

Abstract
In mammals, the non-heme iron enzyme cysteine dioxygenase (CDO) helps regulate cysteine (Cys) levels through converting Cys to cysteine sulfinic acid (CSA). Its activity is in part modulated by the formation of a Cys93-Tyr157 crosslink that increases its catalytic efficiency over 10-fold. Here, 21 high-resolution mammalian CDO structures are used to gain insight into how the Cys-Tyr crosslink promotes activity and how select competitive inhibitors bind. Crystal structures of crosslink-deficient C93A and Y157F variants reveal similar ~1.0 Å shifts in the side chain of residue 157, and both variant structures have a new chloride ion coordinating the active site iron. Cys binding is also different than for wild-type CDO and no Cys-persulfenate forms in the C93A or Y157F active sites at either pH=6.2 or 8.0. We conclude that the crosslink enhances activity by positioning the Tyr157 hydroxyl for enabling proper Cys binding, proper oxygen binding, and optimal chemistry. In addition, structures are presented for homocysteine, thiosulfate and azide bound as competitive inhibitors. The observed binding modes of homocysteine and D-Cys make clear why they are not substrates, and the binding of azide shows that, in contrast to what has been proposed, it does not bind in these crystals as a superoxide mimic.
Graphical abstract

Introduction
Cysteine dioxygenase (CDO) is a mononuclear non-heme iron protein that catalyzes the first and committed step of cysteine catabolism, converting cysteine (Cys) to cysteine sulfinic acid (CSA) (Scheme 1) by incorporating both oxygen atoms of molecular oxygen to form CSA (reviewed in1; 2). The regulation of CDO is crucial for animals to maintain cellular cysteine levels within a narrow range,3; 4 as high levels of Cys are toxic,5; 6; 7 and imbalances of Cys metabolism have been associated with several neurological disorders.8; 9; 10; 11 In a CDO knockout mouse, Cys levels are only slightly elevated, but an increased catabolism of Cys though desulfhydration pathways gives rise to increased thiosulfate and H2S toxicity12.
Scheme 1.
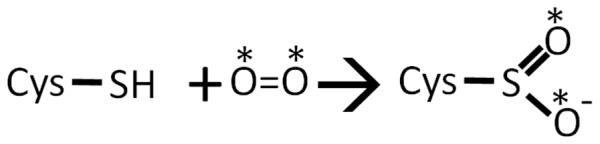
Reaction Catalyzed by Cysteine Dioxygenase
In mammals, CDO activity is upregulated in two ways in response to high cysteine levels. First is an over ~20-fold increase in CDO through a decrease in its degradation by the ubiquitin-proteosome system,13; 14 and second is an over 10-fold increase in specific activity derived in a yet unknown manner from the autocatalyzed formation of a Cys93-Tyr157 crosslink.15; 16; 17; 18 This crosslink is not present in newly transcribed CDO but builds up over time as the result of a reaction that occurs during occasional turnovers.15
Structurally, mammalian CDOs have a cupin fold, with three (neutral19) His residues coordinating the iron.20; 21 The Tyr157-OH of the Cys93-Tyr157 crosslink is located near the iron and is proposed to be a catalytic acid/base that is activated via a Ser153-His155-Tyr157 catalytic triad.20; 22 Spectroscopic studies show Cys binds to the enzyme first and creates the oxygen binding site,23; 24; 25 but except for the recent evidence for a fleeting UV-absorbing oxygen-bound intermediate,26 spectroscopic evidence for discrete intermediates has been unobtainable,23; 24; 25; 27 and the mechanism of CDO remains uncertain (e.g. 28,29 and 30; 31; 32; 33 and 25; 30). A high resolution crystal structure of a Cys-persulfenate/persulfenic acid complex with wild-type mammalian CDO revealed geometries for how Cys and O2 may coordinate the iron and a central role for Tyr157-OH (Figure 1).22 pH-dependent analyses of these same crystals showed that an important change takes place around pH=7.5, as Cys-persulfenate is formed in the active site from pH 5.5 to 7.0 but at pH values of 8.0 and 9.0, at which CDO is more active, Cys binds to the active site but no persulfenate forms.28 We proposed that the changes were due to deprotonation of Tyr157, as it is located close to both oxygen atoms of the persulfenate where it could influence oxygen binding. As the persulfenate formed in the crystal is not turned over to make CSA,29 and calculations indicate other pathways are of lower energy, 31; 32; 33; 34 the persulfenate/persulfenic acid may be an off-pathway complex rather than a true intermediate of the reaction.
Figure 1.

Standard view of the wild-type rat CDO active site with bound cysteine persulfenate/persulfenic acid. Stick model from PDB code 3ELN22 is shown with standard atom coloring (oxygen: red, nitrogen: blue, sulfur: yellow, iron: orange, and carbons: grey). Iron ligation bonds (solid) are shown and inferred hydrogen bonds (dashed with distances) are indicated. The Cys position matches that seen for Cys alone28 and is thought to be catalytically relevant. The dioxygen moiety of the Cys-persulfenate is also thought to roughly occupy the catalytically relevant oxygen binding site that is created upon Cys binding.
Interestingly, bacterial CDOs have similar turnover rates to mammalian CDO,35 but have a Gly in place of Cys9335; 36 proving that the Cys-Tyr crosslink is not essential for catalysis and raising questions about how its formation enhances catalysis. We have shown that bacterial CDO has the same binding mode for Cys and the same relative position of the Tyr157-OH as crosslinked mammalian CDO,36 and another recent study showed a conserved Tyr157-OH position in a C93G variant of mammalian CDO.37 Kinetics of the variant and low-crosslinked preparations of wild-type mammalian CDO showed higher pH optima, consistent with thioether formation decreasing the pKa of Tyr157, but still not revealing how crosslink formation enhances activity.21; 24; 37; 38 Davies et al.37 proposed that the advantage of crosslink formation was not to directly increase activity, but to remove side-reactions that could be caused by the free Cys93 thiolate at higher pH.37 However, a shortcoming of C93G as a model for non-crosslinked CDO is that it is missing the non-covalent steric interaction that must exist between non-crosslinked Cys93 and Tyr157 sidechains.
Here, we further investigate the roles of the Cys93-Tyr157 crosslink and the putative acid/base Tyr157 by defining high resolution structures of the crosslink deficient C93A and Y157F variants in the presence and absence of Cys. As the active sites of C93A and Y157F are similarly disrupted due to the absence or the shift of Y157-OH, we conclude it is the proper positioning of this hydroxyl upon crosslink formation that directly enhances the enzyme activity by influencing Cys binding and active site chemistry. We also provide insight into the specificity of the enzyme through determining structures of wild-type CDO and/or the variants in the presence of the competitive inhibitors homocysteine, azide, thiosulfate and D-Cys.19; 39; 40
Results
In all, we describe in this report 21 new CDO structures at resolutions between 1.25 and 1.65 Å (Tables 1, 2, S1, and S2). Of these, 16 are of C93A and Y157F CDO either alone or bound to L-Cys, D-Cys or the inhibitor homocysteine; the other five are of wild-type CDO bound to homocysteine, azide, or thiosulfate. Based on our previous study of Cys-soaked wild-type CDO crystals,28 pH values of both 6.2 and 8.0 were studied to probe possible pH-dependent differences in active site chemistry (except three structures solved at pH ~7). Generally, the structures of the CDO variants at pH 6.2 and 8.0 are qualitatively similar, and because the enzyme is more active at pH ~8,24; 29; 38 we primarily focus on the higher pH structures in the main presentation and include the pH 6.2 structures as supplemental information.
Table 1.
Data collection and Refinement Statistics for CDOsa
| CDO form | C93A | Y157F | Y157F | Wild-type | C93A | Y157F | Y157F |
|---|---|---|---|---|---|---|---|
| soak | none | none | dt | thiosulfat | Cys | Cys | Cys + dt |
| nominal pH | 8.0 | 8.0b | 8.0 |
e
8.0b |
8.0 | 8.0 | 8.0 |
|
Data
collection |
|||||||
| Resolution (Å) |
41-1.35 (1.37 −1.35) |
34-1.30 (1.32 −1.30) |
26-1.25 (1.27 −1.25) |
31-1.37 (1.39 −1.37) |
34-1.40 (1.42 −1.40) |
34-1.55 (1.58 −1.55) |
34-1.55 (1.58 −1.55) |
| Unique Obs. | 45533 (1757) |
47533 (1331) |
53790 (1261) |
44485 (2135) |
37823 (1109) |
31437 (1539) |
31232 (1519) |
| Multiplicity | 23.3 (9.1) |
40.7 (18.0) |
22.2 (6.8) |
27.3 (21.5) | 28.1 (26.4) |
25.9 (16.3) |
26.0 (16.3) |
| Completenes s |
97.9 (78.6) |
91 (52) | 92.3 (45.2) |
100 (100) | 91.8 (54.1) |
100 (100) |
100 (100) |
| <I/ρ> | 22.5 (1.0) |
27.8 (1.5) |
27.1 (1.4) |
15.9 (0.7) | 22.5 (1.8) |
21.4 (0.6) |
17.3 (0.6) |
| Rmeasc (%) | 10.3 (257) |
9.1 (247) | 7.0 (129) | 19.0 (538) | 15.4 (366) |
7.6 (596) | 9.0 (500) |
| CC1/2d (%) | 1.0 (0.29) |
1.0 (0.44) |
1.0 (0.56) |
1.0 (0.20) | 1.0 (0.26) |
1.0 (0.24) |
1.0 (0.23) |
| Refinement | |||||||
| Rcryst / Rfree (%) |
16.1/19. 1 |
16.2/18. 8 |
12.1/16. 2 |
16.9/20.0 | 15.5/18. 6 |
18.3/22. 4 |
18.7/22. 2 |
| No. Obs | 45445 | 47060 | 53704 | 44155 | 37752 | 30548 | 30399 |
| No. residues | 186 | 186 | 186 | 186 | 186 | 186 | 186 |
| No. waters | 333 | 289 | 354 | 313 | 302 | 143 | 133 |
| No. atoms | 1952 | 1888 | 1935 | 1914 | 1869 | 1717 | 1702 |
| rmsd angles (°) |
1.30 | 1.32 | 1.25 | 1.07 | 1.28 | 1.27 | 1.30 |
| rmsd lengths (Å) |
0.014 | 0.011 | 0.011 | 0.008 | 0.010 | 0.012 | 0.014 |
| φ,ψ-favored (%)e |
99.5 | 99.0 | 99.0 | 98.0 | 99.0 | 98.5 | 99.0 |
| <B> protein (Å2) |
18 | 20 | 17 | 23 | 20 | 46 | 46 |
| <B> ligand (Å2) |
18 | 21 | 15 | 19 | 14 | 33 | 38 |
| PDB code | 4XFB | 4XET | 4XEZ | 5I0T | 4XF0 | 4XF1 | 4XF3 |
All refinements used space group P43212 with a=b=57.60 Å and c= 122.40 Å. Numbers in parentheses refer to the highest resolution bin.
Soak solution at pH 8, but pH sensitive loop conformation indicates pH ~ 7 (see27 and methods)
Rmeas is the multiplicity-weighted merging R-factor65
CC1/2 is the correlation between two datasets each based on half of the data as defined in Karplus & Diederichs.62
Ramachandran statistics as defined by Molprobity64, no φ,ψ-outliers in any structures
Table 2.
Data collection and Refinement Statistics for inhibitor-complexed CDOsa
| CDO form | Wild-type | C93A | Y157F | Wild-type | C93A |
|---|---|---|---|---|---|
| soak | Hcy | Hcy | Hcy | azide | D-Cys |
| nominal pH | 8.0 | 8.0 | 8.0 | 6.2 | pH 8.0 |
| Data collection | |||||
| Resolution (Å) | 41-1.35 (1.37-1.35) |
34-1.30 (1.32-1.30) |
33-1.65 (1.68-1.65) |
34-1.50 (1.58-1.50) |
42-1.35 (1.37-1.35) |
| Unique Obs. | 43919 (1521) |
43282 (896) |
25597 (1236) |
33827 (4812) |
46343 (2257) |
| Multiplicity | 23.8 (10.3) | 26.7 (17.9) | 27.4 (22.8) | 11.8 (11.9) | 53.5 (36.7) |
| Completeness | 94.9 (68) | 84.2 (36.2) | 98.9 (95.2) | 100 (100) | 100 (100) |
| <I/ρ> | 23.7 (1.0) | 21.3 (1.2) | 26.3 (2.0) | 8.6 (0.6) | 17.4 (0.8) |
| Rmeasc (%) | 8.0 (245) | 10.5 (251) | 9.1 (199) | 18.2 (538) | 25.0 (614) |
| CC1/2d | 1.0 (0.26) | 1.0 (0.33) | 1.0 (0.60) | 1.0 (0.17) | 1.0 (0.22) |
| Refinement | |||||
| Rcryst / Rfree (%) | 16.2/19.1 | 15.7/18.6 | 16.9/21.0 | 17.2/21.0 | 16.9/20.0 |
| No. Obs | 43838 | 43245 | 25071 | 33667 | 46118 |
| No. residues | 186 | 186 | 186 | 186 | 186 |
| No. waters | 280 | 302 | 182 | 214 | 321 |
| No. atoms | 1914 | 1907 | 1749 | 1785 | 1914 |
| rmsd angles (°) | 1.36 | 1.27 | 1.33 | 1.31 | 1.01 |
| rmsd lengths (Å) | 0.011 | 0.010 | 0.013 | 0.012 | 0.008 |
| φ,ψ-favored (%)e | 99.0 | 99.0 | 99.5 | 99.5 | 99.0 |
| <B> protein (Å2) | 23 | 20 | 40 | 28 | 23 |
| <B> ligand (Å2) | 40 | 33 | 30 | 47 | 16 |
| PDB code | 4XF4 | 4XF9 | 4XFA | 4PJY | 5I0R |
All refinements used space group P43212 with a=b=57.60 Å and c= 122.40 Å. Numbers in parentheses refer to the highest resolution bin.
Soak solution at pH 8, but pH sensitive loop conformation indicates pH ~ 7 (see27 and methods)
Rmeas is the multiplicity-weighted merging R-factor65
CC1/2 is the correlation between two datasets each based on half of the data as defined in Karplus & Diederichs.62
Ramachandran statistics as defined by Molprobity64, no φ,ψ-outliers in any structures
Also, some of these crystal structures have relatively low occupancy (≤0.3) features along with the high occupancy features that are of primary interest. Such low occupancy features can be difficult to accurately interpret, and result in models with higher coordinate uncertainties. While we have modeled certain ones we felt confident about, we have left others uninterpreted. We emphasize that all of the main conclusions of this work are based on clear higher occupancy features and so would not change even if the details of the low occupancy features have not been fully or correctly interpreted.
The C93A and Y157F unliganded active sites
As purifying native, non-crosslinked wild-type mammalian CDO has been difficult,17 to gain insight into its structure we used the C93A and Y157F mutants that are known to have some activity but remain non-crosslinked.15; 24 The Y157F mutant can provide insight into how the un-reacted Cys93 and the phenyl group of Tyr157 would pack, and the C93A mutant can provide a control for distinguishing which active site changes seen in Y157F CDO are due to the loss of the crosslink and which are due to the loss of the Y157 hydroxyl group. Some evidence for Y157F having a somewhat perturbed structure is that Y157F crystals were more difficult to grow and only a fraction of the crystals diffracted to high resolution (see methods).
Ligand-free C93A and Y157F structures were solved at ~1.3 Å resolution at pH 8 (Table 1) or at pH 6.2 (Table S1). For C93A (Figure 2A and S1A), the active site iron was penta-coordinate with the three His ligands plus one peak with even higher electron density than the sulfurs of Met and Cys residues (~17 ρrms vs. ~15 ρrms), and another with density appropriate for a water/hydroxide. The strong density is well modeled as a chloride ion, which is present at 15 mM in the crystallization buffer, is known to bind to iron,41; 42; 43; 44 and was also recently similarly modeled in a crystal structure of a CDO Cys164Ser mutant.45 The fully occupied water/hydroxide ligand is opposite to His140, and both the chloride and water appear stabilized by reasonably well-aligned bifurcated hydrogen-bonds from the Tyr157 hydroxyl (Figure 2A).
Figure 2.

High resolution views of the Y157F and C93A active sites and of thiosulfate binding. All 2Fo-Fc density is contoured at 1.4 ρrms, and stick models and interactions are shown as in Figure 1 and with chloride atoms green. (A) unliganded C93A at pH 8.0 (PDB code 4XFB). (B) unliganded Y157F at pH ~7.0 with the Cys93 side chain in two conformations at 0.8 and 0.2 occupancy (PDB code 4XET). The dominant Cys93-Sγ position is 3.7 Å from the chloride, a potential hydrogen-bonding distance.46 (C) Y157F soaked in dithionite at pH 8.0 showing thiosulfate bound at occupancy ~0.3 (PDB code 4XEZ). (D) wild-type CDO soaked in thiosulfate at pH 6.8 with thiosulfate bound differently and at occupancy ~0.4 (PDB code 5I0T).
In Y157F (Figure 2B and S1B), the iron was largely four-coordinate with a chloride also present but slightly shifted so it blocks water from binding opposite to His140. The density indicates that a low (~0.3) occupancy water/hydroxide ligand is found opposite His86. This weak water appears unreasonably close to both the Fe (~1.7 Å), and the Cys93-Sγ (~2.5 Å), possibly indicating it is a hydroxide, but we otherwise have no good explanation for this. It is apparently not associated with the minor conformation adopted by the Cys93 side chain in the pH 8 structure (Figure 2B), since this conformation is not seen at pH 6.2 (Figure S1B). Nevertheless, the reliable information we infer from these structures is that the major conformation of Y157F has the Cys93 side chain in a similar orientation as in the wild-type enzyme and no fifth iron ligand. A further feature of this structure is that the Arg60 side chain mostly (~0.6) adopts a different conformation, which we will refer to as position II, in which its guanidino hydrogen bonds with Tyr58 and a water that in turn hydrogen bonds (at a typical 3.3 Å distance46) to the chloride (Figure 2B). This Arg60 position matches that seen previously in CDO forms having a disulfide modification at Cys16418; 20; 47 and is not compatible with Cys binding.28
To assess whether the chloride binding seen in these non-crosslinked structures could be due to the iron being present in the ferric state, we soaked C93A and Y157F crystals with dithionite and methyl viologen. Although most crystals did not survive this treatment, two crystals of Y157F allowed for 1.25 Å resolution analyses at pH 8 (Table 1) and 6.2 (Table S1). In both cases, the active site largely still had chloride bound, confirming that the coordination seen represents the Fe(II) active site. A minor difference in the active site was the presence of some weak density that thanks to the high resolution, we could interpret as a low occupancy thiosulfate (S-SO3), a decomposition product of dithionite48 (Figures 2C and S1C). The thiosulfate bound with the terminal sulfur ligating the iron at roughly the same position as the chloride, and with the SO3 moiety hydrogen bonding with Arg60 (in position II). We tested whether thiosulfate could also bind to wild-type crosslinked CDO and found it bound at ~40% occupancy at both pH 8 and 6.2 (Figure 2D and S1D). Despite the fairly clear electron density, an implausible feature of these complexes as modeled is an apparent ~2.1 Å approach of one thiosulfate oxygen with a guanidine nitrogen (Fig 2D). We do not have a good explanation for this, but suggest that there is some level of disorder not easily seen in the electron density that allows the separation to actually be a still short but plausible ~2.4-2.5 Å.
Comparing the two non-crosslinked mutant structures with the crosslinked wild-type structure reveals that they undergo similar changes (Figure 3). For C93A, the Tyr157 side chain shifts ~1.0 Å away from the iron apparently due to the steric interaction with residue 93-Cβ that is just 3.65 Å from 157-CD and –CE. The chloride ion is near the position of the iron-bound water in wild-type CDO, and as noted above, is stabilized by a hydrogen-bond from the shifted Tyr157 (Figure 3). No protein atoms outside of this region experience noteworthy shifts meaning that His155 is no longer well aligned with the Tyr157 hydroxyl. For Y157F, the shift of the Phe157 side chain matches remarkably well with that seen for Tyr157 in C93A CDO (Figure 3). The missing hydroxyl of residue 157 opens space in the active site, and the chloride is slightly shifted into this space, and this in turn opens space for the partially occupied coordinating water near Cys93.
Figure 3.

Stereoview comparison of the wild-type, Y157F and C93A unliganded active sites. Ligand-free crosslinked wild-type CDO (PDB entry 2B5H)20 has green carbons/waters, chloride-bound C93A (PDB entry 4XFB) has orange carbons/waters/chloride, and chloride-bound Y157F (PDB entry 4XET) has tan carbons/water/chloride. Chlorides are represented as larger spheres than the waters.
C93A and Y157F in the presence of cysteine
Crystals of C93A and Y157F soaked with 100 mM Cys at pH 8.0, analyzed at ~1.5 Å resolution (Table 1), show Cys binding with no molecular oxygen binding or Cys-persulfenate formation (Figure 4). The position of residue 157 in all complexes is unchanged from that seen in the unliganded variants suggesting it is well fixed in that position. For C93A, Cys has an estimated occupancy of 90% based on residual electron density representing the chloride at occupancy ~0.1 (Figure 4A). The overall binding mode is quite similar to that seen in wild-type CDO, with the Sγ and α-amino atoms both coordinating the iron (2.35 and 2.3 Å, respectively). Compared to the wild-type CDO complex, a small shift (~0.6 Å) of the α-carboxylate appears to accommodate the altered position of the Tyr157 hydroxyl (Figure 5). The active site Arg60 and Tyr58 that interact with the α-carboxylate shift a bit less (~0.3 Å), leading to small variations in the hydrogen bonds between them and the α-carboxylate. The most notable difference from the wild-type complex is a new strong water site between the iron and the Tyr157 hydroxyl, roughly matching the position seen for the distal oxygen (i.e. the OE atom) of Cys-persulfenate (Figure 5).
Figure 4.

High resolution views of the cysteine-complexes of Y157F and C93A CDO. 2Fo-Fc electron density and models and interactions are shown as in Figure 2 with occupancies of the bound chloride and Cys provided in parentheses. (A) Cys soak of C93A at pH 8.0 (PDB code 4XF0), the water near Ala93 receives a hydrogen bond from His155 (not shown). (B) Cys soak of Y157F at pH 8.0 (PDB code 4XF1). A Cys plus dithionite soak of Y157F at pH 8.0 yielded virtually identical results (PDB code 4XF3).
Figure 5.

Stereoview comparison of Cys binding to wild-type, Y157F and C93A CDO. Wild-type Cys-persulfenate (PDB code 3ELN22) has green carbons/waters, C93A CDO (PDB code 4XF0) has orange carbons/waters and for simplicity the 0.1 occupancy chloride is not shown, and Y157F CDO (PDB code 4XF1) has tan carbons/waters and the lower (~0.3) occupancy water and alternate conformation of Cys93 are not shown.
For Y157F, Cys binds mostly (70%) in the canonical binding mode, coordinating the iron via both its thiol and α-amino group (Figure 4B).28 In addition, a minor (~30%) binding mode places the Cys-Sγ at the site of chloride binding rather than in the typical position (Figure 4B and Figure 5). This minor binding mode plausibly represents Cys molecules having a protonated α-amino group (which cannot coordinate the iron), but this is not certain. Arg60 and Tyr58 are in their standard positions, forming hydrogen bonds to the α-carboxylate oxygens (Figure 4B). Interestingly, Cys93 switches its dominant conformation (Figure 4B), which we suggest is enabled by the iron shift and small domain motions that occur upon Cys binding in its standard position.28 A control soak of Y157F crystals with both Cys and dithionite (Table 2) gave virtually identical results (PDB code 4XF3), indicating that the binding modes seen are relevant to Fe(II)-CDO.
Cys soaks with both variants at the lower pH of 6.2 were solved at ~1.4 Å resolution (Table S1 and Figure S2). For C93A, the same Cys binding mode is seen, but the occupancy is lower at 0.65. For Y157F, none of the standard bi-dentate Cys binding mode is seen, and thus no iron shift and alternative Cys93 conformation is observed. Instead, the minor site seen in the pH 8.0 analysis is still present as a minor site, and another mode is seen that also places the Sγ-atom at the chloride position and has no coordination by the α-amino group, consistent with this group being fully protonated at pH 6.2 (Figure S2B).
Homocysteine binding to CDO
Soaks of C93A, Y157F and wild-type CDO at pH 8.0 with homocysteine (Hcy), a known competitive inhibitor,19; 39; 49 were solved at resolutions between 1.3 and 1.65 Å (Table 2). All of the complexes showed a very similar binding mode with only the thiol coordinating the iron (~2.20 Å) and the α-amino group being ~3.5 Å from the iron (Figure 6A-C). The thiol sits roughly in the position of the chloride rather than the position thought to be catalytically relevant (Figure 7A). The Tyr157-hydroxyl is interacting with the thiol and α-amino groups of Hcy (Figure 6A,B), and Arg60 only hydrogen bonds with one carboxylate oxygen rather than both. Tyr58 and Arg60 shift slightly relative to their positions in the Cys-bound active site to accommodate the α-carboxylate of Hcy, which extends further from the iron (Figure 7A). In both the C93A and Y157F complexes, residue 157 remains in the position seen in the unliganded and Cys-bound structures. Structures determined at pH 6.2 (Table S2) are very similar (Figure S3).
Figure 6.

High resolution views of the homocysteine (Hcy), D-Cys, and azide complexes of wild-type and/or mutant CDOs. 2Fo-Fc electron density and models and interactions are as in Figure 2. (A) Hcy soak of wild-type CDO at pH 8.0 (PDB code 4XF4). (B) Hcy soak of C93A at pH 8.0 (PDB code 4XF9). (C) Hcy soak of Y157F at pH 8.0 (PDB code 4XFA). (D) D-Cys soak of C93A at pH 8.0 (PDB code 5I0R). The orientation is rotated slightly relative to the other figures to better show the His86 alternative conformation. The bound D-Cys α-amino group would collide with His86 in its iron ligating position (being ~1.6 Å from His86-Nε2). (E) Azide soak of wild-type CDO at pH 6.2 (PDB code 4PJY).
Figure 7.

Stereoview comparisons of Cys-persulfenate, Hcy and azide binding to wild-type CDO. (A) Overlaid complexes of wild-type crosslinked CDO with bound Cys-persulfenate (green carbons/waters) and Hcy (tan-carbons/waters). (B) Overlaid complexes of wild-type crosslinked CDO with bound Cys-persulfenate (green carbons/waters) and azide (orange carbons/waters/azide). Also, visible in the overlays is that neither Hcy nor azide cause the same shift in the iron as does binding the Cys-persulfenate.
D-Cys binding to CDO
D-Cys, which causes 50% inhibition of CDO at 12 mM,24 has been shown to uncouple CDO catalysis so that CDO consumes molecular oxygen readily, but with only approximately 4% being converted into CSA, compared with ~80% incorporation of O2 into CSA for L-Cys.24 For soaks of D-Cys into wild-type and C93A CDO crystals, the electron density was complex and included evidence of some displacement of His86 from its iron ligating position, and a partial disulfide modification of Cys16418; 20 that is known to disrupt the active site.18; 20; 47;28 For the wild-type crystals, we could not place D-Cys with confidence, and so are not reporting that structure. However for C93A, despite the complexities, the density could be modeled as a 45% occupied dominant and clear D-Cys position, and a 10% occupied minor position (Figure 6D). Both coordinate the iron through their thiol and α-amino groups, with the thiol of the dominant D-Cys sitting in the position of the chloride, and that of the minor D-Cys at what is considered the canonical L-Cys binding site. The α-amino group for both binding modes is close to His86 with the clash apparently leading His86 to adopt an alternate position (at 0.55 occupancy) (Figure 6D). Also, the D-Cys carboxylate points up to interact with Tyr58 and does not directly interact with Arg60.
Azide binding to CDO
Azide, which causes 50% inhibition of CDO at 1.4 mM,39 is considered a superoxo mimic, despite its ability to bind to CDO even in the absence of Cys.40 Crystal soaks with 100 mM azide at pH 6.2 showed no binding to the C93A or Y157F variants, but yielded a 1.5 Å resolution full occupancy azide-bound structure of wild-type CDO (Figure 6E and Table 2). In the azide-bound structure, a terminal nitrogen coordinates the iron and the azide extends at a 105° angle from the iron, similar to what has been seen in 3-His 1-carboxylate mono-iron enzymes (e.g. PDB code 1AVM50). Consistent with spectroscopic work,40 a fifth water/hydroxide iron ligand is also present. The azide also interacts with the Tyr157-OH though its iron-proximal nitrogen, perhaps explaining why it does not bind as well to C93A or Y157F CDO, and Arg60 becomes rather disordered. The position of the coordinating nitrogen roughly matches that of the iron-proximal oxygen of the Cys-persulfenate complex and the middle nitrogen atom sits close to the normal Cys-bound sulfur position (Figure 7B). Interestingly, wild-type CDO crystals soaked with both 100 mM azide and 100 mM Cys, gave no evidence of azide binding implying that they do not readily bind simultaneously and that at the equal concentrations used, Cys outcompetes azide.
Discussion
Disruption of the Cys-Tyr crosslink by mutation of either Cys93 to Ala or Tyr157 to Phe results in the phenyl ring of residue 157 undergoing, in a wide variety of contexts, a nearly identical shift of ~ 1.0 Å relative to crosslinked enzyme (Figure 8). That the shift is so similar in both mutants allows us to conclude that Cys93 does not push away residue 157 any more than does an Ala, so that the main cause of the shift must be the steric approach of residue 93 Cβ to residue 157 atoms. This implies that a reliable model for what would exist in non-crosslinked wild-type CDO can be derived by combining the Cys93 side chain as seen in Y157F with the rest of the C93A structure. It also implies that we can make inferences about how crosslink formation changes the active site from the mutant structures. The importance of the Tyr-OH position for catalysis is further supported by the observation that even the bacterial CDO36 and the fully active non-crosslinked C93G mutant of rat CDO37 also have the Tyr-OH in a position that is closely similar to that in wild-type crosslinked CDO (Figure 8).
Figure 8.

Positional conservation of the Ser-His-Tyr catalytic triad among wild-type CDO structures. Side chains equivalent to rat CDO residues 93, 153, 155 and 157 are shown for wild-type rat CDO structures (green carbons; six inhibitor-bound structures from this work plus the previously published unliganded–PDB code 2B5H20 – and Cys-persulfenate-bound – PDB code 4IEU28 – structures), its C93A variant (orange carbons; 6 structures from this work), its Y157F variant (tan carbons; 10 structures from this work), along with previously published structures of its C93G variant (cream carbons; PDB code 4UBG37) and the non-crosslinked, but fully active Bacillus subtilis CDO (purple carbons; PDB code 4QM936).
The positioning of Tyr157 influences iron coordination
In wild-type crosslinked CDO without substrate bound, the Tyr157 hydroxyl makes a rather short (~2.6 Å) hydrogen bond to the iron bound water. The importance of this interaction for maintaining that coordination is demonstrated in C93A CDO by the ~1 Å shift in Tyr157-OH that allows a chloride to displace the water. Because O…Cl hydrogen bonds are in the 3.0 - 3.7 Å range,46; 51 the native position of the Tyr157 hydroxyl excludes the chloride from binding to the wild-type crosslinked enzyme. Human intracellular chloride concentration is 5 to 60 mM,52; 53 and conceivably chloride might act as a competitive inhibitor and as an indirect reason for lower activity in non-crosslinked CDO. Although non-crosslinked CDO in vivo may not have a bound chloride, it is certain that its iron coordination will be different from that of the crosslinked CDO.
The positioning of Tyr157 enables proper Cys binding
Exposing crystals of C93A and Y157F variants of CDO to Cys results in direct coordination of Cys to the active-site iron, consistent with EPR changes observed for Cys binding to the Fe(III) C93A and Y157F variants,24 and to non-crosslinked wild-type enzyme.17 In the crosslinked enzyme, Cys was seen to bind to the iron from pH 5.5 to 9.0 in the now canonical bidentate mode via its α-amino and thiol groups, indicating that in the active site environment the α-amino group is deprotonated over this entire range.28 The structures presented here define Tyr157 as a key factor that lowers this pKa from its normal value around 8 or 9 to be near 5. Assuming that loss of bidentate binding is due to α-amino group protonation, the Y157F bi-dentate Cys binding levels being ~70% and 0% occupancy at pH=8 and 6.2, respectively, implies that the Cys α-amino pKa increases to between 7.5 and 8.0 in the absence of the Tyr157-OH. When the Tyr157-OH is shifted (i.e. in the C93A mutant) the impact is not as dramatic, but the weaker bidentate Cys binding at pH 6.2 (65% instead of 90% occupancy) is consistent with the likelihood that the α-amino group is already partially protonated at that pH. That the Cys93-Tyr157 crosslink is especially important for promoting the Fe-N(Cys) coordination is consistent with EPR results showing that the most pronounced increase in g strain upon the disruption of the C93-Y157 crosslink by mutation (C93A or Y157F) is along the Fe-N(Cys) bond (g1 axis).24
The positioning of Tyr157 influences its activation and dioxygen binding and chemistry
The ~1.0 Å shift in Tyr157 in the non-crosslinked enzyme also can be inferred to impair the ability of Tyr157 to serve as a catalytic acid/base. Importantly, as Ser153 and His155 do not change positions, the shift in Tyr157 causes a misalignment of the catalytic triad (Figure 8). The His155-NE2 to Tyr157-OH hydrogen bonding interaction goes from an N…O distance of ~2.8 to ~3.2 Å and an N-H…O angle of ~145° to ~130°. The substantial nature of this disruption is underscored by the binding in the C93A:Cys complex of a new ordered water (Figure 3A); it both accepts a hydrogen bond from His155 and blocks the expected dioxygen binding site.22; 23; 24; 27; 28 Finally, the exquisitely central location of the Tyr157 hydroxyl at or below van der Waals distances from both oxygens of the Cys persulfenate and from the Cys α-amino group (Figure 1), imply that it is highly involved in the chemistry of CSA formation. Even without knowing the mechanistic details, one can surmise that the 1 Å shift in the hydroxyl away from the iron is bound to be deleterious to the chemistry it normally facilitates. Indeed, all high activity CDOs (cross-linked or not) have a well-aligned catalytic triad with a remarkable positional conservation of the Tyr157 equivalent residue, distinct from those CDOs with diminished activity (Figure 8).
Among the four published studies of Y157F variants, all show impaired catalytic activity (ranging from no activity24 to 2-47 or 20-54 or 100-fold15 lower than wild-type), illustrating the importance of the Y157-OH in catalysis.24 Interestingly, the roughly 20-fold lower activity of C93A compared with the crosslinked wild-type enzyme24 is similar to the 10-or-more-fold lower activity reported for the non-crosslinked enzyme, consistent with C93A being a reasonable model for that form. Also, a CDO H155A variant had ~100-fold reduced catalytic activity compared with pure crosslinked wild-type CDO, and a spectroscopic and computational investigation implicated the His155-Tyr157 interaction in optimally positioning the Tyr157-hydroxyl, and through this influencing iron coordination.24; 55 Finally, a recent kinetics study of the CDO homolog 3-mercaptoproprionate dioxygenase led to a proposal that the residue equivalent to Tyr157 must be protonated for the enzyme to be active.56
Binding of Inhibitors and insights into CDO specificity
In Alzheimer’s disease high levels of Hcy are observed together with elevated levels of cysteine,11 raising the possibility that CDO inhibition by Hcy, with an IC50 of 6.5 mM,57 is physiologically significant. Although attempts to crystallize human CDO in the presence of Hcy were unsuccessful,47 we have captured Hcy in the active sites of wild-type, C93A and Y157F rat CDO through soaking experiments. As Hcy is not a substrate for CDO, it was speculated that there must be an energetic reason why the iron(II) CDO-Hcy complex either does not bind oxygen or does not react with it.19 However, the structures reveal that the inability of CDO to turnover Hcy is not subtle, but is simply due to its thiol binding in an incorrect location that blocks the putative oxygen binding site (Figure 7A). It was predicted that Hcy has the capacity to coordinate iron via dual S- and N-ligation. 58 However in the limited space of the active site, the Hcy α-amino group and thiol groups cannot simultaneously bind to the iron as they do for Cys, and this dramatically alters the binding mode and hinders catalysis. The equivalent binding of Hcy to the non-crosslinked CDO forms implies that, in vivo, in addition to directly preventing Cys conversion to CSA, Hcy could inhibit CDO crosslink formation and keep the enzyme in its low efficiency form.
For D-Cys we were able to obtain an interpretable complex with C93A (Figure 6D), but not wild type CDO. With its Sγ-atom mostly binding similarly to the thiol of Hcy (comparing Figures 6A and D), this implies that D-Cys would be an inhibiter rather than an uncoupler of non-crosslinked CDO (for which C93A is a model). Also, the lesser occupied binding mode explains how D-Cys could act as an uncoupler of wild-type CDO (promoting the turnover of oxygen24; 58) rather than as an inhibitor or substrate. This is because its Sγ-atom being positioned similarly to that of a productively bound L-Cys would help create the oxygen binding site, but its different geometry of approach would not properly orient the Sγ orbitals for reaction with oxygen. Recently, study of a “Gln-type” 36 CDO homolog which shows specificity for 3-mercaptopropionic acid as substrate,59 reported that its non-preferred substrates such as cysteine or cystamine bound less tightly to the iron and in at least two different conformations (i.e. heterogenously).59 These results all support the idea that the high specificity of CDO is not related to intrinsic differences in the chemistry of potential alternate substrates, but simply due to discrimination against molecules different from L-Cys because they do not bind the iron in a proper orientation to promote catalysis.
The structure of azide bound to crosslinked CDO is quite informative. In a recent EPR study azide was shown to form an inner-sphere OH−/N3−-Fe(III)CDO complex in the absence of Cys.40 Also, based on very weak spectral changes that were said to be due to “a small subset” of the active sites that bound both Cys and azide, inferences were made about how azide binds as a superoxide mimic in the presence of Cys, and modeling was carried out to predict the binding mode.40 The azide complex seen here is consistent with the experimental observations, but not their interpretation. It shows that azide does coordinate the iron at roughly the same position as the putative oxygen docking site, but rather than binding as a superoxide mimic, the remainder of the molecule extends in a direction that directly blocks the Cys binding site (Figure 7B). This competition provides a rationale for why azide could not be bound at high occupancy in the presence of Cys, and calls into question the accuracy of the density functional theory model modeling of a CDO-Cys-azide ternary complex in which the azide coordinates the iron at a similar position, but then extends in a completely different direction than is seen in the crystal structure (Figure S4). Our conclusion that azide binds competitively with Cys rather than being a true molecular oxygen/superoxide analog is also consistent with azide being able to bind in the absence of Cys, while oxygen and other known oxygen mimics do not.40
Outlook
Much remains to be learned about the CDO mechanism, but the structures reported here advance the field by providing reliable high resolution structures of complexes that can be used to guide theoretical studies of CDO catalysis, and by providing insight into two key open questions. First, regarding the origins of the exquisite specificity of the enzyme for L-Cys, the inhibitor-binding studies show that it is a combination of sterics and hydrogen-bonding groups that allow L-Cys to uniquely bind in a manner that places both its thiolate and α-amino groups in proper positions to create the oxygen binding site at the right place and with the right geometry for reacting with the thiolate. Second, regarding the mechanism by which crosslink formation in mammalian CDO enhances activity, the structures here provide compelling support for the conclusion that the importance of crosslink formation is primarily about properly positioning the Tyr157 hydroxyl. In all, we see four ways the shifted Tyr157 in the non-crosslinked form would be less catalytically effective: (i) proper Cys binding should be diminished due to it having more spatial freedom and a higher α-amino group pKa; (ii) Tyr157 should not be as effectively activated by the catalytic triad; (iii) room would be present in the Cys-complex for a bound water that could compete with dioxygen binding; and (iv) even with Cys and dioxygen bound properly, the shifted Tyr157 hydroxyl would not be as well placed to properly influence the binding and reaction of molecular oxygen. Consistent with this, all high activity CDOs (cross-linked or not) have a well-aligned catalytic triad with a remarkable positional conservation of the equivalent Tyr157-OH (Figure 8). Key open questions still to be answered are what are the exact steps the lead from a complex with Cys and O2 bound to the CSA product and what exactly is the role of Tyr157 in promoting these steps.
Materials and Methods
Expression, purification and crystallization
Expression, purification and crystallization of wild-type, C93A and Y157F recombinant rat CDO was done as described previously for rat CDO,22; 28; 60; 61 with seeding being especially important for consistent crystal growth for the Y157F variant. The enzymes were stored at 6-10 mg/mL in 10 mM Tris pH=7.4 buffer and crystallized at room temperature in hanging drops formed from 0.5 μL seed stock in reservoir buffer, 1 μL protein stock and 1 μL of reservoir buffer composed of 0.1 M tri-sodium citrate pH=5.6, 24% PEG 4000, and 0.15 M ammonium acetate (final pH=6.2).28 The crystals of all variants were isomorphous with those previously described,22; 28; 60 having space group P43212 and one chain in the asymmetric unit.
Crystal soaks and data collection
Soaks were done at room temperature using the reservoir condition as an artificial mother liquor (AML) that had its pH adjusted to a final value of 6.2 or 8.0. For data collection, crystals were flash frozen by plunging into liquid nitrogen. For cysteine, D-cysteine, thiosulfate, homocysteine and azide soaks, crystals were incubated for 10 min in solutions with 100 mM cysteine, D-cysteine, thiosulfate, homocysteine, or azide added to AML. Ligand-free structures at pH 6.2 were scooped directly from the hanging drop. As previously,32 for ligand-free structures at pH 8.0, crystals were incubated for 10 min in solutions that contained 100 mM alanine to serve as a buffer that was similar to cysteine. For preparing the dithionite-reduced form, crystals were incubated for 45 min under argon in an solution of 5 mM dithionite and 0.5 mM methyl viologen in AML with or without 100 mM Cys which had been degassed and flushed with argon. The blue color of reduced methyl viologen was present during the course of the incubation, indicating that the reducing power of the solution was not exhausted.
Synchrotron data sets were collected at beamlines 5.0.1, 5.0.2, and 5.0.3 at the Advanced Light Source (Lawrence Berkeley National Laboratory). Data were processed and scaled using iMosflm62 and Aimless. 63 For all refinements, the same 10% of the data as used previously22; 28 were flagged for use in Rfree.
Crystallographic refinement
For each of the 21 structures reported here, PHENIX-refine64 was used to test eight refinement strategies using either the unique unit cell constants for individual crystals (within the ranges a=b=57.6-58.2 Å and c=121.7-122.9 Å) or using a=b=57.60 Å and c=122.40 Å. Six of the refinement strategies tested were similar to the automated protocol used previously28; 65 and began with the 1.5 Å resolution unliganded CDO model (PDB code 2b5h) or with the 1.55 Å resolution unliganded CDO model (PDB code 4ieo) either with isotropic temperature factors, with one TLS group defined by the entire chain, or with individual anisotropic temperature factors. Two additional refinement strategies began with PDB code 4ieo with waters retained and a simple minimization either with one TLS group defined by the entire chain, or with individual anisotropic temperature factors. The anisotropic B-factors were only accepted if their use resulted in an Rfree improvement of at least 1% compared with the TLS refinements. The best refinement strategy used the same unit cell for all refinements (a=b=57.60 Å and c=122.40 Å) with 4ieo as the starting model with the automated protocol. For refinement against all but two of the datasets, the best strategy used TLS. The anisotropic B-factors were only used for Y157F dithionite-treated structures at pH 6.2 and 8.0 which at 1.25 Å resolution were the best resolved structures.
After the automated stage, each refinement was completed using manual rebuilding in Coot66 and PHENIX-refine minimization. Some water sites were removed and others were added using standard criteria (>1 ρrms density in the 2Fo −Fc map, >2.4 Å distance from nearest contact), and all difference map peaks above 5*ρrms were checked. Alternate conformations were only added when electron density features required it and when those features could be interpreted in a way that made chemical sense. For instance, for cases of partially occupied ligands, atoms expected to be present in an alternate unliganded conformation were not included simply based on the principle that they must be present if the ligand is not fully occupied. For those refinements using TLS, the temperature factors of the iron were refined anisotropically, and this resulted in an improvement of ~0.5% in Rfree. Molprobity67 was used to find problems with model geometry. Data collection and refinement statistics are in Tables 1 and 2 for structures reported in the main paper, and in Tables S1, S2 and S3 for structures reported in the supplemental information.
Supplementary Material
Highlights.
In mammalian CDO, the Cys-Tyr crosslink formation properly positions the Tyr-OH
Proper Tyr-OH position guides Cys-binding and aligns Ser-His-Tyr catalytic triad
Homocysteine and D-Cys binding modes make clear why they are not substrates
Azide does not bind as a superoxide mimic
Diverse high-accuracy CDO structures can ground future simulations and experiments
Acknowledgements
We thank Dale Tronrud for useful discussions and help with methods. This project was supported in part by Grant NIH-DK-056649 to MHS and PAK from the National Institute of Diabetes and Digestive and Kidney Diseases. Synchrotron data were collected at the Advanced Light Source supported by Contract DE-AC02-98CH10886 from the Office of Basic Energy Sciences of the U.S. Department of Energy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession Numbers. Coordinates and structure factors for the CDO models have been deposited in the Protein Data Bank with accession numbers as follows: unliganded C93A variant at pH 8.0 (PDB code 4XFB), Fe-Cl bound Y157F variant at pH ~7 in the presence of azide (PDB code 4XET), Y157F variant at pH 8.0 with dithionite (PDB code 4XEZ), C93A variant at pH 8.0 with cysteine (PDB code 4XF0), Y157F variant at pH 8.0 with cysteine (PDB code 4XF1), Y157F variant at pH 8.0 with cysteine and dithionite (PDB code 4XF3), CDO at pH 8.0 in complex with homocysteine (PDB code 4XF4), C93A variant at pH 8.0 in complex with homocysteine (PDB code 4XF9), Y157F variant at pH 8.0 in complex with homocysteine (PDB code 4XFA), Azide bound wild-type CDO at pH 6.2 (PDB code 4PJY), C93A variant at pH 6.2 (PDB code 4PIX), Y157F variant at pH 6.2 (PDB code 4XFC), Y157F variant at pH 6.2 with dithionite (PDB code 4XFF), C93A variant at pH 6.2 with cysteine (PDB code 4XFG), Y157F variant at pH 6.2 with cysteine (PDB code 4XFH), homocysteine bound wild-type CDO at pH 6.2 (PDB code 4PIZ), homocysteine bound C93A variant at pH 6.2 (PDB code 4PIY), Y157F variant at pH 6.2 with homocysteine (PDB code 4XFI), thiosulfate bound Cysteine Dioxygenase at pH 6.8 (PDB code 5I0T), thiosulfate bound Cysteine Dioxygenase at pH 6.2 (PDB code 5I0S), and D-cysteine bound C93A mutant of Cysteine Dioxygenase at pH 8 (PDB code 5I0R).
References
- 1.Stipanuk MH, Simmons CR, Karplus PA, Dominy JE., Jr. Thiol dioxygenases: unique families of cupin proteins. Amino Acids. 2011;41:91–102. doi: 10.1007/s00726-010-0518-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Driggers CM, Stipanuk MH, Karplus PA. Encyclopedia of Inorganic and Bioinorganic Chemistry. John Wiley & Sons, Ltd.; 2015. Mammalian Cysteine Dioxygenase. [Google Scholar]
- 3.Stipanuk MH, Ueki I, Dominy JE, Jr., Simmons CR, Hirschberger LL. Cysteine dioxygenase: a robust system for regulation of cellular cysteine levels. Amino Acids. 2009;37:55–63. doi: 10.1007/s00726-008-0202-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dominy JE, Jr., Hwang J, Stipanuk MH. Overexpression of cysteine dioxygenase reduces intracellular cysteine and glutathione pools in HepG2/C3A cells. Am J Physiol Endocrinol Metab. 2007;293:62–9. doi: 10.1152/ajpendo.00053.2007. [DOI] [PubMed] [Google Scholar]
- 5.Park S, Imlay JA. High levels of intracellular cysteine promote oxidative DNA damage by driving the Fenton reaction. Journal of Bacteriology. 2003;185:1942–1950. doi: 10.1128/JB.185.6.1942-1950.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olney JW, Zorumski C, Price MT, Labruyere J. L-Cysteine, a Bicarbonate-Sensitive Endogenous Excitotoxin. Science. 1990;248:596–599. doi: 10.1126/science.2185543. [DOI] [PubMed] [Google Scholar]
- 7.Hogins J, Crawford DC, Zorumski CF, Mennerick S. Excitotoxicity Triggered by Neurobasal Culture Medium. PLoS One. 2011;6 doi: 10.1371/journal.pone.0025633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deth R, Muratore C, Benzecry J, Power-Charnitsky VA, Waly M. How environmental and genetic factors combine to cause autism: A redox/methylation hypothesis. Neurotoxicology. 2008;29:190–201. doi: 10.1016/j.neuro.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 9.James SJ, Cutler P, Melnyk S, Jernigan S, Janak L, Gaylor DW, Neubrander JA. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am J Clin Nutr. 2004;80:1611–7. doi: 10.1093/ajcn/80.6.1611. [DOI] [PubMed] [Google Scholar]
- 10.Heafield MT, Fearn S, Steventon GB, Waring RH, Williams AC, Sturman SG. Plasma cysteine and sulphate levels in patients with motor neurone, Parkinson's and Alzheimer's disease. Neurosci Lett. 1990;110:216–20. doi: 10.1016/0304-3940(90)90814-p. [DOI] [PubMed] [Google Scholar]
- 11.McCaddon A, Hudson P, Hill D, Barber J, Lloyd A, Davies G, Regland B. Alzheimer's disease and total plasma aminothiols. Biological Psychiatry. 2003;53:254–260. doi: 10.1016/s0006-3223(02)01451-8. [DOI] [PubMed] [Google Scholar]
- 12.Ueki I, Roman HB, Valli A, Fieselmann K, Lam J, Peters R, Hirschberger LL, Stipanuk MH. Knockout of the murine cysteine dioxygenase gene results in severe impairment in ability to synthesize taurine and an increased catabolism of cysteine to hydrogen sulfide. Am J Physiol Endocrinol Metab. 2011;301:E668–84. doi: 10.1152/ajpendo.00151.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stipanuk MH, Hirschberger LL, Londono MP, Cresenzi CL, Yu AF. The ubiquitin-proteasome system is responsible for cysteine-responsive regulation of cysteine dioxygenase concentration in liver. American Journal of Physiology-Endocrinology and Metabolism. 2004;286:E439–E448. doi: 10.1152/ajpendo.00336.2003. [DOI] [PubMed] [Google Scholar]
- 14.Dominy JE, Jr., Hirschberger LL, Coloso RM, Stipanuk MH. Regulation of cysteine dioxygenase degradation is mediated by intracellular cysteine levels and the ubiquitin-26 S proteasome system in the living rat. Biochem J. 2006;394:267–73. doi: 10.1042/BJ20051510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dominy JE, Jr., Hwang J, Guo S, Hirschberger LL, Zhang S, Stipanuk MH. Synthesis of amino acid cofactor in cysteine dioxygenase is regulated by substrate and represents a novel post-translational regulation of activity. J Biol Chem. 2008;283:12188–201. doi: 10.1074/jbc.M800044200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siakkou E, Rutledge MT, Wilbanks SM, Jameson GN. Correlating crosslink formation with enzymatic activity in cysteine dioxygenase. Biochim Biophys Acta. 2011;1814:2003–9. doi: 10.1016/j.bbapap.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 17.Njeri CW, Ellis HR. Shifting Redox States of the Iron Center Partitions CDO between Crosslink Formation or Cysteine Oxidation. Arch Biochem Biophys. 2014 doi: 10.1016/j.abb.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Kleffmann T, Jongkees SA, Fairweather G, Wilbanks SM, Jameson GN. Mass-spectrometric characterization of two posttranslational modifications of cysteine dioxygenase. J Biol Inorg Chem. 2009;14:913–21. doi: 10.1007/s00775-009-0504-x. [DOI] [PubMed] [Google Scholar]
- 19.Tchesnokov EP, Wilbanks SM, Jameson GN. A strongly bound high-spin iron(II) coordinates cysteine and homocysteine in cysteine dioxygenase. Biochemistry. 2012;51:257–64. doi: 10.1021/bi201597w. [DOI] [PubMed] [Google Scholar]
- 20.Simmons CR, Liu Q, Huang Q, Hao Q, Begley TP, Karplus PA, Stipanuk MH. Crystal structure of mammalian cysteine dioxygenase. A novel mononuclear iron center for cysteine thiol oxidation. J Biol Chem. 2006;281:18723–33. doi: 10.1074/jbc.M601555200. [DOI] [PubMed] [Google Scholar]
- 21.McCoy JG, Bailey LJ, Bitto E, Bingman CA, Aceti DJ, Fox BG, Phillips GN., Jr. Structure and mechanism of mouse cysteine dioxygenase. Proc Natl Acad Sci U S A. 2006;103:3084–9. doi: 10.1073/pnas.0509262103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simmons CR, Krishnamoorthy K, Granett SL, Schuller DJ, Dominy JE, Jr., Begley TP, Stipanuk MH, Karplus PA. A putative Fe2+-bound persulfenate intermediate in cysteine dioxygenase. Biochemistry. 2008;47:11390–2. doi: 10.1021/bi801546n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pierce BS, Gardner JD, Bailey LJ, Brunold TC, Fox BG. Characterization of the nitrosyl adduct of substrate-bound mouse cysteine dioxygenase by electron paramagnetic resonance: electronic structure of the active site and mechanistic implications. Biochemistry. 2007;46:8569–78. doi: 10.1021/bi700662d. [DOI] [PubMed] [Google Scholar]
- 24.Li W, Blaesi EJ, Pecore MD, Crowell JK, Pierce BS. Second-sphere interactions between the C93-Y157 cross-link and the substrate-bound Fe site influence the O(2) coupling efficiency in mouse cysteine dioxygenase. Biochemistry. 2013;52:9104–19. doi: 10.1021/bi4010232. [DOI] [PubMed] [Google Scholar]
- 25.Crawford JA, Li W, Pierce BS. Single turnover of substrate-bound ferric cysteine dioxygenase with superoxide anion: enzymatic reactivation, product formation, and a transient intermediate. Biochemistry. 2011;50:10241–53. doi: 10.1021/bi2011724. [DOI] [PubMed] [Google Scholar]
- 26.Tchesnokov EP, Faponle AS, Davies CG, Quesne MG, Turner R, Fellner M, Souness RJ, Wilbanks SM, de Visser SP, Jameson GN. An iron-oxygen intermediate formed during the catalytic cycle of cysteine dioxygenase. Chem Commun (Camb) 2016;52:8814–7. doi: 10.1039/c6cc03904a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gardner JD, Pierce BS, Fox BG, Brunold TC. Spectroscopic and computational characterization of substrate-bound mouse cysteine dioxygenase: nature of the ferrous and ferric cysteine adducts and mechanistic implications. Biochemistry. 2010;49:6033–41. doi: 10.1021/bi100189h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Driggers CM, Cooley RB, Sankaran B, Hirschberger LL, Stipanuk MH, Karplus PA. Cysteine dioxygenase structures from pH4 to 9: consistent cyspersulfenate formation at intermediate pH and a Cys-bound enzyme at higher pH. J Mol Biol. 2013;425:3121–36. doi: 10.1016/j.jmb.2013.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Souness RJ, Kleffmann T, Tchesnokov EP, Wilbanks SM, Jameson GB, Jameson GN. Mechanistic implications of persulfenate and persulfide binding in the active site of cysteine dioxygenase. Biochemistry. 2013;52:7606–17. doi: 10.1021/bi400661a. [DOI] [PubMed] [Google Scholar]
- 30.Crowell JK, Li W, Pierce BS. Oxidative uncoupling in cysteine dioxygenase is gated by a proton-sensitive intermediate. Biochemistry. 2014;53:7541–8. doi: 10.1021/bi501241d. [DOI] [PubMed] [Google Scholar]
- 31.Aluri S, de Visser SP. The mechanism of cysteine oxygenation by cysteine dioxygenase enzymes. J Am Chem Soc. 2007;129:14846–7. doi: 10.1021/ja0758178. [DOI] [PubMed] [Google Scholar]
- 32.Kumar D, Thiel W, de Visser SP. Theoretical study on the mechanism of the oxygen activation process in cysteine dioxygenase enzymes. J Am Chem Soc. 2011;133:3869–82. doi: 10.1021/ja107514f. [DOI] [PubMed] [Google Scholar]
- 33.de Visser SP, Straganz GD. Why do cysteine dioxygenase enzymes contain a 3-His ligand motif rather than a 2His/1Asp motif like most nonheme dioxygenases? J Phys Chem A. 2009;113:1835–46. doi: 10.1021/jp809700f. [DOI] [PubMed] [Google Scholar]
- 34.Alberto ME. A trispyrazolylborato iron cysteinato complex efficiently mimics the cysteine dioxygenation process: mechanistic insights. Chem Commun (Camb) 2015;51:8369–72. doi: 10.1039/c5cc00813a. [DOI] [PubMed] [Google Scholar]
- 35.Dominy JE, Jr., Simmons CR, Karplus PA, Gehring AM, Stipanuk MH. Identification and characterization of bacterial cysteine dioxygenases: a new route of cysteine degradation for eubacteria. J Bacteriol. 2006;188:5561–9. doi: 10.1128/JB.00291-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Driggers CM, Hartman SJ, Karplus PA. Structures of Arg- and Gln-type bacterial cysteine dioxygenase homologs. Protein Sci. 2014 doi: 10.1002/pro.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davies CG, Fellner M, Tchesnokov EP, Wilbanks SM, Jameson GN. The Cys-Tyr Cross-Link of Cysteine Dioxygenase Changes the Optimal pH of the Reaction without a Structural Change. Biochemistry. 2014;53:7961–8. doi: 10.1021/bi501277a. [DOI] [PubMed] [Google Scholar]
- 38.Fellner M, Doughty LM, Jameson GN, Wilbanks SM. A chromogenic assay of substrate depletion by thiol dioxygenases. Anal Biochem. 2014;459C:56–60. doi: 10.1016/j.ab.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 39.Chai SC, Bruyere JR, Maroney MJ. Probes of the catalytic site of cysteine dioxygenase. J Biol Chem. 2006;281:15774–9. doi: 10.1074/jbc.M601269200. [DOI] [PubMed] [Google Scholar]
- 40.Blaesi EJ, Fox BG, Brunold TC. Spectroscopic and Computational Investigation of Fe(III) Cysteine Dioxygenase: Implications for the Nature of the Putative Superoxo-Fe(III) Intermediate. Biochemistry. 2014 doi: 10.1021/bi500767x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gonzalez-Ovalle LE, Quesne MG, Kumar D, Goldberg DP, de Visser SP. Axial and equatorial ligand effects on biomimetic cysteine dioxygenase model complexes. Org Biomol Chem. 2012;10:5401–9. doi: 10.1039/c2ob25406a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Witten EH, Reiff WM, Lazar K, Sullivan BW, Foxman BM. The Ferric Chloride-Alpha-Diamine System .3. X-Ray Crystallographic, Magnetic-Susceptibility, and Zero-Field and High-Field Mossbauer-Spectroscopy Investigation of [Fe(2,2'bpy)2cl2][Fecl4] - Slow Paramagnetic Relaxation and Magnetic-Ordering of Complex Bimetallic Salts. Inorganic Chemistry. 1985;24:4585–4591. [Google Scholar]
- 43.Vaillancourt FH, Yeh E, Vosburg DA, Garneau-Tsodikova S, Walsh CT. Nature's inventory of halogenation catalysts: Oxidative strategies predominate. Chemical Reviews. 2006;106:3364–3378. doi: 10.1021/cr050313i. [DOI] [PubMed] [Google Scholar]
- 44.Blasiak LC, Drennan CL. Structural Perspective on Enzymatic Halogenation. Accounts of Chemical Research. 2009;42:147–155. doi: 10.1021/ar800088r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fellner M, Siakkou E, Faponle AS, Tchesnokov EP, de Visser SP, Wilbanks SM, Jameson GN. Influence of cysteine 164 on active site structure in rat cysteine dioxygenase. J Biol Inorg Chem. 2016;21:501–10. doi: 10.1007/s00775-016-1360-0. [DOI] [PubMed] [Google Scholar]
- 46.Steiner T. Hydrogen-bond distances to halide ions in organic and organometallic crystal structures: Up-to-date database study. Acta Crystallographica Section B-Structural Science. 1998;54:456–463. [Google Scholar]
- 47.Ye S, Wu X, Wei L, Tang D, Sun P, Bartlam M, Rao Z. An insight into the mechanism of human cysteine dioxygenase. Key roles of the thioether-bonded tyrosine-cysteine cofactor. J Biol Chem. 2007;282:3391–402. doi: 10.1074/jbc.M609337200. [DOI] [PubMed] [Google Scholar]
- 48.Kilroy WP. Anaerobic Decomposition of Sodium Dithionite in Alkaline-Solution. Journal of Inorganic & Nuclear Chemistry. 1980;42:1071–1073. [Google Scholar]
- 49.Blaesi EJ, Fox BG, Brunold TC. Spectroscopic and computational investigation of iron(III) cysteine dioxygenase: implications for the nature of the putative superoxo-Fe(III) intermediate. Biochemistry. 2014;53:5759–70. doi: 10.1021/bi500767x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meier B, Scherk C, Schmidt M, Parak F. pH-dependent inhibition by azide and fluoride of the iron superoxide dismutase from Propionibacterium shermanii. Biochem J. 1998;331:403–7. doi: 10.1042/bj3310403. (Pt 2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goldsmith CR, Jonas RT, Cole AP, Stack TD. A spectrochemical walk: single-site perturbation within a series of six-coordinate ferrous complexes. Inorg Chem. 2002;41:4642–52. doi: 10.1021/ic025616z. [DOI] [PubMed] [Google Scholar]
- 52.Ishiguro H, Naruse S, Kitagawa M, Mabuchi T, Kondo T, Hayakawa T, Case RM, Steward MC. Chloride transport in microperfused interlobular ducts isolated from guinea-pig pancreas. J Physiol. 2002;539:175–89. doi: 10.1113/jphysiol.2001.012490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Glykys J, Dzhala V, Egawa K, Balena T, Saponjian Y, Kuchibhotla KV, Bacskai BJ, Kahle KT, Zeuthen T, Staley KJ. Local impermeant anions establish the neuronal chloride concentration. Science. 2014;343:670–5. doi: 10.1126/science.1245423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Njeri CW, Ellis HR. Shifting redox states of the iron center partitions CDO between crosslink formation or cysteine oxidation. Arch Biochem Biophys. 2014;558:61–9. doi: 10.1016/j.abb.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 55.Blaesi EJ, Fox BG, Brunold TC. Spectroscopic and Computational Investigation of the H155A Variant of Cysteine Dioxygenase: Geometric and Electronic Consequences of a Third-Sphere Amino Acid Substitution. Biochemistry. 2015;54:2874–84. doi: 10.1021/acs.biochem.5b00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Crowell JK, Sardar S, Hossain MS, Foss FW, Jr., Pierce BS. Non-chemical proton-dependent steps prior to O2-activation limit Azotobacter vinelandii 3-mercaptopropionic acid dioxygenase (MDO) catalysis. Arch Biochem Biophys. 2016;604:86–94. doi: 10.1016/j.abb.2016.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chai SC, Jerkins AA, Banik JJ, Shalev I, Pinkham JL, Uden PC, Maroney MJ. Heterologous expression, purification, and characterization of recombinant rat cysteine dioxygenase. J Biol Chem. 2005;280:9865–9. doi: 10.1074/jbc.M413733200. [DOI] [PubMed] [Google Scholar]
- 58.Li W, Pierce BS. Steady-state substrate specificity and O2-coupling efficiency of mouse cysteine dioxygenase. Arch Biochem Biophys. 2015;565:49–56. doi: 10.1016/j.abb.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 59.Pierce BS, Subedi BP, Sardar S, Crowell JK. The "Gln-Type" Thiol Dioxygenase from Azotobacter vinelandii Is a 3-Mercaptopropionic Acid Dioxygenase. Biochemistry. 2015;54:7477–90. doi: 10.1021/acs.biochem.5b00636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Simmons CR, Hao Q, Stipanuk MH. Preparation, crystallization and X-ray diffraction analysis to 1.5 A resolution of rat cysteine dioxygenase, a mononuclear iron enzyme responsible for cysteine thiol oxidation. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2005;61:1013–6. doi: 10.1107/S1744309105033737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Simmons CR, Hirschberger LL, Machi MS, Stipanuk MH. Expression, purification, and kinetic characterization of recombinant rat cysteine dioxygenase, a non-heme metalloenzyme necessary for regulation of cellular cysteine levels. Protein Expr Purif. 2006;47:74–81. doi: 10.1016/j.pep.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 62.Leslie A. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4+ ESF-EAMCB newsletter on protein crystallography. 1992;26 [Google Scholar]
- 63.Evans PR. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr D Biol Crystallogr. 2011;67:282–92. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–3. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Word JM, Lovell SC, LaBean TH, Taylor HC, Zalis ME, Presley BK, Richardson JS, Richardson DC. Visualizing and quantifying molecular goodness-of-fit: small-probe contact dots with explicit hydrogen atoms. J Mol Biol. 1999;285:1711–33. doi: 10.1006/jmbi.1998.2400. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.