acknowledgments
We thank other Wang Lab members for technical support and advice. This work was supported by the National Natural Science Foundation of China grants 91439129 and 31430047, the National Basic Research Program of China (973 Programs 2013CB531101 and 2012CB517801), a Natural Science Key Program of Hubei Province (2014CFA074), the Hubei Province’s Outstanding Medical Academic Leader program, a Key Project in the National Science & Technology Pillar Program during 395 the Twelfth Five-year Plan Period (2011BAI11B19), Specialized Research Fund for the Doctoral Program of Higher Education from the Ministry of Education, the “Innovative Development of New Drugs” Key Scientific Project (2011ZX09307-001-09), and NIH R01 HL121358 and R01 HL126729.
Footnotes
Author Contributions J.Y., Y.Y., Q.S. and Q.K.W. conceived and designed the study; J.Y., Y.Y., Q.S., S.L., Z.H., C.H., X.D., H.L. and Y.Y. performed experiments; J.Y., Y.Y., Q.S., S.L., Q.C. and Q.K.W. analyzed data; J.Y. and Y.Y. drafted the manuscript; Q.C. and Q.K.W. critically revised the manuscript; Q.K.W. supervised the study; all authors reviewed the manuscript.
References
- Song P. et al. Biomarkers: evaluation of screening for and early diagnosis of hepatocellular carcinoma in Japan and china. Liver cancer 2, 31–39 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asia-Pacific Working Party on Prevention of Hepatocellular, C. Prevention of hepatocellular carcinoma in the Asia-Pacific region: consensus statements. Journal of gastroenterology and hepatology 25, 657–663 (2010). [DOI] [PubMed] [Google Scholar]
- Yuen M. F., Hou J. L., Chutaputti A. & Asia Pacific Working Party on Prevention of Hepatocellular, C. Hepatocellular carcinoma in the Asia pacific region. Journal of gastroenterology and hepatology 24, 346–353 (2009). [DOI] [PubMed] [Google Scholar]
- Kansagara D., Papak J. & Jou J. H. Screening for hepatocellular carcinoma in chronic liver disease. Ann Intern Med 162, 240 (2015). [DOI] [PubMed] [Google Scholar]
- El-Serag H. B. & Rudolph K. L. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 132, 2557–2576 (2007). [DOI] [PubMed] [Google Scholar]
- Abbas T. & Dutta A. p21 in cancer: intricate networks and multiple activities. Nature reviews. Cancer 9, 400–414 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper J. W., Adami G. R., Wei N., Keyomarsi K. & Elledge S. J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75, 805–816 (1993). [DOI] [PubMed] [Google Scholar]
- Deng C., Zhang P., Harper J. W., Elledge S. J. & Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 82, 675–684 (1995). [DOI] [PubMed] [Google Scholar]
- Moldovan G. L., Pfander B. & Jentsch S. PCNA, the maestro of the replication fork. Cell 129, 665–679 (2007). [DOI] [PubMed] [Google Scholar]
- Umar A. et al. Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell 87, 65–73 (1996). [DOI] [PubMed] [Google Scholar]
- Mortusewicz O., Schermelleh L., Walter J., Cardoso M. C. & Leonhardt H. Recruitment of DNA methyltransferase I to DNA repair sites. Proceedings of the National Academy of Sciences of the United States of America 102, 8905–8909 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zirbes T. K. et al. Prognostic impact of p21/waf1/cip1 in colorectal cancer. Int J Cancer 89, 14–18 (2000). [DOI] [PubMed] [Google Scholar]
- Anttila M. A. et al. p21/WAF1 expression as related to p53, cell proliferation and prognosis in epithelial ovarian cancer. Br J Cancer 79, 1870–1878 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiya T. et al. p21 expression as a predictor for favorable prognosis in squamous cell carcinoma of the lung. Clin Cancer Res 3, 1831–1835 (1997). [PubMed] [Google Scholar]
- Li Y., Dowbenko D. & Lasky L. A. AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival. The Journal of biological chemistry 277, 11352–11361 (2002). [DOI] [PubMed] [Google Scholar]
- Hwang C. Y. & Kwon K.-S. p21Cip1 regulation by ERK2: A post-translational mechanism that relays a proliferation signal. Cell Cycle 8, 3625–3626 (2014). [DOI] [PubMed] [Google Scholar]
- Lee J. Y., Yu S. J., Park Y. G., Kim J. & Sohn J. Glycogen synthase kinase 3beta phosphorylates p21WAF1/CIP1 for proteasomal degradation after UV irradiation. Molecular and cellular biology 27, 3187–3198 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossig L., Badorff C., Holzmann Y., Zeiher A. M. & Dimmeler S. Glycogen synthase kinase-3 couples AKT-dependent signaling to the regulation of p21Cip1 degradation. The Journal of biological chemistry 277, 9684–9689 (2002). [DOI] [PubMed] [Google Scholar]
- Bartel D. P. MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio M. V. & Croce C. M. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. EMBO molecular medicine 4, 143–159 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram P. & Santosh K. K. Down-regulation of miRNA-106b inhibits growth of melanoma cells by promoting G1-phase cell cycle arrest and reactivation of p21/WAF1/Cip1 protein. Oncotarget 5, 10636–10649 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashita Y. et al. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer research 65, 9628–9632 (2005). [DOI] [PubMed] [Google Scholar]
- Fan B. et al. Downregulation of miR-95-3p inhibits proliferation, and invasion promoting apoptosis of glioma cells by targeting CELF2. Int J Oncol 47, 1025–1033 (2015). [DOI] [PubMed] [Google Scholar]
- Huang X. et al. MiRNA-95 mediates radioresistance in tumors by targeting the sphingolipid phosphatase SGPP1. Cancer research 73, 6972–6986 (2013). [DOI] [PubMed] [Google Scholar]
- Huang Z. et al. MicroRNA-95 promotes cell proliferation and targets sorting Nexin 1 in human colorectal carcinoma. Cancer research 71, 2582–2589 (2011). [DOI] [PubMed] [Google Scholar]
- Hwang S. J. et al. Overexpression of microRNA-95-3p suppresses brain metastasis of lung adenocarcinoma through downregulation of cyclin D1. Oncotarget 6, 20434–20448 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun K. H. et al. The rationality of N3 classification in the 7th edition of the International Union Against Cancer TNM staging system for gastric adenocarcinomas: a case-control study. International journal of surgery 12, 893–896 (2014). [DOI] [PubMed] [Google Scholar]
- Tian X. L. et al. Identification of an angiogenic factor that when mutated causes susceptibility to Klippel-Trenaunay syndrome. Nature Protocols 427, 640–645 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S. et al. Genomic variant in CAV1 increases susceptibility to coronary artery disease and myocardial infarction. Atherosclerosis 246, 148–156 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q. et al. Angiogenic factor AGGF1 promotes therapeutic angiogenesis in a mouse limb ischemia model. PLoS One 7, e46998 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y. et al. Role of microRNA-27a in down-regulation of angiogenic factor AGGF1 under hypoxia associated with high-grade bladder urothelial carcinoma. Biochimica et biophysica acta 1842, 712–725 (2014). [DOI] [PubMed] [Google Scholar]
- Huang Y. et al. Molecular Basis of Gene-Gene Interaction: Cyclic Cross-Regulation of Gene Expression and Post-GWAS Gene-Gene Interaction Involved in Atrial Fibrillation. PLoS genetics 11, e1005393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B. et al. MicroRNA-503 targets FGF2 and VEGFA and inhibits tumor angiogenesis and growth. Cancer Lett 333, 159–169 (2013). [DOI] [PubMed] [Google Scholar]
- Fan C. et al. Novel roles of GATA1 in regulation of angiogenic factor AGGF1 and endothelial cell function. The Journal of biological chemistry 284, 23331–23343 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic acids research 33, e179 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen T. D. & Livak K. J. Analyzing real-time PCR data by the comparative CT method. Nature Protocols 3, 1101–1108 (2008). [DOI] [PubMed] [Google Scholar]
- Zhao Y. et al. Post-transcriptional regulation of cardiac sodium channel gene SCN5A expression and function by miR-192-5p. Biochimica et biophysica acta 1852, 2024–2034 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J. et al. Cardiac sodium channel regulator MOG1 regulates cardiac morphogenesis and rhythm. Scientific reports 6, 21538 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S. et al. MicroRNA-192 targeting retinoblastoma 1 inhibits cell proliferation and induces cell apoptosis in lung cancer cells. Nucleic acids research 39, 6669–6678 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B. et al. SCF-mediated mast cell infiltration and activation exacerbate the inflammation and immunosuppression in tumor microenvironment. Blood 112, 1269–1279 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyzas P. A., Stefanou D., Batistatou A. & Agnantis N. J. Hypoxia-induced tumor angiogenic pathway in head and neck cancer: an in vivo study. Cancer letters 225, 297–304 (2005). [DOI] [PubMed] [Google Scholar]
- Xu F. Y. et al. The antitumor activity study of ginsenosides and metabolites in lung cancer cell. American journal of translational research 8, 1708–1718 (2016). [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.