

Abstract
Exposure to organophosphates (OPs) often results in seizures and/or status epilepticus (SE) that produce neural damage within the central nervous system (CNS). Early control of SE is imperative for minimizing seizure-related CNS neuropathology. Although standard therapies exist, more effective agents are needed to reduce OP-induced SE and neuronal loss, particularly therapies with efficacy when administered 10’s of minutes after the onset of SE. To evaluate novel antiseizure compounds, animal models should simulate the CNS effects of OP exposure observed in humans. We characterized in rats the effects of the OP, diisopropyl flourophosphate (DFP) as a function of dose and route of administration of supporting agents (pyridostigmine, 2-PAM, atropine); outcome measures were mortality, electrographic seizure activity during SE, and subsequent CNS neuropathology. Doses of DFP between 3 and 7 mg/kg consistently caused SE, and the latency to behavioral tremors and to subsequent initiation of SE were dose related. In distinction, all doses of DFP that resulted in electrographic SE (3–7 mg/kg) produced seizures of similar intensity and duration, and similar CNS neuropathology (i.e., the effects were all-or-none). Although SE was similar across doses, mortality progressively increased with higher doses of DFP. Mortality was significantly lower when the route of administration of therapeutic agents was intramuscular compared to intraperitoneal. This rodent model of OP poisoning demonstrates pathological characteristics similar to those observed in humans, and thus begins to validate this model for investigating potential new therapeutic approaches.
Keywords: seizures, status epilepticus, EEG, antiseizure, neuroprotection
1. Introduction
Organophosphates (OPs) comprise the active components of many insecticides including parathion and malathion, as well as the nerve agents (NA) soman, tabun and sarin. Accidental or deliberate exposure to these OP-containing compounds is a major, worldwide health problem. OPs are potent inhibitors of cholinesterase (Lotti, 2001); therefore, OP exposure induces the accumulation of acetylcholine in peripheral synapses, which results in muscle tremors, cardiovascular dysfunction, hypotension, and bronchial spasms (Hulse et al., 2014, Lotti, 2001). OP-related morbidity most frequently results from respiratory failure due to brochospasm, bronchorrhea, and paralysis of respiratory muscles (Karalliedde and Senanayake, 1989, Lotti, 2001).
In addition to peripheral effects, OPs, such as diisopropyl flourophosphate (DFP) (Deshpande et al., 2010, Todorovic et al., 2012), parathion (Garcia et al., 2003, Hoffmann and Papendorf, 2006), dimethoate (Hoffmann and Papendorf, 2006), paraoxon (Deshpande et al., 2014, Shrot et al., 2014), dichlorvos (Gaspari and Paydarfar, 2007), and NAs (McDonough J.H. Jr. and Shih, 1997, Nozaki et al., 1995) all can act in the central nervous system (CNS) to induce seizures and/or status epilepticus (SE). SE is characterized by continuous or recurrent seizure activity that often results in neuronal injury in the CNS and mortality (Fujikawa, 2005, Fujikawa et al., 2000, Wasterlain et al., 1993). This central neuropathology produced by SE can result in long-term neurological and behavioral disorders (Apland et al., 2010, Prager et al., 2014). The CNS neuronal damage can occur after only 20 min of continuous seizure activity, and worsens as the duration of the SE increases (McDonough J.H. et al., 1995). Consequently, early control of SE is critical for survival and CNS neuroprotection following OP exposure (Shih et al., 2003).
Benzodiazepines are the first-line standard-of-care pharmacotherapy for OP-induced SE (Alldredge et al., 2001, Newmark, 2007, Rotenberg and Newmark, 2003, Treiman, 2007, Walton and Treiman, 1988). Although these agents can be effective if an appropriate dose is administered shortly after initiation of SE, these drugs fail to terminate SE in many cases of OP poisoning, and become less effective when administered >30 min after exposure, as SE becomes increasingly resistant to benzodiazepines as the duration of the SE increases (Todorovic et al., 2012). Furthermore, studies in animal models found that these agents do not prevent brain damage or behavioral deficits resulting from SE if administration is delayed (Apland et al., 2014, Myhrer et al., 2005, Todorovic et al., 2012). In the case of a mass civilian exposure to NA or other OP, it is unlikely that first responders would be on site and administer these standard therapeutic agents within the relatively short post-exposure time period needed for maximum efficacy. Consequently, development of novel therapies that can effectively control OP-induced SE and reduce CNS neuropathology, when administered following a significant time period after exposure, is essential.
In order to experimentally evaluate potential therapies, the development of a new, delayed treatment, animal model of OP exposure is required. Although both kainic acid and pilocarpine induce SE and neuropathology, their mechanisms of action are different than OPs. Consequently, the optimal animal model should use an OP and/or NA to induce SE of adequate intensity and duration to induce neuropathology, but not result in rapid mortality that would preclude determination of the complete time course and duration of SE, or prevent appropriate processing of CNS tissue for histopathology. The initial phase of developing a new, delayed treatment model of OP poisoning, which would mimic a mass civilian exposure is to determine the dose of an OP that induces seizure activity and neuropathology with an experimentally acceptable rate of mortality. These studies were designed to determine the optimal dose of the prototypical OP DFP that meets these criteria.
We characterized behavioral and electrographic indicators of SE activity, CNS neuropathology, and mortality following administration of DFP (2–7 mg/kg), along with clinically appropriate supporting compounds (pyridostigmine, atropine, and 2-PAM) (Bajgar, 2010, Jokanovic and Stojiljkovic, 2006, Leadbeater et al., 1985, Voicu et al., 2010, Wetherell et al., 2002) in rats. Appropriate adjustment of OP dose and route of administration of supporting agents resulted in an animal model producing SE and CNS neuropathology similar to effects reported in patients (Holstege and Dobmeier, 2005, Shetty, 2014, Xiang et al., 2014), with an experimentally tolerable level of mortality.
2. Materials and methods
2.1. Animals
Male Sprague-Dawley rats (200–250 g) were purchased from Charles River, were housed in the temperature-controlled vivarium, and maintained on a 12hr:12hr light:dark cycle with ad libitum access to food and water. The University of Utah Institutional Animal Care and Use Committee approved all surgical and experimental procedures used in these studies.
2.2 Drug Preparation
DFP was dissolved in 0.9% sterile saline to a concentration of 10 ul/ml that was stored frozen (−80° C) until the day of the experiment. DFP was kept on ice until injected. Pyridostigmine was prepared in 0.9% sterile saline (0.052 mg/ml) and refrigerated until used. Atropine methyl nitrate and 2-PAM were combined and dissolved in sterile 0.9% saline to final concentrations of 4 mg/ml and 50 mg/ml, respectively. Pyridostigmine, atropine, and 2-PAM were prepared on the day of testing.
2.3. Surgical Procedures
Animals were implanted with electrodes for electroencephalographic (EEG) recording. The rats were anesthetized with 2% isoflourane and placed in a stereotaxic instrument. An incision was then made on the midline and the scalp was retracted laterally. Six holes were drilled in the skull, three for support screws and three for the electrode wires. Bipolar recording electrodes (MS333-3-B, Plastics One, Roanoake, VA) were placed right of midline, while the ground electrode was positioned on the left. All electrode wires were trimmed for epidural, differential recording of EEG activity. Dental cement was used to secure the support screws and electrode pedestal, and the wound was sutured shut. All animals were then returned to their home cages and allowed at least 7 days to recover from these procedures prior to testing.
2.4. Video and EEG Recordings
Following recovery, the conscious, unrestrained rats were placed into Plexiglas recording chambers with swivel communicators for testing. The implanted electrodes were connected to spring-covered EEG cables (Plastics One, Roanoke, VA). EEG100 amplifiers (high-pass filter, 1 Hz, low-pass, 100 Hz, notch filter at 60 Hz, 5000x gain) amplified the signals, which were then digitized at 500 Hz with an MP150 digital-to-analog converter. The EEG signals were recorded using AcqKnowledge software (BioPac Systems, Inc. Santa Barbara, CA).
During testing, the rats were continuously monitored using an infrared surveillance system whose output was recorded by DVD player/recorders for subsequent evaluation (DMR-ES20, Panasonic).
2.5. Experimental Protocols
2.5.1. Experiment 1
Fig. 1 is a schematic representation of the protocol used in these studies. To attenuate the peripheral lethal effects of OP and decrease mortality, animals were pretreated with pyridostigmine bromide (0.026 mg/kg, IM) 30 min prior to DFP administration, and atropine methyl nitrate (2 mg/kg, IM) plus 2-PAM (25 mg/kg, IM) 1 min after the injection of DFP. Pyridostigmine is a competitive cholinesterase inhibitor that reduces binding of the irreversible inhibitor DFP, and consequently is beneficial following DFP exposure. Doses of DFP were 2.0, 3.0, 4.0, 4.5, 6.5 or 7.0 mg/kg administered subcutaneously (SC). Video-EEG monitoring was continued for 24 h after DFP injections. The percentage mortality was calculated as the proportion of animals that did not survive 24-hr after DFP exposure.
Figure 1.

Schematic summary of protocol.
2.5.2. Experiment 2
These studies used a similar protocol, but compared SE and mortality following 4.5 mg/kg DFP (SC) in animals given pyridostigmine, atropine, and 2-PAM IM or IP.
2.6. Behavioral Analysis
Animals were continuously observed for behavioral indicators of seizure activity until the first electrographic seizure occured following DFP administration. The latency between DFP administration and the initial appearance of behaviors associated with seizure activity, including tremors, was noted.
2.7. EEG Analysis
The EEG data were analyzed using a quantitative algorithm (Lehmkuhle et al., 2009), where data from 0–16 h were band-pass filtered (20–70 Hz) and the power spectral density was calculated. Mean power was computed by band-pass filtering the signal over 1-h intervals, and then computing the mean power in the gamma band (20–70 Hz) for each hour after DFP injection and plotted over time. Spike frequencies were computed by counting the number of spikes occurring within sequential 15-min windows, and then dividing the total number by the time duration to give the average frequency. Spikes were characterized by a fast rising phase, and thus the amplitude of the slope of the rising phase was used as a measure to discriminate spikes from artifacts. Single spikes were quantified by first dividing the total time into 16-msec intervals, which is assumed to be the maximum duration of a single spike, then finding the maximum magnitude of the increasing slope within that interval and then comparing that slope to a threshold.
2.8. Histopathology Analysis
Twenty-four hr following DFP injections, rats were deeply anesthetized and perfused with buffered formalin. The fixed brains were cryoprotected with 30% sucrose, subsequently frozen, and 30-μm coronal sections were cut and stained with Flouro-Jade B (FJB, Histo-chem Inc, Jefferson, AR)(Schmued and Hopkins, 2000). For staining, sections were first incubated in 0.06% KMnO4, and then in 0.001% F-J-B (Schmued and Hopkins, 2000). Previous studies suggest all primary neurons affected by SE demonstrate FJB staining characteristic of neuronal degeneration within 4 hr (Hopkins et al., 2000). Whole slide imaging was performed using a Nanozoomer 2.0 (Hamamatsu).
Observers blinded to the experimental treatment of the rats performed the histopathological analyses. An unbiased random sampling technique quantifying the number of FJ B positive neurons in selected sections was used to estimate the neuropathological effects of DFP-induced SE. Specifically, four sections from each brain area, separated by a minimum of 70 um, were randomly selected for evaluation. Then, within each of these selected sections, three 175 um2 counting boxes were randomly chosen. The following regions were analyzed: dorsal and ventral hippocampus (CA1, CA3), parietal cortex, amygdala, thalamus, piriform and entorhinal cortices. These brain areas were selected for analysis, as they have been implicated in contributing to seizure initiation and/or progression induced by NAs or other OPs (McDonough J.H. et al., 1987, Myhrer et al., 2010, Skovira et al., 2010).
2.9. Statistical Analyses
Data are presented as means + SEM. Multiple means were compared using an ANOVA, and a Bonferroni post hoc test. The mortality data were analyzed with Fisher’s exact test. Results were considered significant when p<0.05.
3. Results
3.1. Experiment 1. SE and Mortality following DFP
We conducted studies to assess the relationships between increasing doses of DFP, the resulting intensity of SE activity, and subsequent mortality. Figure 2 shows the latency from the injection of DFP to the first behavioral tremor (Fig. 2A), and the time from the initial tremor to first EEG activity characteristic of seizures (Fig. 2B). While all doses of DFP elicited behavioral tremors, electrographic seizures were not observed in animals given 2 mg/kg. The first behavioral signs and the initial evidence of seizure activity were evident significantly sooner in animals administered the higher doses of DFP (6.5 mg/kg and 7 mg/kg) than in rats treated with lower doses.
Figure 2.

Latency from DFP to first tremor (Panel A) and latency from first tremor to the initial electrographic seizure (Panel B) in animals administered 2–7 mg/kg DFP. p<0.05 re: lowest dose in panel. (Ns= 2 mg/kg, 12; 3 mg/kg, 10; 4 mg/kg, 10; 4.5 mg/kg, 15; 6.5 mg/kg, 14; 7.0 mg/kg, 12), Panel A, *p<0.05 re: 2.0 mg/kg; Panel B *p<0.05 re: 3.0 mg/kg, †p<0.05 re: 4 mg/kg.
The latencies to the initial electrographic seizures shown in Fig. 2, Panel B represent the time to the first seizure activity. In the majority of all animals exhibiting seizures (3–7 mg/kg DFP) this marked the initiation of continuous seizure activity. However, some animals (n=2–5/group) exhibited a brief (<2 min) period of normal EEG activity following the initial seizure. There were no dose related differences in either the number of animals exhibiting this period of normal EEG, nor the duration of normal EEG activity prior to continuous seizures. Furthermore, since SE is characterized by either continuous seizure activity, or by repetitive seizures separated by less than 30 min, this latency is considered to represent the time to SE.
Figure 3 illustrates representative EEG recordings from animals injected with either 7 mg/kg (Fig 3A) or 2 mg/kg (Fig 3B) DFP. Complete 16-hr evaluations of both gamma power and spike rates for all doses of DFP are shown in Figure 4. Although behavioral tremors were present in animals after administration of all doses of DFP, we observed no significant EEG seizure activity in animals given 2 mg/kg. However, all higher doses of DFP (3–7 mg/kg) elicited SE, and there were no differences in either intensity or duration of the SE induced by these higher doses (Fig. 4)
Figure 3.

Representative EEG recordings from rats administered either 7 mg/kg (Panel A) or 2 mg/kg (Panel B) DFP. Recordings from within the dashed boxes are shown below each EEG with an expanded time scale. The arrows denote DFP administration.
Fig. 4.
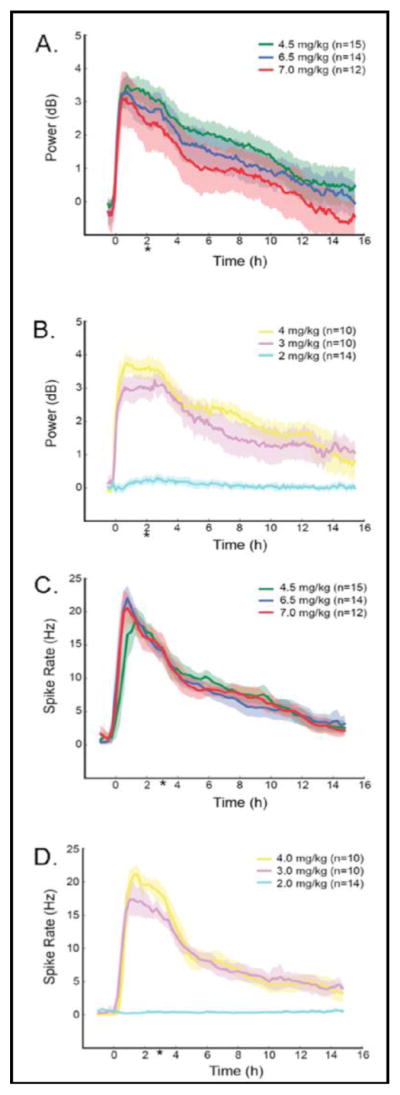
SE analysis using power (bandwidth filtered 20–70 Hz) (Panels A and B) and spike rate (Panels C and D) following administration of 2.0, 3.0, 4.0, 4.5, 6.5, or 7.0 mg/kg DFP. Traces represent means + the 95% confidence intervals. *p<0.05, 2 mg/kg DFP compared to all other groups.
Table 1 shows the percentage of animals administered each dose of DFP that exhibited SE (middle column), and the percentage that did not survive for 24 hr after treatment. No animals administered 2 mg/kg DFP exhibited any electrographic seizure activity. Above this minimal dose, there was a steep dose-response relationship between DFP and the proportion of animals exhibiting electrographic SE that showed a rapid rise and plateau approaching 100% at 4 mg/kg DFP (Center Column of Table 1).
Table 1.
Percentage of animals exhibiting electrographic seizures (middle column) and mortality at 24 hr (right column) following administration of 2–7 mg/kg DFP.
| Dose DFP (mg/kg) | % Animals with EEG Seizures | % Mortality at 24 hr |
|---|---|---|
| 2.0 | 0% (0/16) | 0% (0/16) |
| 3.0 | 62% (10/16) | 0% (0/16) |
| 4.0 | 93% (15/16) | 12% (2/16) |
| 4.5 | 100% (21/21) | 23% (5/21) |
| 5.0 | 90% (10/11) | 27% (3/11) |
| 6.0 | 100% (10/10) | 30% (3/10) |
| 6.5 | 100% (24/24) | 33% (8/24) |
| 7.0 | 100% (24/24) | 50% (12/24) |
In contrast, there was a continuous, more gradual increase in mortality over the tested dose range of 3–7 mg/kg DFP (Right Column of Table 1). Even at doses that evoked seizures in 100% of the animals (i.e. 4.5 mg/kg and 6 mg/kg), mortality was lower compared to animals given 7.0 mg/kg DFP.
3.2. Histopathology
Next, we examined the histopathological effects of increasing doses of DFP on SE-induced neuronal death in several brain regions. Fig. 5 shows representative brain sections obtained from the amygdala of animals administered either 2 mg/kg (Fig 5, upper left) or 7 mg/kg (Fig 5, upper right). No FJB positive neurons were detected in any brain region obtained from animals administered 2 mg/kg DFP. Furthermore, all animals that were administered 3–7 mg/kg DFP had significant FJB staining in several brain areas (Fig. 5, middle and lower panels), including the hilus and the parietal, piriform, and entorhinal cortices (data not shown). Finally, the number of FJB positive neurons was not related to dose in any brain region of animals administered 3–7 mg/kg DFP. Animals that did not exhibit SE showed no DFP stained neurons in any brain region examined.
Fig. 5.

Top Panels - Representative sections stained with Flouro-Jade B from the amygdalae of animals given 2 mg/kg (left panel) or 7 mg/kg (right panel) DFP. Middle and Lower Panels - Summary data showing the numbers of FJB stained cells in representative brain regions from animals given 3–7 mg/kg DFP. (N’s= 3.0 mg/kg, 10; 4.0 mg/kg, 9; 4.5 mg/kg, 10; 6.5 mg/kg, 9; 7.0 mg/kg, 10)
3.3. Experiment 2. IM vs. IP Administration of Supporting Agents
To further characterize the DFP model, we compared the effects of IM versus IP administration of the supporting agents on seizure intensity and duration, and subsequent mortality. Figure 6 shows the analysis of power in the gamma band and change in mean spike frequency during DFP (4.5 mg/kg)-induced SE in animals administered pyridostigmine, atropine, and 2-PAM IM or IP. Neither intensity nor duration of SE were different at any time point between the two routes of administration in these groups of animals.
Fig. 6.
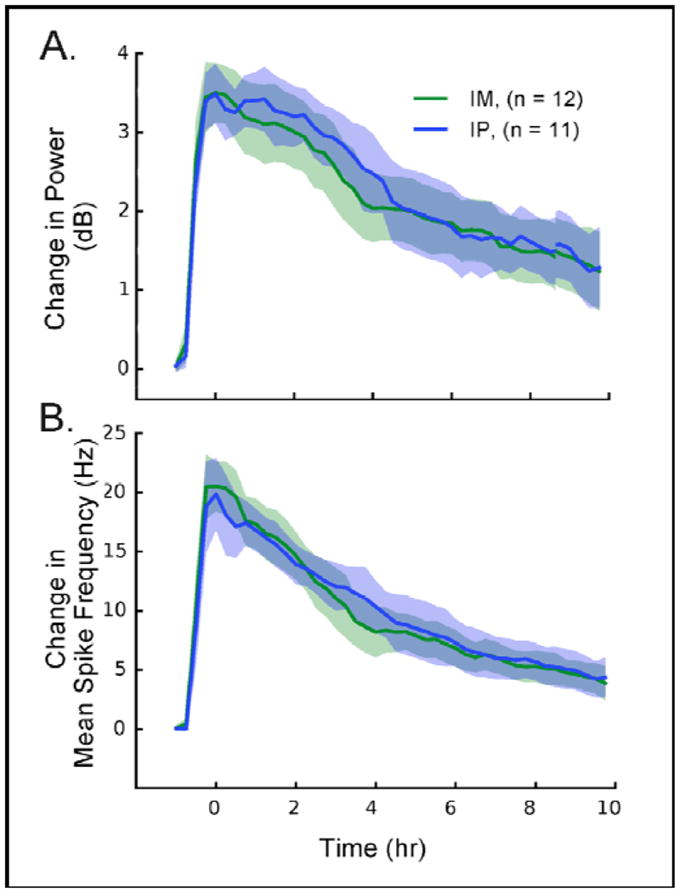
Seizure intensity, measured by EEG power in the gamma band (20–70 Hz)(top panel), and mean spike frequency (bottom panel) in rats given therapeutic agents IM vs. IP with DFP (4.5 mg/kg).
Although neither SE duration nor intensity were significantly different when supporting compounds were given IM or IP, the route of administration did alter mortality. Table 2 shows the percentage of animals exhibiting electrographic seizures and mortality when the agents were administered IP versus IM. Similar to intensity and duration of SE, seizures were evoked in a similar proportion of animals in both groups, but mortality was significantly lower in animals receiving compounds IM.
Table 2.
Percentages and numbers of animals demonstrating electrographic seizures and mortality 24 hr following 4.5 mg/kg DFP (SC) in rats administered supporting agents IP or IM. p<0.05 re: IP
| Route | Electrographic Seizures (%) | Mortality 24 hr (%) |
|---|---|---|
| Intraperitoneal (IP) | 94% (34/36) | 44% (16/36) |
| Intramuscular (IM) | 100% (26/26) | 11% (3/26)* |
4. Discussion
The primary purpose of this study was to initiate development of a model of OP exposure using DFP-induced SE in order to eventually screen potential antiseizure and neuroprotective agents. Ultimately, in the long term, the model would aim to simulate the critical features of NA and/or OP exposure in a large civilian population, where first responders would not be on-site to treat patients for an extended period of time, and treatments would typically be administered IM versus IV. The goal of the present experiments was to determine a dose of DFP that resulted in intense SE and CNS neuropathology with a moderate rate of mortality. We found that moderate doses of DFP (4–7 mg/kg), in conjunction with accepted antidote agents administered IM, produced consistent SE and CNS neuropathology with moderate mortality. The findings reported here provide the rationale for further model development in subsequent studies that will allow quantitative analysis of drug effects, both on SE activity and on neuronal death.
4.1 Dose response data: all-or-none effect
The dose-response curve to DFP was extremely steep; the lowest dose tested (2 mg/kg) did not produce either electrographic SE or neuropathology, while all higher doses (3–7 mg/kg) resulted in intense SE and substantial neuronal death. However, while increasing the dose of DFP above 3 mg/kg did not increase SE intensity or FJB staining, a progressive dose-related, increase in mortality occurred across all doses tested. Consequently, for further studies, we postulate that a mid-range dose of DFP selected from the doses we tested will be optimal to ensure consistent SE and neuropathology, with minimal mortality. Although no difference in seizure activity or FJB staining was observed after any dose of DFP >3 mg/kg, the latency to SE onset was reduced at the highest doses (6.5–7.0 mg/kg). This reduced latency to onset of seizure activity presumably results from the higher peripheral concentration of DFP, and the more rapid accumulation of DFP in the CNS after administration of the higher doses.
4.2 EEG Evaluation of Seizures
All of the data relevant to our analysis of this model are based on EEG activity, since behavioral analyses of SE often have little or no bearing on actual seizure activity in the brain. The behavioral tremors could reflect direct peripheral actions of increased cholinergic stimulation of the neuromuscular junctions, rather than centrally mediated seizures. Consistent with this interpretation, we found all doses of DFP elicited behavioral responses indicative of seizure activity, but 2 mg/kg DFP did not produce either electrographic seizure activity or detectable FJB staining. Furthermore, in previous studies, we found examples of compounds that appeared to differentially alter pilocarpine-induced behavioral and electrographic measures of seizure activity during SE. Specifically, in the pilocarpine model, behavioral analyses of valnoctamide resulted in a false negative; in particular, valnoctamide did not appear to affect behavioral indications of seizures, but was highly effective in reducing electrographic seizure activity evaluated more directly and rigorously with quantitative measures of the EEG (cf. Pouliot et al, 2013 versus White et al., 2012). Taken together, these studies suggest that behavioral measures do not accurately reflect electrographic seizure activity. These findings underscore the importance of monitoring electrographic activity, particularly since behavioral observations of seizure activity are difficult if not impossible to quantify in an efficient or automated manner, as necessary for a screening model. Consequently, EEG recordings are absolutely essential to define precisely the relationship between seizures and CNS neuropathology.
Addition of spike-rate analysis as a second measure of seizure activity, in addition to spectral density analysis, has improved the ability to quantify SE. Although these measures are only partially independent, presumably both reflect EEG spiking intensity. The use of two quantitative measures will improve the ability to evaluate the efficacies of antiseizure and neuroprotective compounds.
4.3 Localization of neuronal damage after DFP-induced SE
Results from histopathological evaluation of FJB-stained brain sections demonstrated that DFP caused neural damage throughout the brain with emphasis on the CA1 and CA3 areas of the hippocampus, as well as the amygdala, thalamus, and several areas of the limbic cortex. The areas of damage after DFP were qualitatively similar to other models of SE, such as pilocarpine and kainic acid (Covolan and Mello, 2000, Dube et al., 1998, Hopkins et al., 2000, Peterson et al., 2005), suggesting that seizure activity preferentially causes neuronal death in these brain areas. We emphasize that the similarity is only qualitative and that it would be quite difficult to undertake a quantitative analysis of neuronal damage without recording electrical activity in each structure under neuropathological investigation. Presumably the variance of neuronal damage across brain structures is related, at least in part to the relative intensity of the electrical activity in these sites, although this is only speculation. No particular area seemed especially sensitive to the SE-inducing effects of DFP, although again this would take a more rigorous quantitative analysis.
4.4 Intramuscular versus intraperitoneal injection of antidote compounds
The accepted treatment for the potentially lethal, peripheral effects of OP is administration of a competitive antagonist of cholinesterase (e.g., pyridostigmine), an oxime (e.g., 2-PAM), and atropine (Jokanovic and Stojiljkovic, 2006, Leadbeater et al., 1985, Voicu et al., 2010, Wetherell et al., 2002). In the present study, we utilized these agents to reduce the mortality following DFP in order to quantify EEG activity for 10 hr and evaluate neuropathology at 24 hr after administration. Our protocol was designed to use these agents to produce optimal survival, not reproduce the on-site, first responder emergency situation. Since the efficacy of these compounds is partially dependent upon the route of administration, we examined mortality in DFP-treated rats given the supporting agents IM and IP. We found that IM administration (versus IP) produced significantly less mortality. One potential mechanism for differential survival based depending on route of delivery may be that IM administration results in an increased duration of therapeutic blood levels, compared to IP delivery. Importantly, the route of administration of these compounds in our protocol did not alter either the SE or resulting FJB staining produced by DFP. Consequently, if future use of this model aims to optimize the similarity to a mass exposure of OP, then the model should incorporate IM administration of these supporting compounds.
5. Conclusion
Assessment of the dose-response relationships between DFP dose, EEG analysis of SE, and mortality in these studies led us to select a dose of 6.0 mg/kg for future experiments. A strength of this model is our evaluation of two measures of electrographic activity from direct EEG recordings, power spectral density in the gamma band and spike-rate frequency. Although we expect that either measure would be satisfactory, use of both increases the rigor of this model for drug screening. Use of quantitative EEG appears far superior to behavioral measures of seizure activity, as behaviors are quite distinct from the electrographic activity. In terms of localization of neuronal injury, we observed DFP-induced FJB staining in structures that typically exhibit neuropathology after SE induced by drugs, such as pilocarpine and kainic acid, including the CA3 and CA1 areas of hippocampus, amygdala, thalamus, and limbic cortex. Neuronal death has also been observed in these areas in patients undergoing severe SE. Taken together, these data imply that it is SE that induces neuronal death, and that this damage is not unique to DFP. This collection of data provide the rationale for further development of a delayed-treatment, rodent model of DFP-induced SE that will allow more efficient screening of new antiseizure and neuroprotective compounds.
HIGHLIGHTS.
DFP consistently caused status epilepticus with concomitant neuropathology. (75)
Between 3 and 7 mg/kg, the effects of DFP were all-or-none. (59)
Mortality increased with dose, and was lower with intramuscular vs intraperitoneal. (83)
Electrographic and pathological characteristics in rat were similar to human. (78)
Acknowledgments
Acknowledgment of Support
The views expressed in this paper are those of the authors and do not reflect the official policy of the Countermeasures Against Chemical Threats (CounterACT) program, National Institutes of Health (NIH), Department of Health and Human Services, Department of Army, Department of Defense (DoD), or the U.S. Government. The experimental protocol was approved by the Animal Care and Use Review Office of the United States Army Medical Research and Material Command and the Institute Animal Care and Use Committee of the University of Utah and all procedures were conducted in accordance with the principles stated in the Guide for the Care and Use of Laboratory Animals and the Animal Welfare Act of 1966 (P.L. 89-544), as amended. This research was supported by the NIH Office of the Director through an Inter-Agency Agreement (Y1-O6-9613-01) between the National Institute of Allergy and Infectious Diseases (NIAID) and DoD USAMRICD (A120-B.P2009-2) and under subcontract W81XWH-14-C-0119 to the University of Utah.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alldredge BK, Gelb AM, Isaacs SM, Corry MD, Allen FA, Ulrich S, Gottwald MD, O’Neil N, et al. A comparison of lorazepam, diazepam, and placebo for the treatment of out-of-hospital status epilepticus. N Eng J Med. 2001;345:631–637. doi: 10.1056/NEJMoa002141. [DOI] [PubMed] [Google Scholar]
- Apland JP, Figueiredo TH, Qasha F, Aroniadou-Anderjaska V, Souza AP, Braga MF. Higher susceptibility of the ventral versus the dorsal hippocampus and the posteroventral versus anterodorsal amygdala to soman-indiced neuropathology. Neurotoxicology. 2010;31:485–492. doi: 10.1016/j.neuro.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apland JP, Aroniadou-Anderjaska V, Figueiredo TH, Rossetti F, Miller SL, Braga MF. Limitations of diazepam as a treatment for nerve agent-induced seizures and neuropathology in rats: comparison with UBP 302. J Pharmacol Exp Ther. 2014;351:359–372. doi: 10.1124/jpet.114.217299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajgar J. Optimal choice of acetylcholinesterase reactivators for antidotal treatment of nerve agent intoxication. Acta Medica. 2010;53:207–211. doi: 10.14712/18059694.2016.78. [DOI] [PubMed] [Google Scholar]
- Covolan L, Mello LE. Temporal profile of neuronal injury following pilocarpine or kainic acid-incuded status eilepticus. Epilepsy Res. 2000;39:133–152. doi: 10.1016/s0920-1211(99)00119-9. [DOI] [PubMed] [Google Scholar]
- Deshpande LS, Carter DS, Blair RE, DeLorenzo RJ. Development of a prolonged calcium plateau in hippocampal neurons in rats surviving status epilepticus induced by the organophosphate diisopropylflourophosphate. Toxicol Sci. 2010;116:623–631. doi: 10.1093/toxsci/kfq157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande LS, Carter DS, Phillips KF, Blair RE, DeLorenzo RJ. Development of status epilepticus, sustained calcium elevations and neuronal injury in a rat survival model of lethal paraoxon intoxication. Neurotoxicology. 2014;44:17–26. doi: 10.1016/j.neuro.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube C, Andre V, Covolan L, Ferranson A, Marescaux C, Nehlig A. C-fos, Jun-D and HSP72 immunoreactivity, and neuroal injury following lithium-pilocarpine induced status epilepticus in immature and adult rats. Brain Res Mol Brain Res. 1998;63:139–154. doi: 10.1016/s0169-328x(98)00282-4. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG. Prolonged seizures and cellular injury: understanding the connection. Epilepsy and Behavior. 2005;7(Suppl 3):S3–11. doi: 10.1016/j.yebeh.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG, Itabashi HH, Wu A, Shinmei SS. Status epilepticus-induced neuronal loss in humans without complications or epilepsy. Epilepsia. 2000;41:981–991. doi: 10.1111/j.1528-1157.2000.tb00283.x. [DOI] [PubMed] [Google Scholar]
- Garcia SJ, Abu-Qare AW, Meeker-O’Connell WA, borton AJ, Abou-Donia MB. Methyl parathion: a review of health effects. J Toxicol Environ Health B, Crit Rev. 2003;6:185–210. doi: 10.1080/10937400306471. [DOI] [PubMed] [Google Scholar]
- Gaspari RJ, Paydarfar D. Pathophysiology of respiratory failure following acute dichlorvos poisoning in a rodent model. Neurotoxicology. 2007;28:664–671. doi: 10.1016/j.neuro.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann U, Papendorf T. Organophosphate poisonings with parathion and dimethoate. Intensive Care Medicine. 2006;32:464–468. doi: 10.1007/s00134-005-0051-z. [DOI] [PubMed] [Google Scholar]
- Holstege CP, Dobmeier SG. Nerve agent toxicity and treatment. Curr Treatment Options Neurol. 2005;7:91–98. doi: 10.1007/s11940-005-0018-y. [DOI] [PubMed] [Google Scholar]
- Hopkins KJ, Wang G-J, Schmued LC. Temporal progression of kainic acid induced neuronal and myelin degeneration in the rat forebrain. Brain Res. 2000;864:69–80. doi: 10.1016/s0006-8993(00)02137-5. [DOI] [PubMed] [Google Scholar]
- Hulse EJ, Davies JOJ, Simpson AJ, Sciuto AM, Eddeston M. Respiratory complications of organophosphorus nerve agent and insecticide poisoning. Am J Resp Critical Care Med. 2014;190:1342. doi: 10.1164/rccm.201406-1150CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokanovic M, Stojiljkovic MP. Current understanding of the application of pyridinium oximes as cholinesterase reactivators in treatment of organophosphate poisoning. Eur J Pharmacol. 2006;553:10–17. doi: 10.1016/j.ejphar.2006.09.054. [DOI] [PubMed] [Google Scholar]
- Karalliedde L, Senanayake K. Organophosphorus insceticide poisoning. Br J Anaesth. 1989;63:736–750. doi: 10.1093/bja/63.6.736. [DOI] [PubMed] [Google Scholar]
- Leadbeater L, Inns RH, Rylands JM. Treatment of poisoning by soman. Fundam Appl Toxicol. 1985;5:S225–S231. doi: 10.1016/0272-0590(85)90132-0. [DOI] [PubMed] [Google Scholar]
- Lehmkuhle MJ, Thomson KE, Scheerlinck P, Pouliot W, Greger B, Dudek FE. A simple quantitative method for analyzing electrographic status epilepticus in rats. J Neurophysiol. 2009;101:1660–1670. doi: 10.1152/jn.91062.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotti M. Clinical toxicology of anticholinesterase agents in humans. Handbook of Pesticide Toxicology. 2001;2:1043–1085. [Google Scholar]
- McDonough JH, McLeod CG, Nipwoda MT. Direct microinjection of soman or VX into the amygdala produces repetivive limbic convulsions and neuropathology. Brain Res. 1987;435:123–137. doi: 10.1016/0006-8993(87)91593-9. [DOI] [PubMed] [Google Scholar]
- McDonough JH, Dochterman LW, Smith CD, Shih TM. Protection against nerve agent-induced neuropathology, but not cardiac pathology, is associated with the anticonvulsant action of drug treatment. Neurotoxicology. 1995;16:123–132. [PubMed] [Google Scholar]
- McDonough JHJ, Shih T-M. Neuropharmacological mechanisms of nerve agent-induced seizure and neurorpathology. Neurosci Biobehav Rev. 1997;21:559–579. doi: 10.1016/s0149-7634(96)00050-4. [DOI] [PubMed] [Google Scholar]
- Myhrer T, Enger S, Aas P. Roles of perirhinal and posterior corticies in control and generation of seizures: a microinfusion study in rats exposed to soman. Neurotoxicology. 2010;31:147–153. doi: 10.1016/j.neuro.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Myhrer T, Andersen JM, Nguyen NH, Aas P. Soman-induced convulsions in rats terminated with pharmacological agents after 45 min: neuropathology and cognitive performance. Neurotoxicology. 2005;26:39–48. doi: 10.1016/j.neuro.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Newmark J. Nerve Agents. Neurologist. 2007;13:20–32. doi: 10.1097/01.nrl.0000252923.04894.53. [DOI] [PubMed] [Google Scholar]
- Nozaki H, Aikawa N, Shinozawa Y, Fujishima S, Takuma K, Sagoh M. Sarin poisoning in Tokyo subway. Lancet. 1995;345:980–981. [PubMed] [Google Scholar]
- Peterson SL, Purvis RS, Griffith JW. Comparison of neuroprotective effects induced by alpha-phenyl-N-tert-butyl nitrone (PBN) and N-tert-butyl-alpha-(2 sulfophenyl) nitrone (S-PBN) in lithium-pilocarpine status epilepticus. Neurotoxicology. 2005;26:969–979. doi: 10.1016/j.neuro.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Pouliot W, Bailer M, Hen N, Shekh-Ahmad T, Kaufmann D, Yagen B, Ricks K, Roach B, Nelson C, Dudek FE. A comparative electrophysiological analysis of the effect of sec-butyl-propylacetamide on pharmacoresistant status epilepticus. Neuroscience. 2013;231:145–156. doi: 10.1016/j.neuroscience.2012.11.005. [DOI] [PubMed] [Google Scholar]
- Prager EM, Aroniadou-Anderjaska V, Almeida-Suhett CP, Figueiredo TH, Apland JP, Rossetti F, Olsen CH, Braga MF. The recovery of acetylcholinesterase activity and the progression of neuropathological and pathophysiological alteratons in the rat basolateral amygdala after soman-induced status-epilepticus: relation to anxiety-like behavior. Neuropharmacology. 2014;81:64–74. doi: 10.1016/j.neuropharm.2014.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotenberg JS, Newmark J. Nerve agent attacks on children: Diagnosis and management. Pediatrics. 2003;112:648–658. doi: 10.1542/peds.112.3.648. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Flouro-Jade B: a high affinity flourescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- Shetty AK. Hippocampal injury-induced cognitive and mood dysfunction, altered neurogenesis, and epilepsy: Can early neural stem cell grafting intervention provide protection. Epilepsy and Behavior. 2014;38:117–124. doi: 10.1016/j.yebeh.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih TM, Duniho SM, McDonough JH. Control of nerve agent-induced seizures is critical for neuroprotection and survival. Toxicol Appl Pharmacol. 2003;188:69–80. doi: 10.1016/s0041-008x(03)00019-x. [DOI] [PubMed] [Google Scholar]
- Shrot S, Ramaty E, Biala Y, Bar-Klein G, Daninos M, Lamintsky L, Makarovshy I, Statlender L, Rosman Y, Krivoy A, Lavon O, Kassirer M, Friedman A, Yaari Y. Prevention of organophosphate-induced chronic epilepsy by early benzodiazepine treatment. Toxicology. 2014;323:19–25. doi: 10.1016/j.tox.2014.05.010. [DOI] [PubMed] [Google Scholar]
- Skovira JW, McDonough JH, Shih T-M. Protection against sarin-induced seizures in rats by direct brain microinjection of scopolamine, midazolam or MK-801. J Molecular Neurosci. 2010;40:56–62. doi: 10.1007/s12031-009-9253-0. [DOI] [PubMed] [Google Scholar]
- Todorovic M, Cowan M, Balint C, Sun C, Kapur J. Characterization of status epilepticus induced by two organophosphates in rats. Epilepsy Res. 2012;101:268–276. doi: 10.1016/j.eplepsyres.2012.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treiman DM. Treatment of convulsive status epilepticus. Int Rev Neurobiology. 2007;81:273–285. doi: 10.1016/S0074-7742(06)81018-4. [DOI] [PubMed] [Google Scholar]
- Voicu VA, Bajgar J, Medvedovici A, Radulescu FS, Miron DS. Pharmacokinetics and pharmacodynamics of some oximes and associated therapeutic consequenses: a critical review. J App Toxicol. 2010;30:719–729. doi: 10.1002/jat.1561. [DOI] [PubMed] [Google Scholar]
- Walton NY, Treiman DM. Response of status epilepticus induced by lithium and pilocarpine to treatment with diazepam. Exp Neurol. 1988;101:267–175. doi: 10.1016/0014-4886(88)90010-6. [DOI] [PubMed] [Google Scholar]
- Wasterlain CG, Fujikawa DG, Penix L, Sankar R. Pathophysiological mechanisms of brain damage from status epilepticus. Epilepsia. 1993;34:S37–S53. doi: 10.1111/j.1528-1157.1993.tb05905.x. [DOI] [PubMed] [Google Scholar]
- Wetherell J, Hall T, Passingham S. Physostigmine and hyoscine improves protection against the lethal and incapaciting effects of nerve agent poisoning in the guinea-pig. Neurotoxicology. 2002;23:341–349. doi: 10.1016/s0161-813x(02)00082-7. [DOI] [PubMed] [Google Scholar]
- White HS, Alex AB, Pollock A, Hen N, Shekh-Ahmad T, Wilcox KS, McDonough JH, Stables JP, Kaufmann D, Yagan B, Bialer M. A new derivative of valproic acid amide possesses a broad-spectrum antiseizure profile and unique activity against status epilepticus and organophosphate neuronal damage. Epilepsia. 2012;53:134–146. doi: 10.1111/j.1528-1167.2011.03338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang T, Li G, Zhou J. A wide spectrum of variability periictal MRI abnormalities induced by a single or cluster of seizures. J Neurol Sci. 2014;343:167–172. doi: 10.1016/j.jns.2014.06.001. [DOI] [PubMed] [Google Scholar]