

ABSTRACT
Oral and oropharyngeal squamous cell carcinoma (OOSCC) have a low survival rate, mainly due to metastasis to the regional lymph nodes. For optimal treatment of these metastases, a neck dissection is required; however, inaccurate detection methods results in under- and over-treatment. New DNA prognostic methylation biomarkers might improve lymph node metastases detection. To identify epigenetically regulated genes associated with lymph node metastases, genome-wide methylation analysis was performed on 6 OOSCC with (pN+) and 6 OOSCC without (pN0) lymph node metastases and combined with a gene expression signature predictive for pN+ status in OOSCC. Selected genes were validated using an independent OOSCC cohort by immunohistochemistry and pyrosequencing, and on data retrieved from The Cancer Genome Atlas. A two-step statistical selection of differentially methylated sequences revealed 14 genes with increased methylation status and mRNA downregulation in pN+ OOSCC. RAB25, a known tumor suppressor gene, was the highest-ranking gene in the discovery set. In the validation sets, both RAB25 mRNA (P = 0.015) and protein levels (P = 0.012) were lower in pN+ OOSCC. RAB25 mRNA levels were negatively correlated with RAB25 methylation levels (P < 0.001) but RAB25 protein expression was not. Our data revealed that promoter methylation is a mechanism resulting in downregulation of RAB25 expression in pN+ OOSCC and decreased expression is associated with lymph node metastasis. Detection of RAB25 methylation might contribute to lymph node metastasis diagnosis and serve as a potential new therapeutic target in OOSCC.
KEYWORDS: DNA methylation, epigenetic regulation, head and neck cancer, metastasis, MethylCap-Seq, oral squamous cell carcinoma
Introduction
Oral and oropharyngeal squamous cell carcinoma (OOSCC) are the most common subtypes of head and neck squamous cell carcinomas (HNSCC) and are characterized by an overall 5-year survival < 50%.1 This low survival rate is greatly impacted by the presence of lymph node (LN) metatasis.2 Patients with metastases in the regional lymph nodes of the neck have a 5-year survival that is half that of those who do not present regional metastases.3,4 Therefore, for treatment decision-making, it is important to accurately detect the presence LN metastasis. Currently, diagnosis consists only of clinical examination and imaging, which are known to have low sensitivity and low specificity for LN metastasis detection.5-8 When LN metastases are detected, a neck dissection is required, but this surgical procedure is accompanied by neck and shoulder morbidity. As a result, under- and over-treatment of OOSCC patients occurs frequently.7,9 Currently, appropriate clinical and tumor biomarkers that predict the presence of LN metastasis are lacking.
DNA methylation is a mechanism of epigenetic modification that impacts cellular phenotypes by regulating gene expression and is known to affect carcinogenesis by altering proliferation rates and DNA repair.10,11 As a result, DNA methylation screening has been used as a tool to predict clinical outcome and therapy response in cancer patients.10,12 Moreover, DNA methylation of several genes has been reported to have a predictive value for nodal metastasis in HNSCC, including TWIST1,13 IGF2,14,15 CDKN2A, MGMT, MLH1, and DAPK.16,17 However, the discovery of these tumor markers has not improved clinical LN metastasis detection rate.
Recently, we have reported on the identification of new DNA methylation markers that predict LN status by MethylCap-Seq.18 The combination of enrichment of methylated DNA fragments and next generation sequencing has been established as a true genome-wide assay compared to other DNA methylation screening techniques (see ref19 for a review). Using a quantitative ranking of genomic loci by likelihood of differential methylation between OOSCC with metastasis negative LN (pN0) and OOSCC with metastasis positive LN (pN+), we identified WISP1 as a hypomethylation marker associated with pN+ OOSCC.18 In the present study, we report on a new approach tailored toward identifying potentially epigenetically downregulated genes in the metastatic OOSCC phenotype. Epigenetically downregulated genes are more suitable for opening up new clinical options, as hypermethylation can be more easily detected in an unmethylated background. In addition, methylated regions are potentially suited as therapeutic targets, thanks to the emergence of epigenetic editing and demethylating agents.19
For this purpose, we used a set of 696 genes that were previously reported to be differentially expressed between 143 pN0 and 79 pN+ OOSCC. This gene signature has a validated negative predictive power of 89% for LN metastases.20-22 We combined the expression levels of the genes in this predictive gene signature with DNA methylation data acquired by MethylCap-Seq analysis.18 Using this approach, we identified 14 genes that were simultaneously hypermethylated and downregulated in pN+ OOSCC. In this manuscript, we report on the identification of RAB25 as the highest-ranking gene and analyze the association between expression and methylation of RAB25 and the presence of LN metastases.
Materials and methods
Patient selection
We selected treatment-naive OOSCC patients who underwent a neck dissection for primary tumor resection resulting in free resection margins upon histopathological examination at the University Medical Center Groningen (UMCG), between 1997 and 2008. Pathological revision was performed for all original hematoxylin and eosin (HE)-slides formalin-fixed, paraffin embedded (FFPE) tissue blocks. All pN0 tumors were histologically confirmed or had pN0 status with >2 y LN metastasis-free follow-up. All patient and tumor characteristics are available in Supplemental Table 1. For the immunohistochemical study, 227 OOSCC tumors were used for 5 tissue-microarrays (TMA) in triplicate, as described previously.23 All TMA contained 7 different normal tissues that served as control. Human papilloma virus (HPV) status was tested by p16 immunohistochemistry followed by high-risk HPV PCR, as previously reported.24 Out of 197 OOSCC patients, 5 were HPV16 positive. A total of 192 HPV-negative patients (102 pN0 and 90 pN+) were included for further analysis. For the MethylCap-Seq study, 6 pN+ and 6 pN0 tumors matched for age and primary tumor site were selected from the total cohort. Leukocytes were acquired from healthy women for endogenous methylation and methylation background estimation.25,26 This study was performed in accordance with the Code of Conduct for proper secondary use of human tissue in the Netherlands (www.federa.org), and relevant institutional and national guidelines were followed.
DNA isolation
DNA isolation was performed as previously reported.18 Briefly, 2 10-μm thick FFPE sections were deparaffinized in xylene and incubated in 300 µl 1% SDS-proteinase K at 60°C overnight. DNA extraction was performed using phenol-chloroform and ethanol precipitation. The acquired DNA pellets were then washed with 70% ethanol, dissolved in 50 µl TE-4 (10 mM Tris/HCl; 0.1 mM EDTA, pH 8.0), and stored at 4°C. To check the DNA structural integrity, genomic DNA was amplified by multiplex PCR according to the BIOMED-2 protocol.27 Cases with products ≥200 bp were selected for further analyses. DNA used in MethylCap-Seq was measured by Quant-iT™ PicoGreen® dsDNA Assay Kit, according to manufacturer's protocol (Invitrogen). The DNA used for pyrosequencing was measured using the Nanodrop ND-1000 Spectrophotometer (Thermo Scientific). Only samples with an absorbance ratio 260/280 nm > 1.8 were selected for further testing. The number of tumor cells required for this study was set at 60%, as estimated by HE-staining of 3-μm thick sections.
MethylCap-Seq
MethylCap-Seq analysis was performed as reported previously.18 Briefly, genome-wide methylation was assessed using 500 ng of DNA fragmented by Covaris S2 (Covaris) and obtained from 6 pN0 OOSCC, 6 pN+ OOSCC, and 2 pools of leukocytes. Methylated DNA was enriched using the methyl binding domain protein MeCP2 (MethylCap-kit, Diagenode) and followed by paired-end next generation sequencing on the Illumina GA II Sequencer (Illumina). Subsequently, the enriched, captured, and sequenced reads were mapped to the human reference genome (NCBI build 37.3) using the BOWTIE software.28 Only the reads that mapped to unique loci were included. Reads that exactly overlapped with each other were excluded, as identical reads are most likely the result of amplification of the same DNA fragment. Additionally, the mapped distance between the paired-ends could not be longer than 400 bp. Finally, all the mapped reads were compared to the “Map of the Human Methylome” build 2 [http://www.biobix.be/map-of-the-human-methylome/, BIOBIX (Lab of Bioinformatics and Computational Genomics), University of Ghent, Ghent, Belgium 2014], which consists of an in-house developed summary of all experimentally assessed genomic sites of potential differential methylation [called Methylation Cores (MC)]
To identify a candidate set of genomic regions differentially methylated between pN0 and pN+ OOSCC, all MC located 2,000 bp upstream to 500 bp downstream of the transcription start site (TSS) or in the first exon of an Ensemble (v65) gene were statistically compared using R35 with R-package Bayseq.29 The sequencing experiment proved to be underpowered in terms of sequencing depth and number of biological replicates, precluding any definite conclusions. Therefore, we focused on the identification of the most interesting set of putatively differentially methylated regions that could be validated in a subsequent setup. This led to the following 2-step MC selection method: In the first step, the number of methylated samples was determined for both groups (pN+ and pN0). A sample was called [unmethylated] if there were no reads and [methylated] if there were one or more reads. A Fisher exact test was performed to rank the MCs for differential number of methylated samples between both groups. Ties in P-values, due to the limited number of samples, were broken by secondary ranking on log fold-change methylation between groups (average methylation incremented by 1 in both groups). In contrast to our previous quantitative ranking based on differential methylation,18 this pre-selection is unaffected by the variability of the signal in the methylated group. In the second step, the Mann-Whitney-U test was applied to the 5,000 highest-ranked MCs from the first step. MCs with a P-value < 0.05 (a total of 1,709) were retained for further consideration. Finally, only the MCs associated with genes that have an annotated function in the UniProtKB/Swiss-Prot database30 were selected for further analyses.
Positive and negative predictive value for the methylation status of all MCs was calculated. For each MC all OOSCC with a read count of ≥3 reads were considered as methylated and OOSCC with a read count <3 reads were considered as unmethylated. The positive predictive value was then calculated as follows: (true positive pN+ OOSCC) / (true positive pN+ OOSCC + false positive pN0 OOSCC). The negative predictive value was calculated as: (true negative pN0 OOSCC) / (true negative pN0 OOSCC + false negative pN+ OOSCC).
Gene selection
To identify epigenetically downregulated genes in pN+ OOSCC, a validated gene signature predictive of pN-status in OOSCC (published by Hooff et al.22) was combined with MethylCap-Seq data (Fig. 1). This gene signature is based on a diagnostic microarray consisting of 696 genes and was validated on 222 OOSCC from 8 different medical centers in The Netherlands.20-22 Genes that were found by MethylCap-Seq to be hypermethylated in pN+ OOSCC and found to be downregulated in pN+ OOSCC by microarray were selected for further analyses.
Figure 1.
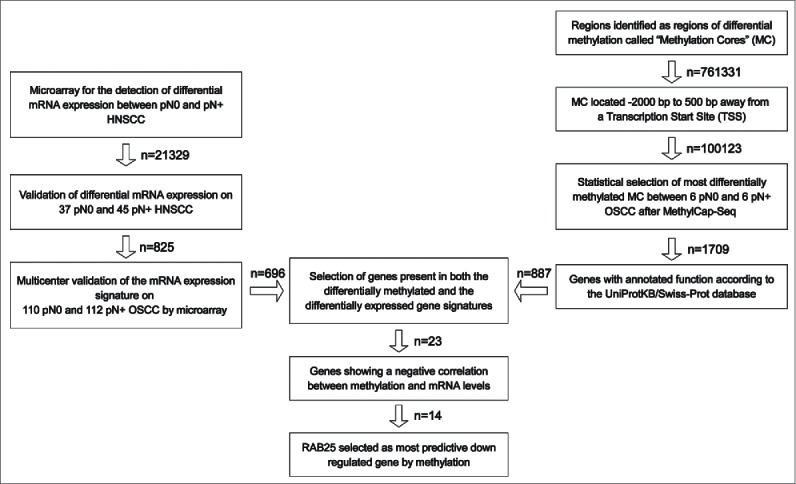
Strategy to identify epigenetically downregulated genes in pN+ OSCC. On the left: published gene signatures predictive of pN-status in OSCC were used to identified significantly downregulated genes in pN+ OSCC.20-22 On the right: MethylCap-Seq was performed on 6 pN0 OSCC and pN+ OSCC.18 All reads of MCs in gene promoter regions were ranked according to the likelihood of differential methylation and an approximate FDR. The 5,000 MCs with the lowest FDR were further tested by Mann-Whitney-U. The MC associated with genes without annotated gene functions were excluded. In the middle: the gene signature and methylation data were compared to select epigenetically regulated genes in pN+ OSCC (n = 23). From these 23 genes, epigenetically downregulated genes in pN+ OSCC were selected. Based on the amount of mRNA downregulation, statistical differences in methylation between pN0 and pN+ OSCC, and positive and negative predictive value, RAB25 was selected as the most significantly epigenetically downregulated gene in pN+ OSCC compared to pN0 OSCC.
The Cancer Genome Atlas data analysis
The Cancer Genome Atlas (TCGA) validation was performed as reported previously.18 Clinical data for all HNSCC patients (n = 423) was downloaded from the TCGA data portal (https://tcga-data.nci.nih.gov/tcga/) on April 7th 2013. Patients with a tumor located in “Floor of Mouth,” “Oral Cavity,” or “Oral Tongue,” with known pathological N-status, available methylation status, and mRNA data were selected (n = 147). Patient and tumor characteristics of the selected TCGA cases are presented in Supplemental Table 1. All pathological N-statuses were dichotomized for further analyses.
For methylation analysis, level 3 methylation Illumina Infinium HumanMethylation450 (450K) data was downloaded for the previously selected oral SCC (OSCC) patients from the TCGA data portal (https://tcga-data.nci.nih.gov/tcga/) on April 7th 2013. Additional Infinium 450K probe information was acquired from the gene expression omnibus (GEO) accession number GSE42409, including, distance to TSS, associated CpG island, and chromosomal localization.9 All probes located up to 2,000 bp upstream and 500 bp downstream of a TSS were selected for further analyses. R (version 3.0.3),35 Rstudio (RStudio, Inc.), and the Lumi package31 were used to convert the 450K probe β-values to M-values using the beta2m function. Subsequently, all M-values were quantile-normalized by the normalizeBetweenArrays function of R package Limma.32 Using the eBayes function of the Lumi R package, all 450K probes located 2,000 bp upstream to 500 bp downstream of the RAB25 TSS (a total of 3) were statistically compared between pN0 OSCC (n = 61) and pN+ OSCC (n = 86).31
For expression analysis, all mRNA expression z-scores (RNA Seq V2 RSEM) from the HNSCC TCGA “provisional cancer study” were downloaded from the cBioportal public portal (http://www.cbioportal.org/public-portal/)33,34 on April 30th 2014 and statistically compared between pN0 and pN+ OSCC by Mann-Whitney test using R. The optimal cut-off value for RAB25 mRNA levels between pN0 and pN+ OSCC was determined to be z-score: −0.4250 by ROC-curve analysis, using SPSS version 22.0.1 (IBM). For copy number and mutation analyses, all RAB25 mutation and GISTIC data from the HNSCC TCGA “provisional cancer study” were downloaded from cBioportal public portal on April 6th 2015. TCGA survival data were incomplete and varied between testing labs and were therefore not analyzed.
Spearman rank correlations between RAB25 mRNA z-scores and normalized M-values of all RAB25 probes were calculated by the basic R function cor.test.35
Putative RAB25-regulating miRNAs were identified using the miRDB database (http://mirdb.org/miRDB/) on April 6th 2015 (n = 12).36 Subsequently, for all miRNAs with available data (n = 6), all RNA Seq V2 RSEM (z-score threshold of ± 2), mutation, and gene copy number data for the miRNA and RAB25 were downloaded from the cBioportal public portal (http://www.cbioportal.org/public-portal/)33,34 on April 17th 2015. In total, 5 different types of gene copy number alterations were distinguished: -2 (homozygous deletion), -1 (hemizygous deletion), 0 (no gene copy number alteration), 1 (gain), and 2 (high-level amplification).
Bisulfite pyrosequencing
Extracted genomic DNA (1 µg/sample) was sodium bisulfite-treated using the EZ DNA methylation kit (Zymo, BaseClear, Leiden, The Netherlands) according to the manufacturer's protocol. RAB25 bisulfite pyrosequencing PCR and sequencing primers were designed using Pyromark Assay design version 2.0.1.15 (Qiagen). All primer sequences and PCR conditions are available in Supplemental Table 2. Bisulfite treated DNA was amplified using the Pyromark PCR kit according to the company protocol (Qiagen). Each reaction was performed with 12.5 µl 2x PCR master mix, 200 nmol forward primer, and 200 nmol reverse primer. PCR was performed as follows: 15 min 95°C, 50 cycles of (30 sec 94°C, 30 sec 59°C, 30 sec 72°C), and 10 min 72°C. PCR products were checked on a 2% agarose gel containing 15 µl ethidium bromide. Biotinylated PCR product (15 µl ) was captured using 1 µl Streptavidin-coated Sepharose High Performance beads (GE Healthcare). Captured amplicons were then purified using the Q24 Vacuum Workstation (Qiagen), according to the manufacturer's protocol, washed with 70% alcohol, denatured using PyroMark Denaturation Solution (Qiagen), and washed with PyroMark Wash Buffer (Qiagen). The purified PCR product was then added to 25 µl 0.3 µM RAB25 sequence primers and followed by bisulfite pyrosequencing analysis using the Pyromark Q24 (Qiagen). Pyrosequencing results were analyzed using the provided Pyromark Q24 software version 2.0.6 (Qiagen). Each pyrosequencing run included 3 control samples: leukocyte DNA from healthy patients as control for normal/endogenous methylation levels; in vitro methylated (SssI digested) leukocyte DNA as hypermethylation control; and whole-genome amplified (WGA) leukocyte DNA, amplified using the Illustra Ready-To-Go GenomiPhi HY DNA Amplification Kit (GE Healthcare), as a control for unmethylated DNA.
Immunohistochemistry
FFPE tumor tissue sections (3-μm thick) were deparaffinized in xylol and rehydrated using decreasing ethanol concentrations (100%, 96%, 80%, 70%, and 50%). Antigen retrieval was performed using a citrate buffer (10 mM citric acid, 0.05% Tween 20, pH 6.0) and heated in a microwave oven for 15 min at 300 W. Endogenous peroxidase was blocked with a 0.3% H2O2 solution for 30 min at room temperature, followed by incubation with a mouse monoclonal antibody to human RAB25 clone 3F12F3 (Santa Cruz), diluted 1:50 in PBS with 1% bovine serum albumin, overnight at 4°C. Subsequently, primary antibody detection was achieved by incubation with Envision+ (Dako) horseradish peroxidase for 30 min at room temperature and developed with 3,3-diaminobenzidine solution (Dako) containing 0.03% H2O2 and counterstained with hematoxylin for 2 min. Mammary epithelial cells were used as a control for positive RAB25 expression.37 The percentage of positive tumor cells was scored as previously reported.38,39 Three RAB25 immunoreactivity intensities were also recorded: 0 (no staining); 1 (moderate staining); and 2 (strong staining). The level of intensity of the staining was scored independently by 2 blinded observers (MJAMC and MFM). Discordant results were discussed until consensus was reached or decided by an experienced HNSCC pathologist (BvdV). The optimal cut-off between high and low RAB25 positive neoplastic cells, defined as the percentage of neoplastic cells with tumors with any level of expression (moderate/strong), was determined by ROC curve analysis relative to pN-status and established as 33% RAB25 positive tumor cells. A total of 178 out of the 192 HPV-negative HNSCC were used for RAB25 immunoreactivity analysis.
Statistical analysis
Statistical analysis was performed using SPSS (IBM) and R (version 3.0.3). Associations between RAB25 expression and clinico-pathological characteristics were tested using the χ2 test. Survival was defined as the number of days between the first treatment and disease specific death (DSS) or disease recurrence (DFS) and analyzed by Kaplan-Meier curves and log rank test. All tests were performed as 2-tailed and a P-value <0.05 was considered statistically significant.
Results
RAB25 is the highest-ranking differentially methylated and expressed gene in pN+ OOSCC
To identify genes whose expression is regulated by methylation, a validated gene expression signature and methylation data were combined using a stepwise selection approach as outlined in Fig. 1. After combining the gene signature and methylation data, 23 genes were found to be present in both the differentially methylated gene panel and the differentially expressed gene panel (Supplemental Table 3).
Out of these 23 potentially epigenetically regulated genes, 20 genes were hypermethylated in the pN+ OOSCC of the UMCG panel, as identified by MethylCap-Seq. Finally, 14 of these 20 genes (ACTA1, BRUNOL4, COBLL1, GFRA1, H2AFY, IL22RA1, KRT17, LAMP3, MALL, MAST4, NDUFA10, RAB25, S100A9, and WDR13) showed both promoter hypermethylation as well as expression downregulation in pN+ OOSCC (Table 1). Of these 14 genes, RAB25 showed the highest downregulation of expression and concomitant highest rate of hypermethylation in pN+ OOSCC (Table 1). Moreover, the RAB25 read count distribution between pN0 and pN+ OOSCC showed the highest positive and negative predictive value for pN-status (Table 1 and Supplemental Table 3). Therefore, RAB25 was studied in more detail as an epigenetically downregulated gene in pN+ OOSCC.
Table 1.
Epigenetically downregulated genes in pN+ OSCC. All 14 potentially epigenetically downregulated genes in pN+ OSCC compared to pN0 OSCC after cross-reference of expression microarray and MethylCap-Seq data (see Fig. 1). The positive and negative predictive value of the reads for pN+ status, associated hypermethylation, read distribution between pN0 and pN+ OSCC, and predictive value of the methylation data are illustrated. P-value for the differential DNA methylation was calculated using the Mann-Whitney-U test. Positive and negative predictive value for the methylation status of all MCs were calculated as follows: OOSCC with a read count of ≥ 3 reads were considered true positives and OOSCC with a count read<3 were considered true negatives. Subsequently, the positive predictive value was then calculated as: (true positive pN+ OOSCC) / (true positive pN+ OOSCC + false positive pN0 OOSCC). Finally, the negative predictive value was calculated as: (true negative pN0 OOSCC) / (true negative pN0 OOSCC + false negative pN+ OOSCC).
| Methylation Core data |
DNA Methylation data |
mRNA data | Epigenetic regulation | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene name | Chr | Distance TSS (bp) | length (bp) | P-Value | Hypermeth in | Pos. Pred. | Neg Pred. | expression pN+ | mRNA & Meth |
| RAB25 | 1 | −108 | 233 | 0.02 | pN+ | 100% | 86% | −0.15 | ↓ Negative |
| COBLL1 | 2 | −1247 | 191 | 0.02 | pN+ | 100% | 75% | −0.14 | ↓ Negative |
| GFRA1 | 10 | −809 | 120 | 0.04 | pN+ | 100% | 67% | −0.11 | ↓ Negative |
| S100A9 | 1 | 490 | 125 | 0.04 | pN+ | 100% | 60% | −0.1 | ↓ Negative |
| LAMP3 | 3 | 0 | 284 | 0.05 | pN+ | 80% | 71% | −0.09 | ↓ Negative |
| ACTA1 | 1 | −756 | 273 | 0.01 | pN+ | 100% | 67% | −0.08 | ↓ Negative |
| KRT17 | 17 | −296 | 1 | 0.02 | pN+ | 100% | 55% | −0.08 | ↓ Negative |
| MAST4 | 5 | −271 | 57 | 0.03 | pN+ | 0% | 50% | −0.06 | ↓ Negative |
| IL22RA1 | 1 | 114 | 229 | 0.05 | pN+ | 75% | 63% | −0.04 | ↓ Negative |
| BRUNOL4 | 18 | −1543 | 24 | 0.03 | pN+ | 100% | 67% | −0.03 | ↓ Negative |
| NDUFA10 | 2 | −1155 | 9 | 0.02 | pN+ | 100% | 55% | −0.01 | ↓ Negative |
| MALL | 2 | 413 | 152 | 0.03 | pN+ | 100% | 55% | −0.01 | ↓ Negative |
| WDR13 | X | 0 | 54 | 0.05 | pN+ | 100% | 55% | −0.01 | ↓ Negative |
| H2AFY | 5 | −1065 | 90 | 0.03 | pN+ | 100% | 55% | −0.01 | ↓ Negative |
Validation of epigenetic regulation of RAB25 in the independent TCGA cohort
Our data revealed a strong association between decreased mRNA expression and increased methylation of the RAB25 gene in pN+ OOSCC compared to pN0 OOSCC. To confirm this association, we selected all 147 OSCC available in the public TCGA database with available RAB25 mRNA levels, RAB25 methylation data, and pN-status data. Among the Illumina Infinium 450K probes, 5 probes were associated with the RAB25 gene (Supplemental Table 4). In total, 3 probes (cg15896939, cg09243900, and cg19580810) were located in the RAB25 promoter region (Supplemental Fig. 1). Methylation status of these 3 RAB25 promoter probes (cg15896939, P = 0.003; cg09243900, P = 0.023; and cg19580810, P < 0.001) was significantly higher in the OSCC with low RAB25 mRNA levels (Fig. 2A). Additionally, methylation levels of all 3 RAB25 probes showed a significant negative correlation with RAB25 mRNA levels (cg15896939, R = −0.230, P = 0.005; cg09243900, R = −0.162, P = 0.049; and cg19580810, R = −0.390, P < 0.001; Fig. 2B). Analysis of TGCA database confirmed that methylation of RAB25 is associated with decreased expression levels. Additionally, the location of 2 of these 3 probes (cg15896939 and cg09243900) overlapped with the RAB25 MCs annotated by MethylCap-Seq (Supplemental Fig. 1).
Figure 2.

RAB25 mRNA levels in relation with the 3 RAB25 TSS 450K probes (cg09243900, cg15896939, and cg19580810) methylation levels in the TCGA OSCC cohort. (A) RAB25 methylation levels compared between OSCC with high RAB25 mRNA levels and OSCC with low RAB25 mRNA levels. The M-values of the 3 RAB25 Infinium 450K promoter probes were significantly higher in OSCC with low RAB25 mRNA z-scores compared to OSCC with high RAB25 mRNA z-scores. (B) Spearman correlations between RAB25 methylation and RAB25 mRNA levels. All 3 RAB25 promoter probes showed a significant negative correlation between RAB25 promoter probe M-values and RAB25 mRNA z-scores.
Association between RAB25 methylation and lymph node status
To determine whether RAB25 promoter methylation is associated with pN-status in OSCC, we analyzed the methylation levels of the 3 RAB25 promoter probes (cg09243900, cg15896939, and cg19580810) in 61 pN0 and 86 pN+ OSCC in the TCGA database. No significantly different methylation was found for any of the 3 RAB25 promoter probes between pN0 and pN+ OSCC (Supplemental Fig. 2A). Additionally, RAB25 methylation was measured in an independent UMCG OOSCC cohort (n = 47) using 3 different bisulfite pyrosequencing assays of the promoter region containing the annotated RAB25 MCs (bisulfite primer locations are shown in Supplemental Fig. 1). No significant differences in RAB25 methylation levels were found between pN0 and pN+ OOSCC for any of the 9 CpG sites (Supplemental Fig. 2B). These data suggest that DNA methylation of RAB25 promoter region is not directly related to LN metastasis in OOSCC.
Association between RAB25 expression and lymph node status
To determine the association between RAB25 expression and LN status in OOSCC, we analyzed RAB25 mRNA levels in OSCC using data available in the public TCGA database. Analyses of RAB25 mRNA levels in 147 OSCC revealed significantly lower (P = 0.015) RAB25 expression in pN+ (n=86) than in pN0 OSCC (n=61) (Fig. 3A). High RAB25 mRNA expression was found to be significantly associated with pN0-status (P = 0.006) (Table 2A). High RAB25 mRNA expression was also associated with decreased lympho-vascular invasion (P = 0.029) (Table 2A).
Figure 3.
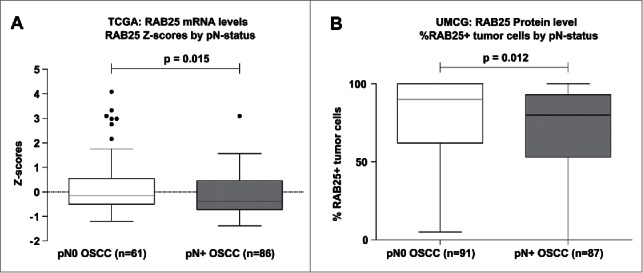
RAB25 expression levels between pN0 and pN+ OSCC in the UMCG and TCGA OSCC cohort. (A) pN+ OSCC in the TCGA cohort (n = 86) express significantly less RAB25 mRNA than pN0 OSCC (n = 61), as revealed by Mann-Whitney-U test. (B) pN + OSCC in the UMCG cohort (n = 87) have significantly less RAB25-positive tumor cells than pN0 OSCC (n = 91), as revealed by Mann-Whitney-U test.
Table 2.
Correlations between RAB25 expression and tumor characteristics. A) Associations between RAB25 mRNA expression and the clinical characteristics of the TCGA OSCC cohort. B) Associations between RAB25 protein expression and the clinical characteristics of the UMCG OSCC cohort.
| A) RAB25 mRNA in TCGA cohort |
B) RAB25 protein in UMCG cohort |
|||||
|---|---|---|---|---|---|---|
| Low RAB25 mRNA | High RAB25 mRNA | P-value | Low RAB25 protein | High RAB25 protein | P-value | |
| N (%) | N (%) | N (%) | N (%) | |||
| Total tumors | 58 (40) | 89 (60) | 18 (10) | 160 (90) | ||
| Total patients | 58 (40) | 89 (60) | 18 (10) | 160 (90) | ||
| Gender | ||||||
| Male | 17 (36) | 30 (64) | 0.576 | 15 (14) | 96 (86) | 0.053 |
| Female | 41 (41) | 59 (59) | 3 (5) | 64 (95) | ||
| Age at diagnosis (yrs) | ||||||
| Median | 61 | 60 | 0.412 | 59 | 64 | 0.197 |
| Range | 26–85 | 19–87 | 38–80 | 25–94 | ||
| Site | ||||||
| OSCC | [Not Applicable] | 14 (9) | 142 (91) | 0.18 | ||
| Other | 4 (18) | 18 (82) | ||||
| pT status | ||||||
| 1–2 | 24 (41) | 35 (59) | 0.804 | 12 (10) | 106 (90) | 0.972 |
| 3–4 | 34 (39) | 54 (61) | 6 (10) | 54 (90) | ||
| pN status | ||||||
| 0 | 16 (26) | 45 (74) | 0.006 | 3 (3) | 88 (97) | 0.002 |
| + | 42 (49) | 44 (51) | 15 (17) | 72 (83) | ||
| Extranodal spread (only pN+) | ||||||
| No | 19 (48) | 21 (52) | 0.154 | 9 (19) | 38 (81) | 0.61 |
| Yes | 17 (65) | 9 (35) | 6 (15) | 34 (85) | ||
| Perineural invasion | ||||||
| No | 18 (35) | 33 (65) | 0.289 | 10 (9) | 106 (91) | 0.595 |
| Yes | 31 (45) | 38 (55) | 5 (11) | 39 (89) | ||
| Lymphovascular invasion | ||||||
| No | 27 (32) | 56 (68) | 0.029 | 12 (10) | 112 (90) | 0.573 |
| Yes | 17 (55) | 14 (45) | 3 (14) | 19 (86) | ||
| Histological differentiation | ||||||
| Well | 4 (22) | 14 (78) | 0.11 | 2 (5) | 38 (95) | 0.181 |
| Moderate or Poor | 54 (42) | 75 (58) | 16 (13) | 112 (87) | ||
| Infiltration depth (mm) | 0.537 | |||||
| Median | [Not Available] | 9 | 15 | |||
| Range | 3.1 – 22 | 0.07 – 40 | ||||
| Infiltration depth (mm) | ||||||
| <4 mm | [Not Available] | 3 (11) | 24 (89) | 0.823 | ||
| >4 mm | 13 (10) | 121 (90) | ||||
To validate whether RAB25 protein expression was also associated with lymph node status in our UMCG OSCC cohort, immunohistochemistry was performed on 192 HPV-negative OOSCC. We could score RAB25 immunoreactivity in 178 OOSCC. RAB25 immunohistochemistry (example in Fig. 4) revealed a significant lower number of neoplastic cells showing RAB25 protein expression in pN+ OOSCC (P = 0.012; Fig. 3B). Using a cut-off of 33% RAB25-positive neoplastic cells to define [low] and [high] expression, low RAB25 expression was significantly associated with pN+ OOSCC (P = 0.002; Table 2B). The association between low RAB25 expression and pN+ status is in good agreement with the TCGA analysis (Table 2 and Fig. 3).
Figure 4.
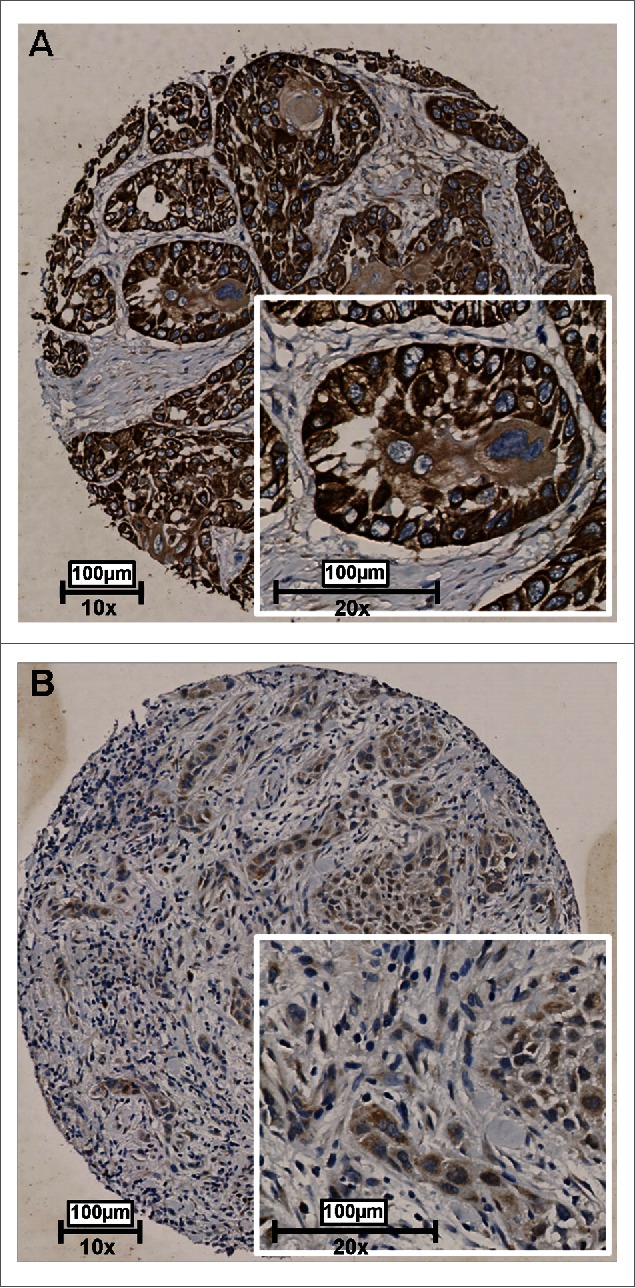
Representative examples of RAB25 expression in 2 OSCC as detected by immunohistochemistry. Tissues were scored by the amount of RAB25-positive cells. (A) Example of a well-differentiated OSCC with a high amount of RAB25-expressing cells. (B) Example of a poorly differentiated OSCC with a very low amount of RAB25-positive cells.
RAB25 protein level of expression was not associated with other clinical characteristics (Table 2B), DSS (P = 0.232), or DFS-survival (P = 0.260). These data support an anti-invasive function of RAB25 expression in OOSCC. Analysis of RAB25 protein levels and RAB25 MC levels revealed no associations between RAB25 methylation and RAB25 protein expression in the UMCG cohort (data not shown).
Analysis of RAB25 gene copy number alterations, mutations, and miRNAs
RAB25 mRNA expression is significantly associated with the methylation status of the RAB25 promoter (Table 1 and Fig. 2). Additionally, both RAB25 mRNA and RAB25 protein expression are associated with pN-status (Fig. 3, Table 2). However, RAB25 methylation status (Supplemental Fig. 2) is not associated with pN+ status. Therefore, DNA methylation only partly explains the regulation of RAB25 protein expression. To assess the frequency of other (epi)genetic changes that might regulate RAB25 protein expression and a possible association with LN status, the frequency of RAB25 gene mutations and gene copy number alterations were assessed in 147 OSCC cases selected from the TCGA database. We found a single OSCC case (1/147) carrying a RAB25 mutation (RAB25-Q98H). We observed RAB25 copy number gain in 29 OSCC cases (1 case with high level amplification) and hemizygous RAB25 deletions (none homozygous) were detected in 15 OSCC. RAB25 mRNA levels were significantly higher in OSCC with RAB25 gene copy number increase (P = 0.024), but RAB25 mRNA levels were not associated with hemizygous deletions of RAB25 (P = 0.330). Additionally, pN-status was not associated with RAB25 copy number gain (P = 0.540), RAB25 copy number loss (P = 0.785), or with RAB25 mRNA levels, RAB25 copy number gain (P = 0.143), or RAB25 copy number loss (P = 0.584).
The miRDB database contains 12 miRNAs putatively targeting RAB25 mRNA (hsa-miR-504-5p, hsa-miR-4725-5p, hsa-miR-608, hsa-miR-4651, hsa-miR-185-3p, hsa-miR-4520-3p, hsa-miR-4447, hsa-miR-8071, hsa-miR-4761-3p, hsa-miR-1296-3p, hsa-miR-6862-5p, hsa-miR-4253). For 6 of those miRNAs, expression, mutation, and copy number data were available from the TCGA database. None of the 6 miRNAs displayed aberrant gene expression, mutations, or copy number alterations in the 530 HNSCC present in the TCGA database (data not shown).
Discussion
We used a combination of genome-wide methylation analysis and a validated gene signature predictive for pN+ status in OOSCC to identify potential epigenetically regulated genes in the OOSCC metastatic phenotype. Of all analyzed genes, RAB25 is the most likely epigenetically regulated and predictive for pN+ OOSCC. RAB25 is reported to be a tumor suppressor gene lost in HNSCC subtypes,38,39 that is also hypermethylated in HNSCC cell lines compared to healthy tissue,38,39 underlining its importance and epigenetic inhibition of RAB25 protein expression during carcinogenesis.
The RAB25 protein is a member of the RAB11 subfamily of small GTPases. These GTPases are emerging as novel and important regulators of cancer development and progression (for a review see ref. 38). Aberrant expression of small GTPases in general, and RAB25 specifically,40,41 has been detected in various cancers,42,43 including HNSCC and OSCC.38,39 Interestingly, changes in RAB25 expression are correlated with tumor invasiveness in almost all cancer types,44-47 but only in triple-negative breast and HNSCC, RAB25 functions as a tumor suppressor gene, and loss of RAB25 leads to increased migration and invasion.38,47-49
Epigenetic downregulation of RAB25 was reported in ovarian cancer compared to normal ovarian tissue,50 esophageal cancer cell lines, compared to paired normal esophageal tissue,38 and in HNSCC cell lines.38,39 This supports the hypothesis that loss of RAB25 expression in pN+ OSCC is caused by hypermethylation, since both increased hypermethylation51 and metastasis are associated with progressive cancer52 and, specifically, HNSCC.53 Additionally, epigenetic regulation of the expression of other small GTPases, to which RAB25 belong, has been shown in metastatic lung cancer54 and in colon cancer.55
We confirmed that loss of RAB25 protein expression correlated with the presence of LN metastasis in HNSCC and OOSCC, specifically,38,39,48 and could be used to predict LN metastasis in OOSCC.20-22 Because of our selection method, genes selected were not only associated with the presence of nodal metastases, but, more specifically, were downregulated in pN+ cases. These selection criteria are reflected in the fact that RAB25 mRNA and protein expression showed the best predictive value for nodal metastases in patients where expression is low (a high positive predictive value). Consequently, the negative predictive value is lower. This effect is independent of T-stage (data not shown), but should be taken into account in future studies into the clinical application of RAB25 expression. Additionally, MethylCap-Seq identified RAB25 as differentially methylated between pN0 OOSCC and pN+ OOSCC. These data suggest that RAB25 is epigenetically regulated and lost during cancer progression as a result of hypermethylation. However, we could not confirm differential methylation on a larger independent cohort using bisulfite pyrosequencing and Illumina Infinium 450K TCGA data, although we did find significant correlation between RAB25 mRNA levels and RAB25 DNA methylation levels. These data suggest that RAB25 is regulated by DNA methylation, but also potentially subjected to other forms of epigenetic regulation, such as histone modification or miRNAs. However, previous reports show no relation between histone modifications and RAB25 expression in esophageal cancer38 and alterations of 6 miRNAs that regulate RAB25 were found to be almost non-existent in the TCGA OSCC database (this paper). Most RAB25 gene copy number alterations were amplifications and can thus not be responsible for downregulated RAB25 protein expression. The frequency of RAB25 loss was, however, very low in the TCGA OSCC database to draw firm conclusions.
In summary, our data suggest that epigenetic silencing of RAB25 contributes to LN metastasis in OOSCC patients. Therefore, RAB25 protein expression assessment might contribute to better patient diagnosis and RAB25 epigenetic editing might open new therapeutic options for the treatment of LN metastasis through epigenetic editing or using demethylating agents in order to increase OOSCC patient prognosis and care. Genome-wide methylation analysis using the MethylCap-Seq is a promising approach to identify important epigenetically regulated genes in carcinogenesis.
Supplementary Material
Disclosure of potential conflicts of interest
The CTMM Air Force Consortium is a private/public consortium with involvement of academia, private companies, and the government. It is not a commercial source of funding. M Clausen and M Mastik are funded by CTMM consortium to perform the research project (global methylation screening in HNSCC) that is a part of Clausen's thesis. These funds had no role in study design, data collection and analysis, decision to publish and preparation of the manuscript. No other external funding sources for this study. W van Criekinge is an employee for MDXHealth. The other authors have declared no conflicts of interest.
Funding
This work was funded by the CTMM Air Force consortium (http://www.ctmm.nl). CTMM pays for the salaries of M Clausen and M Mastik (partly), S Denil is supported by an IWT grant (SB101371).
References
- 1.Van Dijk BA, Gatta G, Capocaccia R, Pierannunzio D, Strojan P, Licitra L, Group RW. Rare cancers of the head and neck area in Europe. Eur J Cancer 2012; 48:783-96; PMID:22051735; http://dx.doi.org/ 10.1016/j.ejca.2011.08.021 [DOI] [PubMed] [Google Scholar]
- 2.Gourin CG, Conger BT, Porubsky ES, Sheils WC, Bilodeau PA, Coleman TA. The effect of occult nodal metastases on survival and regional control in patients with head and neck squamous cell carcinoma. Laryngoscope 2008; 118:1191-4; PMID:18391764; http://dx.doi.org/ 10.1097/MLG.0b013e31816e2eb7 [DOI] [PubMed] [Google Scholar]
- 3.Layland MK, Sessions DG, Lenox J. The influence of lymph node metastasis in the treatment of squamous cell carcinoma of the oral cavity, oropharynx, larynx, and hypopharynx: N0 versus N+. Laryngoscope 2005; 115:629-39; PMID:15805872; http://dx.doi.org/ 10.1097/01.mlg.0000161338.54515.b1 [DOI] [PubMed] [Google Scholar]
- 4.Howlader N, Noone AM, Krapcho M, Garshell J, Miller D, Altekruse SF, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ CK SEER Cancer Statistics Review (CSR), 1975-2012 [Internet]. 2015; Available from: http://seer.cancer.gov/archive/csr/1975_2012/ [Google Scholar]
- 5.Baatenburg de Jong RJ, Rongen RJ, Laméris JS, Harthoorn M, Verwoerd CD, Knegt P. Metastatic neck disease. Palpation vs ultrasound examination. Arch Otolaryngol Head Neck Surg 1989; 115:689-90; PMID:2655666; http://dx.doi.org/ 10.1001/archotol.1989.01860300043013 [DOI] [PubMed] [Google Scholar]
- 6.Schöder H, Carlson DL, Kraus DH, Stambuk HE, Gönen M, Erdi YE, Yeung HWD, Huvos AG, Shah JP, Larson SM, et al.. 18F-FDG PET/CT for detecting nodal metastases in patients with oral cancer staged N0 by clinical examination and CT/MRI. J Nucl Med 2006; 47:755-62; PMID:16644744. [PubMed] [Google Scholar]
- 7.Liao LJ, Lo WC, Hsu WL, Wang CT, Lai MS. Detection of cervical lymph node metastasis in head and neck cancer patients with clinically N0 neck-a meta-analysis comparing different imaging modalities. BMC Cancer 2012; 12:236; PMID:22691269; http://dx.doi.org/ 10.1186/1471-2407-12-236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu HR, Kim JH, Yoon DY, Hwang HS, Rho Y-S. Additional diagnostic value of (18)F-FDG PET-CT in detecting retropharyngeal nodal metastases. Otolaryngol Head Neck Surg 2009; 141:633-8; PMID:19861203; http://dx.doi.org/ 10.1016/j.otohns.2009.08.008 [DOI] [PubMed] [Google Scholar]
- 9.Melchers LJ, Schuuring E, van Dijk BAC, de Bock GH, Witjes MJH, van der Laan BFAM, van der Wal JE, Roodenburg JLN. Tumour infiltration depth ≥4 mm is an indication for an elective neck dissection in pT1cN0 oral squamous cell carcinoma. Oral Oncol 2012; 48:337-42; PMID:22130455; http://dx.doi.org/ 10.1016/j.oraloncology.2011.11.007 [DOI] [PubMed] [Google Scholar]
- 10.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev 2011; 11:726-34; PMID:21941284; http://dx.doi.org/22836879 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev 2012; 13:679-92; PMID:22945394; http://dx.doi.org/22836879 10.1038/nrg3270 [DOI] [PubMed] [Google Scholar]
- 12.Roossink F, de Jong S, Wisman GB, van der Zee AG, Schuuring E. DNA hypermethylation biomarkers to predict response to cisplatin treatment, radiotherapy or chemoradiation: the present state of art. Cell Oncol (Dordr) 2012; 35:231-41; PMID:22836879; http://dx.doi.org/ 10.1007/s13402-012-0091-7 [DOI] [PubMed] [Google Scholar]
- 13.Kwon MJ, Kwon JH, Nam ES, Shin HS, Lee DJ, Kim JH, Rho YS, Sung CO, Lee WJ, Cho SJ. TWIST1 promoter methylation is associated with prognosis in tonsillar squamous cell carcinoma. Hum Pathol 2013; 44:1722-9; PMID:23664538; http://dx.doi.org/ 10.1016/j.humpath.2013.03.004 [DOI] [PubMed] [Google Scholar]
- 14.Zhang P, Wang J, Gao W, Yuan BZ, Rogers J, Reed E. CHK2 kinase expression is down-regulated due to promoter methylation in non-small cell lung cancer. Mol Cancer 2004; 3:14; PMID:15125777; http://dx.doi.org/ 10.1186/1476-4598-3-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao T, He B, Pan Y, Gu L, Chen L, Nie Z, Xu Y, Li R, Wang S. H19 DMR methylation correlates to the progression of esophageal squamous cell carcinoma through IGF2 imprinting pathway. Clin Transl Oncol 2013; 16:410-7; PMID:23943562; http://dx.doi.org/ 10.1007/s12094-013-1098-x [DOI] [PubMed] [Google Scholar]
- 16.Pierini S, Jordanov SH, Mitkova AV, Chalakov IJ, Melnicharov MB, Kunev KV, Mitev VI, Kaneva RP, Goranova TE. Promoter hypermethylation of CDKN2A, MGMT, MLH1 and DAPK genes in laryngeal squamous cell carcinoma and their associations with clinical profiles of the patients. Head Neck 2013; 36:1103-8; PMID:23804521; http://dx.doi.org/ 10.1002/hed.23413 [DOI] [PubMed] [Google Scholar]
- 17.Melchers LJ, Clausen MJAM, Mastik MF, Slagter-Menkema L, van der Wal JE, Wisman GBA, Roodenburg JLN, Schuuring E. Identification of methylation markers for the prediction of nodal metastasis in oral and oropharyngeal squamous cell carcinoma. Epigenetics 2015; 10:850-60; PMID:26213212; http://dx.doi.org/ 10.1080/15592294.2015.1075689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clausen MJAM, Melchers LJ, Mastik MF, Slagter-Menkema L, Groen HJM, van der Laan BFAM, van Criekinge W, de Meyer T, Denil S, Wisman GBA, et al.. Identification and validation of WISP1 as an epigenetic regulator of metastasis in oral squamous cell carcinoma. Gene Chromosomes Canc er 2016; 55:45-59; PMID:26391330; http://dx.doi.org/16485345 10.1002/gcc.22310 [DOI] [PubMed] [Google Scholar]
- 19.Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov 2006; 5:37-50; PMID:16485345; http://dx.doi.org/ 10.1038/nrd1930 [DOI] [PubMed] [Google Scholar]
- 20.Roepman P, Wessels LF, Kettelarij N, Kemmeren P, Miles AJ, Lijnzaad P, Tilanus MG, Koole R, Hordijk GJ, van der Vliet PC, et al.. An expression profile for diagnosis of lymph node metastases from primary head and neck squamous cell carcinomas. Nat Genet 2005; 37:182-6; PMID:15640797; http://dx.doi.org/ 10.1038/ng1502 [DOI] [PubMed] [Google Scholar]
- 21.Roepman P, Kemmeren P, Wessels LF, Slootweg PJ, Holstege FC. Multiple robust signatures for detecting lymph node metastasis in head and neck cancer. Cancer Res 2006; 66:2361-6; PMID:16489042; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-3960 [DOI] [PubMed] [Google Scholar]
- 22.van Hooff SR, Leusink FK, Roepman P, Baatenburg de Jong RJ, Speel EJ, van den Brekel MW, van Velthuysen ML, van Diest PJ, van Es RJ, Merkx MA, et al.. Validation of a gene expression signature for assessment of lymph node metastasis in oral squamous cell carcinoma. J Clin Oncol 2012; 30:4104-10; PMID:23045589; http://dx.doi.org/ 10.1200/JCO.2011.40.4509 [DOI] [PubMed] [Google Scholar]
- 23.Melchers LJ, Bruine de Bruin L, Schnell U, Slagter-Menkema L, Mastik MF, de Bock GH, van Dijk BA, Giepmans BN, van der Laan BF, van der Wal JE, et al.. Lack of claudin-7 is a strong predictor of regional recurrence in oral and oropharyngeal squamous cell carcinoma. Oral Oncol 2013; 49:998-1005; PMID:23953778; http://dx.doi.org/ 10.1016/j.oraloncology.2013.07.008 [DOI] [PubMed] [Google Scholar]
- 24.Melchers LJ, Mastik MF, Samaniego Cameron B, van Dijk BA, de Bock GH, van der Laan BF, van der Vegt B, Speel EJ, Roodenburg JL, Witjes MJ, et al.. Detection of HPV-associated oropharyngeal tumours in a 16-year cohort: more than meets the eye. Br J Cancer 2015; 112:1349-57; PMID:25867270; http://dx.doi.org/ 10.1038/bjc.2015.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang N, Eijsink JJ, Lendvai A, Volders HH, Klip H, Buikema HJ, van Hemel BM, Schuuring E, van der Zee AG, Wisman GB. Methylation markers for CCNA1 and C13ORF18 are strongly associated with high-grade cervical intraepithelial neoplasia and cervical cancer in cervical scrapings. Cancer Epidemiol Biomarkers Prev 2009; 18:3000-7; PMID:19843677; http://dx.doi.org/ 10.1158/1055-9965.EPI-09-0405 [DOI] [PubMed] [Google Scholar]
- 26.Eijsink JJ, Lendvai A, Deregowski V, Klip HG, Verpooten G, Dehaspe L, de Bock GH, Hollema H, van Criekinge W, Schuuring E, et al.. A four-gene methylation marker panel as triage test in high-risk human papillomavirus positive patients. Int J cancerJournal Int du cancer 2012; 130:1861-9; PMID:21796628; http://dx.doi.org/14671650 10.1002/ijc.26326 [DOI] [PubMed] [Google Scholar]
- 27.van Dongen JJ, Langerak AW, Bruggemann M, Evans PA, Hummel M, Lavender FL, Delabesse E, Davi F, Schuuring E, Garcia-Sanz R, et al.. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003; 17:2257-317; PMID:14671650; http://dx.doi.org/ 10.1038/sj.leu.2403202 [DOI] [PubMed] [Google Scholar]
- 28.Langmead B, Trapnell C, Pop M, Salzberg SL Ultrafast and memory efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10:R25; PMID:19261174; http://dx.doi.org/ 20698981 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hardcastle TJ, Kelly KA. baySeq: empirical Bayesian methods for identifying differential expression in sequence count data. BMC Bioinformatics 2010; 11:422-35; PMID:20698981; http://dx.doi.org/ 10.1186/1471-2105-11-422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uniport Consortium . Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res 2014; 42:D191-8; PMID:24253303; http://dx.doi.org/ 10.1093/nar/gkt1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics 2008; 24:1547-8; PMID:18467348; http://dx.doi.org/ 10.1093/bioinformatics/btn224 [DOI] [PubMed] [Google Scholar]
- 32.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015; 43(7):e47; PMID:25605792; http://dx.doi.org/22588877 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al.. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2:401-4; PMID:22588877; http://dx.doi.org/ 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al.. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6:pl1; PMID:23550210; http://dx.doi.org/ 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.R Development Core Team R: A Language and Environment for Statistical Computing [Internet]. 2015; Available from: http://www.R-project.org/ [Google Scholar]
- 36.Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res 2015; 43:D146-52; PMID:25378301; http://dx.doi.org/ 10.1093/nar/gku1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng JM, Ding M, Aribi A, Shah P, Rao K. Loss of RAB25 expression in breast cancer. Int J cancer 2006; 118:2957-64; PMID:16395697; http://dx.doi.org/22991305 10.1002/ijc.21739 [DOI] [PubMed] [Google Scholar]
- 38.Tong M, Chan KW, Bao JY, Wong KY, Chen JN, Kwan PS, Tang KH, Fu L, Qin YR, Lok S, et al.. Rab25 is a tumor suppressor gene with antiangiogenic and anti-invasive activities in esophageal squamous cell carcinoma. Cancer Res 2012; 72:6024-35; PMID:22991305; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-1269 [DOI] [PubMed] [Google Scholar]
- 39.Tellez-Gabriel M, Arroyo-Solera I, Leon X, Gallardo A, Lopez M, Cespedes MV, Casanova I, Lopez-Pousa A, Quer M, Mangues MA, et al.. High RAB25 expression is associated with good clinical outcome in patients with locally advanced head and neck squamous cell carcinoma. Cancer Med 2013; 2:950-63; PMID:24403269; http://dx.doi.org/ 10.1002/cam4.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chua CE, Tang BL. The role of the small GTPase Rab31 in cancer. J Cell Mol Med 2015; 19:1-10; PMID:25472813; http://dx.doi.org/ 10.1111/jcmm.12403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mitra S, Cheng KW, Mills GB. Rab25 in cancer: a brief update. Biochem Soc Trans 2012; 40:1404-8; PMID:23176489; http://dx.doi.org/ 10.1042/BST20120249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng KW, Lahad JP, Gray JW, Mills GB. Emerging role of RAB GTPases in cancer and human disease. Cancer Res 2005; 65:2516-9; PMID:15805241; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-0573 [DOI] [PubMed] [Google Scholar]
- 43.Recchi C, Seabra MC. Novel functions for Rab GTPases in multiple aspects of tumour progression. Biochem Soc Trans 2012; 40:1398-403; PMID:23176488; http://dx.doi.org/ 10.1042/BST20120199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng KW, Lahad JP, Kuo WL, Lapuk A, Yamada K, Auersperg N, Liu J, Smith-McCune K, Lu KH, Fishman D, et al.. The RAB25 small GTPase determines aggressiveness of ovarian and breast cancers. Nat Med 2004; 10:1251-6; PMID:15502842; http://dx.doi.org/ 10.1038/nm1125 [DOI] [PubMed] [Google Scholar]
- 45.Zhang J, Wei J, Lu J, Tong Z, Liao B, Yu B, Zheng F, Huang X, Chen Z, Fang Y, et al.. Overexpression of Rab25 contributes to metastasis of bladder cancer through induction of epithelial-mesenchymal transition and activation of Akt/GSK-3beta/Snail signaling. Carcinogenesis 2013; 34:2401-8; PMID:23722651; http://dx.doi.org/ 10.1093/carcin/bgt187 [DOI] [PubMed] [Google Scholar]
- 46.Cao C, Lu C, Xu J, Zhang J, Zhang J, Li M. Expression of Rab25 correlates with the invasion and metastasis of gastric cancer. Chin J Cancer Res 2013; 25:192-9; PMID:23592900; http://dx.doi.org/ 10.3978/j.issn.1000-9604.2013.03.01 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yin YX, Shen F, Pei H, Ding Y, Zhao H, Zhao M, Chen Q. Increased expression of Rab25 in breast cancer correlates with lymphatic metastasis. Tumour Biol 2012; 33:1581-7; PMID:22644676; http://dx.doi.org/ 10.1007/s13277-012-0412-5 [DOI] [PubMed] [Google Scholar]
- 48.Amornphimoltham P, Rechache K, Thompson J, Masedunskas A, Leelahavanichkul K, Patel V, Molinolo A, Gutkind JS, Weigert R. Rab25 regulates invasion and metastasis in head and neck cancer. Clin Cancer Res 2013; 19(6):1375-88; PMID:23340300; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng JM, Volk L, Janaki DK, Vyakaranam S, Ran S, Rao KA Tumor suppressor function of Rab25 in triple-negative breast cancer. Int J Cancer 2010; 126(12):2799-81; PMID:19795443; http://dx.doi.org/22174824 10.1002/ijc.24900 [DOI] [PubMed] [Google Scholar]
- 50.Wrzeszczynski KO, Varadan V, Byrnes J, Lum E, Kamalakaran S, Levine DA, Dimitrova N, Zhang MQ, Lucito R. Identification of tumor suppressors and oncogenes from genomic and epigenetic features in ovarian cancer. PLoS One 2011; 6:e28503; PMID:22174824; http://dx.doi.org/ 10.1371/journal.pone.0028503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Esteller M. Epigenetics in cancer. N Engl J Med 2008; 358:1148-59; PMID:18337604; http://dx.doi.org/ 10.1056/NEJMra072067 [DOI] [PubMed] [Google Scholar]
- 52.Azad N, Zahnow CA, Rudin CM, Baylin SB. The future of epigenetic therapy in solid tumours-lessons from the past. Nat Rev Oncol 2013; 10:256-66; PMID:23546521; http://dx.doi.org/24656975 10.1038/nrclinonc.2013.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arantes LM, de Carvalho AC, Melendez ME, Carvalho AL, Goloni-Bertollo EM. Methylation as a biomarker for head and neck cancer. Oral Oncol 2014; 50:587-92; PMID:24656975; http://dx.doi.org/ 10.1016/j.oraloncology.2014.02.015 [DOI] [PubMed] [Google Scholar]
- 54.Wu CY, Tseng RC, Hsu HS, Wang YC, Hsu MT. Frequent down-regulation of hRAB37 in metastatic tumor by genetic and epigenetic mechanisms in lung cancer. Lung Cancer 2009; 63:360-7; PMID:18687502; http://dx.doi.org/ 10.1016/j.lungcan.2008.06.014 [DOI] [PubMed] [Google Scholar]
- 55.Tanaka T, Arai M, Wu S, Kanda T, Miyauchi H, Imazeki F, Matsubara H, Yokosuka O. Epigenetic silencing of microRNA-373 plays an important role in regulating cell proliferation in colon cancer. Oncol Rep 2011; 26:1329-35; PMID:21785829; http://dx.doi.org/ 10.3892/or.2011.1401 [DOI] [PubMed] [Google Scholar]
- 56.Price ME, Cotton AM, Lam LL, Farre P, Emberly E, Brown CJ, Robinson WP, Kobor MS. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin 2013; 6:4; PMID:23452981; http://dx.doi.org/ 10.1186/1756-8935-6-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.