

ABSTRACT
T cell Ig mucin-3 (Tim-3), an immune checkpoint inhibitor, shows therapeutic potential. However, the molecular mechanism by which Tim-3 regulates immune responses remains to be determined. In particular, very little is known about how Tim-3 works in innate immune cells. Here, we demonstrated that Tim-3 is involved in the development of tumor-promoting M2 macrophages in colon cancer. Manipulation of the Tim-3 pathway significantly affected the polarization status of intestinal macrophages and the progression of colon cancer. The Tim-3 signaling pathway in macrophages was explored using microarray, co-immunoprecipitation, gene mutation, and high-content analysis. For the first time, we demonstrated that Tim-3 polarizes macrophages by directly binding to STAT1 via residue Y256 and Y263 in its intracellular tail and inhibiting the STAT1-miR-155-SOCS1 signaling axis. We also identified a new signaling adaptor of Tim-3 in macrophages, and, by modulating the Tim-3 pathway, demonstrated the feasibility of altering macrophage polarization as a potential tool for treating this kind of disease.
KEYWORDS: Colon cancer, macrophage polarization, miR-155, STAT1, Tim-3
Abbreviations
- Arg-1
arginase 1
- CAC
colitis-associated cancer
- LPS
lipopolysaccharide
- SOCS1
suppressor of cytokine signaling 1
- STAT1
signal transducer and activator of transcription 1
- Tim-3
T cell Ig mucin-3
Introduction
The gut is constantly exposed to commensal microbes, ingested antigens, and potential pathogens, and regulation of intestinal tolerance is therefore the main task of the immune system of the gut mucosa. Intestinal macrophages play a crucial role in monitoring the environment and in controlling the cellular and molecular networks of the immune system to maintain tissue homeostasis.1 Dysregulated macrophage function has emerged as an important aspect of the progression of intestine disorders, including inflammatory bowel diseases2,3 and colon cancer,4,5 raising the possibility of a therapeutic approach involving the targeting of intestinal macrophages. However, the mechanisms involved in regulating macrophage function in the physiopathology of intestinal diseases are not well understood. Macrophages are categorized as classically activated (M1, pro-inflammatory), which promote antitumor inflammation, or alternatively activated (M2, anti-inflammatory), which support tumor growth.6,7 Modulation of macrophage polarization is a feasible strategy to treat intestinal diseases, such as colon cancer.8,9
The signals controlling intestinal macrophage polarization are not well defined and their identification will be of great value in understanding how these cells control the local microenvironment and affect intestinal homeostasis. The polarization state of macrophages is regulated, at least in part, by local concentrations of cytokines and chemokines, as well as by interactions of macrophages with normal and degraded components of the extracellular matrix.10 As for the intracellular mechanisms, some transcriptional factors have been shown to play a role in regulating macrophage differentiation and polarization. In particular, signal transducer and activator of transcription (STAT) family members, such as STAT1, and suppressor of cytokine signaling 1 (SOCS1) are reported to be important regulators of macrophage polarization.11,12 In addition, dynamically modulated expression of specific microRNAs (miRs), such as miR-155 and miR-146, regulates the physiological differentiation of macrophages.13-15 The targeting of molecular pathways regulating macrophage polarization therefore holds great promise for immunotherapy.
T cell Ig mucin-3 (Tim-3) is an immune regulator first identified on activated T effector cells, including Th1, Th17, and Tc1 cells. By inducing T cell tolerance or exhaustion, Tim-3 negatively regulates the responses of these T effector cells.16-18 Recent reports by ourselves19 and others have demonstrated that Tim-3 is also a negative regulator of innate immune cells, such as macrophages and dendritic cells. As Tim-3 is involved in the physiopathology of many immune disorders, including tumors and chronic virus infections, it is now considered as an immunotherapeutic target.19-21 However, the molecular mechanism by which Tim-3 maintains immune homeostasis remains largely unclear, and, in particular, very little is known about the mechanism of Tim-3 signaling in innate immune cells.
We previously found that Tim-3 is involved in regulating macrophage polarization,19,22 but the precise molecular mechanism and the therapeutic potential remain to be determined. In the present study, we found that Tim-3 may promote tumorigenesis of colon cancer by promoting M2 macrophage polarization. We also identified STAT1 as a new signaling adaptor of Tim-3, through which Tim-3 determines the phenotype and function of macrophages. A better understanding of the molecular mechanisms by which Tim-3 controls intestinal macrophage homeostasis will help in establishing new therapeutic strategies to treat colon cancer.
Results
Dysregulated upregulation of Tim-3 is associated with biased M2 macrophage polarization in colon cancer
To test the possible roles of Tim-3 in the physiopathology of colon cancer, Tim-3 mRNA expression was examined in colon tissues from colon cancer patients and from C57BL/6 mice with a mouse model of colitis-associated cancer (CAC). The results showed that, compared to controls, levels were significantly higher in both the patients (Fig. 1A) and the CAC mice (Fig. 1B). We then examined Tim-3 protein expression and function on intestinal macrophages from CAC mice. As shown in Fig. 1C, immunohistochemical analysis showed colocalization of Tim-3 with F4/80, a macrophage marker, in intestinal tissues from CAC mice. Interestingly, we found that upregulation of Tim-3 expression was associated with biased M2 macrophage polarization in CAC, as shown by increased expression of dectin-1 (M2) and decreased expression of CD16/32 (M1) on macrophages (Fig. 1D) and increased levels of Arg-1 and IL-10 mRNAs (M2) and decreased levels of NOS2 and IL-12 mRNAs (M1) in macrophages (Fig. 1E).
Figure 1.
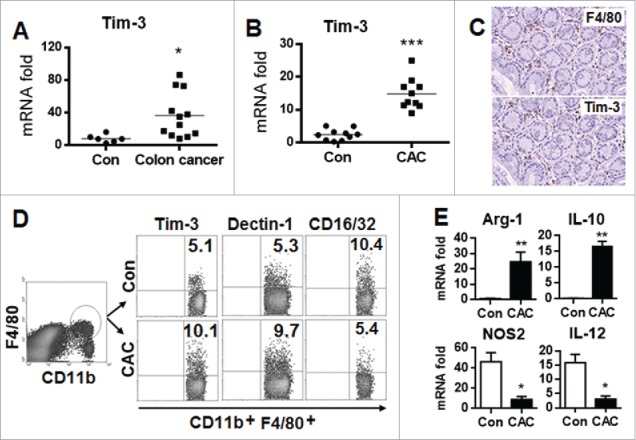
Increased Tim-3 expression is associated with biased M2 macrophage polarization in colon cancer. (A) Tim-3 mRNA levels were measured in tumor tissues from colon cancer patients or a control intestine tissue biopsy by real-time PCR. (B–E) The CAC model was established in wild-type C57BL/6 mice as described in the Materials and methods, then, on day 100, Tim-3 mRNA levels were measured in intestinal tissues from CAC mice and control mice (B), tumor tissue from CAC mice was examined for F4/80 and Tim-3 co-expression (C), total tumor-infiltrating cells were analyzed for Tim-3, dectin-1, and CD16/32 expression on gated CD11b+F4/80+macrophages by flow cytometry (D), and tumor-infiltrating macrophages were isolated using FACS sorting and analyzed for cytokine mRNA levels by real-time PCR. (E) In (B–E), the data are representative of those obtained in three independent assays, each using 8–10 mice per group. In B & E, the results are expressed as the mean ± SD; *p <0.05; **p < 0.01; ***p < 0.001.
Tim-3 overexpression polarizes macrophages toward the M2 type and promotes tumor growth in mice with CAC
To further test the role of Tim-3 in the physiopathology of colon cancer and in intestinal macrophage polarization, we generated Tim-3 transgenic (TG) mice by overexpressing Tim-3 under the control of the CMV promoter; incorporation of Tim-3 DNA in the recipient was confirmed by PCR and Tim-3 expression on macrophages and T cells was confirmed by flow cytometry (Fig. S1), then established our CAC model in both wild-type (WT) and the Tim-3 TG mice. We found that Tim-3-TG mice with CAC showed a significantly decreased survival rate (Fig. 2A) and increased tumor growth (Fig. 2B) and tumor burden and tumor numbers (Fig. 2C). When the effects of Tim-3 overexpression on macrophage polarization in CAC mice were examined, intestinal macrophages isolated from Tim-3-TG mice showed significantly increased Tim-3 protein expression compared to those from WT mice and enhanced M2 polarization, as demonstrated by increased dectin-1 and decreased CD16/32 expression on macrophages (Fig. 2D) and increased Arg-1 and IL-10 mRNA levels and decreased NOS2 and IL-12 mRNA levels in macrophages (Fig. 2E). These data suggest that Tim-3 may promote the growth of colon cancer by polarizing macrophages toward the M2 type.
Figure 2.
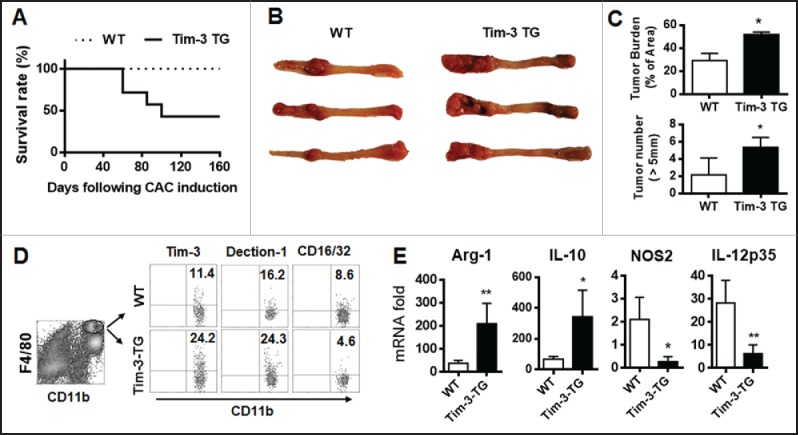
Tim-3 transgenic expression enhances tumor growth and promotes M2 macrophage polarization in mice with CAC. The CAC model was established in wild type and Tim-3-TG mice. (A) Survival rate. (B–E) At day 160, the surviving mice were sacrificed and tumor growth (B), tumor burden (C, top panel), and number of tumors > 5 mm diameter (C, bottom panel) measured, tumor-infiltrating cells were analyzed for Tim-3, dectin-1, and CD16/32 expression on gated CD11b+F4/80+ macrophages (D), and tumor-infiltrating macrophages were isolated using FACS sorting and analyzed for gene expression by real-time PCR (E). The data are representative of those obtained in three independent assays, each using 8–10 mice per group. In C and E, the results are expressed as the mean ± SD; *p < 0.05 and **p < 0.01.
Blockade of the Tim-3 pathway alters macrophage polarization and inhibits colon cancer growth
We then examined whether blockade of the Tim-3 pathway altered progression of colon cancer by altering macrophage polarization. To test this, a different colon cancer model was established by transplanting CT-26 cells into Balb/C mice with or without blockade of the Tim-3 pathway by injection of a soluble form of Tim-3 (sTim-3-Ig). Interestingly, as shown in Figs. 3A and B, sTim-3-Ig inhibited the growth of the transplanted tumor in a dose-dependent manner and led to an enhanced pro-inflammatory response, as shown by increased levels of mRNAs for IFNγ, TNF-α, IL-6, and IL-1β in the total tumor-infiltrating cells (Fig. 3C). When tumor-infiltrating macrophages were isolated and analyzed, they showed significantly increased surface expression of Tim-3 compared to intestinal macrophages from naive mice (Fig. 3D). Blockade of the Tim-3 pathway in mice with CAC by injection of sTim-3 resulted in inhibition of the M2 macrophage response, as shown by decreased dectin-1 expression and increased CD16/32 expression on macrophages (Fig. 3E) and decreased levels of Arg-1 and IL-10 mRNAs and increased levels of IL-12 and NOS2 mRNAs in macrophages (Fig. 3F).
Figure 3.
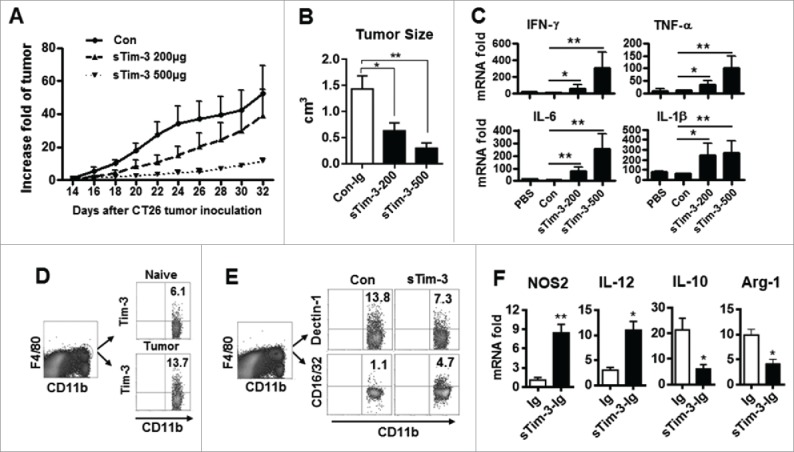
Tim-3 pathway blockade inhibits tumor growth and the M2 response in CT26-bearing mice. Mice were inoculated with CT26 cells as described in the Materials and methods. Starting on the day of tumor inoculation, sTim-3-Ig or Ig (Con) was injected (i.p.) every other day till the mice were sacrificed at day 32 and the tumors dissected out. (A) Daily tumor size (fold increase in volume). (B–E) Results at day 32: (B) Tumor size; (C) Isolated tumor-infiltrating cells examined for cytokine expression by real-time PCR; (D) Tim-3 expression on tumor-infiltrating macrophages (tumor) and on peritoneal macrophages from naive mice (naive) examined by flow cytometry (E) Total tumor-infiltrating cells were isolated and analyzed for dectin-1 and CD16/32 expression on gated CD11b+F4/80+ macrophages; (F) Tumor-infiltrating macrophages isolated using FACS sorting were analyzed for NOS2, IL-12, IL-10, or Arg-1 gene expression by real-time PCR. The data are representative of those obtained in three independent experiments, each with 6–8 mice per group. In A–C and F, the results are expressed as the mean ± SD; *p < 0.05, **p < 0.01.
Tim-3 signaling controls macrophage polarization in vitro
The effects of Tim-3 signaling on macrophage polarization were then examined in vitro, using control RAW264.7 cells or RAW264.7 macrophages stably overexpressing Tim-3 or with Tim-3 knockdown or peritoneal macrophages from WT or Tim-3-TG mice; Fig. 4A shows Tim-3 mRNA levels in these different cell populations. Tim-3-overexpressing RAW264.7 macrophages (Fig. 4B) and peritoneal macrophages from Tim-3-TG mice (Fig. 4C) both showed an M2 macrophage phenotype, as evidenced by decreased levels of NOS2 mRNA and increased levels of Arg-1 mRNA compared to controls (left panels), and showed increased IL-4-induced IL-10 secretion (center panels) and a decrease in the LPS-induced increase in IL-12 mRNA levels (right panels), while RAW264.7 macrophages silenced for Tim-3 expression showed M1 polarization, as demonstrated by increased NOS2 mRNA levels, decreased Arg-1 mRNA levels, decreased IL-4-induced IL-10 secretion, and an increase in the LPS-induced increase in IL-12 mRNA levels (Fig. 4D). In addition, in vitro blockade of Tim-3 signaling in control RAW264.7 macrophages using sTim-3-Ig resulted in a biased M1 phenotype, as shown by increased NOS2 mRNA levels and decreased Arg-1 mRNA levels (Fig. 4E). Finally, peritoneal macrophages from untreated WT C57BL/6 mice incubated with IL-4 expressed higher levels of Tim-3 and Dectin-1 than those incubated with LPS and IFNγ (Fig. 4F). These in vitro data provide additional evidence that Tim-3 polarizes macrophages toward the M2 type.
Figure 4.
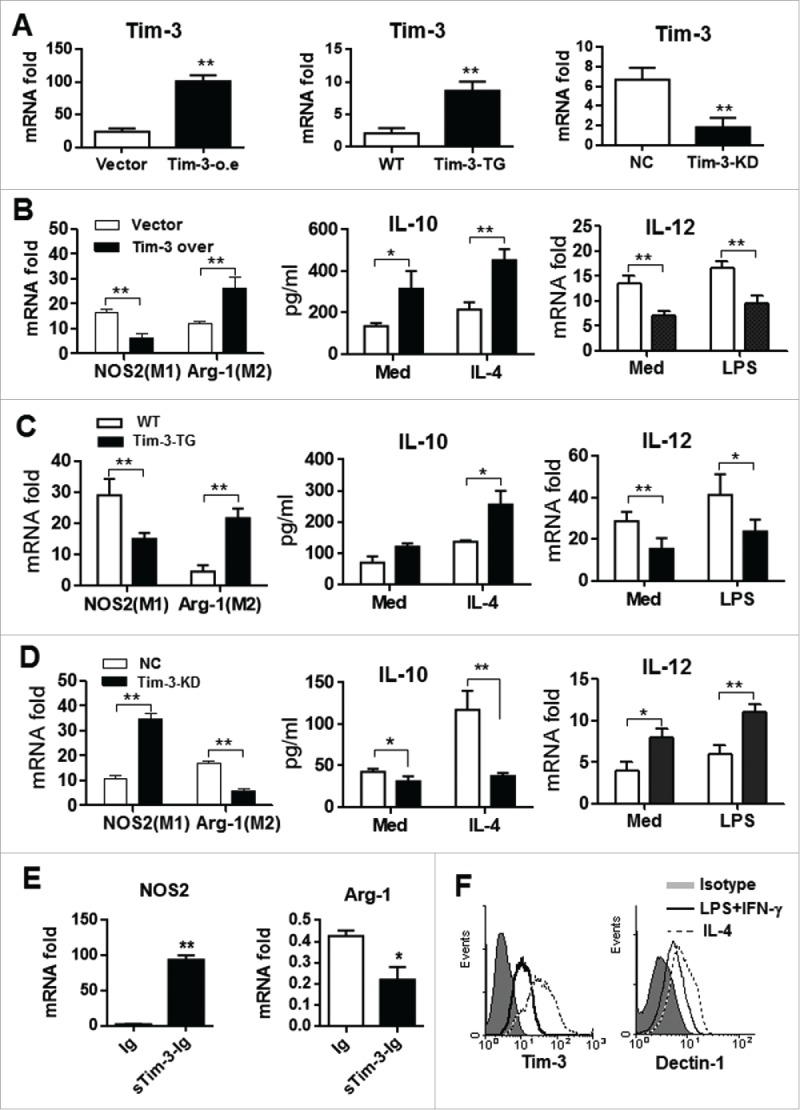
Tim-3 signaling regulates macrophage polarization in vitro. (A) Expression of Tim-3 in Tim-3- or vector control transfected RAW264.7 cells (left panel), in peritoneal macrophages isolated from wild type (WT) or Tim-3-TG mice(central panel), and in Tim-3 knockdown RAW264.7 cells (right panel) examined by real-time PCR. (B–D) RAW264.7 macrophages stably overexpressing Tim-3 (B), peritoneal macrophages from Tim-3-TG mice (C), Tim-3 knockdown (KD) RAW264.7 macrophages (D), or controls were tested for levels of NOS2 and Arg-1 mRNAs by real-time PCR (left panels) or were incubated with or without IL-4 (50 ng/mL) or LPS (100 ng/mL) for 12 h, then IL-10 secretion (center panels) or IL-12 mRNA levels (right panels) were measured by ELISA or real-time PCR, respectively. (E) RAW264.7 cells were incubated with 10 μg/mL of sTim-3-Ig or Ig (Con) for 6 h, then NOS2 (left panel) or Arg-1 (right panel) mRNA levels were measured by real-time PCR. (F) Peritoneal macrophages from control C57BL/6 mice were incubated for 24 h with either LPS (100 ng/mL) plus IFN γ (100 ng/mL) or with IL-4 (50 ng/mL), then expression of Tim-3 (left panel) or Dectin-1 (right panel) was measured by flow cytometry. The data shown are representative of those obtained in three independent experiments. *p < 0.05, **p < 0.01. Isotype represents the isotype control for the antibodies used.
Tim-3 inhibits miR-155 expression in vitro and in CAC mice in vivo
To explore the molecular mechanisms by which Tim-3 polarizes macrophages, a miRs array assay was used to compare miRNA profiles in control and Tim-3 knockdown RAW264.7 cells, and more than 100 differentially expressed miRNAs, including miR-155, miR-146, and miR-27a, were identified (Fig. 5A) (series entry, GSE76839, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE76839). We then selected miR-155 for further studies, as it has been reported to be involved in regulation of macrophage polarization.15 Real-time PCR data confirmed upregulation of miR-155 expression in RAW264.7 cells either stably knocked-down for Tim-3 or treated with sTim-3-Ig (Fig. 5B) and significant downregulation in RAW264.7 cells stably transfected with Tim-3 or in peritoneal macrophages isolated from Tim-3-TG mice (Fig. 5C). These data show that Tim-3 signaling inhibits miR-155 expression both in vitro and in vivo. We then examined whether blockade of miR-155 activity affected Tim-3-induced polarization of RAW264.7 cells by transfecting the cells with a miR-155 inhibitor or control inhibitor, then treating them with Ig or sTim-3-Ig. As shown in Fig. 5D, incubation with sTim-3-Ig resulted in a significant increase in NOS2 mRNA levels and a significant decrease in Arg-1 mRNA levels in control inhibitor-transfected cells, whereas the effect on NOS2 mRNA levels, but not Arg-1 mRNA levels, was significantly lower in miR-155 inhibitor-treated cells. These data show that miR-155 plays critical roles in Tim-3 signaling-mediated macrophage polarization.
Figure 5.
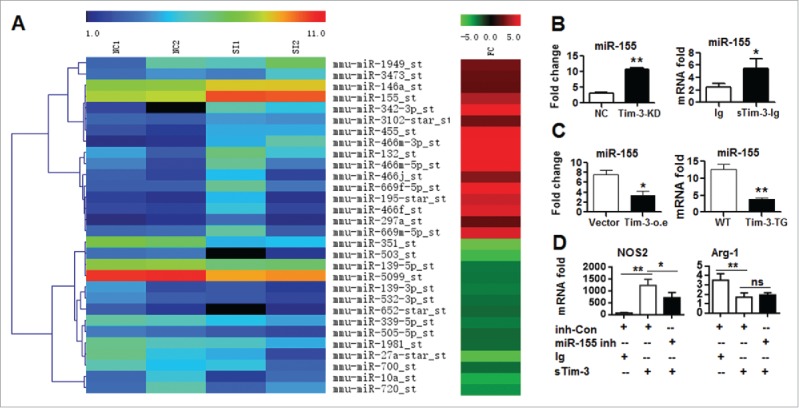
mi-R155 is involved in Tim-3-induced macrophage polarization. (A) Microarray data showing microRNAs differentially expressed in control RAW264.7 cells (NC1 and NC2) or RAW264.7 cells with stable knockdown of Tim-3 (SI1 and SI2) (left column) and the fold change (FC) in the knockdown sample compared to the control sample (right column). (B and C) miR-155 levels measured by real-time PCR in RAW264.7 cells with stable knockdown of Tim-3 and controls (B, left panel), RAW264.7 cells treated with 10 μg/mL of sTim-3-Ig or Ig (B, right panel), RAW264.7 cells stably overexpressing Tim-3 or controls (C, left), or peritoneal macrophages isolated from wild type or Tim-3-TG mice (C, right). (D) RAW264.7 cells transfected with miR-155 inhibitor (miR-155 inh) or inhibitor control (inh-Con) were incubated with Ig or sTim-3-Ig (10 μg/mL) for 10 h, then NOS2 (left panel) and Arg-1 (right panel) mRNA levels were measured by real-time PCR. In B–D, the data are representative of those obtained in three independent experiments, each performed in triplicate. ns: no significance; *p < 0.05, **p < 0.01.
We then examined miR-155 expression in the intestinal tissues of colon cancer patients by real-time PCR and found that it was lower in tumor tissues than in normal biopsy tissue controls (Fig. S2A). Decreased miR-155 expression was also seen in intestinal macrophages from Tim-3-TG mice with CAC compared to those from WT mice with CAC (Fig. S2B). These data show causative relations between increased Tim-3 expression, decreased miR-155 levels, and progression of colon cancer.
STAT1 acts as a key signaling adaptor linking Tim-3 signaling to miR-155 during macrophage polarization
To find the structural basis of the link between Tim-3, miR155, and the downstream signaling cascade, we generated several Tim-3 mutants, Y218A, Y256A, Y263A, Y272A, and Y256A/Y263A, in which tyrosine mutant has previously been shown to prevent phosphorylation.23,24 As shown in Fig. S3, in response to Gal-9 stimulation, HET293T cells transfected with control Tim-3 or the Y218A or Y272A mutant showed significantly decreased miR-155 expression compared to cells transfected with vector, while cells transfected with Y256A, Y263A, or the double mutant Y256A/Y263A did not. These mutagenesis studies show that Y256 and Y263 are required for the inhibitory effects of Tim-3 on miR-155 expression.
Next, we investigated the signaling adaptor linking Tim-3 to miR-155. We focused on STAT1 as (1) it is an upstream transcriptional factor involved in miR-155 regulation25; (2) it is involved in regulating macrophage polarization12, 26; and (3) it contains an SH2 domain, while the intracellular tail of Tim-3 contains a highly conserved tyrosine- containing src homology 2 (SH2)-binding motif, and tyrosine residues within this motif can be constitutively phosphorylated.23,24
We first examined phosphorylation of Y256 and Y263 in HEK293T cells transfected with Tim-3 or the Y256/263A Tim-3 mutant or co-transfected with Gal-9 and found that co-transfection with Gal-9 led to enhanced tyrosine phosphorylation on Tim-3 compared to transfection of Tim-3 alone; while mutant of Y256AY263A resulted in decreased phosphorylation (Fig. S4).
We then tested the hypothesis that Tim-3 might interact with STAT1 and regulate a downstream signaling cascade by examining STAT1 expression and phosphorylation (1) in control RAW264.7 cells and Tim-3 knockdown RAW264.7 cells in the presence or absence of LPS stimulation or in control RAW264.7 cells in the presence or absence of sTim-3-Ig and (2) in peritoneal macrophages from WT or Tim-3-TG C57BL/6 mice in the presence or absence of LPS stimulation. Our results showed that knockdown of Tim-3 (Fig. 6A) enhanced the LPS-induced phosphorylation of STAT1 (Fig. 6B) and blockade of Tim-3 signaling increased STAT1 phosphorylation (Fig. 6C), while TG expression of Tim-3 (Fig. 6D) significantly inhibited LPS-induced phosphorylation of STAT1 (Fig. 6E). To examine whether there was a direct interaction between Tim-3 and STAT1, and, if so, whether residues Y256 and Y263 of Tim-3 played a role, HEK293T cells were co-transfected with STAT1 and either Tim-3 or the Y256/Y263 double mutant, then the cell lysate was immunoprecipitated (IP) with an anti-Tim-3 antibody that binds to extracellular Tim-3, and the precipitate immunoblotted (IB) with anti-STAT1 antibody and the results showed that STAT1 bound to Tim-3, but not Y256A/Y263A-Tim-3 (Fig. 6F, left panel). Furthermore, when HEK293T cells were transfected with either Tim-3 or Y256A/Y263A-Tim-3, Tim-3, but not Y256A/Y263A-Tim-3, bound to endogenous STAT1 (Fig. 6F, central panel). Finally, we examined whether galectin-9 (Gal-9), the Tim-3 ligand, played a role in the interaction between Tim-3 and STAT1 by transecting HEK-293T cells with Tim-3, then incubating the cells in the presence or absence of recombinant Gal-9 and the results showed that ligation of Tim-3 by Gal-9 increased the binding betweenTim-3- and STAT1 (Fig. 6F, right panel). Together, these data show that (1) Tim-3 inhibits STAT1 phosphorylation; (2) Tim-3 binds to STAT1 and this requires residues Y256 and Y263; and (3) Tim-3 engagement by Gal-9 enhances the binding between Tim-3 and STAT1.
Figure 6.
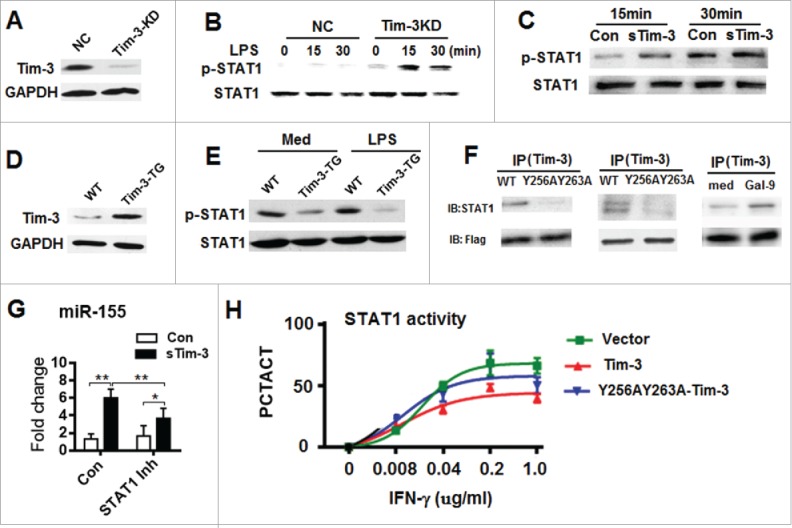
Tim-3 binds to STAT1 through residue Y256/263 and inhibits the STAT1-miR-155 axis. (A) Efficiency of Tim-3 knock down in RAW264.7 cells examined by Western blotting. (B and C) STAT1 expression and phosphorylation in control (NC) and Tim-3 knockdown RAW264.7 cells treated with LPS for the indicated time (B) or in RAW264.7 cells incubated with 10 μg/mL of sTim-3-Ig or Ig (Con) for the indicated time (C). (D) Overexpression of Tim-3 in macrophages from Tim-3 TG-mice examined by Western blotting. (E) STAT1 expression and phosphorylation in peritoneal macrophages from wild type or Tim-3-TG C57BL/6 mice incubated in the absence or presence of 100 ng/mL of LPS for 15 min. (F) HEK293T cells were co-transfected with STAT1 and Flag-tagged Tim-3 or the Flag-tagged- Y256/263 Tim-3 mutant (left panel), transfected with Flag-tagged Tim-3 or Flag-tagged-Y256/263 Tim-3 mutant (central panel), or transfected with Flag-tagged Tim-3 and incubated with or without Gal-9 (50 nM) for 30 min (right panel), then immunoprecipitation was carried out using anti-mouse Tim-3 antibody, and the precipitated protein tested for STAT1 or Flag by immunoblotting. In A–F, the data are representative of those obtained in three independent experiments. (G) RAW264.7 cells were incubated with 10 μg/mL of sTim-3-Ig or Ig in the presence or absence of a STAT1 inhibitor for 12 h, then miR-155 levels were measured by real-time PCR. **p < 0.01, *p < 0.05. (H) U2OS cells stably expressing STAT1-GFP transfected with Tim-3 (Tim-3), the double Tim-3 mutant (Y256A/Y263A-Tim-3), or vector alone were incubated with the indicated concentration of IFNγ for 30 min, then were analyzed on a GEIN Cell Analyzer 2000. PCTACT = percent activation and is calculated as the ratio of the GFP signal intensity in the nucleus / cytosol. Each dot is the mean ± SD for triplicate wells.
STAT1 acts upstream of miR-155 and promotes miR-155 transcription15 and it as therefore reasonable to hypothesize that STAT1 acts as an adaptor mediating the Tim-3-induced inhibition of miR-155 expression. To test this, we examined the effect of sTim-3-Ig blockade of Tim-3 signaling in cultured RAW264.7 cells on miR-155 expression in the presence or absence of the STAT1 inhibitor fludarabine. As shown in Fig. 6G, the increase in miR-155 expression caused by blockade of the Tim-3 pathway was partially inhibited by fludarabine. These data show that STAT1 is involved in the Tim-3-induced decrease in miR-155 expression.
To examine whether binding of STAT1 to Tim-3 mediates the downstream signaling cascade of Tim-3, U2OS cells transfected with a STAT1-EGFP vector were used. As shown in Fig. S5, STAT1 was found throughout the cytoplasm in untreated cells (top row) and translocated into the nucleus in response to IFNγ stimulation (center row) and this translocation was inhibited by a JAK inhibitor (bottom row).
To examine whether Tim-3 affected the nuclear translocation of STAT1, control Tim-3 or Y256A/Y263A-Tim-3 fused to a red marker protein was transfected into STAT1-tranfected U2OS cells and the results, shown in Fig. 6H, showed that, following IFNγ stimulation, STAT1 nuclear translocation in Tim-3-transfected cells was considerably lower than that in vector-transfected and Y256A/Y263A-Tim-3 transfected cells. These data show a suppressive effect of Tim-3 on the nuclear translocation of STAT1.
Finally, we examined STAT1 phosphorylation in intestinal macrophages from mice with CAC or controls, and found that it was lower in cells from CAC mice (Fig. S6A). In addition, STAT1 phosphorylation was lower in Tim-3-TG mice with CAC than that in WT mice with CAC (Fig. S6B). These data show that STAT1 is a critical transcriptional factor affecting the status of macrophages in colon cancer in vivo and that this process can be regulated by Tim-3.
SOCS1 is involved in a signaling cascade downstream of Tim-3-STAT1-miR-155 and mediates macrophage polarization
Finally, the signaling cascade downstream of the Tim-3-STAT1-miR-155 axis was investigated. We focused on SOCS1, as it is a critical mediator promoting the production of the immunoregulatory cytokine IL-10 and enhancing M2 macrophage polarization12,27 and is reported to be a target of miR-155.28 Silencing of Tim-3 in Tim-3 knockdown RAW264.7 cells and blockade of Tim-3 signaling in RAW264.7 cells both decreased SOCS1 protein expression (Figs. 7A and B) and decreased SOCS1 mRNA expression (Figs. 7C and D), while TG expression of Tim-3 in mice increased SOCS1 mRNA levels (Fig. 7E). Furthermore, in response to Gal-9 stimulation, SOCS1 mRNA levels were higher in HET293T cells transfected with Tim-3 than in cells transfected with Y256A/Y263A-Tim-3 (Fig. 7F), showing that residues 256 and 263 of Tim-3 are involved in Tim-3-enhanced SOCS1 expression.
Figure 7.
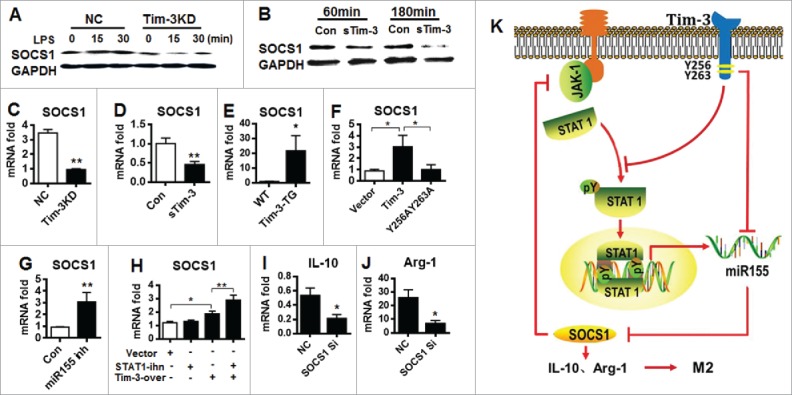
STAT1-miR-155 control of Tim-3-induced M2 macrophage polarization is mediated by SOCS1. (A and B) Control or Tim-3 knockdown RAW264.7 cells were treated with LPS (100 pg/mL) for the indicated time (A) or control RAW264.7 cells were incubated with 10 μg/mL of sTim-3-Ig or Ig for the indicated time (B), then SOCS1 levels were examined using Western blotting. (C–G) SOCS1 mRNA levels were measured by real-time PCR in Tim-3 knockdown or control (NC) RAW264.7 cells (C), RAW264.7 cells incubated with 10 μg/mL of sTim-3-Ig or Ig (Con) for 12 h (D), peritoneal macrophages from wild type and Tim-3-TG mice (E), HEK293T cells transfected with Tim-3, Tim-3 mutated at Y256/263, or vector alone incubated with Gal-9 (50 nM) for 12 h (F), RAW264.7 cells transfected with miR-155 inhibitor (miR-155 inh) or inhibitor control (Con) (G), or RAW264.7 cells transfected with Tim-3 (Tim-3-over) or vector alone incubated for 12 h with or without STAT1 inhibitor (STAT1 inh) (H). (I and J). Peritoneal macrophages from Tim-3-TG mice were transfected with SOCS1 siRNA or negative control, then IL-10 (I) or Arg-1 (J) mRNA levels were measured by real-time PCR. (K) Schematic diagram of how Tim-3 promotes M2 macrophage polarization. Tim-3 binds to SH2-containing STAT1 through residues Y256 and Y263 in C-terminal region of Tim-3, then inhibits the phosphorylation and nuclear translocation of STAT1, leading to decreased miR-155 expression, which exerts less inhibition on SOCS1 expression, leading to increased expression of IL-10 and Arg-1, and, finally, polarization of macrophages to the M2 phenotype.
To determine whether there was a direct interaction between miR-155 and SOCS1 in macrophages, RAW264.7 cells were transfected with miR-155 inhibitor or control inhibitor and the effects on SOCS1 mRNA levels examined by real-time PCR. As shown in Fig. 7G, transfection with miR-155 inhibitor increased SOCS1 mRNA levels, suggesting that Tim-3 increases SOCS1 expression by inhibiting miR-155 expression. In addition, Tim-3-overexpressing RAW264.7 cells incubated with the STAT1 inhibitor showed significantly increased SOCS1 mRNA levels (Fig. 7H), suggesting that STAT1 is involved in the Tim-3-mediated upregulation of SOCS1. We also noted a negative regulatory effect of SOCS1 on STAT1 following Tim-3 signaling, as the enhanced STAT1 phosphorylation seen in RAW264.7 cells after Tim-3 blockade (Fig. 6C) was inhibited by overexpression of SOCS1 (data not shown). Finally, in peritoneal macrophages from Tim-3-TG mice, which showed increased SOCS1 expression (Fig. 7E), transfection with SOCS1 siRNA resulted in a significant decrease in mRNA levels for IL-10 (Fig. 7I) and Arg-1 (Fig. 7J).
Discussion
In the present study, we demonstrated that increased Tim-3 expression was associated with biased M2 macrophage polarization in colon cancer and promoted tumor growth. We then focused on exploring the molecular mechanisms by which Tim-3 polarizes macrophages. As summarized in Fig. 7K, Tim-3 promotes M2 macrophage polarization by binding STAT1 via residues Y256 and Y263, then inhibits the STAT1-miR-155 signaling axis, resulting in increased SOCS1 activity and Arg-1 and IL-10 expression. This is the first report demonstrating that STAT1 acts as a signaling adaptor of Tim-3 in macrophages. More importantly, our results showed that blockade of the Tim-3 pathway inhibited both the polarization of tumor-supporting macrophages and colon cancer growth, demonstrating the feasibility of modulating the Tim-3 pathway and altering macrophage polarization to treat this kind of disease.
The effects of Tim-3 on macrophage polarization and tumor pathogenesis were first examined in vivo. Using CAC and CT-26 tumor models, we demonstrated that Tim-3 promoted the polarization of tumor-promoting M2 macrophages in colon cancer. Although Tim-3 is also expressed on T cells and we cannot exclude the possibility that Tim-3 induces tumor tolerance by suppressing the function of T cells29, our data did demonstrate that Tim-3-induced macrophage tolerance in tumor models in vivo. Our findings therefore shed new light on the mechanisms by which Tim-3 induces tumor tolerance in colon cancer. A previous report showed that PDL1 expression by tumor-associated macrophages is a key mechanism for disarming the T cell antitumor response in hepatocellular carcinoma30 and we argue that Tim-3-mediated M2 macrophage polarization in colon cancer may have a similar effect.
Biased M2 macrophage polarization also contributes to tumor progression and metastasis in colon cancer and other tissues31-33 and macrophage re-education has been considered as a microenvironment-targeted therapy.34,35 For this purpose, factors determining the diversity of macrophages are under intensive investigation. For example, Wang et al.36 showed that Notch signaling determines the M1/M2 polarization of macrophages in the antitumor immune response, and Pyonteck et al.10 demonstrated that the CSF-1/CSF-1R pathway mediates macrophage polarization and that inhibition of this pathway reduces M2 macrophage levels and blocks glioma progression. Here, we demonstrated that Tim-3, an immune checkpoint inhibitor, is involved in macrophage polarization. Although a recent report by Yan eta al.37 showed that Tim-3 is involved in the activation and function of tumor-infiltrating macrophages in hepatocellular carcinoma, a finding consistent with our previous finding that Tim-3 is involved in macrophage polarization,22 the molecular mechanism by which this is achieved remains largely unclear. Local factors leading to increased Tim-3 expression in colon cancer were not investigated in the present study. However, manipulation of the Tim-3 pathway altered colon cancer progression, suggesting that Tim-3 can be used as a new therapeutic target to re-program tumor-associated intestinal macrophages to an antitumor phenotype and restrict colon cancer growth.
The therapeutic potential of Tim-3 called for the definition of the precise molecular mechanism of Tim-3-mediated immune suppression. Recently, Kuchroo et al. showed that CEACAM1 is a heterophilic ligand of Tim-3 and is required for Tim-3 to mediate T cell inhibition38 and that Bat3 acts as a safety catch, which blocks Tim-3-mediated inhibitory signals.39 However, these studies focused on the mechanism by which Tim-3 inhibits T cell activity and very little is known about how Tim-3 signals in innate immune cells and no adaptor(s) mediating the inhibitory effects of Tim-3 in innate immune cells have been previously identified. Here, for the first time, we have demonstrated that STAT1 functions as a signaling adaptor of Tim-3 in innate immune cells. We suggest a scenario in which Tim-3 binds to STAT1 through residues Y256 and Y263 and inhibits STAT1 phosphorylation and nuclear translocation, resulting in inhibition of the STAT1-miR-155 signaling axis in macrophages.
We also examined the relationship between Tim-3 dysregulation and miR-155 expression and STAT1 activity in colon cancer in vivo. Our data in Figs. S2 and S6 support causative relations between increased Tim-3 expression, inhibited miR-155 expression, and suppressed STAT1 activation in vivo. A previous report showing that tumor growth is enhanced in miR155−/− mice13 supports the tumor suppressing role of miR-155 in vivo. In addition, both STAT1 and miR-155 play critical roles in polarizing macrophages,11,13 and our present findings support a scenario in which Tim-3 signaling alters the progression of colon cancer by modulating macrophage polarization in vivo.
Finally, our data demonstrated that SOCS1 is involved in the signaling cascade downstream of the Tim-3-STAT1-miR-155 axis in macrophages. SOCS1 is a transcriptional factor controlling the activity of M2 macrophages, in part, by inhibiting the JAK-STAT pathway.28 Our finding that transfection with SOCS1 inhibited Tim-3 blockade-induced STAT1 phosphorylation (data not shown) suggests negative feedback regulation by SOCS1 of the JAK-STAT pathway. As Tim-3-induced upregulation of Arg-1 and IL-10 was prevented by SOCS knockdown, our data explain how Tim-3 signaling controls macrophage polarization. In such a scenario, Tim-3 may regulate macrophage activity according to the following scheme: (1) in the constitutive state, Tim-3 competes with JAK for binding to STAT1 through the constitutively phosphorylated residues Y256 and Y263, then inhibits STAT1 phosphorylation and nuclear translocation and (2) this leads to decreased miR-155 expression, a lower inhibitory effect on SOCS1 expression, a subsequent increase in IL-10 and Arg-1 expression, and, finally, polarization of macrophages to the M2 phenotype.
In summary, this study identifies mechanisms by which Tim-3 determines intestinal homeostasis in colon cancer and how Tim-3 signaling operates in innate immune cells. We identified STAT1 is a signaling adaptor of Tim-3 in macrophages and demonstrated that Tim-3 controls macrophage polarization by inhibiting the STAT1-miR-155 signaling axis. These findings have potential clinical implications. We demonstrated the feasibility of re-educating macrophage polarization by modulating the Tim-3 pathway for treating this kind of disease.
Materials and methods
Mice
Male (6- to 8-week-old) C57BL/6 mice and Balb/C mice (both from Jackson Laboratory (Bar Harbor, ME, USA)) and Tim-3-TG mice, developed by Cyagen Biosciences Inc. China, were used under specific pathogen-free conditions.
Induction of colitis-associated cancer
CAC was induced as described previously.40 Briefly, WT or Tim-3-TG C57BL/6 mice were injected intraperitoneally (i.p.) with azoxymethane on day 0, then, starting on day 5, underwent four cycles of drinking water containing 2% DSS (MP Biochemicals) for 1 week and plain water for 2 weeks, ending on day 84, then normal drinking water was provided until day 100, when they were killed and macrophages analyzed by flow cytometry.
Immunohistochemistry
Paraffin-embedded slides were deparaffinized and immersed overnight in a 80°Cwater bath in 10 mM sodium citrate buffer containing 0.1% Tween 20 for antigen unmasking. Slides were incubated for 1 h at 37°C with primary antibody against Tim-3 (Abcam) or F4/80 (BD Biosciences) in PBS containing1% BSA and 10% goat serum, then with biotinylated secondary antibody (Dako) at room temperature for 1 h. Streptavidin-HRP (BD PharMingen) was then added, and, after 40 min the sections were stained with DAB substrate and counterstained with hematoxylin.
Cell culture and transfection
The mouse macrophage cell line RAW264.7, human embryonic kidney cell line HEK-293T, and human osteosarcoma cell line U2OS were obtained from ATCC (Manassas, VA). RAW264.7 cells stably overexpressing Tim-3 were generated in our laboratory, as described previously.19 The U2OS cell line stably expressing Stat1-EGFP was a kind gift from Prof. Lili Wang, State Key Laboratory of Toxicology and Medical Countermeasures, Beijing, China. Mouse colon tumor-infiltrating macrophages were elicited as described previously.8 All cells were maintained in complete DMEM medium in a humidified 5% CO2 atmosphere at 37°C. For cell transfection, Tim-3 cDNA was cloned into pcDNA3.1 to generate Tim-3-wt-Flag and Tim-3-wt-RFP. Gal-9 cDNA was cloned into pcDNA3.1. Overlap PCR was used to generate the point mutation constructs Y218A-Tim-3, Y256A-Tim-3, Y263A-Tim-3, Y272A-Tim-3, and Y256A/Y263A-Tim-3, in which the indicated tyrosine residues in the tail region of Tim-3 were replaced by alanine. To transiently express SOCS1, mouse SOCS1 cDNA was cloned into pcDNA3.1 and transfected into RAW264.7 cells. The recombinant fusion protein mouse Tim-3-Ig (sTim-3-Ig) and control Ig were prepared as described previously.24 Recombinant Gal-9 protein was purchased from R&D. Corp (USA). All lines were grown according to the supplier's recommendation.
Tumor transplantation and Tim-3 blockade
The mouse colon carcinoma cell line CT26 was obtained from ATCC (Manassas, VA) and the cells injected (1 × 106/mouse) subcutaneously into one flank of 6-week-old male nude mice (Jackson Laboratory), then, after 2 weeks, the mice were sacrificed and the tumor tissues collected and cut into several similar-sized pieces, which were transplanted subcutaneously into 6-week-old male Balb/C mice.
In some studies, starting on the day of tumor transplantation, sTim-3-Ig or Ig (0, 200, or 500 μg/mouse) was injected i.p. every other day till day 32, and tumor size was measured every 2 d.
FACS analysis and macrophage sorting
Tumor-infiltrating cells were stained with allophycocyanin (APC)-conjugated rat anti-mouse CD11b mAb (clone M1/70), FITC-conjugated rat anti-mouse F4/80 mAb (clone BM8), and/or phycoerythrin (PE)-conjugated rat anti-mouse Tim-3 mAb (clone GL3) (all from eBioScience) diluted in 2% FBS in PBS, then, after two washes with PBS/2% FBS, were analyzed by flow cytometry in a FACS Calibur (BD Biosciences). To isolate macrophages from tumor-infiltrating cells, antibodies against mouse F4/80 and CD11b (eBioScience) and FACS cell sorting were used.
miRNA microarray analysis
Differentially expressed miRNAs in Tim-3 siRNA- or control siRNA-transfected RAW264.7 cells were profiled using Affymetrix miRNA microarray chips following the manufacturer's protocol. Briefly, collected samples was subjected to Gene Chip miRs array analysis using a GeneChip Scanner 3000 with GeneChip Operating Software (GCOS) and analyzed. Candidate miR(s) was further identified by quantitative real-time PCR analysis.
Quantitative real-time RT-PCR
For human studies, the study protocol was approved by the Ethics Committee of the General Hospital of the PLA, Beijing, China, and all patients gave their written informed consent. In both humans and mice, gene expression was analyzed by two-step QRT-PCR. The relative expression of a gene was determined using the 2−δδCt method, with GAPDH as the internal control. The primers for miR-155 were provided by RiboBio (Guangzhou, China). Other primers used, including those for human and mouse Tim-3, are listed in Table S1.
ELISA
The concentration of IL-10 in cell-free supernatants was measured using a sandwich ELISA according to the manufacturer's protocol (eBiosciences, San Diego, CA).
Western blots
Western blotting was performed as described previously19 to evaluate levels of phospho-STAT1, STAT1, SOCS1, and GAPDH using rabbit primary antibodies (CST, USA), horseradish peroxidase-conjugated anti-rabbit IgG antibodies (KPL, Gaithersburg, MD, USA), and enhanced chemiluminescence kits (Amersham Biosciences).
Co-immunoprecipitation of STAT1 withTim-3
HEK-293T cells were cotransfected with the STAT1 construct and either the control Tim-3 or Y256A/Y263A-Tim-3 construct using Lipofectamine 2000 (Invitrogen), then Tim-3 immunoprecipitation with anti-Tim-3 antibodies was performed at 48 h post-transfection and the precipitated protein immunoblotted for STAT1 as described previously.35
High-content analysis
U2OS cells stably expressing STAT1-GFP were transfected with the Tim-3-RFP or Y256A/Y263A-Tim-3-RFP construct on 96-well assay plates (Corning) using Lipofectamine 2000 (Invitrogen) and incubated with or without IFNγ for 30 min at 48 h post-transfection, then were fixed for 20 min in 4% formaldehyde at room temperature, the nuclei stained with Hoechst 33342 at 4°Covernight, and the cells imaged and analyzed using a GE IN Cell Analyzer 2000 High-Content Cellular Analysis System (GE Healthcare Bio-Sciences Corp.). To measure STAT1 translocation, the ratio of the GFP signal intensity in the nucleus/cytosol was calculated.
Statistical analysis
SPSS software (version 20.0) was used for statistical procedures. Data are expressed as the mean ± standard deviation. Differences between groups were analyzed using the Kruskal–Wallis test and ANOVA. Survival rate and tumor growth were analyzed using a log-rank test. A p value less than 0.05 was considered significant.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Prof. Zhengfan Jiang, School of Life Sciences, Peking University, Beijing, China; Dr Wenwen Zheng, Tsinghua University School of Medicine, Beijing, China; and Dr Jingran Zhou, Department of Infectious Diseases, St Jude Children's Research Hospital, Memphis, Tennessee, USA, for critical reading.
Funding
This work was supported by the National “973” Fund, China (2013CB530506, 2015CB553704), the National Natural Sciences Foundation of China (grants no. 81471540, 81471529, and 81472647), and the Key program of the Beijing Natural Sciences Foundation (grant no. 7141007)
References
- 1.Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 2011; 474:298-306; PMID:21677746; http://dx.doi.org/ 10.1038/nature10208 [DOI] [PubMed] [Google Scholar]
- 2.Zhu W, Yu J, Nie Y, Shi X, Liu Y, Li F, Zhang XL. Disequilibrium of M1 and M2 macrophages correlates with the development of experimental inflammatory bowel diseases. Immunol Invest 2014; 43:638-52; PMID:24921428; http://dx.doi.org/ 10.3109/08820139.2014.909456 [DOI] [PubMed] [Google Scholar]
- 3.Weisser SB, Brugger HK, Voglmaier NS, McLarren KW, van Rooijen N, Sly LM. SHIP-deficient, alternatively activated macrophages protect mice during DSS-induced colitis. J Leukoc Biol 2011; 90:483-92; PMID:21685246; http://dx.doi.org/ 10.1189/jlb.0311124 [DOI] [PubMed] [Google Scholar]
- 4.Yang M, Shao JH, Miao YJ, Cui W, Qi YF, Han JH, Lin X, Du J. Tumor cell-activated CARD9 signaling contributes to metastasis-associated macrophage polarization. Cell Death Differ 2014; 21:1290-302; PMID:24722209; http://dx.doi.org/ 10.1038/cdd.2014.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edin S, Wikberg ML, Dahlin AM, Rutegard J, Oberg A, Oldenborg PA, Palmqvist R. The distribution of macrophages with a M1 or M2 phenotype in relation to prognosis and the molecular characteristics of colorectal cancer. PloS One 2012; 7:e47045; PMID:23077543; http://dx.doi.org/ 10.1371/journal.pone.0047045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol 2013; 14:986-95; PMID:24048120; http://dx.doi.org/ 10.1038/ni.2705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature 2013; 496:445-55; PMID:23619691; http://dx.doi.org/ 10.1038/nature12034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tai SK, Chang HC, Lan KL, Lee CT, Yang CY, Chen NJ, Chou TY, Tarng DC, Hsieh SL. Decoy receptor 3 enhances tumor progression via induction of tumor-associated macrophages. J Immunol 2012; 188:2464-71; PMID:22287720; http://dx.doi.org/22647600 10.4049/jimmunol.1101101 [DOI] [PubMed] [Google Scholar]
- 9.Arranz A, Doxaki C, Vergadi E, Martinez de la Torre Y, Vaporidi K, Lagoudaki ED, Ieronymaki E, Androulidaki A, Venihaki M, Margioris AN et al.. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc Natl Acad Sci U S A 2012; 109:9517-22; PMID:22647600; http://dx.doi.org/ 10.1073/pnas.1119038109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V et al.. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 2013; 19:1264-72; PMID:24056773; http://dx.doi.org/ 10.1038/nm.3337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol 2011; 11:750-61; PMID:22025054; http://dx.doi.org/ 10.1038/nri3088 [DOI] [PubMed] [Google Scholar]
- 12.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol 2007; 7:454-65; PMID:17525754; http://dx.doi.org/ 10.1038/nri2093 [DOI] [PubMed] [Google Scholar]
- 13.Cai X, Yin Y, Li N, Zhu D, Zhang J, Zhang CY, Zen K. Re-polarization of tumor-associated macrophages to pro-inflammatory M1 macrophages by microRNA-155. J Mol Cell Biol 2012; 4:341-3; PMID:22831835; http://dx.doi.org/ 10.1093/jmcb/mjs044 [DOI] [PubMed] [Google Scholar]
- 14.Nazari-Jahantigh M, Wei Y, Noels H, Akhtar S, Zhou Z, Koenen RR, Heyll K, Gremse F, Kiessling F, Grommes J et al.. MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in macrophages. J Clin Invest 2012; 122:4190-202; PMID:23041630; http://dx.doi.org/ 10.1172/JCI61716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Squadrito ML, Etzrodt M, De Palma M, Pittet MJ. MicroRNA-mediated control of macrophages and its implications for cancer. Trends Immunol 2013; 34:350-9; PMID:23498847; http://dx.doi.org/ 10.1016/j.it.2013.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanchez-Fueyo A, Tian J, Picarella D, Domenig C, Zheng XX, Sabatos CA, Manlongat N, Bender O, Kamradt T, Kuchroo VK et al.. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol 2003; 4:1093-101; PMID:14556005; http://dx.doi.org/ 10.1038/ni987 [DOI] [PubMed] [Google Scholar]
- 17.Hastings WD, Anderson DE, Kassam N, Koguchi K, Greenfield EA, Kent SC, Zheng XX, Strom TB, Hafler DA, Kuchroo VK. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur J Immunol 2009; 39:2492-501; PMID:19676072; http://dx.doi.org/ 10.1002/eji.200939274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu W, Shi Y, Li S, Zhang Y, Liu Y, Wu Y, Chen Z. Blockade of Tim-3 signaling restores the virus-specific CD8(+) T-cell response in patients with chronic hepatitis B. Eur J Immunol 2012; 42:1180-91; PMID:22539292; http://dx.doi.org/ 10.1002/eji.201141852 [DOI] [PubMed] [Google Scholar]
- 19.Yang X, Jiang X, Chen G, Xiao Y, Geng S, Kang C, Zhou T, Li Y, Guo X, Xiao H et al.. T cell Ig mucin-3 promotes homeostasis of sepsis by negatively regulating the TLR response. J Immunol 2013; 190:2068-79; PMID:23365080; http://dx.doi.org/22842346 10.4049/jimmunol.1202661 [DOI] [PubMed] [Google Scholar]
- 20.Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, Fujioka Y, Ohba Y, Gorman JV, Colgan JD et al.. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol 2012; 13:832-42; PMID:22842346; http://dx.doi.org/ 10.1038/ni.2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuchroo VK, Anderson AC, Freeman GJ. Comment on “Tim-3 directly enhances CD8 T cell responses to acute Listeria monocytogenes infection”. J Immunol 2014; 193:467; PMID:24994907; http://dx.doi.org/24945079 10.4049/jimmunol.1401123 [DOI] [PubMed] [Google Scholar]
- 22.Zhao Z, Jiang X, Kang C, Xiao Y, Hou C, Yu J, Wang R, Xiao H, Zhou T, Wen Z et al.. Blockade of the T cell immunoglobulin and mucin domain protein 3 pathway exacerbates sepsis-induced immune deviation and immunosuppression. Clin Exp Immunol 2014; 178:279-91; PMID:24945079; http://dx.doi.org/ 10.1111/cei.12401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vega-Carrascal I, Bergin DA, McElvaney OJ, McCarthy C, Banville N, Pohl K, Hirashima M, Kuchroo VK, Reeves EP, McElvaney NG. Galectin-9 signaling through TIM-3 is involved in neutrophil-mediated Gram-negative bacterial killing: an effect abrogated within the cystic fibrosis lung. J Immunol 2014; 192:2418-31; PMID:24477913; http://dx.doi.org/17069754 10.4049/jimmunol.1300711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van de Weyer PS, Muehlfeit M, Klose C, Bonventre JV, Walz G, Kuehn EW. A highly conserved tyrosine of Tim-3 is phosphorylated upon stimulation by its ligand galectin-9. Biochem Biophy Res Commun 2006; 351:571-6; PMID:17069754; http://dx.doi.org/ 10.1016/j.bbrc.2006.10.079 [DOI] [PubMed] [Google Scholar]
- 25.Gracias DT, Stelekati E, Hope JL, Boesteanu AC, Doering TA, Norton J, Mueller YM, Fraietta JA, Wherry EJ, Turner M et al.. The microRNA miR-155 controls CD8(+) T cell responses by regulating interferon signaling. Nat Immunol 2013; 14:593-602; PMID:23603793; http://dx.doi.org/ 10.1038/ni.2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ et al.. TLR4, but not TLR2, mediates IFN-β-induced STAT1alpha/β-dependent gene expression in macrophages. Nat Immunol 2002; 3:392-8; PMID:11896392; http://dx.doi.org/ 10.1038/ni774 [DOI] [PubMed] [Google Scholar]
- 27.Wilson HM. SOCS Proteins in Macrophage Polarization and Function. Front Immunol 2014; 5:357; PMID:25120543; http://dx.doi.org/ 10.3389/fimmu.2014.00357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yan XL, Jia YL, Chen L, Zeng Q, Zhou JN, Fu CJ, Chen HX, Yuan HF, Li ZW, Shi L et al.. Hepatocellular carcinoma-associated mesenchymal stem cells promote hepatocarcinoma progression: role of the S100A4-miR155-SOCS1-MMP9 axis. Hepatology 2013; 57:2274-86; PMID:23316018; http://dx.doi.org/ 10.1002/hep.26257 [DOI] [PubMed] [Google Scholar]
- 29.Kang CW, Dutta A, Chang LY, Mahalingam J, Lin YC, Chiang JM, Hsu CY, Huang CT, Su WT, Chu YY et al.. Apoptosis of tumor infiltrating effector TIM-3+CD8+ T cells in colon cancer. Sci Rep 2015; 5:15659; PMID:26493689; http://dx.doi.org/ 10.1038/srep15659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, Zheng L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med 2009; 206:1327-37; PMID:19451266; http://dx.doi.org/ 10.1084/jem.20082173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol 2010; 11:889-96; PMID:20856220; http://dx.doi.org/ 10.1038/ni.1937 [DOI] [PubMed] [Google Scholar]
- 32.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell 2010; 141:39-51; PMID:20371344; http://dx.doi.org/ 10.1016/j.cell.2010.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruffell B, Affara NI, Coussens LM. Differential macrophage programming in the tumor microenvironment. Trends Immunol 2012; 33:119-26; PMID:22277903; http://dx.doi.org/ 10.1016/j.it.2011.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, Robinson SC, Balkwill FR. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med 2008; 205:1261-8; PMID:18490490; http://dx.doi.org/ 10.1084/jem.20080108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saccani A, Schioppa T, Porta C, Biswas SK, Nebuloni M, Vago L, Bottazzi B, Colombo MP, Mantovani A, Sica A. p50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res 2006; 66:11432-40; PMID:17145890; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-1867 [DOI] [PubMed] [Google Scholar]
- 36.Wang YC, He F, Feng F, Liu XW, Dong GY, Qin HY, Hu XB, Zheng MH, Liang L, Feng L et al.. Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res 2010; 70:4840-9; PMID:20501839; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-0269 [DOI] [PubMed] [Google Scholar]
- 37.Yan W, Liu X, Ma H, Zhang H, Song X, Gao L, Liang X, Ma C. Tim-3 fosters HCC development by enhancing TGF-β-mediated alternative activation of macrophages. Gut 2015; 64:1593-604; PMID:25608525; http://dx.doi.org/ 10.1136/gutjnl-2014-307671 [DOI] [PubMed] [Google Scholar]
- 38.Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, Dougan SK, Petersen BS, Melum E, Pertel T et al.. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 2015; 517:386-90; PMID:25363763; http://dx.doi.org/ 10.1038/nature13848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rangachari M, Zhu C, Sakuishi K, Xiao S, Karman J, Chen A, Angin M, Wakeham A, Greenfield EA, Sobel RA et al.. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3-mediated cell death and exhaustion. Nat Med 2012; 18:1394-400; PMID:22863785; http://dx.doi.org/ 10.1038/nm.2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Han G, Wang K, Liu G, Wang R, Xiao H, Li X, Hou C, Shen B, Guo R et al.. Tumor-derived GM-CSF promotes inflammatory colon carcinogenesis via stimulating epithelial release of VEGF. Cancer Res 2014; 74:716-26; PMID:24366884; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-1459 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.