

ABSTRACT
Despite their central function in tumor immunity, dendritic cells (DCs) can respond to inhibitory signals and become tolerogenic, curtailing T cell responses in vivo. Here, we provide the evidence for an inhibitory function of signal regulatory protein (SIRP) α in DC survival and activation. In tumors from human liver cancer patients, infiltrative DCs expressed elevated levels of SIRPα, which is correlated with the induction of immune tolerance within the tumors. Silencing of SIRPα resulted in a significant increase in the longevity of antigen-pulsed DCs in the draining lymph nodes. In addition, SIRPα controls the activation and output of DCs. Silencing of DC-expressed SIRPα induced spontaneous and enhanced production of IL12 and costimulatory molecules, resulting in more potent cytotoxic T lymphocyte responses, including the eradication of previously established solid tumors. SIRPα exerted such effects, at least in part, via the association and sequestration of p85 subunit of PI3K. Thus, SIRPα is a critical regulator of DC lifespan and activity, and its inhibition might improve the clinical efficacy of DC-based tumor vaccines.
KEYWORDS: Akt, antitumor immunity, dendritic cells, SIRPα
Abbreviations
- BMDCs
bone marrow-derived dendritic cells
- CCR
chemokine receptor
- CFSE
carboxyfluorescein succinimidyl ester
- CTL
cytotoxic T lymphocyte
- DCs
dendritic cells
- GM-CSF
granulocyte macrophage colony-stimulating factor
- IgSF
immunoglobulin superfamily
- MHC
major histocompatibility complex
- NF-κB
nuclear factor-κB
- PBDCs
peripheral blood-derived dendritic cells
- PI3K
phosphoinositide 3-kinase
- SHP2
SH2-domain-containing protein tyrosine phosphatase 2
- SIRPα
signal regulatory protein α
- TLRs
Toll-like receptors
- TRANCE
tumor necrosis factor–related activation-induced cytokine
- TRP-2
tyrosinase-related protein-2
Introduction
DCs are characterized by a high capacity for antigen capture and processing, migration to lymphoid organs and the expression of various costimulatory molecules for antigen-specific lymphocyte activation, which endows them with key roles in the adaptive immune system.1 The unique machinery of DCs makes them ideal natural adjuvants for cancer vaccines. Nevertheless, tumors do not induce an inflammatory response conducive for optimal activation of DCs, and as a result, the ensuing immune response is weak and ineffective.2,3
The immunostimulatory properties of DC-based vaccines are limited by their short lifespan and transient activation state within lymphoid tissues.4 The lifespan of activated antigen-bearing DCs has been estimated to be as short as 3 d. Prolonging the lifespan of DCs by means of anti-apoptosis can increase the availability of antigen for T cell stimulation and promote immune responses.5-7 The survival of activated DCs is partly regulated by pathogen-derived molecules, acting through one or more evolutionarily conserved Toll-like receptors (TLRs),8-10 as well as T cell-expressed costimulatory molecules, such as CD40 ligand (CD40L)11 and TRANCE (tumor necrosis factor–related activation-induced cytokine) whose anti-apoptotic activities depend on Bcl-2 family members and nuclear factor (NF)-κB subunits.12 Although the importance of TLRs and CD40 in DC longevity and activity is well documented, rapid turnover of mature DCs in secondary lymphoid organs suggests that DCs possess several negative feedback mechanisms that might allow them to control the magnitude and duration of adaptive immunity beyond the point of maturation.13 Therefore, inhibition of key intrinsic negative regulatory signaling is likely to enable development of a general strategy to augment DC longevity and activity, thus enhancing DC-based vaccines.
SIRPα, a plasma-membrane protein expressed mainly by myeloid cells, including DCs, contains putative tyrosine phosphorylation sites in its cytoplasmic region that conform loosely to inhibitory immunoreceptor tyrosine-based inhibition motifs (ITIMs). These motifs mediate the association of SIRPα with the phosphatase SH2-domain-containing protein tyrosine phosphatase 2 (SHP2).14 The extracellular region consists of three immunoglobulin superfamily (IgSF) domains which bind to either widely expressed transmembrane ligand CD4715 or soluble ligands, such as the surfactant proteins A and D, which are copious in the lungs.16 Interaction of CD47 with SIRPα negatively regulates phagocytosis of host cells by macrophages.17-21 Consistently, anti-CD47 antibody or engineered SIRPα variants with increased affinity for CD47 can induce potent tumoricidal activity of macrophage by antagonizing CD47 on cancer cells.22,23 In lungs, ligation of SIRPα to macrophages by surfactant proteins is required to keep the activity of alveolar macrophages in check, thus preventing damage to the airways caused by pro-inflamatory responses.16,24 These data strongly suggest that SIRPα induces inhibitory signals in immune response. Here, we demonstrate that the expression level of SIRPα represents a threshold and self-limiting factor for DC activation, most likely by regulating the lifespan and adjuvant capacity of these cells.
Results
SIRPα is upregulated in tumor-associated DCs
To investigate the role of SIRPα in DC activation, we treated mouse bone marrow-derived DCs (BMDCs) and human peripheral blood-derived DCs (PBDCs) with LPS, CpG and polyI:C and then measured the levels of SIRPα protein. A transient downregulation and a subsequent restoration of SIRPα expression were observed in both cells after stimulation (Fig. 1A and B), suggesting that downregulation of SIRPα is integral in DC activation. We then explored whether SIRPα expression is altered in tumor-associated DCs. The expression levels of SIRPα in infiltrative CD11c+ DCs in 10 hepatocellular carcinomas (HCCs) were compared with those in CD11c+ PBDCs obtained from the same patients. PBDCs from five normal healthy people were used as a control. In PBDCs from patients with HCC, SIRPα was marginally upregulated compared with SIRPα expression in the PBDC control (Fig. 1C). However, its expression was significantly higher in tumor-associated CD11c+ DCs than in autologous CD11c+ PBDCs. These results indicate that the expression level of SIRPα seems to be inversely linked to DC activation both in vitro and in vivo (Fig. 1C).
Figure 1.
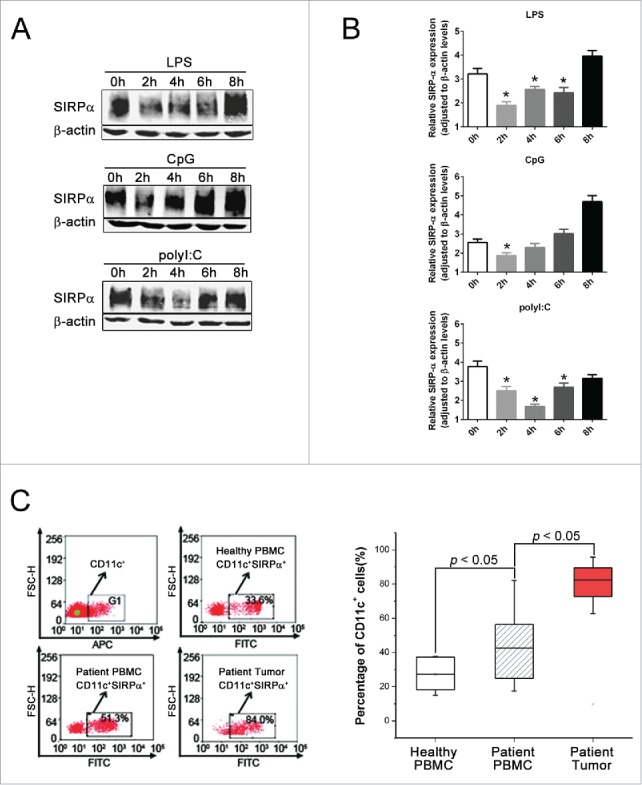
SIRPα is upregulated in tumor-associated DCs. (A & B) Mouse BMDCs from C57BL/6 mice were incubated with 100 ng/mL LPS, 5 μg/mL CpG or 0.25 μg/mL polyI:C for the indicated time, and SIRPα protein expression levels were ascertained by Western blotting. (C) FACS analysis of surface expression of SIRPα in CD11c+ DCs infiltrated in HCC and in autologous CD11c+ PBDCs. PBDCs from five normal healthy people were used as control.
SIRPα controls the maturation and cytokine production of DCs
To elucidate the role of SIRPα in DC function, we generated a recombinant lentiviral vector expressing an intefering miRNA that effectively silences SIRPα expression (LV-miSIRPα) in mouse BMDCs (mDCs) (Fig. 2A). mDCs transduced with LV-miSIRPα spontaneously produced large amounts of IL-12 as compared with LV-GFP-transduced DCs or PBS controls (Fig. 2B). We also observed a higher expression of maturation markers, including CD80, CD86 and major histocompatibility complex (MHC) class II, in LV-miSIRPα-DCs or LV-shSIRPα-DCs (Fig. 2C, Fig. S1 and S2), suggesting an enhanced maturation in SIRPα-silenced DCs. One important outcome of the maturation process is that DCs acquire the capacity to home to lymph nodes.25 SIRPα silencing in DCs resulted in elevated expression of chemokine receptor CCR7 (Fig. 2D), which directs DCs to secondary lymphoid nodes.26 In contrast, the chemokine receptor CCR5, which is thought to be involved in recruitment of immature DC to tissues,27 had reduced expression in DCs transduced with LV-miSIRPα compared with GFP control. Consistent with that observation, mDCs expressing exogenous SIRPα (adenovirally directed overexpression of SIRPα, AV-SIRPα) showed considerably decreased IL-12 production and maturation marker expression (Fig. 2E and F). Taken together, these data suggest that SIRPα could negatively regulate DC activation and maturation.
Figure 2.
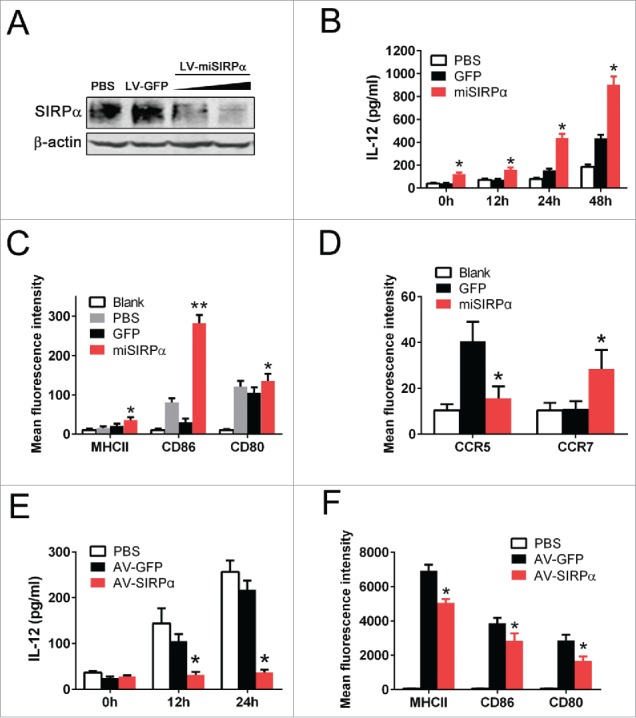
SIRPα controls the maturation and cytokine production of DCs. (A) Mouse BMDCs were infected with lentiviral vectors expressing miRNA targeting SIRPα or GFP control. The expression levels of SIRPα were analyzed 72 h after infection at an MOI of 50 or 80. (B) ELISA measurement of IL-12 production at the indicated times after lentiviral infection. Experiments were repeated three times yielding similar results. *p < 0.05 versus GFP controls. (C) FACS analysis of surface expression of costimulatory factors and MHC class II molecules on DCs 48 h after lentiviral infection. (D) FACS analysis of surface expression of CCR7 or CCR5 on DCs 48 h after lentiviral infection. Experiments were repeated three times yielding similar results. (E) ELISA measurement of IL-12 production at the indicated times after adenoviral infection. AV-GFP, adenoviral vector expressing GFP control; AV-SIRPα, adenoviral vector expressing exogenous SIRPα. (F) FACS analysis of surface expression of costimulatory factors and MHC class II molecules on DCs 48 h after adenoviral infection. Experiments were repeated three times with similar results.
SIRPα restrains the survival and homeostasis of DCs in lymphoid tissues
In addition to enhancing mDC maturation and cytokine production, silencing of SIRPα significantly prolonged DC lifespan after GM-CSF deprivation (Fig. 3A). By contrast, adenovirally directed overexpression of SIRPα (AV-SIRPα) significantly reduced the survival of DCs relative to adenoviral GFP transduction (Fig. 3B), revealing an inhibitory function for SIRPα in DC survival. To determine whether SIRPα contributes to the survival and maturation of DCs in vivo, we delivered peptide-pulsed, CFSE-stained and LPS-treated miSIRPα-DCs or GFP-DCs subcutaneously to syngeneic mice. We found that the percentage of CFSE+ miSIRPα-DCs in draining popliteal lymph nodes was significantly higher than that of CFSE+ GFP-DCs (Fig. 3C). This disproportionate representation was sustained for at least 10 d after DC delivery. Moreover, the average volume of draining lymph nodes exposed to miSIRPα-DCs was enlarged at least 80–100% compared with that of control mice, indicating that LV-miSIRPα transduction enhanced the number and longevity of leukocytes resident in secondary lymphoid tissues (Fig. 3D). Collectively, these results indicate that the increased survival and activation of miSIRPα-DCs resulted in increases in the size, cellularity and functions of secondary lymphoid organs.
Figure 3.
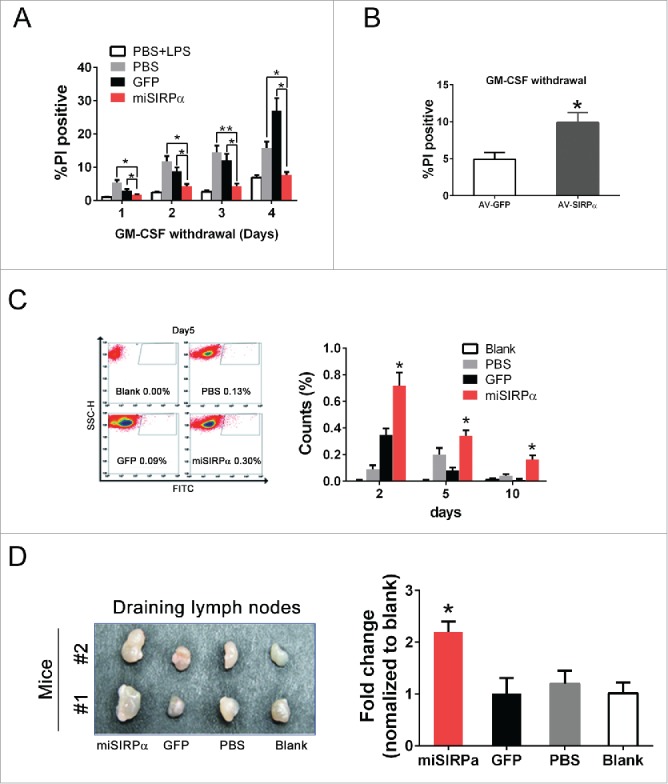
SIRPα restrains the survival and homeostasis of DCs in lymphoid tissues. (A & B) DC survival was negatively regulated by SIRPα. Viabilities of LV-miSIRPα-DCs, LV-GFP-DCs and other controls (A) or AV-SIRPα-DCs and AV-GFP control (B) were assayed by propidium iodide staining after 1–4 d of GM-CSF deprivation culture followed by analysis with flow cytometry. (C) BMDCs were either transduced with LV-miSIRPα or controls followed by LPS treatment. BMDCs were then stained with CFSE and injected into hind legs of syngeneic mice. Draining popliteal lymph nodes were harvested at the indicated times after DC injection, and PtdIns-negative populations were analyzed by flow cytometry. Experiments were repeated three times yielding similar results. (D) The secondary lymphoid organs were examined on day 10 after DC injection. Quantification of lymphoid organ volumes is shown in the right panel. *p < 0.05 vs. GFP controls.
SIRPα-silent DCs prime enhanced T cell responses
The enhanced survival and activation exhibited by miSIRPα-DCs led us to hypothesize that they might also promote antigen-specific cytotoxic T lymphocyte (CTL) responses. Therefore, we evaluated the CD8+ OT-I T cell proliferative response to ovalbumin (OVA)-pulsed DCs by thymidine incorporation or CFSE labeling in vitro. As expected, silencing of SIRPα or anti-CD47 Ab blockage significantly enhanced the proliferation of CD8+ T cells relative to LV-GFP or PBS controls (Fig. 4A and B).
Figure 4.

SIRPα-silent DCs prime enhanced T cell responses. (A & B) Stimulatory effect of LV-miSIRPα-DCs on CD8+ T cell proliferation in vitro. CD8+ OT-I-specific T cells isolated from the spleens of OT-I-transgenic mice were CFSE stained and co-cultured with irradiated DCs pulsed with H-2Kb OVA-I peptide in triplicate or CD47 antibody. [3H]TdR incorporation rates were then measured (A), and CFSE positive cells were counted microscopically (B); those shown are representative of three independent experiments. (C & D) IFNγ ELISPOT assays of splenocytes, CD4+ and CD8+ T cell responses to OVA (C) or TRP-2. (D) C57BL/6 mice were immunized with OVA or TRP2-pulsed (100 mg/mL), lentivirally transduced DCs ex vivo that had been matured with LPS, followed by no stimulation (left) or in vivo stimulation with polyI:C (50 mg/mouse i.p; right) daily for three consecutive days. Splenocytes pooled from immunized mice (two or three per group) or isolated CD8+ T cells or CD4+ T cells were subjected to IFNγ ELISPOT assays. Experiments were repeated three times with similar results. p < 0.05, miSIRPα-DCs compared with GFP-DCs. (E) Stimulatory effect of LV-miSIRPα-DCs on antigen-specific CD8+ T proliferation in vivo. Mice were immunized three times at 2-week intervals with LPS matured LV-miSIRPα-DCs or controls that had been pulsed with TRP2 ex vivo. Two months later, 1 week after a booster DC vaccine, data for TRP-2-APC-Pentamer+ and CD8+ T cells were collected from one of three independent experiments. *p < 0.05 versus GFP and PBS controls.
To further test whether the immunostimulatory potency of DCs is regulated by SIRPα in vivo, we transduced mDCs with LV-miSIRPα or LV-GFP lentiviral vectors, pulsed the transduced cells with OVA (Fig. 4C) or self-tumor associated antigen tyrosinase-related protein-2 (TRP-2) (Fig. 4D), matured the cells ex vivo with LPS and then subcutaneously injected them into syngeneic C57BL/6 mice. The functional status of the T cells was evaluated using interferon-γ (IFNγ) enzyme-linked immunosorbent spot (ELISPOT) assays. Mice injected with miSIRPα-DCs had significantly greater numbers of antigen-specific IFNγ+CD4+, IFNγ+CD8+ T cells and IFNγ+ splenocytes than did mice injected with GFP-DCs. Moreover, in vivo ligand (polyI:C) stimulation more effectively boosted antigen-specific CTL responses in miSIRPα-DC recipients than in GFP-DC recipients.
We next tested the ability of SIRPα-silent DCs to prime an antigen-specific response in vivo by directly immunizing mice with TRP-2-pulsed, transduced DCs with ex vivo maturation by LPS. Pentamer staining showed that 28.8% of total CD8+ T cells were positive for TRP-2-pentamer in mice immunized with LV-miSIRPα-DCs, compared with only 15.7% and 13.5% in mice immunized with LV-GFP-DCs or PBS-DCs, respectively (Fig. 4E). These data indicate that silencing of SIRPα likely reduces the threshold of DC responsiveness, resulting in a high magnitude of T cell responses.
SIRPα inhibits DC activation and survival through sequestration of p85
The PI3K/Akt pathway is a key regulator of DC lifespan and is required for DC maturation and survival.28 As SIRPα has been shown to recruit and signal via p85 subunit of PI3K, we thus investigated whether PI3K mediated SIRPα regulation of DC function. As shown in Fig. 5A, endogenous SIRPα was constitutively bound to p85 in DCs. Silencing of SIRPα led to a significant activation of PI3K as indicated by in vitro PIP3 production (Fig. 5B) while overexpression of SIRPα inhibited LPS-induced Akt phosphorylation, which normally occurs as a consequent event of PI3K activation (Fig. 5C). These results indicate that SIRPα might compete with the p110 catalytic subunit to bind p85, thus inhibiting PI3K activity. Likewise, overexpression of a dominant negative mutant of Akt (AV-AKT-T308A/S473A) significantly reduced IL-12 production in DCs transduced with LV-miSIRPα relative to AV-GFP controls (Fig. 5D). We also tested the effects of selective inhibitors against PI3K (wortmannin, Ly294002) on LV-miSIRPα-mediated DC activation and survival. Inhibition of their activities abrogated LV-miSIRPα-mediated IL-12 production (Fig. 5E) and DC maturation (Fig. 5F), suggesting that the PI3K/Akt pathway is involved in LV-miSIRPα-mediated DC activation. PI3K inhibitors also substantially prevented the LV-miSIRPα-mediated rescue of DC death following GM-CSF deprivation (Fig. 5G). Furthermore, adenovirally directed overexpression of a dominant-negative mutant of Akt (AV-AKT-T308A/S473A) greatly reduced the survival of DCs transduced with LV-miSIRPα relative to AV-GFP control (Fig. 5H). These results suggest an essential role of the PI3K/Akt pathway in SIRPα regulation of DC survival and activation.
Figure 5.
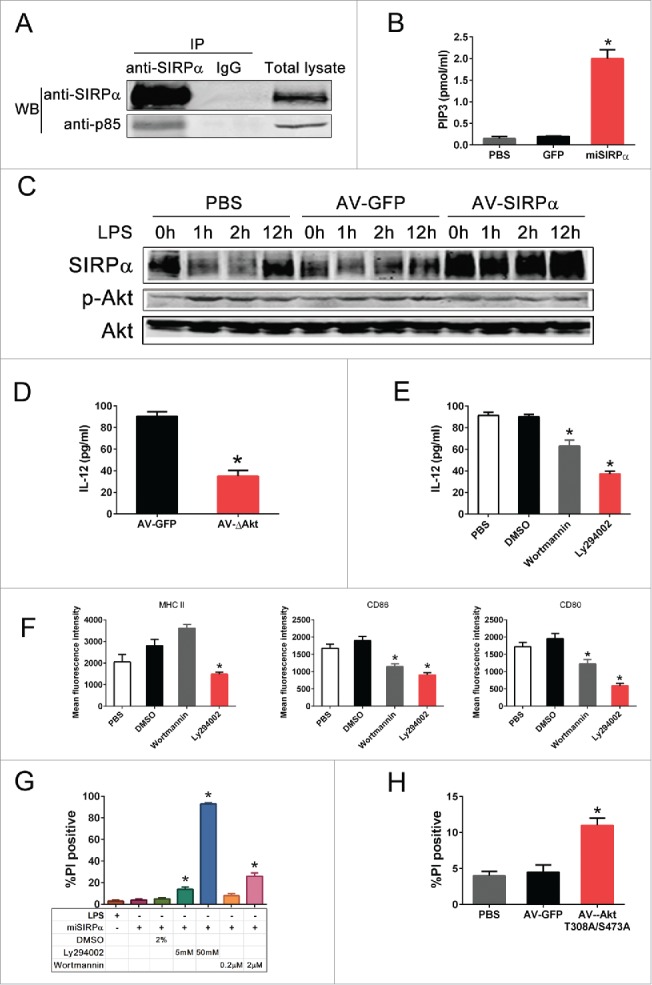
SIRPα inhibits DC activation and survival through sequestration of p85. (A) BMDC lysates were immunoprecipitated for endogenous SIRPα and immunoprecipitates were probed with anti-SIRPα and anti-p85 antibodies. (B) PI3-Kinase activity was negatively regulated by SIRPα. BMDC lysates were collected 48 h after infection with LV-miSIRPα, LV-GFP or PBS control and assayed for PI3-Kinase activity by ELISA. (C) BMDCs were infected with adenoviral vectors (AV-GFP and AV-SIRPα) for 24 h and the level of total and phospho-Akt were ascertained by Western blotting after incubation with 100 ng/mL LPS for the indicated times. (D) Overexpression of dominant-negative Akt prevented the LV-miSIRPα-mediated IL-12 production by DCs. After infection with LV-miSIRPα for 24 h, BMDCs were infected with AV-AKT-T308A/S473A or the controls for 48 h, and the amounts of IL-12 secreted were measured by ELISA. The experiments were repeated three times yielding similar results. (E & F) Effects of inhibitors of MAPK or PI3K pathway on LV-miSIRPα-mediated IL-12 production and DC maturation. After infection with LV-miSIRPα for 24 h, BMDCs were incubated with the indicated concentrations of PI3K inhibitors or DMSO control for 24 h. ELISA measurement of IL-12 secreted by DCs and FACS analysis of surface expression of costimulatory and MHC class II molecules are shown from one of three independent experiments. *p < 0.05 vs. DMSO controls. (G) In the presence of PI3K-Akt pathway inhibitors, knockdown of SIRPα expression failed to enhance DC survival. After infection with LV-miSIRPα or incubation with 100ng/mL LPS for 24 h, BMDCs were treated with the indicated concentrations of PI3K inhibitor Ly294002, Wortmannin or DMSO control. Cell viability was assessed 2 d later using PI staining. Experiments were repeated three times yielding similar results. (H) LV-miSIRPα-transduced BMDCs were further infected with adenoviral vectors expressing dominant-negative mutant of Akt (AV-AKT-T308A/S473A) or GFP control. The survival of DCs was examined as described above. *p < 0.05 versus GFP and PBS controls. Experiments were repeated three times with similar results.
SIRPα silencing in DCs enhances antitumor immune effects
The observed regulatory role of SIRPα in DC activation prompted us to test whether SIRPα-silent DCs might induce potent antitumor immunity against subcutaneous E.G7-OVA lymphomas to correlate in vitro findings. Groups of 8 to 10 mice were primed by subcutaneous injection of 2 × 106 DCs that had been pulsed with OVA and matured ex vivo with LPS. One week after vaccination, animals were challenged by local injection of 1 × 106 (E.G7) tumor cells, and tumor growth or overall survival was assessed at regular intervals by a blinded observer to determine therapeutic efficacy. As expected, immunization with a single dose of OVA-pulsed miSIRPα-DCs completely blocked E.G7 tumor growth in all mice tested, whereas inoculation with GFP-DCs or PBS-DCs failed to inhibit tumor growth in most animals (Fig. 6A). In addition, an IFNγ ELISPOT assay showed that vaccination with peptide-pulsed miSIRPα-DCs significantly increased the numbers of antigen-specific IFNγ+CD8+ T cells compared to GFP-DCs and PBS-DCs (Fig. 6B). These data demonstrate that silencing SIRPα is an effective means of priming DCs for the prevention of primary autologous tumor outgrowth after local challenge.
Figure 6.
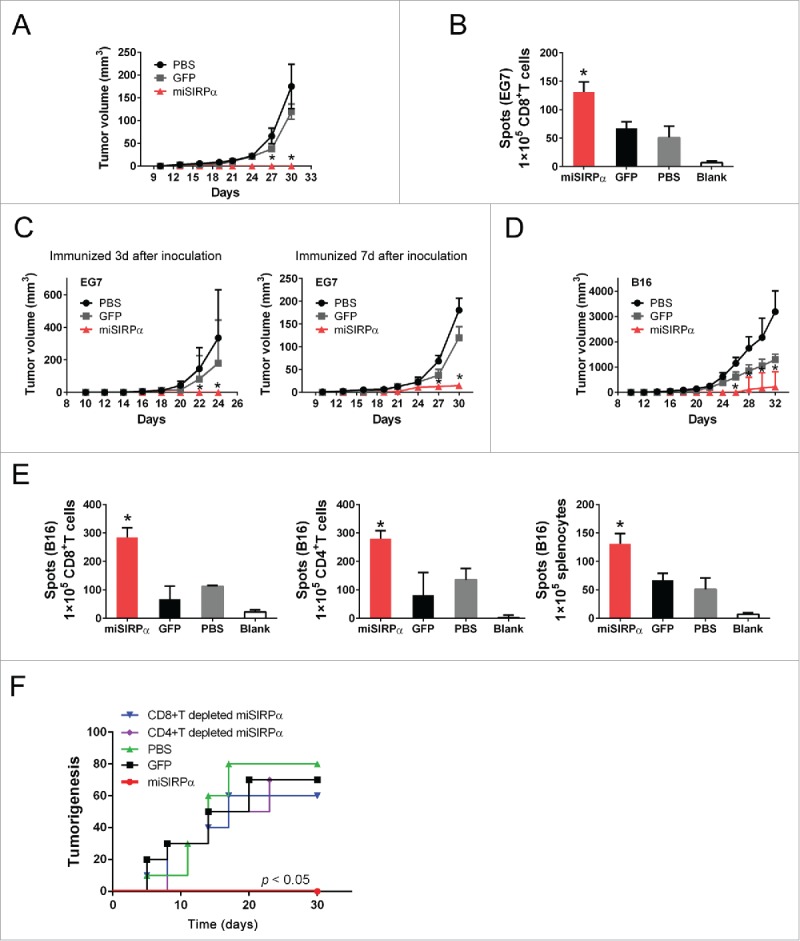
SIRPα silencing in DCs enhances antitumor immune effects. (A & B) Antitumor immunosurveillance activity against EG7. Syngeneic C57BL/6 mice were vaccinated with 2 × 106 LV-miSIRPα–DCs or controls pulsed with OVA peptide 7 d before challenge with 1 × 106 OVA positive EG7 cells. Tumor sizes were measured (A) and CD8+ responses to OVA were assessed by IFNγ ELISPOT assay (B). Tumor growth curves (n = 6 mice per group) represent one of two repeated experiments. p < 0.01, LV-miSIRPα-DCs compared with GFP-DCs. (C & D). Antitumor activity against pre-established or established tumors. C57BL/6 mice were inoculated subcutaneously with 1 × 106 EG7-OVA or 1× 105 B16 tumor cells and 3 d or 7 d later were immunized with 2 × 106 OVA or TRP2-pulsed LV-miSIRPα-DCs or control DCs ex vivo matured with LPS twice at a weekly interval, followed by in vivo polyI:C stimulation i.p. after each DC immunization. Tumor growth curves (n = 6 mice per group) represent one of two repeated experiments. *p < 0.01, LV-miSIRPα-DCs compared with GFP-DCs. (E) IFNγ ELISPOT assays of CD8+ T cell, CD4+ T cell or splenocytes responses to TRP-2 were recorded. *p < 0.01 vs. GFP controls. (F) Characterization of the antitumor immune response induced by LV-miSIRPα-DCs. 1 × 107 pooled splenocytes or 1 × 107 CD8+ or CD4+ T cells isolated from the pooled splenocytes (two or three per group) of different groups of mice were transferred to host mice 1 d before B16 melanoma cell inoculation. Tumorigenesis was monitored for 30 d. Shown are the pooled results of two independent experiments (n = 10).
To determine the efficacy of SIRPα-silent DCs for the treatment of established disease, we exploited the well-established E.G7-OVA lymphomas and poorly immunogenic B16 metastases models. Groups of 10 animals with established melanoma or metastases were inoculated weekly with DCs for 3 weeks. In both the pre-established E.G7 experiments and the B16 model, local tumor outgrowth was significantly inhibited by vaccination with SIRPα-silent DCs, whereas GFP-DC or PBS-DC vaccines failed to inhibit tumor growth in most animals (Fig. 6C and D). Furthermore, we observed that immunization of mature miSIRPα-DCs pulsed with TRP-2 peptide also significantly increased the numbers of antigen-specific IFNγ+CD4+, IFNγ+CD8+ T cells and IFNγ+ splenocytes relative to vaccination with control GFP-DCs (Fig. 6E).
To further evaluate the role of CTLs in miSIRPα-DC-induced tumor protection, we inoculated 10 naive mice with B16 melanoma cells 7 d after injection with DCs. Splenocytes from miSIRPα-DC-treated mice or from control groups were then transferred to these mice 1 d before challenge with B16 melanoma cells. As expected, almost all of the mice that had received splenocytes from the miSIRPα-DC-treated mice were efficiently protected from tumor growth, whereas mice that had received control splenocytes were not (Fig. 6F). To determine the role of CD4+ and CD8+ T cells in tumor protection, we repeated this experiment using splenocytes depleted of CD4+ or CD8+ T cells from miSIRPα-DC-treated mice. One day after T cells infusion, these mice were injected with B16 melanoma cells. As shown in Fig. 5F, both CD4+ T and CD8+ T cells were required to mediate the antitumor effects.
Discussion
Here, we provide the evidence for an inhibitory function of SIRPα in DC survival and activation. The silencing of SIRPα resulted in a significant increase in the number of antigen-pulsed DCs in the draining LNs. The effect of SIRPα knockdown on DC survival in vivo suggests that it might not only increase the survival of DCs at the site of injection but also enhance the longevity of Ag-bearing DCs after migration to the LNs. SIRPα knockdown also led to an upregulation of CCR7, which favor the entry of DCs into afferent lymphatics and homing to the T cell areas.26 These mechanisms might operate together to enhance the longevity of mature DCs in secondary lymphoid tissues and thereby increase adaptive immune responses. Interestingly, SIRPα has also been shown to be necessary for DC homeostasis in lymphoid tissues. The number of CD11c+ DCs was reduced in secondary lymphoid tissues of mice expressing a mutant form of SIRPα that lacks the cytoplasmic region.29 It is of note that this mutant SIRPα is structurally similar to SIRPγ, another member of the SIRP family which is functionally involved in T-cell proliferation and activation.30 As the mutant SIRPα is still able to ligate CD47 on T cells, it is not yet known if this mutant can represent a functionally deficient SIRPα in DCs.
In addition to restraining the survival of DCs, SIRPα also controls the activation and output of DCs. Activation of DCs occurs in two phases: maturation and licensing. We found that SIRPα-silent DCs undergo a maturation process even in the absence of inflammatory stimuli, such as LPS. Furthermore, DCs with SIRPα knockdown acquired an enhanced capacity to home to lymph nodes, one important consequence of the maturation process. On the other hand, miSIRPα-DCs spontaneously produced large amounts of IL-12, a licensing cytokine that mediates the polarization of activated CD4+ T cells to a Th1 phenotype such that they aid in eliciting potent CD8+ CTL responses.3 Thus, by prolonging DC lifespans, inducing optimal maturation and DC activation, accompanied by IL-12 production, silencing of SIRPα could, as was expected, lead to much greater DC-mediated T cell proliferation and IFNγ production. These results suggest that SIRPα-silent DCs might elicit CTL responses via at least two sequential mechanisms: the enhanced DC maturation facilitates initiation of CTL responses and the increased IL-12 production intensifies the magnitude of CTL responses. Moreover, SIRPα-silent DCs might be capable of turning off regulatory T cells by enhancing DC maturation and cytokine production. A recent finding indicates that a thymic SIRPα+ but not SIRPα− conventional DC subset can support the development of central tolerance against circulating peripheral Ags (CCR2) and induce CD4+CD25+ Foxp3+ T reg cells both in vitro and in vivo.31
Signaling events downstream of SIRPα and their functional impact on DC maturation and activation are incompletely understood. In DCs, TLR signaling induced by inflammatory stimuli has been shown to activate the PI3K/Akt pathway to promote DC longevity.32 We show that activation of TLR signaling also induces a transient downregulation of SIRPα and a subsequent restoration of its expression. More importantly, we find that SIRPα is constitutively associated with the p85 subunit of PI3K in DC, which might interfere with activation of the p85/p110 PI3K heterodimer. Indeed, as seen in the PI3K kinase assay, the silencing of SIRPα resulted in a significant increase in PI3K activity, whereas overexpression of SIRPα inhibited PI3K activation. Consistent with the known role of the PI3K/Akt pathway in DC survival, SIRPα-silent DCs failed to exhibit a prolonged lifespan when treated with specific small inhibitors of the PI3K/Akt pathway or when expressing the dominant-negative form of Akt. It was previously demonstrated that Akt deficiency results in defective DC maturation and activation.28,33 In light of the dependence of Akt on PI3K, that finding agrees with our results showing that the recruitment of the PI3K p85 subunit by SIRPα impaired activation of the PI3K/Akt pathway, thus endowing DCs with tolerogenic properties. These results provide strong evidence for a key role of SIRPα in regulating Akt-dependent DC survival and maturation. Our data also suggest that the disappearance and reappearance of SIRPα during DC maturation might reflect an intrinsic process that is inversely linked to DC lifespan and plays a key role in controlling immune responses.
The efficacy of the DC-based tumor vaccine we tested was significantly improved by silencing SIRPα. This was reflected in the substantially retarded tumor growth associated with supra-physiological T cell expansion in C57BL/6 mice bearing E.G7-OVA lymphoma or highly aggressive and poorly immunogenic B16 melanoma cells. Silencing of SIRPα resulted in more robust and extended activation of DCs and achieved greater T cell proliferation, IFNγ production and cytolytic activity than was observed when DCs were treated with LPS. SIRPα-silent DCs also efficiently induced long-lived CD8+ memory responses without detectable signs of autoimmunity. The superior and unique immunostimulatory effect of miSIRPα-DCs may be due to the broad roles of SIRPα in negatively regulating many proinflammatory signal transduction pathways, as indicated by the higher surface expression of costimulatory molecules, the more selective but increased production of licensing cytokines, and the enhanced capacity to initiate antitumor immunity.34-36 Taken together, our study uncoveres a critical role for SIRPα in restricting the DC-mediated adaptive immune response. The silencing of SIRPα in DCs might tip the balance from immune suppression to antitumor immunity as a collective outcome of the cells' effects on interactive networks of pro- and anti-inflammatory factors and cells in tumor-bearing mice. SIRPα inhibition might therefore be clinically useful for augmenting anti-infection and anticancer immunity.
Materials and methods
Antibodies and reagents
The following antibodies were used: anti-MHC class II, anti-CD80, anti-CD86, anti-CCR7 and anti-CCR5, anti-SIRPα and anti-CD11c (eBioscience, San Diego, CA); antibodies specific for AKT, Myc-tag, p85 (Cell Signaling Technology). Rabbit polyclonal SIRPα antibody was generated in our laboratory against residues in the cytoplasmic domain.37 LPS and OVA were from Sigma-Aldrich. The CpG oligodeoxynucleotide 1826 was purchased from Invivogen. The PI3-Kinase inhibitors Ly294002 and wortmannin were purchased from Calbiochem. CFSE was purchased from Molecular Probes. DC viability was assessed using propidium iodide (PI; Sigma). TRP2 peptide (VYDFFVWL) was purchased from SANGON.
Animals and cell lines
Male C57BL/6 mice (6–8 weeks) were purchased from Shanghai Experimental Center (Chinese Science Academy) and maintained in the barrier facility under pathogen-free conditions. OT-1 TCR transgenic mice were purchased from Jackson laboratory. B16 melanoma cells and OVA-expressing EL4 (EG7) cells were grown in 1640 medium (Gibco BRL, Gaithersburg, MD), containing 10% FBS. 293T and 293A cells were maintained in DMEM supplemented with 10% FBS.
Human PBMCs and tumor samples
The peripheral blood cells and tumor tissues were obtained from healthy volunteers or patients in Eastern Heptobiliary Surgery Hospital. All procedures were performed according to investigator's protocols approved by the Second Military Medical University Research Ethics Committee.
Lentiviral vector production
The lentiviral vectors used in this study were constructed using the BLOCK-iT™ lentiviral Pol II miR RNAi expression kits (Invitrogen). Predesigned miRNA targeted against SIRPα was provided by Invitrogen, with the following sequences: miSIRPα: 5′-TGCTGTCT ATGAGCAGATGAGTTCACGTTTTGGCCACTGACTGACGTGAACTCCTGCTC ATAGA - 3′.
Transduction of bone marrow-derived DC with lentiviral vectors
Mouse BMDCs were prepared as described previously. Lentiviral infections of BMDCs were performed on six-well plates. After 5 d of culture in mGM-CSF (10 ng/mL) and mIL-4 (1 ng/mL, PeproTech), immature DCs were removed to serum-free 1640. The transductions were conducted by adding concentrated supernatant at an MOI of 50 or 80 in the presence of 8 μg/mL polybrene(Sigma).
Cytokine and PI3-Kinase activity assays
Cytokine or PIP3 levels in culture-derived supernatants were determined using commercial ELISA kits for IL-12 (eBiosource). For PI3-Kinase Activity (Echelon Biosciences Inc. ), BMDC lysates were collected 48 h after infection with LV-miSIRPα, LV-GFP or PBS control. PI3 kinase was immunoprecipitated with 5 μL of rabbit antibody against PI3 kinase (which coprecipitates the p85 subunit of PI3 kinase) and 60 μL of Protein A/G PLUS-Agarose beads (Santa Cruz Biotechnology, Inc.). PI3 kinase activity in the immunoprecipitates was analyzed with PI3 kinase enzyme-linked immunosorbent assay (ELISA) (from Echelon Biosciences, Salt Lake City, UT) according to the manufacturer's instructions.
Western blotting analysis
Samples of cell lysates were separated by 8–12% SDS-PAGE gels and transferred onto polyvinyldifluoride membranes. Duplicate membranes were probed with polyclonal or monoclonal antibodies (mAbs) such as anti-SIRPα, anti-Akt or Akt, and then incubated with anti-mouse or rabbit HRP (Santa Cruz, CA). Specific bands were detected using an IRDye 800CW-conjugated secondary antibody and LI-COR imaging system (LI-COR Biosciences).
Flow cytometry analysis
1 × 105 cells were suspended in PBS containing 1% BSA and were stained with various fluorochrome-conjugated mAbs for 30 min on ice. Flow cytometry analysis was performed using Moflo-XDP (Beckman Coulter, Fullerton, CA) equipped with Summit 5.1 Software. The frequency of TRP-2-specific CD8+ T cells was determined by staining lymph node cells with TRP-2-APC-pentamer (Pro-Immune, Bradenton, FL, USA). Briefly, We stained cells with anti-CD8+-FITC (clone 53-6.7; BD Biosciences) and APC-H-2Kb/TRP-2 (SVYDFFVWL, SVL) Pro5 pentamer (ProImmune) for 20 min at room temperature and then lysed erythrocytes with red blood cell lysing buffer (Sigma-Aldrich) for 7 min. TRP-2-specific CD8+ T cells were then determined by analysis by flow cytometry.
Proliferation of CD8+ T cells
Spleens of OT-1 were collected, and CD8+ T lymphocytes were enriched by immunomagnetic kit following the manufacturer's protocol (StemCell Technologies, Inc.) and seeded onto 96-well plates at 2 × 105 cells/well. Bone marrow DCs derived from 8-d culture of lentivirus-transduced or control groups were irradiated at 3 Gy and seeded onto the 96-well plates with T cells in triplicates at the stimulator (DC)-to-effector (T cells) ratios shown in Fig. 4A. Three days later, the wells were pulsed with 1 μCi/well [3H]thymidine and harvested 18–20 h later with a Packard Micromate cell harvester. The 3H incorporation was determined as counts per minute through a Packard Matrix 96 direct β counter.
IFNγ ELISPOT assay
C57BL/6 mice were immunized with OVA-pulsed or TRP2-pulsed DCs (100 μg/mL) that had been transduced with lentiviral vectors and matured with LPS ex vivo, and then stimulated in vivo with polyI:C (50 μg/mouse i.p.) daily for three consecutive days. Splenocytes pooled from immunized mice (two or three per group) and single-cell suspensions were obtained by crushing and passing through nylon mesh. CD8+ or CD4+ T cells were isolated by positive selection using a CD8+ or CD4+ T cell isolation kit (StemCell Technologies, Inc.). IFNγ ELISPOT assays were conducted according to the manufacturer's protocol (Quick Spot™ IFNγ ELISPOT kit, Dakewe).
DC immunization and tumor models
E.G7-OVA lymphoma , B16 melanoma cells (ATCC) were expanded in culture and injected subcutaneously into the right flanks of C57BL/6 mice (1 × 106 E.G7-OVA cells/mouse or 1 × 105 B16 melanoma cells/mouse). For prophylactic or therapeutic DC vaccines, miSIRPα-DCs or control DCs were pulsed with OVA proteins (100 μg/mL) for E.G7-OVA cells or TRP-2 peptide (10 mg/mL) for B16 or B16F10 cells and injected subcutaneously via footpads (2 × 106 DCs/mouse). Prophylactic DC vaccine treatment was done 7 d before tumor injection or therapeutic DC vaccination was done 3 d after tumor injection with administration of polyI: C (50 μg/mouse) in 7, 14 and 21 d after tumor injection. Tumor volumes were recorded every 2 d.
Statistical analysis
All values are expressed as the mean ± SEM of data obtained from at least three experiments. Comparisons between groups were made with the Student's t test and ANOVA test. A significance level of p < 0.05 was used.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Shan-Hua Tang, Lin-Na Guo, Dan Cao and Dong-Ping Hu for their helpful technical assistance. We also thank kind help of Prof. Fubin Li from Shanghai Institute of Immunology, Shanghai Jiao Tong University School of Medicine.
Funding
This work was supported by National Natural Science Foundation of China (81370061, 31371440 and 31571477), Shanghai Eastern Scholarship (2012-32), Program of Shanghai Academic/Technology Research Leader (16XD1403300), Shanghai Municipal Commission of Health and Family Planning (20134044) and Natural Science foundation of Shanghai Municipal (13ZR1418500).
References
- 1.Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol 2012; 30:1-22; PMID:22136168; http://dx.doi.org/10412980 10.1146/annurev-immunol-100311-102839 [DOI] [PubMed] [Google Scholar]
- 2.Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol 2005; 5:296-306; PMID:15803149; http://dx.doi.org/10412980 10.1038/nri1592 [DOI] [PubMed] [Google Scholar]
- 3.Gilboa E. DC-based cancer vaccines. J Clin Invest 2007; 117:1195-203; PMID:17476349; http://dx.doi.org/10412980 10.1172/JCI31205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol 2007; 7:19-30; PMID:17170756; http://dx.doi.org/10412980 10.1038/nri1996 [DOI] [PubMed] [Google Scholar]
- 5.Nopora A, Brocker T. Bcl-2 controls dendritic cell longevity in vivo. J Immunol 2002; 169:3006-14; PMID:12218115; http://dx.doi.org/10412980 10.4049/jimmunol.169.6.3006 [DOI] [PubMed] [Google Scholar]
- 6.Wang J, Zheng L, Lobito A, Chan FK, Dale J, Sneller M, Yao X, Puck JM, Straus SE, Lenardo MJ. Inherited human Caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type II. Cell 1999; 98:47-58; PMID:10412980; http://dx.doi.org/ 10.1016/S0092-8674(00)80605-4 [DOI] [PubMed] [Google Scholar]
- 7.Ouaaz F, Arron J, Zheng Y, Choi Y, Beg AA. Dendritic cell development and survival require distinct NF-kappaB subunits. Immunity 2002; 16:257-70; PMID:11869686; http://dx.doi.org/ 10.1016/S1074-7613(02)00272-8 [DOI] [PubMed] [Google Scholar]
- 8.Park Y, Lee SW, Sung YC. Cutting Edge: CpG DNA inhibits dendritic cell apoptosis by up-regulating cellular inhibitor of apoptosis proteins through the phosphatidylinositide-3′-OH kinase pathway. J Immunol 2002; 168:5-8; PMID:11751939; http://dx.doi.org/9062191 10.4049/jimmunol.168.1.5 [DOI] [PubMed] [Google Scholar]
- 9.Prins RM, Craft N, Bruhn KW, Khan-Farooqi H, Koya RC, Stripecke R, Miller JF, Liau LM. The TLR-7 agonist, imiquimod, enhances dendritic cell survival and promotes tumor antigen-specific T cell priming: relation to central nervous system antitumor immunity. J Immunol 2006; 176:157-64; PMID:16365406; http://dx.doi.org/9062191 10.4049/jimmunol.176.1.157 [DOI] [PubMed] [Google Scholar]
- 10.Rescigno M, Martino M, Sutherland CL, Gold MR, Ricciardi-Castagnoli P. Dendritic cell survival and maturation are regulated by different signaling pathways. J Exp Med 1998; 188:2175-80; PMID:9841930; http://dx.doi.org/9062191 10.1084/jem.188.11.2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanks BA, Jiang J, Singh RA, Song W, Barry M, Huls MH, Slawin KM, Spencer DM. Re-engineered CD40 receptor enables potent pharmacological activation of dendritic-cell cancer vaccines in vivo. Nat Med 2005; 11:130-7; PMID:15665830; http://dx.doi.org/9062191 10.1038/nm1183 [DOI] [PubMed] [Google Scholar]
- 12.Bachmann MF, Wong BR, Josien R, Steinman RM, Oxenius A, Choi Y. TRANCE, a tumor necrosis factor family member critical for CD40 ligand-independent T helper cell activation. J Exp Med 1999; 189:1025-31; PMID:10190893; http://dx.doi.org/9062191 10.1084/jem.189.7.1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen M, Wang J. Programmed cell death of dendritic cells in immune regulation. Immunol Rev 2010; 236:11-27; PMID:20636805; http://dx.doi.org/9062191 10.1111/j.1600-065X.2010.00916.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kharitonenkov A, Chen Z, Sures I, Wang H, Schilling J, Ullrich A. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature 1997; 386:181-6; PMID:9062191; http://dx.doi.org/ 10.1038/386181a0 [DOI] [PubMed] [Google Scholar]
- 15.Jiang P, Lagenaur CF, Narayanan V. Integrin-associated protein is a ligand for the P84 neural adhesion molecule. J Biol Chem 1999; 274:559-62; PMID:9872987; http://dx.doi.org/14531999 10.1074/jbc.274.2.559 [DOI] [PubMed] [Google Scholar]
- 16.Gardai SJ, Xiao YQ, Dickinson M, Nick JA, Voelker DR, Greene KE, Henson PM. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell 2003; 115:13-23; PMID:14531999; http://dx.doi.org/ 10.1016/S0092-8674(03)00758-X [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez PL, Harada T, Christian DA, Pantano DA, Tsai RK, Discher DE. Minimal [Self] peptides that inhibit phagocytic clearance and enhance delivery of nanoparticles. Science 2013; 339:971-5; PMID:23430657; http://dx.doi.org/ 10.1126/science.1229568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okazawa H, Motegi S, Ohyama N, Ohnishi H, Tomizawa T, Kaneko Y, Oldenborg PA, Ishikawa O, Matozaki T. Negative regulation of phagocytosis in macrophages by the CD47-SHPS-1 system. J Immunol 2005; 174:2004-11; PMID:15699129; http://dx.doi.org/24215318 10.4049/jimmunol.174.4.2004 [DOI] [PubMed] [Google Scholar]
- 19.Yamao T, Noguchi T, Takeuchi O, Nishiyama U, Morita H, Hagiwara T, Akahori H, Kato T, Inagaki K, Okazawa H et al.. Negative regulation of platelet clearance and of the macrophage phagocytic response by the transmembrane glycoprotein SHPS-1. J Biol Chem 2002; 277:39833-9; PMID:12167615; http://dx.doi.org/24215318 10.1074/jbc.M203287200 [DOI] [PubMed] [Google Scholar]
- 20.Barclay AN, Van den Berg TK. The interaction between signal regulatory protein α (SIRPalpha) and CD47: structure, function, and therapeutic target. Annu Rev Immunol 2014; 32:25-50; PMID:24215318; http://dx.doi.org/ 10.1146/annurev-immunol-032713-120142 [DOI] [PubMed] [Google Scholar]
- 21.Mukhopadhyay S, Pluddemann A, Hoe JC, Williams KJ, Varin A, Makepeace K, Aknin ML, Bowdish DM, Smale ST, Barclay AN et al.. Immune inhibitory ligand CD200 induction by TLRs and NLRs limits macrophage activation to protect the host from meningococcal septicemia. Cell Host Microbe 2010; 8:236-47; PMID:20833375; http://dx.doi.org/ 10.1016/j.chom.2010.08.005 [DOI] [PubMed] [Google Scholar]
- 22.Weiskopf K, Ring AM, Ho CC, Volkmer JP, Levin AM, Volkmer AK, Ozkan E, Fernhoff NB, van de Rijn M, Weissman IL et al.. Engineered SIRPalpha variants as immunotherapeutic adjuvants to anticancer antibodies. Science 2013; 341:88-91; PMID:23722425; http://dx.doi.org/ 10.1126/science.1238856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tseng D, Volkmer JP, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, Seita J, Inlay MA, Weiskopf K, Miyanishi M et al.. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci U S A 2013; 110:11103-8; PMID:23690610; http://dx.doi.org/23547101 10.1073/pnas.1305569110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soroosh P, Doherty TA, Duan W, Mehta AK, Choi H, Adams YF, Mikulski Z, Khorram N, Rosenthal P, Broide DH et al.. Lung-resident tissue macrophages generate Foxp3+ regulatory T cells and promote airway tolerance. J Exp Med 2013; 210:775-88; PMID:23547101; http://dx.doi.org/ 10.1084/jem.20121849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Randolph GJ, Angeli V, Swartz MA. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol 2005; 5:617-28; PMID:16056255; http://dx.doi.org/20682853 10.1038/nri1670 [DOI] [PubMed] [Google Scholar]
- 26.Yanagihara S, Komura E, Nagafune J, Watarai H, Yamaguchi Y. EBI1/CCR7 is a new member of dendritic cell chemokine receptor that is up-regulated upon maturation. J Immunol 1998; 161:3096-102; PMID:974337620682853 [PubMed] [Google Scholar]
- 27.Vecchi A, Massimiliano L, Ramponi S, Luini W, Bernasconi S, Bonecchi R, Allavena P, Parmentier M, Mantovani A, Sozzani S et al.. Differential responsiveness to constitutive vs. inducible chemokines of immature and mature mouse dendritic cells. J Leukoc Biol 1999; 66:489-94; PMID:1049632020682853 [DOI] [PubMed] [Google Scholar]
- 28.Park D, Lapteva N, Seethammagari M, Slawin KM, Spencer DM. An essential role for Akt1 in dendritic cell function and tumor immunotherapy. Nat Biotechnol 2006; 24:1581-90; PMID:17143278; http://dx.doi.org/20682853 10.1038/nbt1262 [DOI] [PubMed] [Google Scholar]
- 29.Saito Y, Iwamura H, Kaneko T, Ohnishi H, Murata Y, Okazawa H, Kanazawa Y, Sato-Hashimoto M, Kobayashi H, Oldenborg PA et al.. Regulation by SIRPalpha of dendritic cell homeostasis in lymphoid tissues. Blood 2010; 116:3517-25; PMID:20682853; http://dx.doi.org/ 10.1182/blood-2010-03-277244 [DOI] [PubMed] [Google Scholar]
- 30.Barclay AN, Brown MH. The SIRP family of receptors and immune regulation. Nat Rev Immunol 2006; 6:457-64; PMID:16691243; http://dx.doi.org/20351312 10.1038/nri1859 [DOI] [PubMed] [Google Scholar]
- 31.Baba T, Nakamoto Y, Mukaida N. Crucial contribution of thymic Sirp α+ conventional dendritic cells to central tolerance against blood-borne antigens in a CCR2-dependent manner. J Immunol 2009; 183:3053-63; PMID:19675159; http://dx.doi.org/20351312 10.4049/jimmunol.0900438 [DOI] [PubMed] [Google Scholar]
- 32.Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG et al.. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010; 115:4742-9; PMID:20351312; http://dx.doi.org/ 10.1182/blood-2009-10-249540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hammer GE, Ma A. Molecular control of steady-state dendritic cell maturation and immune homeostasis. Annu Rev Immunol 2013; 31:743-91; PMID:23330953; http://dx.doi.org/23690610 10.1146/annurev-immunol-020711-074929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chao MP, Majeti R, Weissman IL. Programmed cell removal: a new obstacle in the road to developing cancer. Nat Rev Cancer 2012; 12:58-67; PMID:22158022; http://dx.doi.org/23690610 10.1038/nrc3171 [DOI] [PubMed] [Google Scholar]
- 35.Tseng D, Volkmer JP, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, Seita J, Inlay MA, Weiskopf K, Miyanishi M et al.. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci U S A 2013; 110:11103-8; PMID:23690610; http://dx.doi.org/ 10.1073/pnas.1305569110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jaiswal S, Chao MP, Majeti R, Weissman IL. Macrophages as mediators of tumor immunosurveillance. Trends Immunol 2010; 31:212-9; PMID:20452821; http://dx.doi.org/ 10.1016/j.it.2010.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kong XN, Yan HX, Chen L, Dong LW, Yang W, Liu Q, Yu LX, Huang DD, Liu SQ, Liu H et al.. LPS-induced down-regulation of signal regulatory protein {α} contributes to innate immune activation in macrophages. J Exp Med 2007; 204:2719-31; PMID:17954568; http://dx.doi.org/ 10.1084/jem.20062611 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.