

Abstract
3-Nitrobenzanthrone (3-NBA), a byproduct of diesel exhaust, is highly present in the environment and poses a significant health risk. Exposure to 3-NBA results in formation of N-(2′-deoxyguanosin-8-yl)-3-aminobenzanthrone (dGC8-N-ABA), a bulky DNA lesion that is of particular importance due to its mutagenic and carcinogenic potential. If not repaired or bypassed during genomic replication, dGC8-N-ABA can stall replication forks, leading to senescence and cell death. Here we used pre-steady-state kinetic methods to determine which of the four human Y-family DNA polymerases (hPolη, hPolκ, hPolι, or hRev1) are able to catalyze translesion synthesis of dGC8-N-ABA in vitro. Our studies demonstrated that hPolη and hPolκ most efficiently bypassed a site-specifically placed dGC8-N-ABA lesion, making them good candidates for catalyzing translesion synthesis (TLS) of this bulky lesion in vivo. Consistently, our publication (Biochemistry 53, 5323–31) in 2014 has shown that small interfering RNA-mediated knockdown of hPolη and hPolκ in HEK293T cells significantly reduces the efficiency of TLS of dGC8-N-ABA. In contrast, hPolι and hRev1 were severely stalled by dGC8-N-ABA and their potential role in vivo was discussed. Subsequently, we determined the kinetic parameters for correct and incorrect nucleotide incorporation catalyzed by hPolη at various positions upstream, opposite, and downstream from dGC8-N-ABA. Notably, nucleotide incorporation efficiency and fidelity both decreased significantly during dGC8-N-ABA bypass and the subsequent extension step, leading to polymerase pausing and error-prone DNA synthesis by hPolη. Furthermore, hPolη displayed nucleotide concentration-dependent biphasic kinetics at the two polymerase pause sites, suggesting that multiple enzyme•DNA complexes likely exist during nucleotide incorporation.
Keywords: 3-nitrobenzanthrone, translesion synthesis, human DNA polymerase eta, DNA replication, DNA repair
Graphical Abstract
1. INTRODUCTION
DNA damage caused by prevalent chemical agents in the environment poses significant threats to cell survival and proliferation and represents a major concern for occupational and public health. One molecule in particular, 3-nitrobenzanthrone (3-NBA)a, a nitro-polycyclic aromatic hydrocarbon (nitroPAH), was discovered in diesel exhaust as bound to particulate air matter 1. 3-NBA is ubiquitously present in the environment, especially in areas exposed to high levels of diesel emissions, such as cities or areas of heavy construction 2. It has one of the most mutagenic profiles of known pollutants tested in the Ames Salmonella typhimurium (TA98) assay and is responsible for inducing mostly G→T transversions 1; 3. Upon inhalation, 3-NBA is metabolized into various mutagenic and potentially carcinogenic metabolites that can react with purines in DNA 4. These reactions lead to the formation of bulky, aromatic DNA lesions which act as roadblocks to genomic replication. One such product, N-(2′-deoxyguanosin-8-yl)-3-aminobenzanthrone (dGC8-N-ABA), is of particular interest because it is a major lesion formed in mice after intraperitoneal treatment with 3-NBA 5. The bulky lesion dGC8-N-ABA, produced by the electrophilic attack of deoxyguanosine (dG) by a 3-NBA metabolite 4 (Figure S1), has been found to severely block replication fork progression 6.
During normal DNA replication, highly efficient and faithful DNA polymerases perform the bulk of the work. It is known that replicative DNA polymerases possess tight and selective active sites that allow them to stringently choose against incorrect nucleotides but also limit their abilities to catalyze translesion synthesis (TLS) through damaged template nucleotides 7; 8. When a damaged site is encountered, replicative polymerases are stalled due to a significantly slowed nucleotide incorporation activity and a competing 3′→5′ exonuclease activity, resulting in futile cycles of DNA synthesis and excision9. In contrast, Y-family DNA polymerases, which lack proofreading activity and possess comparatively flexible and solvent accessible active sites, are more suited to accommodate DNA lesions and catalyze TLS 10. Notably, the human genome encodes four Y-family members: DNA polymerase eta (hPolη), kappa (hPolκ), iota (hPolι), and hRev1. These human polymerases vary in their lesion bypass capabilities and are differentially recruited to damaged DNA to catalyze TLS depending on the lesion that is present 10. In this study, we sought to determine which of the human Y-family DNA polymerase(s) are able to efficiently bypass dGC8-N-ABA in vitro. Previously we utilized pre-steady-state kinetic methods to study the bypass of dGC8-N-ABA catalyzed by a model Y-family polymerase, Sulfolobus solfataricus DNA Polymerase IV (Dpo4) 11. We observed that Dpo4 is significantly less efficient and faithful during dGC8-N-ABA bypass and the subsequent extension compared to undamaged DNA, implicating the mutagenic effect of this lesion in vivo. Here we applied similar methods to determine the most active human Y-family polymerase for dGC8-N-ABA bypass and establish a kinetic mechanism for TLS of dGC8-N-ABA. Initially, running start assays were performed to study the effect that a site-specifically placed, bulky dGC8-N-ABA lesion had on each human Y-family polymerase. Our results indicate that hPolη is a likely candidate to catalyze TLS of dGC8-N-ABA in vivo. Subsequently, we performed single-turnover kinetic assays to determine the DNA binding affinities (Kd, DNA), maximum nucleotide incorporation rate constants (kp), apparent nucleotide binding affinities (Kd, dNTP), and the substrate specificities (kp/Kd, dNTP) for nucleotide incorporations by hPolη at sites upstream, opposite, and downstream from the lesion. Finally, biphasic kinetic assays were performed to establish a minimal kinetic mechanism for nucleotide incorporation opposite dGC8-N-ABA and the subsequent extension of the lesion bypass product.
2. MATERIALS AND METHODS
2.1 Materials
All reagents were purchased from the following companies: OptiKinase from United States Biochemical, [γ-32P] ATP from Perkin-Elmer, Biospin-6 columns from Bio-Rad Laboratories, and dNTPs from Bioline. Full-length hPolη and truncated fragments of hPolκ (9–518), hPolι (1–420), and hRev1 (341–829) were overexpressed and purified as described previously 12.
2.2 DNA Substrates
A DNA template (26-mer) containing a dGC8-N-ABA lesion at the 21st position was synthesized as described previously 11. DNA containing the dGC8-N-ABA lesion was kept in dark conditions to preserve the light-sensitive substrate. All other synthetic oligonucleotides were purchased from Integrated DNA Technologies (IDT) and gel purified by 17% denaturing polyacrylamide gel electrophoresis (PAGE). Primers were 5′-radiolabeled by incubating with [γ-32P]-ATP and OptiKinase for 3 hr, followed by heat inactivation (95 °C for 2 min), and isolation of radiolabeled DNA using a Biospin-6 column to remove excess, unreacted [γ-32P]-ATP. A primer was annealed to a template at a 1:1.35 ratio. Annealing solutions containing undamaged DNA and dGC8-N-ABA-containing DNA were heated to 95 and 72 °C, respectively, for 5 minutes, followed by slow cooling to room temperature. All DNA substrates used in this study are listed in Table 1.
Table 1.
Sequences of DNA oligonucleotides
| Primers (Position)a | Sequence |
|---|---|
| 17-mer (−4) | 5′-AACGACGGCCAGTGAAT-3′ |
| 18-mer (−3) | 5′-AACGACGGCCAGTGAATT-3′ |
| 19-mer (−2) | 5′-AACGACGGCCAGTGAATTC-3′ |
| 20-mer (−1) | 5′-AACGACGGCCAGTGAATTCG-3′ |
| 21-mer (0) | 5′-AACGACGGCCAGTGAATTCGC-3′ |
| 22-mer (+1) | 5′-AACGACGGCCAGTGAATTCGCG-3′ |
| Templates | |
| 26-mer | 3′-TTGCTGCCGGTCACTTAAGCGCGCCC-5′ |
| 26-mer-dGC8-N-ABA | 3′-TTGCTGCCGGTCACTTAAGCGCGCCC-5′ |
G indicates the dGC8-N-ABA lesion at the 21st position in the template.
Position of primer 3′ terminus relative to the templating dGC8-N-ABA.
2.3 Reaction Buffers and Assay Analysis
All chemical quench assays were performed with a KinTek Rapid Chemical Quench-Flow apparatus at 37 °C in buffer R (50 mM HEPES, pH 7.5 at 37 °C, 50 mM NaCl, 5 mM MgCl2, 0.1 mM EDTA, 10% glycerol, 5 mM DTT, and 0.1 mg/mL Bovine Serum Albumin (BSA)). Electrophoretic mobility shift assays (EMSAs) were performed at 23 °C in buffer S (50 mM Tris-HCl, pH 7.5 at 23 °C, 50 mM NaCl, 5 mM MgCl2, 0.1 mM EDTA, 10% glycerol, 5 mM DTT, and 0.1 mg/mL BSA). All concentrations listed were final upon mixing and all gels were scanned using a TyphoonTrio (GE Healthcare). Quantification was carried out using ImageQuant (GE Healthcare) and Kaleidagraph (Synergy) software.
2.4 Running Start Assays
A pre-incubated solution of hPolη (100 nM), hPolκ (100 nM), hPolι (1 μM), or hRev1 (1 μM) and 5′-[32P]-17/26-mer (100 nM) was rapidly mixed with all four dNTPs (200 μM each). After various incubation periods, the samples were quenched with the addition of 0.37 M EDTA and separated by 17% PAGE (8 M Urea, 1X TBE). Analysis and quantification of gels were performed as described in Reaction Buffers and Assay Analysis. The relative lesion bypass efficiencies (bypass %) were calculated for each polymerase as a function of reaction time. For each time point t, the bypass % was calculated as a ratio of bypass to encounter events (Eq. 1) as described previously 12; 13; 14.
| Eq. 1 |
Quantification of running start assays (Figure 1A–H) was achieved by treating the total encounter (E) events as equivalent to the concentration of 20-mer, and bypass (B) events as equivalent to the sum of the concentrations of all products with sizes greater than or equal to the 21-mer. The bypass % was calculated using Eq. 1 then plotted against reaction times, t. To compare the relative amount of time it took for each polymerase to bypass dGC8-N-ABA, the t50bypass was determined as the time to bypass of 50% of the total lesions encountered. In a similar fashion, the t50 was determined as the time to bypass 50% of the undamaged dG base.
Figure 1. Running start assays at 37 °C.

A pre-incubated solution of 100 nM 5′-[32P]-labeled 17/26-mer DNA (A, C, E, and G) or 17/26-mer-dGC8-N-ABA (B, D, F, and H) and 100 nM hPolη (A and B), 100 nM hPolκ (C and D), 1 μM hPolι (E and F), or 1 μM hRev1 (G and H) was rapidly mixed with all four dNTPs (200 μM each) for various incubation periods before being quenched with the addition of 0.37 M EDTA. Sizes of important products are indicated. The dGC8-N-ABA lesion is located at the 21st position from the 3′ terminus of the DNA template.
2.5 Electrophoretic Mobility Shift Assays
hPolη (2–650 nM) was titrated into solutions containing 5′-[32P]-labeled DNA (10 nM) and allowed to equilibrate in buffer S for 15 min at room temperature (23 °C). The samples were separated using 4.5% native PAGE which were analyzed and quantified as described in Reaction Buffers and Assay Analysis. The concentration of hPolη•DNA complex was plotted against the total concentration of hPolη and the data were fit to a quadratic equation (Eq. 2):
| Eq. 2 |
Where E0 is the initial enzyme concentration, D0 is the initial DNA concentration, and Kd, DNA is the apparent equilibrium dissociation constant for the hPolη•DNA complex.
2.6 Standing Start Assay for hRev1
A pre-incubated solution of hRev1 (200 nM) and 5′-[32P]-labeled undamaged (20/26-mer) or damaged (20/26-mer-dGC8-N-ABA) DNA (200 nM) in buffer R was mixed with dATP, dCTP, dGTP, dTTP, or all four nucleotides (200 μM each). After 10 min, the reaction was quenched with the addition of 0.37 M EDTA and the samples were separated using 17% denaturing PAGE.
2.7 Single-Turnover Assays
A pre-incubated solution of 5′-[32P]-labeled DNA (20 nM) and hPolη (130 nM) in buffer R was rapidly mixed with varying concentrations of dNTP (10 – 1200 μM). After various incubation times, the reaction was quenched with the addition of 0.37 M EDTA. The samples were then analyzed using 17% denaturing PAGE. Product concentration was plotted against time and the data were fit to a single-exponential equation (Eq. 3).
| Eq. 3 |
Where kobs is the observed rate constant of nucleotide incorporation and A is the reaction amplitude. The kobs values obtained using Eq. 3 were plotted against the respective dNTP concentration and the data were fit to a hyperbolic equation (Eq. 4)
| Eq. 4 |
Where kp is the maximum nucleotide incorporation rate constant and Kd, dNTP is the apparent equilibrium dissociation constant for the binding of dNTP to hPol•DNA.
2.8 Biphasic Assays
Biphasic assays were performed by rapidly mixing a pre-incubated solution of hPolη (120 nM) and 5′-[32P]-labeled DNA (30 nM) with various concentrations of dNTP (25–1,000 μM) and excess, unlabeled DNA trap (5 μM). After various incubation periods, reactions were quenched with the addition of 0.37 M EDTA and samples were separated using 17% denaturing PAGE. Product concentration was plotted against time and the data were fit to a double-exponential equation (Eq. 5)
| Eq. 5 |
Where Af and As are the reaction amplitudes of the fast and slow phases, respectively, and kf and ks are the rate constants of the fast and slow phases, respectively.
3. RESULTS
3.1 Measuring Relative Polymerase Efficiency during dGC8-N-ABA Bypass
To test if each individual human DNA polymerase could bypass a single dGC8-N-ABA lesion, running start assays were performed by rapidly mixing pre-incubated hPolη, hPolκ, hPolι, or hRev1 and undamaged 17/26-mer or damaged 17/26-mer-dGC8-N-ABA containing a site-specifically placed dGC8-N-ABA (Table 1) with all four dNTPs. The lesion stalled DNA synthesis by each of the Y-family polymerases, but the degree of stalling as well as the positions at which stalling was observed varied between each polymerase. For hPolη, moderate pausing was observed at the 19-mer (−2), 20-mer (−1), 21-mer (0), and 22-mer (+1) positions. Significant accumulation of the 20-mer product indicated that incorporation of nucleotide at the bypass step (Position −1) was the most problematic for hPolη (Figure 1B). Despite this stalling, the full-length product appeared after 6 s, which was only 2-fold longer than the time required for hPolη to synthesize the full-length product with the undamaged 17/26-mer. hPolκ stalled significantly during the bypass and extension steps, indicated by the 20-mer and 21-mer accumulation at these positions (Figure 1D). hPolκ produced the full length products within 1 s with the undamaged 17/26-mer and took only 3-fold longer (3 s) with the 17/26-mer-dGC8-N-ABA substrate, indicating that hPolκ was similarly efficient as hPolη when bypassing dGC8-N-ABA. The observed accumulation of the 25-mer product is likely caused by polymerase slippage and template repositioning within the 5′-dC-rich region of the template (Table 1), resulting in the “−1” frameshift 15; 16. Additionally, the full length product (25-mer) produced by this mechanism was likely further extended via a blunt-end addition 17; 18. Interestingly, the bulky lesion caused significant stalling of hPolι and hRev1 with no appearance of the full-length product even after long reaction periods (3 and 10.5 hr, respectively). With 17/26-mer-dGC8-N-ABA, hPolι synthesized the bypass and extension products after 300 and 3,900 s, respectively, which are 30- and 65-fold, respectively, longer than with the undamaged 17/26-mer (Figure 1E–F). In comparison, hRev1 synthesized the lesion bypass product within 1 hr but was unable to extend it even after 10.5 hr. The failure of hRev1, a dCTP polymerase, to efficiently catalyze TLS of dGC8-N-ABA was likely caused by multiple template-independent misincorporations of dCTP by hRev1 19, resulting in DNA distortion. This is supported by the fact that hRev1was not able to synthesize the full-length product even with the undamaged DNA substrate (Figure 1H). The results of the running start assay with hRev1 prompted us to perform standing start primer extension assays to qualitatively evaluate the efficiency and fidelity of hRev1 to incorporate a nucleotide directly opposite from dGC8-N-ABA. A pre-incubated solution of hRev1 and 20/26-mer-dGC8-N-ABA (or 20/26-mer) was mixed and reacted with a solution containing either dATP, dCTP, dGTP, dTTP, or all four dNTPs. Interestingly, hRev1 showed greater selectivity with damaged than with undamaged DNA by predominantly incorporating dCTP (Figure S1). With undamaged DNA, hRev1 incorporated all four nucleotides with a preference of dCTP > dATP = dGTP = dTTP.
To provide a comparison for the amount of time it took for each human Y-family polymerase to bypass dGC8-N-ABA, gels from the running start assays (Figure 1 A–H) were quantified and Eq. 1 was used to calculate values for t50 and t50bypass which represent the times it took to elongate 50% of the 20-mer to the 21-mer or longer products with undamaged and damaged DNA, respectively. The t50 and t50bypass times measured for each of the Y-family polymerases is summarized in Table 2. Notably, hPolι and hRev1 did not bypass 50% of the dGC8-N-ABA lesion in the allowed reaction time for the running start assays. In contrast, the relatively short t50bypass times for hPolη (13 s) and hPolκ (7 s) suggest a potential role involving these two polymerases in the bypass and/or extension of dGC8-N-ABA lesions in vivo. Consistently, a recent publication reports ~25 and ~30% efficiency reductions in TLS of dGC8-N-ABA in human embryonic kidney (HEK293T) cells when hPolη and hPolκ, respectively, were subject to gene knockdown 20. Based on the slightly smaller perturbation of DNA synthesis by hPolη in the presence of dGC8-N-ABA, we decided to use pre-steady-state kinetic methods to initially characterize the efficiency and fidelity of TLS of dGC8-N-ABA catalyzed by hPolη. However, it is apparent based on both the running start and HEK293T cell-based assays that hPolκ is also potentially involved in TLS of dGC8-N-ABA in vivo. Accordingly, we are currently performing similar kinetic studies to determine the effect of dGC8-N-ABA on nucleotide incorporation kinetics with hPolκ.
Table 2.
dGC8-N-ABA bypass halftimes of human Y-family polymerases at 37 °C
| Polymerase | t50(s)a | t50bypass(s)b | t50bypass/t50 |
|---|---|---|---|
| hPolη | 0.7 | 13 | 19 |
| hPolκ | 0.3 | 7 | 23 |
| hPolι | 200 | >10,800 | >55 |
| hRev1 | 1,800 | >37,000 | >20 |
Calculated as the time required for traversing 50% of undamaged dG.
Calculated as the time required for bypassing of 50% of dGC8-N-ABA.
3.2 DNA Binding Affinity Estimated through Electrophoretic Mobility Shift Assays
To determine the apparent equilibrium dissociation constant for the binding of hPolη to DNA containing dGC8-N-ABA (Kd, DNA), EMSAs were performed. A representative gel image and a plot for an EMSA experiment are shown in Figure S2 for hPolη binding to 21/26-mer-dGC8-N-ABA. For all DNA substrates tested, the Kd, DNA values were within a 2-fold range for the corresponding undamaged and damaged DNA substrates (Figure S3D and Table 3). Interestingly, the binding affinities of hPolη for 19/26-mer-dGC8-N-ABA and 20/26-mer-dGC8-N-ABA (Kd, DNA =12 ± 2 nM for both) were 2-fold tighter than the corresponding undamaged DNA substrates (Kd, DNA =25 ± 5 nM; 23 ± 2 nM). Based on our ternary crystal structure of hPolη in complex with dGC8-N-ABA-DNA and dCTP21, the slight increase in binding affinity likely arises from positioning of the ABA moiety of dGC8-N-ABA within a hydrophobic cleft in the hPolη active site thereby stabilizing the conformation of the damaged base. Similar effects are observed in the crystal structures of Dpo4 with dGBPDE and dGAP 22; 23. Overall, hPolη binds to dGC8-N-ABA-containing DNA tightly enough such that it does not significantly contribute to the polymerase pausing pattern observed in the running start assay (Figure 1B). Similar results have been obtained for dGAP bypass by hPolη 13 as well as dGAP, dGC8-N-ABA, and abasic site bypass by Dpo4 11; 24; 25.
Table 3.
DNA binding affinity of hPolη at 23 °C
| DNA Substrate | Kd, undamaged DNA (nM) | Kd, damaged DNA (nM)a | Affinity Ratiob |
|---|---|---|---|
| 18/26-mer | 28 ± 3 | 32 ± 5 | 1.1 |
| 19/26-mer | 25 ± 5 | 12 ± 2 | 0.5 |
| 20/26-mer | 23 ± 2 | 12 ± 2 | 0.5 |
| 21/26-mer | 26 ± 1 | 26 ± 7 | 1.0 |
| 22/26-mer | 17 ± 6 | 24 ± 4 | 1.4 |
Damaged DNA refers to template 26-mer containing a dGC8-N-ABA lesion at the 21st position.
Calculated as Kd, damaged DNA/Kd, undamaged DNA.
3.3 Determination of Kinetic Parameters for Nucleotide Incorporations upstream, opposite, and downstream from dGC8-N-ABA
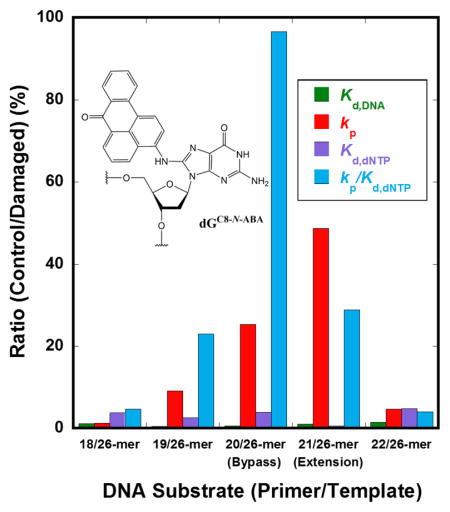
Since DNA binding did not significantly affect TLS of dGC8-N-ABA by hPolη, it is possible that the stalling of hPolη during TLS (Figure 1B) was caused by decreased nucleotide binding affinity and incorporation efficiency. To examine this hypothesis, we utilized single-turnover assays to measure the apparent equilibrium dissociation constant for nucleotide binding (Kd, dNTP) and the maximum rate constant of nucleotide incorporation (kp) at positions upstream, downstream, and opposite from dGC8-N-ABA. By varying the primer length, we were able to measure the kinetic parameters at specific positions along the template strand relative to dGC8-N-ABA, and thus determine the effect of dGC8-N-ABA on the efficiency and fidelity of nucleotide incorporation during each step of TLS catalyzed by hPolη. Briefly, a pre-incubated solution of 5′-[32P]-labeled DNA (20 nM, Table 1) and hPolη (130 nM) was rapidly mixed with a single dNTP at varying concentrations. Reactions were analyzed as described (Materials and Methods), and the data were fit to Eq. 3 to yield an observed rate constant of incorporation (kobs). The kobs values were then plotted against the respective dNTP concentrations and fit to a hyperbolic equation (Eq. 4) to yield the kp and Kd, dNTP (Figure 2). hPolη showed a decrease in nucleotide incorporation efficiency (kp/Kd, dNTP) at positions upstream, opposite, and downstream from the lesion (Position −3, −2, −1, 0, +1). With 18/26-mer-dGC8-N-ABA at Position −3, hPolη correctly incorporated dCTP with a kp of 66 ± 1 s−1, although the dCTP binding affinity was weakened by 4-fold (Kd, dCTP =200 ± 11 μM) relative to undamaged 18/26-mer. As a result, the polymerase efficiency (kp /Kd, dCTP = 3.3 × 10−1 s−1 μM−1) was only slightly reduced compared to the value with 18/26-mer (kp/Kd, dCTP = 1.5 s−1 μM−1). Similar experiments were carried out with both undamaged and damaged DNA substrates containing primers of 19-mer (Position −2), 20-mer (Position −1), 21-mer (Position 0), and 22-mer (Position +1) and the kinetic parameters are listed in Tables 3 and S1. The most significant stalling occurred during incorporation opposite dGC8-N-ABA (Position −1), caused by a 25-fold decrease in kp (1.9 ± 0.1 s−1) and a 4-fold increase in Kd, dCTP (325 ± 44 μM) which resulted in an overall reduction in efficiency by approximately 100-fold (kp/Kd, dCTP = 5.8 × 10−3 s−1 μM−1) with 20/26-mer-dGC8-N-ABA compared to undamaged 20/26-mer (kp/Kd, dCTP = 5.6 × 10−1 s−1 μM−1). At the extension step (Position 0), hPolη displayed a kp that was 49-fold slower (0.8 ± 0.03 s−1) but surprisingly bound the incoming dGTP more tightly (Kd, dGTP = 28 ± 5 μM). With 21/26-mer-dGC8-N-ABA, hPolη showed an approximate 5-fold increase in efficiency (kp/Kd, dGTP = 2.8 × 10−2 s−1 μM−1) compared to the lesion bypass step (Position −1). With 22/26-mer-dGC8-N-ABA (Position +1), the efficiency of hPolη was similar (kp/Kd, dCTP = 4.2 × 10−2 s−1 μM−1) to that measured at position −2 (Table 4) which is indicated by similarly moderate accumulation at these positions (Figure 1B).
Figure 2. Determination of the kinetic parameters for dCTP incorporation into 20/26-mer-dGC8-N-ABA at 37 °C.

(A) A pre-incubated solution of hPolη (120 nM) and 5′-[32P]-labeled 20/26-mer-dGC8-N-ABA (20 nM) was rapidly mixed with increasing concentrations of dCTP (25 μM, ●; 50 μM, ○; 100 μM, ■;200 μM, □; 400 μM, ◆; 600 μM, ◇; and 800 μM, ▲) before being quenched by the addition of 0.37 M EDTA at the indicated time points. Product formation was plotted against time and the data were fit to Eq. 3 to yield the observed rate constant of product formation (kobs). (B) Each kobs value was plotted against its respective dCTP concentration and the data were fit to Eq. 4 to yield the maximum dCTP incorporation rate constant (kp) of 1.9 ± 0.1 s−1 and dCTP equilibrium dissociation constant (Kd, dCTP) of 325 ± 44 μM.
Table 4.
Kinetic parameters for dNTP incorporation into dGC8-N-ABA-containing DNA at 37 °C
| dNTP | Kd, dNTP (μM) | kp(s−1) | kp/Kd, dNTP (μM−1s−1) | Efficiency Ratioa,b | Fidelityc | Fidelity ratiod | Probability e (%) |
|---|---|---|---|---|---|---|---|
| 18/26-mer-dGC8-N-ABA - Template dG (Position −3) | |||||||
| dCTP | 200 ± 11 | 66 ± 1 | 3.3 × 10−1 | 5 | - | - | - |
| dATP | 369 ± 48 | 2.1 ± 0.1 | 5.7 × 10−3 | 0.6 | 1.7 × 10−2 | 7.4 | - |
| 19/26-mer-dGC8-N-ABA - Template dC (Position −2) | |||||||
| dGTP | 150 ± 34 | 6.6 ± 0.4 | 4.4 × 10−2 | 23 | - | - | - |
| dCTP | 90 ± 19 | 0.093 ± 0.0001 | 1.0 × 10−3 | 5 | 2.2 × 10−2 | 4.8 | - |
| 20/26-mer-dG C8-N-ABA - Template dGC8-N-ABA (Position −1) | |||||||
| dCTP | 325 ± 44 | 1.9 ± 0.1 | 5.8 × 10−3 | 97 | - | - | 77 |
| dATP | 161 ± 38 | 0.18 ± 0.01 | 1.1 × 10−3 | 3 | 1.6 × 10−1 | 33 | 15 |
| dGTP | 39 ± 5 | 0.014 ± 0.0003 | 3.6 × 10−4 | 3 | 5.8 × 10−2 | 29 | 5 |
| dTTP | 475 ± 85 | 0.14 ± 0.01 | 2.9 × 10−4 | 23 | 4.8 × 10−2 | 4 | 4 |
| 21/26-mer-dGC8-N-ABA - Template dC (Position 0) | |||||||
| dGTP | 28 ± 5 | 0.8 ± 0.03 | 2.9 × 10−2 | 29 | - | - | 74 |
| dATP | 473 ± 25 | 3.2 ± 0.10 | 6.8 × 10−3 | 2 | 1.9 × 10−1 | 11 | 17 |
| dCTP | 201 ± 20 | 0.4 ± 0.02 | 2.0 × 10−3 | 9 | 6.4 × 10−2 | 3 | 5 |
| dTTP | 392 ± 108 | 0.6 ± 0.01 | 1.5 × 10−3 | 3 | 4.9 × 10−2 | 9 | 4 |
| 22/26-mer-dGC8-N-ABA - Template dG (Position +1) | |||||||
| dCTP | 215 ± 67 | 9.1 ± 1.0 | 4.2 × 10−2 | 4 | - | - | - |
| dGTP | 41 ± 8 | 0.028 ± 0.001 | 6.8 × 10−4 | 0.6 | 1.6 × 10−2 | 6.5 | - |
Calculated as [(kp/Kd, dNTP)undamaged DNA/(kp/Kd, dNTP)damaged DNA].
Kinetic parameters for undamaged DNA from Table S1 of Sherrer et al.13
Calculated as (kp/Kd, dNTP)incorrect/[(kp/Kd, dNTP)incorrect) + (kp/Kd, dNTP)correct].
Calculated as Fidelitydamaged DNA/Fidelityundamaged DNA.
Calculated as [(kp/Kd, dNTP)damaged DNA/Σ(kp/Kd, dNTP)damaged DNA] × 100.
Previously, it has been shown that dGC8-N-ABA bypass carried out by hPolη is error-prone in HEK293T cells 20. Thus, we investigated the effect of dGC8-N-ABA on the fidelity of hPolη during dGC8-N-ABA bypass and the subsequent extension step. The fidelity of hPolη, defined as (kp /Kd, dNTP)incorrect/[(kp/Kd, dNTP)correct + (kp/Kd, dNTP)incorrect], was calculated for the bypass (20/26-mer-dGC8-N-ABA) and extension (21/26-mer-dGC8-N-ABA) steps by using the single-turnover kinetic data of both correct and three incorrect nucleotide incorporations (Table 4). At these two steps, the fidelity of hPolη (Table 4) was determined to be in the ranges of 4.8 × 10−2 to 1.6 × 10−1 and 4.9 × 10−2 to 1.9 × 10−1, respectively. In comparison, the fidelity of hPolη with undamaged DNA was previously determined to be in the range of 10−2 to 10−3 (Table S1) by pre-steady-state kinetics 13 and this range is consistent with the values determined using phage plaque assays 26; 27. Thus, the presence of dGC8-N-ABA significantly reduced the fidelity of hPolη at both the lesion bypass and subsequent extension steps, especially during misincorporations of dGTP (29-fold) and dATP (33-fold) opposite dGC8-N-ABA (Table 4). For these two TLS steps, nucleotide incorporation probabilities, given by [(kp/Kd, dNTP)damaged DNA/Σ(kp/Kd, dNTP)damaged DNA] × 100, were also calculated. The probability of correct incorporation was reduced from 98% and 96% with undamaged DNA (Table S1) to 77% and 74% with damaged DNA (Table 4) for dGC8-N-ABA bypass and the subsequent extension step, respectively. Notably, with damaged DNA, the probability of dATP misincorporation was 15 and 17% at the bypass and extension steps, respectively, and these probabilities are the highest among all misincorporations (Table 4). Consistently, the G→T transversion was found to occur at a high rate during TLS of dGC8-N-ABA in vivo 5; 20; 28.
3.4 Determination of Biphasic Kinetic Parameters at Polymerase Pause Sites
Nucleotide incorporation during TLS has often been shown to follow biphasic kinetics. Thus, we performed biphasic kinetic assays as described previously 13; 24; 25; 29 to test if hPolη followed the same trend during dGC8-N-ABA bypass and the subsequent extension step. A pre-incubated solution of 5′-[32P]-labeled DNA (30 nM) and hPolη (120 nM) was rapidly mixed with increasing concentrations of a correct nucleotide (25 to 1,000 μM) and an unlabeled trap DNA substrate 20/26-mer (5 μM). The trap DNA was added to a 150-fold molar excess over the labeled DNA so that any enzyme that had dissociated from the radiolabeled DNA substrate during the incubation time would be sequestered by the trap DNA. Thus, only labeled DNA product formed during a single enzyme binding event would be observed. The trap DNA concentration (5 μM) was determined to be sufficiently high enough to sequester any hPolη that had dissociated from 5′-[32P]-labeled DNA 24. During the incorporation of dCTP (1,000 μM) onto 20/26-mer-dGC8-N-ABA, hPolη indeed followed biphasic kinetics (Figure 3A). The plot of reaction time versus product concentration was fit to Eq. 5 (Materials and Methods) to yield reaction amplitudes of Af = 5.8 ± 0.8 nM (19%) and As = 8.6 ± 0.8 nM (28%) with rate constants of kf = 7.2 ± 1.4 s−1 and ks = 0.42 ± 0.06 s−1 for the fast and slow phases, respectively (Table 5). To test if the kinetic parameters were affected by dCTP concentration, similar biphasic kinetic experiments were performed individually with 300 and 600 μM dCTP. Indeed, as the dCTP concentration increased, the reaction rate constants (kf and ks) also increased while the reaction amplitudes remained relatively unchanged (Table 5). In addition, the biphasic kinetic experiments were also performed with 21/26-mer-dGC8-N-ABA. As with 20/26-mer-dGC8-N-ABA, a similar biphasic kinetic trend and a dNTP concentration-dependent increase in the rate constants were observed with 21/26-mer-dGC8-N-ABA during the extension step (Figure 3B and Table 5). Interestingly, the dNTP concentration-dependent increase in the rate constants (kf and ks) had been observed previously with Dpo4 when it bypassed the dGC8-N-ABA 11 and dG1,8 lesions 30. However, our current study is the first one to report this concentration-dependence for lesion bypass catalyzed by a human Y-family DNA polymerase. Notably, kinetic parameters for the DNA trap experiments performed with undamaged 20/26-mer and 21/26-mer DNA are reported in Table S2, showing that even with undamaged DNA substrates, hPolη also displays biphasic kinetics of nucleotide incorporation.
Figure 3. Biphasic kinetics of dNTP incorporation in the presence of a DNA trap at 37 °C.

A pre-incubated solution of 120 nM hPolη and 30 nM 20/26-mer-dGC8-N-ABA (A) or 21/26-mer-dGC8-N-ABA (B) was rapidly mixed with 5 μM unlabeled 20/26-mer trap DNA and an indicated concentration of dCTP (A) or dGTP (B). After various incubation periods, the reaction was quenched with the addition of 0.37 M EDTA. Product formation was plotted against time and the data were fit to Eq. 5 to yield Af and As, which are the reaction amplitudes of the fast and slow phases, respectively, as well as kf and ks, which are the rate constants of the fast and slow phases, respectively. The insets are magnifications of shorter time points to show the fast phase of product formation.
Table 5.
Kinetic parameters from biphasic kinetic assays with hPolη at 37 °C
| [dNTP] | Af (nM) | kf (s−1) | As (nM) | ks (s−1) |
|---|---|---|---|---|
| 20/26-mer-dGC8-N-ABA | ||||
| 300 μM dCTP | 5.6 ± 0.8 (19%) | 4.6 ± 1.3 | 8.3 ± 0.8 (28%) | 0.32 ± 0.06 |
| 600 μM dCTP | 5.5 ± 0.7 (18%) | 6.3 ± 1.5 | 8.5 ± 0.7 (28%) | 0.36 ± 0.05 |
| 1000 μM dCTP | 5.8 ± 0.8 (19%) | 7.2 ± 1.4 | 8.6 ± 0.8 (29%) | 0.42 ± 0.06 |
| 21/26-mer-dGC8-N-ABA | ||||
| 25 μM dGTP | 4.4 ± 0.7 (15%) | 3.8 ± 1.5 | 9.2 ± 0.8 (31%) | 0.082 ± 0.002 |
| 50 μM dGTP | 4.4 ± 0.8 (15%) | 5.2 ± 1.7 | 9.1 ± 0.6 (30%) | 0.13 ± 0.02 |
| 90 μM dGTP | 4.3 ± 0.6 (14%) | 11 ± 4 | 9.3 ± 0.7 (31%) | 0.16 ± 0.03 |
Af and As are the reaction amplitudes of the fast and slow phases, respectively, while kf and ks are the rate constants of the fast and slow phases, respectively.
4. DISCUSSION
In this paper, we performed running start assays to determine which of the four human Y-family DNA polymerases were able to most efficiently catalyze TLS of dGC8-N-ABA in vitro. Both hPolη and hPolκ were able to bypass dGC8-N-ABA, while hPolι and hRev1 were completely stalled by the lesion (Figure 1). Consistently, small interfering RNA (siRNA)-mediated knockdown of hPolι had a minimal effect on overall TLS of dGC8-N-ABA in HEK293T cells, suggesting that hPolι plays a very minor role during dGC8-N-ABA bypass in vivo 20. Surprisingly, siRNA-mediated knockdown of hRev1 proved to be detrimental to dGC8-N-ABA bypass in HEK293T cells. Based on its inability to catalyze the bypass of dGC8-N-ABA in vitro, hRev1 likely serves as a scaffold protein that synergistically interacts with proliferating cell nuclear antigen (PCNA) and other TLS polymerases to aid in TLS of dGC8-N-ABA and likely other DNA lesions in vivo 13; 31; 32; 33.
In contrast, hPolη and hPolκ were able to bypass dGC8-N-ABA with similarly small t50bypass values (Table 2), indicating potential roles for these polymerases in dGC8-N-ABA bypass in vivo. Consistently, knockdown of these two polymerases in HEK293T cells significantly reduced the efficiency of TLS of dGC8-N-ABA 20. Knockdown of hPolκ resulted in no change in the percentage of mutagenic dGC8-N-ABA bypass events, suggesting that hPolκ carries out relatively error-free TLS of dGC8-N-ABA 20. On the other hand, knockdown of hPolη resulted in a 39% decrease in mutation frequency, indicating that the bypass of dGC8-N-ABA by hPolη in vivo is error-prone 20. Taken together, the bypass of dGC8-N-ABA by hPolη is likely a mechanism through which 3-NBA promotes lesion-induced mutagenesis and tumorigenesis. To determine a kinetic basis for mutagenic bypass of dGC8-N-ABA by hPolη, we utilized pre-steady-state kinetic methods to investigate the effect of a site-specifically placed dGC8-N-ABA on hPolη-catalyzed nucleotide incorporation at and near the lesion.
4.1 Kinetic basis for hPolη pausing caused by dGC8-N-ABA
Overall, the binding affinity of hPolη to DNA (Table 3) is insignificantly affected by the presence of dGC8-N-ABA and therefore does not significantly contribute to the polymerase stalling observed in Figure 1B. Instead, the decrease of nucleotide incorporation rate constants and the weakened nucleotide binding affinities are likely responsible for the observed accumulation of intermediate products. A similar kinetic trend is seen for the conversion of 18-mer→19-mer→20-mer as well as 19-mer→20-mer→21-mer. Based on the kinetic data in Table 4, the first conversion step has a higher rate constant (kp) and also a higher efficiency ratio (kp/Kd, dNTP) than the second, less efficient conversion step, leading to the observed accumulation of the 19-mer and 20-mer products (Figure 1B). The 19-mer and 20-mer accumulation was absent at later time points (30–1200 s) due to their eventual conversion to the 20-mer and 21-mer products, respectively. For the reaction series of the conversion of 20-mer→21-mer→22-mer and 21-mer→22-mer→23-mer, the first conversion step was less efficient than the second conversion step. This kinetic pattern led to accumulation of 20-mer and 21-mer as seen in Figure 1B. Taken together, it is clear that the conversion of 20-mer→21-mer is less efficient than both the preceding and subsequent reactions leading to the strong accumulation of the 20-mer product. For 23-mer, 24-mer, and 25-mer, their accumulation appeared the same in Figure 1B, indicating that the nucleotide incorporation in the consecutive conversion steps associated with these intermediate products were similarly efficient, and that hPolη likely resumed its normal polymerase efficiency after being several positions downstream from the lesion. Thus, the conversion rate and efficiency of 23-mer→24-mer, although not measured here, are likely as high as the conversion of 18-mer→19-mer (kp = 66 s−1, kp/Kd, dNTP = 0.33 μM−1s−1) but higher than the conversion of 22-mer→23-mer (kp = 9.1 s−1, kp/Kd, dNTP = 0.042 μM−1s−1), contributing to the observed 22-mer accumulation (Figure 1B).
4.2 Kinetic mechanism for dGC8-N-ABA bypass and subsequent extension of the lesion bypass product
Our biphasic kinetic assays revealed that two distinct phases exist when hPolη incorporated correct dNTPs opposite dGC8-N-ABA (Position −1) and one base pair downstream (Position 0) from the lesion. The fast phases were characterized by relatively small reaction amplitudes and fast rate constants while the slow phases had larger reaction amplitudes and slower rate constants (Table 5). Individual contributions of the amplitudes (Af and As) and rate constants (kf and ks) were used to calculate the overall nucleotide incorporation rate constant in the presence of the DNA trap. For example, in the presence of 20/26-mer-dGC8-N-ABA and 1,000 μM dGTP (Table 5), the sum of the individual contributions of the fast phase [(7.2 s−1) × 19%] and slow phase [(0.42 s−1) × 29%] gave an overall rate constant of 1.5 s−1. As expected, 1.5 s−1 is close to the kobs of 1.4 s−1, estimated using Eq. 4, 1.0 mM dCTP, and corresponding measured kp and Kd, dCTP values (Table 4), for dCTP incorporation opposite dGC8-N-ABA under single-turnover reaction conditions. Similarly, in the presence of 21/26-mer-dGC8-N-ABA and 50 μM dGTP, the sum of the individual contributions of the fast phase [(5.2 s−1) × 15%] and slow phase [(0.13 s−1) × 30%] gave an overall nucleotide incorporation rate constant of 0.8 s−1, which is similar to the kobs of 0.5 s−1, calculated using Eq. 4, 50 μM dGTP, and corresponding measured kp and Kd, dGTP values (Table 4), for dGTP incorporation during the extension step. Thus, the biphasic kinetic feature of nucleotide incorporation at Positions −1 and 0 under single-turnover conditions was successfully deconvoluted into a fast and a slow phase through the DNA trap experiments (Figure 3). These two kinetic phases prompted us to propose a kinetic mechanism for dGC8-N-ABA bypass and the subsequent extension by including the formation of productive (E•DNAnP) and non-productive (E•DNAnN) complexes (Scheme 1A). The portion of the hPolη population bound in the E•DNAnP will bind correct dNTP and rapidly elongate DNA with a fast rate constant of kf. The fast phase kf should be dNTP concentration-dependent and can reach its maximum value of k1. The remainder of hPolη bound in the E•DNAnN complex will elongate DNA only after the slow conversion (ke) of E•DNAnN → E•DNAnP. The slow phase rate constant of ks is a function of both ke and k1 but should be limited by ke. The bypass of a bulky dGAP lesion by hPolη as well as the bypass of an abasic site, a cisplatin-d(GG) adduct, and a dGAP lesion by Dpo4 have previously been proposed to follow this mechanism 13; 24; 25; 29. If this mechanism is correct, the E•DNAnN → E•DNAnP conversion rate (ke) and reaction amplitudes should not depend on dNTP concentration. However, both kf and ks increased with dNTP concentration at Positions −1 and 0 while both the fast and slow phase reaction amplitudes remained unchanged. To accommodate these observations, an alternative kinetic mechanism (Scheme 1B) was proposed for hPolη to bypass dGC8-N-ABA and to subsequently extend the lesion bypass product. The same mechanism has been proposed by us for dGC8-N-ABA bypass by Dpo4 11. This mechanism shows that ks is actually a function of an exchange rate (ke) and a slow nucleotide incorporation rate constant (k2). If ks had remained the same as dNTP concentration was increased, this would indicate that ks were solely a function of ke, and the mechanism could be simplified to reflect Scheme 1A. Because ks increased with higher dNTP concentration, especially during the extension step, ks was predominantly a function of k2 during TLS of dGC8-N-ABA by hPolη.
Scheme 1.

Proposed kinetic mechanism for dGC8-N-ABA bypass and subsequent extension.
Notably, the total reaction amplitudes for the bypass and extension steps (48% and 46%, Table 5) are significantly smaller than the corresponding values with undamaged DNA (88% and 86%, Table S2). These differences indicate that hPolη may have bound to damaged DNA in a catalytically incompetent complex (E•DNAnD) where conformational change(s) of hPolη would not be able to produce a catalytically competent complex (Scheme 1B). In instances when any molecules of the E•DNAD complex dissociated, the enzyme would bind to excess, unlabeled trap DNA resulting in no detectable product formation. Thus, the presence of such a dead-end binary complex can only be inferred by the sub-maximal product formation observed over the course of TLS of dGC8-N-ABA by hPolη. Similar results have been observed in previous kinetic studies of lesion bypass under comparable conditions 13; 24; 25; 29.
Although the hPolη•dGC8-N-ABA-DNA binary structures are not available yet, some of their binding conformations could be inferred from an NMR structure of a 12-mer DNA duplex containing an embedded dGC8-N-ABA:dC basepair 34 and crystal structures of yeast Polη (yPolη) in complex with DNA containing a bulky 2-acetylaminofluorine lesion (dGAAF) 35. In the NMR structure, the ABA moiety of dGC8-N-ABA is intercalated in the DNA duplex and takes the place of a base pair, with the damaged guanine in the syn conformation and extruded into the major groove, and with the opposing cytosine also pushed into the major groove 34. Similarly, one yPolη•dGAAF-DNA structure shows that the bulky adduct stacks on the primer/template junction base pair, occupies the polymerase active site, and precludes the incoming nucleotide from binding 35. If such a binding conformation also exists in hPolη•dGC8-N-ABA-DNA, it likely represents E•DNAnD in Scheme 1. Interestingly, in the other structure of yPolη•dGAAF-DNA 35, the AAF adduct is partially rotated out of the DNA helix to allow some excess for incoming dCTP and the structure likely represents the binding conformation of E•DNAnN (Scheme 1). Lastly, our ternary crystal structure of hPolη•dGC8-N-ABA-DNA•dCTP shows that the adducted guanine forms a standard Watson-Crick pair with the incoming dCTP, with the ABA moiety is accommodated inside a hydrophobic cleft to the side of the active site of hPolη21. Such a binding conformation of hPolη•dGC8-N-ABA-DNA is considered to be E•DNAnP (Scheme 1).
4.3 Error-prone bypass of dGC8-N-ABA and subsequent extension of the lesion bypass product
During dGC8-N-ABA bypass, hPolη showed a 10-fold reduced fidelity (4.8 × 10−2–1.6 × 10−1) relative to undamaged DNA (2.0 × 10−3–1.2 × 10−2, Table S1) with nucleotide incorporation preference following the order of dCTP > dATP > dGTP > dTTP (Table 4). Thus, the presence of dGC8-N-ABA decreased the ability of hPolη to discriminate against incorrect nucleotides, especially dATP. Our kinetic studies show that hPolη will misincorporate dATP 15% of the time when bypassing dGC8-N-ABA (Table 4). Interestingly, our previous studies have shown that in NER-proficient HEK293T cells, G→T transversions were the most common mutation resulting from dGC8-N-ABA bypass, followed by G→A transitions and G→C transversions, albeit at much lower rates 20. Furthermore, our previous studies have also demonstrated that siRNA-mediated knockdown of hPolη leads to a 39% decrease in mutation frequency in HEK293T cells, and strongly suggest that hPolη plays a major role in the error-prone bypass of dGC8-N-ABA in vivo 20. Therefore, these observations from our published cell-based assays 20 are consistent with the results of our kinetic studies with hPolη (Table 4).
Supplementary Material
Highlights.
hPolη and hPolκ efficiently bypassed a site-specifically placed dGC8-N-ABA lesion
The fidelity of hPolη decreased during dGC8-N-ABA bypass and extension
hPolη predominantly misincorporated dATP during dGC8-N-ABA bypass
Nucleotide incorporation at hPolη pause sites followed biphasic kinetics
A kinetic basis was established for mutagenic bypass of dGC8-N-ABA in vivo
Acknowledgments
Funding: This work was supported by the National Institutes of Health grant ES009127 to both A.K.B. and Z.S.
Footnotes
Abbreviations: 3-nitrobenzanthrone, 3-NBA; nitro-polycyclic aromatic hydrocarbon, nitroPAH; Salmonella typhimurium strain TA98, TA98; N-(2′-deoxyguanosin-8-yl)-3-aminobenzanthrone, dGC8-N-ABA; translesion DNA synthesis, TLS; human DNA polymerase eta, hPolη; human DNA polymerase kappa, hPolκ; human DNA polymerase iota, hPolι; human Rev1, hRev1; DNA polymerase IV from Sulfolobus solfataricus; polyacrylamide gel electrophoresis, PAGE; electrophoretic mobility shift assay, EMSA; human embryonic kidney cell line 293T, HEK293T; aminobenzanthrone, ABA; trans 10S benzo[a]pyrene-N2-deoxyguanosine, dGBPDE; N-(deoxyguanosin-8-yl)-1-aminopyrene, dGAP; 8-(deoxyguanosin-N2-yl)-1-aminopyrene, dG1,8; small interfering RNA, siRNA; proliferating cell nuclear antigen, PCNA; N-(deoxyguanosin-8-yl)-2-acetylaminofluorine; dGAAF.
Conflict of interest statement: The authors declare that there are no conflicts of interest.
Supplementary data: Supplementary scheme, tables, and figures are included.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
E. John Tokarsky, Email: tokarsky.8@osu.edu.
Varun V. Gadkari, Email: gadkari.3@osu.edu.
Walter J. Zahurancik, Email: zahurancik.2@osu.edu.
Chanchal K. Malik, Email: malikchanchal@gmail.com.
Ashis K. Basu, Email: ashis.basu@uconn.edu.
Zucai Suo, Email: suo.3@osu.edu.
References
- 1.Arlt VM. 3-Nitrobenzanthrone, a potential human cancer hazard in diesel exhaust and urban air pollution: a review of the evidence. Mutagenesis. 2005;20:399–410. doi: 10.1093/mutage/gei057. [DOI] [PubMed] [Google Scholar]
- 2.Taga R, Tang N, Hattori T, Tamura K, Sakai S, Toriba A, Kizu R, Hayakawa K. Direct-acting mutagenicity of extracts of coal burning-derived particulates and contribution of nitropolycyclic aromatic hydrocarbons. Mutat Res. 2005;581:91–5. doi: 10.1016/j.mrgentox.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 3.vom Brocke J, Krais A, Whibley C, Hollstein MC, Schmeiser HH. The carcinogenic air pollutant 3-nitrobenzanthrone induces GC to TA transversion mutations in human p53 sequences. Mutagenesis. 2009;24:17–23. doi: 10.1093/mutage/gen049. [DOI] [PubMed] [Google Scholar]
- 4.Arlt VM, Phillips DH, Reynisson J. Theoretical investigations on the formation of nitrobenzanthrone-DNA adducts. Org Biomol Chem. 2011;9:6100–10. doi: 10.1039/c1ob05570d. [DOI] [PubMed] [Google Scholar]
- 5.Arlt VM, Zhan L, Schmeiser HH, Honma M, Hayashi M, Phillips DH, Suzuki T. DNA adducts and mutagenic specificity of the ubiquitous environmental pollutant 3-nitrobenzanthrone in Muta Mouse. Environ Mol Mutagen. 2004;43:186–95. doi: 10.1002/em.20014. [DOI] [PubMed] [Google Scholar]
- 6.Kawanishi M, Fujikawa Y, Ishii H, Nishida H, Higashigaki Y, Kanno T, Matsuda T, Takamura-Enya T, Yagi T. Adduct formation and repair, and translesion DNA synthesis across the adducts in human cells exposed to 3-nitrobenzanthrone. Mutat Res. 2013;753:93–100. doi: 10.1016/j.mrgentox.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 7.Kunkel TA. DNA replication fidelity. J Biol Chem. 2004;279:16895–8. doi: 10.1074/jbc.R400006200. [DOI] [PubMed] [Google Scholar]
- 8.Pursell ZF, Isoz I, Lundstrom EB, Johansson E, Kunkel TA. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317:127–30. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maxwell BA, Suo Z. Kinetic Basis for the Differing Response to an Oxidative Lesion by a Replicative and a Lesion Bypass DNA Polymerase from Solfolobus solfataricus. Biochemistry. 2012;51:3485–96. doi: 10.1021/bi300246r. [DOI] [PubMed] [Google Scholar]
- 10.Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem. 2005;74:317–53. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 11.Gadkari VV, Tokarsky EJ, Malik CK, Basu AK, Suo Z. Mechanistic investigation of the bypass of a bulky aromatic DNA adduct catalyzed by a Y-family DNA polymerase. DNA Repair (Amst) 2014;21:65–77. doi: 10.1016/j.dnarep.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sherrer SM, Fiala KA, Fowler JD, Newmister SA, Pryor JM, Suo Z. Quantitative analysis of the efficiency and mutagenic spectra of abasic lesion bypass catalyzed by human Y-family DNA polymerases. Nucleic Acids Res. 2011;39:609–22. doi: 10.1093/nar/gkq719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sherrer SM, Sanman LE, Xia CX, Bolin ER, Malik CK, Efthimiopoulos G, Basu AK, Suo Z. Kinetic analysis of the bypass of a bulky DNA lesion catalyzed by human Y-family DNA polymerases. Chem Res Toxicol. 2012;25:730–40. doi: 10.1021/tx200531y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taggart DJ, Fredrickson SW, Gadkari VV, Suo Z. Mutagenic potential of 8-oxo-7,8-dihydro-2'-deoxyguanosine bypass catalyzed by human Y-family DNA polymerases. Chem Res Toxicol. 2014;27:931–40. doi: 10.1021/tx500088e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolfle WT, Washington MT, Prakash L, Prakash S. Human DNA polymerase kappa uses template-primer misalignment as a novel means for extending mispaired termini and for generating single-base deletions. Genes Dev. 2003;17:2191–9. doi: 10.1101/gad.1108603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Yuan F, Xin H, Wu X, Rajpal DK, Yang D, Wang Z. Human DNA polymerase kappa synthesizes DNA with extraordinarily low fidelity. Nucleic Acids Res. 2000;28:4147–56. doi: 10.1093/nar/28.21.4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fiala KA, Brown JA, Ling H, Kshetry AK, Zhang J, Taylor JS, Yang W, Suo Z. Mechanism of Template-independent Nucleotide Incorporation Catalyzed by a Template-dependent DNA Polymerase. J Mol Biol. 2007;365:590–602. doi: 10.1016/j.jmb.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi JY, Zang H, Angel KC, Kozekov ID, Goodenough AK, Rizzo CJ, Guengerich FP. Translesion synthesis across 1,N2-ethenoguanine by human DNA polymerases. Chem Res Toxicol. 2006;19:879–86. doi: 10.1021/tx060051v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown JA, Fowler JD, Suo Z. Kinetic basis of nucleotide selection employed by a protein template-dependent DNA polymerase. Biochemistry. 2010;49:5504–10. doi: 10.1021/bi100433x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pande P, Malik CK, Bose A, Jasti VP, Basu AK. Mutational Analysis of the C8-Guanine Adduct of the Environmental Carcinogen 3-Nitrobenzanthrone in Human Cells: Critical Roles of DNA Polymerases eta and kappa and Rev1 in Error-Prone Translesion Synthesis. Biochemistry. 2014;53:5323–31. doi: 10.1021/bi5007805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patra A, Politica DA, Chatterjee A, Tokarsky EJ, Suo Z, Basu A, Stone MP, Egli M. Mechanism of Error-Free Bypass of the Environmental Carcinogen N-(2-Deoxyguanosin-8-yl)-3-aminobenzanthrone Adduct by Human DNA Polymerase. Chembiochem. 2016 doi: 10.1002/cbic.201600420. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirouac KN, Basu AK, Ling H. Structural mechanism of replication stalling on a bulky amino-polycyclic aromatic hydrocarbon DNA adduct by a y family DNA polymerase. J Mol Biol. 2013;425:4167–76. doi: 10.1016/j.jmb.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bauer J, Xing G, Yagi H, Sayer JM, Jerina DM, Ling H. A structural gap in Dpo4 supports mutagenic bypass of a major benzo[a]pyrene dG adduct in DNA through template misalignment. Proc Natl Acad Sci U S A. 2007;104:14905–10. doi: 10.1073/pnas.0700717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherrer SM, Brown JA, Pack LR, Jasti VP, Fowler JD, Basu AK, Suo Z. Mechanistic Studies of the Bypass of a Bulky Single-base Lesion Catalyzed by a Y-family DNA Polymerase. J Biol Chem. 2009;284:6379–88. doi: 10.1074/jbc.M808161200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fiala KA, Hypes CD, Suo Z. Mechanism of abasic lesion bypass catalyzed by a Y-family DNA polymerase. J Biol Chem. 2007;282:8188–98. doi: 10.1074/jbc.M610718200. [DOI] [PubMed] [Google Scholar]
- 26.Suarez SC, Toffton SM, McCulloch SD. Biochemical analysis of DNA polymerase eta fidelity in the presence of replication protein A. PLoS One. 2014;9:e97382. doi: 10.1371/journal.pone.0097382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCulloch SD, Kokoska RJ, Masutani C, Iwai S, Hanaoka F, Kunkel TA. Preferential cis-syn thymine dimer bypass by DNA polymerase eta occurs with biased fidelity. Nature. 2004;428:97–100. doi: 10.1038/nature02352. [DOI] [PubMed] [Google Scholar]
- 28.Kawanishi M, Enya T, Suzuki H, Takebe H, Matsui S, Yagi T. Mutagenic specificity of a derivative of 3-nitrobenzanthrone in the supF shuttle vector plasmids. Chem Res Toxicol. 1998;11:1468–73. doi: 10.1021/tx9801054. [DOI] [PubMed] [Google Scholar]
- 29.Brown JA, Newmister SA, Fiala KA, Suo Z. Mechanism of double-base lesion bypass catalyzed by a Y-family DNA polymerase. Nucleic Acids Res. 2008;36:3867–78. doi: 10.1093/nar/gkn309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vyas R, Efthimiopoulos G, Tokarsky EJ, Malik CK, Basu AK, Suo Z. Mechanistic Basis for the Bypass of a Bulky DNA Adduct Catalyzed by a Y-Family DNA Polymerase. J Am Chem Soc. 2015;137:12131–42. doi: 10.1021/jacs.5b08027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohashi E, Murakumo Y, Kanjo N, Akagi J, Masutani C, Hanaoka F, Ohmori H. Interaction of hREV1 with three human Y-family DNA polymerases. Genes Cells. 2004;9:523–31. doi: 10.1111/j.1356-9597.2004.00747.x. [DOI] [PubMed] [Google Scholar]
- 32.Sherrer SM, Taggart DJ, Pack LR, Malik CK, Basu AK, Suo Z. Quantitative analysis of the mutagenic potential of 1-aminopyrene-DNA adduct bypass catalyzed by Y-family DNA polymerases. Mutat Res. 2012;737:25–33. doi: 10.1016/j.mrfmmm.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tissier A, Kannouche P, Reck MP, Lehmann AR, Fuchs RP, Cordonnier A. Co-localization in replication foci and interaction of human Y-family members, DNA polymerase pol eta and REVl protein. DNA Repair (Amst) 2004;3:1503–14. doi: 10.1016/j.dnarep.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 34.Politica DA, Malik CK, Basu AK, Stone MP. Base-Displaced Intercalated Structure of the N-(2'-Deoxyguanosin-8-yl)-3-aminobenzanthrone DNA Adduct. Chem Res Toxicol. 2015;28:2253–66. doi: 10.1021/acs.chemrestox.5b00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schorr S, Schneider S, Lammens K, Hopfner KP, Carell T. Mechanism of replication blocking and bypass of Y-family polymerase {eta} by bulky acetylaminofluorene DNA adducts. Proc Natl Acad Sci U S A. 2010;107:20720–5. doi: 10.1073/pnas.1008894107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.