

ABSTRACT
Glioblastoma (GBM) is a fatal brain cancer for which new treatment options are sorely needed. Platinum-based drugs have been investigated extensively for GBM treatment but few have shown significant efficacy without major central nervous system (CNS) and systemic toxicities. The relative success of platinum drugs for treatment of non-CNS cancers indicates great therapeutic potential when effectively delivered to the tumor region(s). New insights into the broad anticancer effects of platinum drugs, particularly immunomodulatory effects, and innovative delivery strategies that can maximize these multi-modal effects and minimize toxicities may promote the re-purposing of this chemotherapeutic drug class for GBM treatment.
KEYWORDS: Carboplatin, cisplatin, glioblastoma immunosuppression, glioblastoma, immunomodulation, immunotherapy, nanomedicine, nanoparticles, oxaliplatin, platinum-based chemotherapy
Abbreviations
- BCNU
bis-chloroethylnitrosourea
- BBB
blood–brain barrier
- CED
convection enhanced delivery
- CIW
chemotherapy-loaded interstitial wafers
- CNS
central nervous system
- CTL
cytotoxic T-lymphocyte
- CTLA4
cytotoxic T-lymphocyte-associated protein 4
- CTR1
copper influx transporter 1
- DCs
dendritic cells
- EPR
enhanced permeability and retention
- FasL
Fas ligand
- FGL2
fibrinogen-like protein-2
- Fn14
fibroblast growth factor inducible-14
- GBM
glioblastoma
- HLA
human leukocyte antigen
- HMGB-1
high mobility group protein-1
- HNSCC
head and neck squamous cell carcinoma
- IFNs
interferons
- M6P
mannose-6-phosphate
- mAbs
monoclonal antibodies
- MDSCs
myeloid derived suppressor cells
- MGMT
methylguanine methyltransferase
- MHC
major histocompatibility complex
- MMP
matrix metalloproteinase
- MMR
mismatch repair
- NER
nucleotide excision repair
- NP
nanoparticle
- NSCLC
non-small cell lung cancer
- PD
programmed death
- PD-L
programmed death ligand
- PEG
polyethylene-glycol
- PGE
prostaglandins
- PLGA
polylactic-co-glycolic acid
- PTEN
phosphatase and tensin homolog
- STAT
signal transducers and activators of transcription
- TGF
transforming growth factor
- TLR
toll-like receptor
- TMZ
temozolomide
- Tregs
regulatory T cells
Introduction
Glioblastoma
Glioblastoma (GBM) is the most common primary brain cancer in adults.1 GBM is characterized by extensive vascularization, a high mitotic index, cellular pleomorphism, genetic instability, tissue necrosis, brain invasion, and immune evasion. The current standard of care for patients with GBM consists of surgery for maximal safe resection or biopsy followed by radiation and oral chemotherapy [temozolomide (TMZ)] and/or implantation of chemotherapy [bis-chloroethylnitrosourea (BCNU)]-loaded interstitial wafers (CIW) into the surgical resection cavity. Without treatment, most patients live fewer than 6 mo. With the most aggressive combination therapies, the mean survival is still less than 18 mo, often with devastating neurological consequences. Thus, GBM remains one of the most lethal tumors and new treatments are needed that will improve patient survival and quality of life. To date, the use of platinum drugs for the treatment of GBM has shown minimal success in large part due to limited delivery to the tumor and extensive off-target toxicities, as will be described below. However, new information is emerging that suggests the broad, multi-faceted therapeutic potential of platinum-based agents, including new insights related to treatment failure and methods to improve the therapeutic ratio. Most notably, newly recognized immunomodulatory properties of platinum compounds have the potential to overcome many of the mechanisms of GBM immune evasion. A detailed understanding of this drug class of compounds may allow for the successful adaptation and re-purposing of these chemotherapeutics for the treatment of GBM.
Platinum-based therapeutics as cytotoxic agents
The discovery that platinum compounds could inhibit cell growth was made after realizing that products from a platinum electrode inhibited the growth of E. coli cells.2 Shortly thereafter, platinum compounds were shown to display anticancer properties.2 To date, three platinum compounds—cisplatin, carboplatin and oxaliplatin—have achieved FDA approval for cancer therapy. Platinum compounds have become an important class of chemotherapeutics used clinically for the treatment of a variety of cancers.3
The majority of research efforts devoted to understanding platinum-based agents have focused on the ability of these compounds to induce cancer cell apoptosis. Platinum compounds accumulate within cells mainly through the copper influx transporter 1 (CTR1) protein, although other mechanisms have been shown to play a minor role.3 Once in the cell, platinum compounds exert cytotoxic effects through a variety of mechanisms. The best-characterized cytotoxic mechanism of platinum drugs is the formation of DNA adducts. The platinum atoms bond with purine nucleotide bases forming intrastrand and interstrand crosslinks, which prevents both DNA replication and gene transcription.4 Platinum-induced DNA damage is detected by the cell, leading to upregulation of nucleotide excision repair (NER), mismatch repair (MMR), and other mechanisms. If cells are unable to repair the DNA damage, the cell initiates a cascade of events culminating in apoptosis.4 The cytotoxic effects of platinum drugs rely on these apoptotic pathways.
Failure considerations for platinum therapy in glioblastoma—limited efficacy and dose-related toxicity
Platinum drugs are used successfully to treat a variety of cancers; however, they have a checkered history in the treatment of GBM patients, featuring hints of success but mostly dose limiting toxicities when delivered systemically or in regions of sensitive tissues. This treatment failure may be due to the limited amount of a given platinum drug dose that crosses the blood–brain barrier (BBB) and/or major systemic toxicities occurring before effective drug concentrations are reached within the tumor.5-7 Early clinical trials with these agents offered great promise, such as a phase II trial which found either partial responses or stable disease in 20 out of 38 patients treated with chemotherapy supplemented with carboplatin and etoposide (a microtubule disruptor).8 However, subsequent trials focusing on platinum drugs in combination with radiation therapy and other chemotherapies showed no significant survival advantage with the addition of a platinum agent.9-11 Systemic toxicity remains a key limitation with administration of platinum chemotherapeutics, including ‘protected’ formulations like Lipoplatin and less toxic forms such as carboplatin.12,13 Hence, the most significant hurdle to the successful application of platinum drugs for advanced brain cancer has been dose-limiting toxicity.6 This is the likely reason why platinum-based therapies for GBM have not led to the same level of success seen in other cancers.
Repurposing platinum drugs: Non-cytotoxic and immunomodulatory effects of platinum compounds
The DNA damaging effect noted above as well as other direct non-cytotoxic platinum drug mechanisms interconnect with pathways related to cell invasion, angiogenesis, chemo- and radio-sensitization, and immunomodulation (Fig. 1). These broad effects may require lower sustained platinum drug doses over longer times to permit extended cell viability and reorganization of complex cellular pathways and the tumor microenvironment. The exploitation of these effects may enable the repurposing of platinum drugs for GBM therapy. Specifically, platinum compounds may hinder the ability of GBM cells to invade the surrounding tissue by downregulating matrix metalloproteinase (MMP) expression.14 Evidence in GBM models also suggests that platinum compounds may have anti-angiogenic effects.15 Additionally, platinum compounds are capable of enhancing the efficacy of the current adjuvant therapies for GBM (TMZ and radiotherapy) by modulating the MGMT DNA repair enzyme 16 and by enhancing radiation effects possibly by increasing the formation of transient anionic molecules.17 Lastly, the relatively unknown non-cytotoxic effect of platinum drugs—immunomodulation (see below)—may hold great promise for the treatment of GBM, because this offers the possibility of reversing GBM-mediated immune evasion.
Figure 1.
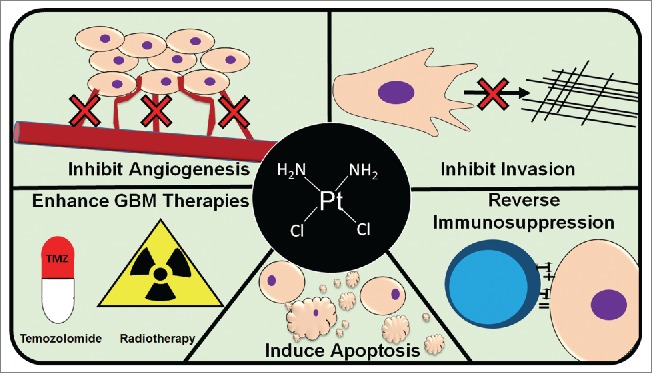
Therapeutic effects of platinum drugs: platinum drugs have several known anticancer effects including: inhibiting cancer cell (brown cells) invasion, inducing apoptosis, inhibiting angiogenesis, reversing immunosuppression (blue cell represents a lymphocyte), and enhancing the current GBM therapies, temozolomide (TMZ) and radiation.
Immune evasion in glioblastoma
Malignant brain tumors, including GBM, develop numerous mechanisms to evade recognition and elimination by the immune system.18-20 A complete description of the mechanisms of glioma-mediated immune evasion is beyond the scope of this review, for a summary of the subject see refs. 18 and 19. However, it is known that GBM cells alter the expression of cell surface proteins such as Human Leukocyte Antigens (HLAs)21 and costimulatory molecules.19 Altered expression of these proteins hampers immune responses against GBM cells. The loss of HLA class I, expressed by most nucleated cells, or the downregulation of tumor antigen expression impairs antigen presentation leading to defective cytotoxic T-lymphocyte (CTL) responses against the glioma cell.18,19 In addition, the absence of critical costimulatory molecules on glioma cells further limits an effective immune response by CTLs.18,19 Glioma cells can also increase the expression of immunosuppressive molecules such as program death ligand-1 and 2 (PD-L1 and PD-L2) leading to diminished T cell responses.18,19 PD-L1, in particular, is highly expressed in many GBM patient samples, likely due to increased PI (3) kinase activity secondary to phosphatase and tensin homolog (PTEN) loss.22,23 Furthermore, gliomas have been shown to upregulate PD-L1 expression on tumor-associated macrophages and circulating monocytes by producing high levels of IL-10.24 In addition to IL-10, glioma cells can produce a variety of factors including prostaglandins (PGE), transforming growth factor (TGF)-β2 and fibrinogen-like protein-2 (FGL2), which can suppress antitumor T cell activity and promote the development of a variety of immunosuppressive cell types. Similarly, increased expression of Fas ligand (FasL), CD70, and numerous immunosuppressive cytokines disrupts immune responses by inducing apoptosis or anergy in lymphocytes.18,19
Major immuno-biochemical signaling hubs controlled by the Signal Transducer and Activator of Transcription (STAT) family of transcription factors also play a major role in regulating immune function in the GBM microenvironment. In particular, STAT3 and STAT6 are constitutively active in several cell types within the microenvironment of many GBM tumors 25 and contribute to inhibition of pro-inflammatory cytokines, induction of regulatory T cells,25 and reduced antitumor antibodies.26 27 This broad chemical and cellular reprogramming of the tumor microenvironment leads to the development of immunosuppressive cells, including M2 macrophages, regulatory T cells (Tregs), and myeloid derived suppressor cells (MDSCs), which combine to allow the tumor to grow and invade the brain.18,19
Recalibrating the balance of immune function for cancer therapy has become a major research focus in recent years leading to the FDA approval of several immunomodulatory cancer therapies including ipilimumab, perbrolizumab, and nivolumab—monoclonal antibodies (mAbs) now referred to as ‘immune checkpoint inhibitors’. Ipilimumab is a cytotoxic T-lymphocyte-associated protein 4 (CTLA4) mAb 28 while pembrolizumab and nivolumab recognize PD-1, the cell surface receptor for PDl-L1 and PD-L2.28 Immune checkpoint inhibitors have found remarkable success, particularly for the treatment of melanoma.29 Specifically, both ipilimumab and nivolumab improve overall survival compared to the first line chemotherapy for metastatic melanoma in a subset of patients.30 To date, most immune checkpoint inhibitor studies have focused on treating melanoma; however, emerging evidence suggests that these therapies can be applied successfully to other cancers.29 The success of immune checkpoint blockade in various cancer types highlights the importance and potential of immunomodulation for GBM.
Platinum drugs modulate immune function in non-CNS cancers
Platinum drugs are capable of modulating a variety of the immunosuppressive features associated with numerous cancers, including colon cancer and head and neck squamous cell carcinoma (HNSCC).19,31 One of the first studies regarding the immunomodulatory potential of platinum drugs revealed that oxaliplatin is more effective in immunocompetent animal models.31 Since that report, it was realized that platinum drugs have dose-dependent immunomodulatory effects, generally most evident at non-cytotoxic, non-lymphotoxic levels.31,32 Treatment of cancer cells with oxaliplatin increases the expression of major histocompatibility complex (MHC) class I (the non-human equivalent of HLA I).33,34 Additionally, treatment of cancer cells with platinum drugs reduced PD-L2 expression, resulting in enhanced T cell activation.33 This reduction of PD-L2 expression may be mediated by STAT6 inhibition as PD-L2 is known to be regulated by STAT6 signaling and platinum drugs have been shown to reduce STAT6 phosphorylation (activation).33 Importantly, patients with HNSCC that overexpressed STAT6 had a better response to treatment with cisplatin and radiotherapy compared to patients whose tumors did not exhibit activated STAT6 signaling.31 In addition to the modulation of STAT6, emerging evidence suggests that platinum drugs may also inhibit STAT3 signaling, possibly by directly binding to STAT3, thereby preventing dimerization and nuclear translocation.35 Platinum drugs have also been shown to alter the profile of circulating immune cells and the profile of tumor-infiltrating immune cells.33 More specifically, oxaliplatin reduces the number of circulating MDSCs,36 increases the number of circulating CTLs, and reduces the number of Treg cells.37 Platinum drugs are also able to directly enhance CTL antitumor activity by upregulating the expression of mannose-6-phosphate (M6P) receptors.38 Expression of M6P receptors modulates CTL killing of cancer cells by increasing cancer cell sensitivity to the pro-apoptotic serine protease granzyme-B.38
Oxaliplatin is also capable of inducing immunogenic cell death, defined as cell death that generates an antitumor adaptive immune response against antigens expressed by the dead cell.39 This results in ‘immunological memory’, which helps to generate durable antitumor immune recognition and control of tumor growth.39 Immunogenic cell death requires several events to occur. First, calreticulin, a protein chaperone normally found in the ER, is expressed on the cell surface. This serves as a signal to dendritic cells (DCs) to engulf the affected cell.39 The cell then releases ATP, which serves as a chemokine that attracts DCs and macrophages to the tumor.39 Pro-inflammatory cytokines and type I interferons (IFNs) are also produced.39 Cells release high mobility group protein-1 (HMGB-1), a nuclear protein that interacts with toll-like receptor (TLR)-2 and −4 expressed on antigen-presenting cells such as DCs in their activation and maturation.39 The importance of immunogenic cell death, and HMGB-1 expression in particular, in mediating the effects of oxaliplatin is highlighted by findings that colorectal cancer patients with mutations in the TLR4 gene have a decreased response to treatment with oxaliplatin.40
Immunogenic cancer cell death driven by oxaliplatin, and in particular the first phase characterized by expression of calreticulin on the cell surface, is dependent on an induction of ER stress.39 ER stress is the result of a disruption in the normal function of the ER, caused by events such as the accumulation of proteins within the ER.41 Notably, platinum compounds cause ER stress by a mechanism independent of the drugs’ DNA-based effects.39 Interestingly, cisplatin does not appear to induce immunogenic cell death,31 despite increasing the release of HMGB-1 and ATP from cells. This is likely due to inability of cisplatin to induce the expression of calreticulin on the cell surface.39 31
These broad and specific immunomodulatory effects on non-CNS cancers suggest that platinum drugs may be capable of modulating a number of aspects of glioma immune evasion (Table 1). Ongoing investigations into the extent to which platinum drugs are capable of modulating the glioma microenvironment and host immune system will shed more light on this possibility. The validation of these immunological effects in future studies could result in the re-purposing of platinum drugs for GBM therapy and could likely become a renewed focus of clinical research in neuro-oncology.
Table 1.
Summary of Immunomodulatory Effects of Platinum Drugs.
| Immunomodulation: | Effect of Modulation: | GBM Significance: | Refs: |
|---|---|---|---|
| Reduce PD-L Expression | Decreases inhibitory signals that hamper anti-tumor immune responses | GBM cells upregulate expression of PD-L, hindering anti-tumor T cell responses | 17,19,31 |
| Increase MHC I Expression | Improves tumor cell recognition by the immune system | GBM cells reduce MHC class I expression, leading to impaired antigen presentation | 17,19,32,33 |
| Inhibit STAT Signaling | Inhibits pathways involved in numerous oncogenic processes, including immunosuppression | Aberrant STAT signaling is found in many GBM tumors and contributes to immunosuppression | 25-27, 32-34 |
| Alter Tumor Microenvironment Immune Cell Profile | Reduces immunosuppressive cells including, Tregs and MDSCs, and increases CTLs | GBM induces numerous immunosuppressive cell types which enhance immunosuppression | 17, 19, 32, 34-36 |
| Increase M6P Expression | Enhances CTL anti-tumor activity by altering cancer cell sensitivity to the pro-apoptotic serine protease, granzyme-B | GBM cells suppress anti-tumor CTL responses | 17,19, 37 |
| Immunogenic Cell Death | Generates ‘immunological memory’ enabling durable anti-tumor immune responses | GBM cells effectively evade and suppress the immune system, preventing durable anti-tumor immune responses | 17,19, 33 |
Abbreviations: CTL, cytotoxic T lymphocyte; GBM, glioblastoma; M6P, mannose 6-Phosphate; MDSC, myeloid derived suppressor cells; MHC, major histocompatibility complex; PD-L, programmed death ligand; STAT, signal transducers and activators of transcription; Treg, regulatory T-cell
Augmenting delivery to improve platinum-based therapy
To successfully repurpose platinum drugs for glioma therapy, effective delivery to the tumor remains a critical issue. The BBB complicates systemic delivery by controlling the passage of most molecules and drugs from the blood circulation to the brain.42,43 The BBB consists of cerebral endothelial cells connected together by tight junctions, a thick basement membrane, and astrocytic end-feet. It has been estimated that >98% of small-molecule drugs, and nearly all biologics (e.g., therapeutic mAbs) minimally cross this barrier.44 Numerous advanced delivery strategies designed to mitigate the BBB have been explored to capture the beneficial effects of platinum drugs while minimizing undesired side effects. These strategies include (a) increasing BBB permeability,45 (b) delivering cisplatin within biodegradable polymer implants in the tumor bed of patients, 46 and (c) bypassing the BBB via delivery under low sustained pressure (‘convection’) directly into the brain through an implanted catheter(s), an approach termed as convection enhanced delivery (CED).47 The expanding field of nanomedicine offers a variety of drug formulation options to improve platinum-based therapies, such as (a) increased solubility and increased blood half-life, (b) reduced side-effects through targeted delivery and broader tissue distribution in mouse intracranial glioma models,48 (c) controlled and sustained drug release, and (d) simultaneous incorporation and delivery of other anticancer drugs for combination therapy.49
Improved solubility and increased blood half-life
Many platinum-based drugs have limited solubility in water, have a short half-life, and are rapidly cleared by the circulatory and lymphatic system. To overcome these challenges, platinum drugs can be encapsulated in or conjugated to nanoparticles (NPs) to improve their water solubility and half-life. Peng et al. 50 observed significantly prolonged blood circulation time (>7-fold) and improved pharmacokinetics and biodistribution of EGFR-targeted heparin–cisplatin NPs compared to free cisplatin after systemic delivery in nude mice bearing H292 cell tumors. Often, platinum agent-encapsulated/conjugated NPs are decorated with low molecular weight polyethylene glycol (PEG), which is a hydrophilic and biocompatible polymer approved for use in humans. PEG reduces the opsonization of the particles and obstructs particle interaction with other biomolecules and cells. This serves to prolong blood circulation, which helps particles passively accumulate into tumors. For instance, Miller et al. 51 observed a >6-fold increase in half-life of Pt(IV)-encapsulated polylactic-co-glycolic acid (PLGA) PEG NPs compared to the free Pt(IV) in a breast cancer xenograft mouse model.
Reduced side-effects through enhanced site-specific delivery and distribution into tumors
As mentioned above, the main challenges in treating brain tumors such as GBM with platinum drugs are dose-limiting toxicities and ineffective methods to deliver these drugs to the target. Indeed, only a limited amount of systemically administered drug reaches the CNS.52 This is a result of the BBB, the extracellular space matrix, and the glialymphatic system of brain tissue which limits the distribution of therapeutics within the CNS.53 NPs may improve the delivery of therapeutics to invasive GBM cells by overcoming such drug delivery challenges.48 Furthermore, a major reason for failure of platinum-based chemotherapeutics in GBM patients is off-target toxicity. NP formulations may effectively address this issue; indeed, NPs have been shown to reduce toxicity compared to free drug.54 Carboplatin NPs engineered using the biodegradable polymer poly (e-caprolactone) were shown to reduce the incidence of carboplatin-induced hemolysis, in addition to being more efficiently taken up by glioma cells.55 In another study, the delivery of carboplatin PLGA NPs had less neuronal toxicity compared to free carboplatin.56 Importantly, free platinum drugs at high doses induce lymphodepletion,57 suggesting that a non-targeted systemic delivery of platinum drugs may actually hinder antitumor immune responses. Therefore, NP encapsulation appears to offer many opportunities to improve platinum drug delivery and significantly reduce toxicity.
A number of drug-NP formulations are under investigation as a means to improve platinum-based chemotherapy. For a comprehensive review of cisplatin NP formulations, see ref 58. Several PEGylated cisplatin NP formulations have advanced to clinical trials for NSCLC, pancreatic, breast, and other cancers.58 Lipoplatin, a PEGylated liposomal cisplatin formulation, has been shown to lower side effects and specifically reduce nephrotoxicity compared to free cisplatin.59 In a phase III clinical trial, patients with NSCLC treated with Lipoplatin had a better response rate and fewer toxicities compared to patients treated with free cisplatin.60 Liposomal formulations of oxaliplatin analogs also have been developed. For example, Lipoxal is a liposomal oxaliplatin formulation that has reached clinical trials for advanced cancer,61 and was produced using similar formulation strategies as Lipoplatin. A recent study using Lipoxel in F98 glioma-bearing rats has shown that the maximum tolerable dose of Lipoxal is as much as 3-fold higher than that of free oxaliplatin.62 Another NP formulation, NC-6004, encapsulates cisplatin in polymeric micelles of PEG-poly (glutamic acid) and has advanced to clinical trials for solid tumors.63 NC-6004 provides a sustained release of cisplatin and consequently has low toxicity.63
Platinum NP delivery approaches often can exploit differences between normal tissues and tumors to increase the selectivity of the drug toward its intended target. Specifically, the enhanced permeability and retention (EPR) effect is based on the increased permeability of macromolecules in the tumor coupled with poor lymphatic clearance and slow venous return in these tissues,64 resulting in increased accumulation of the NPs within the tumors. Platinum drug-loaded NPs may take advantage of the EPR to achieve passive targeting to CNS tumors. This in some cases can be enhanced further by active targeting using ligands or antibodies attached to the NP surface that can selectively bind to tumor-specific moieties displayed on the target cells. Such moieties are generally transporters, antigens, or receptors that are expressed at higher levels in tumors compared to normal tissues. For example, NPs containing cisplatin were targeted to glioma cells using a monoclonal antibody to connexin 43, a protein highly expressed in the tumor. This targeted NP formulation exhibited reduced toxicity and prolonged the survival of glioma-bearing rats.65 Furthermore, a mitochondrial-targeted NP loaded with the cisplatin prodrug, Platin-M, successfully delivered the drug to neuroblastoma cells 66 and has shown very little neurotoxicity in animal models despite a high level of drug accumulation in the brain.67 Another intriguing glioma cell-specific target is the cell surface receptor fibroblast growth factor-inducible 14 (Fn14). NPs targeted to Fn14-positive GBM cells using a monoclonal antibody improved NP tumor localization and internalization.48,68 Thus, a similar targeting strategy may enhance the delivery of platinum compounds specifically to GBM cells, improving efficacy and minimizing toxicity.
Controlled and sustained drug release
Not only do NPs appear to enhance drug delivery to the GBM tumor tissue, they also offer the property of controlled drug release. Depending on the NP encapsulating material, drugs are released either as NPs degrade over time (e.g., PLGA) or simply diffuse from the NP system (e.g., liposome). For example, biodegradable PLGA carboplatin NPs successfully provided sustained release of carboplatin in rat brains.56 Moreover, NP formulations of carboplatin compounds are capable of providing controlled release of the drug for more than a week.69 Although sustained release can be achieved through numerous biomaterial formulation strategies, NPs can be designed to enable brain-penetration and tumor targeting,48,70 potentially improving treatment for invasive brain tumors, like GBM. Furthermore, free platinum drugs have a limited half-life in most tissues. For instance, the half-life of cisplatin is approximately 58h in rodent brains.23 Sustained platinum drug release by nanocarriers may be necessary for sustained inhibition of immunosuppressive features and a prolonged antitumor immune response.
Simultaneous incorporation and delivery of other anticancer treatments for combination therapy
The common observation that many single agent chemotherapeutic treatment regimens fail due to the emergence of resistant sub-clonal tumor cell populations strongly suggests that combinational treatment strategies will be necessary which utilize the diverse mechanisms of actions of multiple therapeutics to reduce the possibility of resistance.71 Combinations of drugs can have a synergistic effect, providing better treatment outcomes than single drug therapy.71-73 As mentioned earlier, platinum compounds are combined commonly with other anticancer agents including fluorouracil, etoposide, paclitaxel, and capecitabin.74 However, the successful simultaneous administration of two anticancer agents can be difficult due to the differences in drug solubility, biodistribution, and pharmacokinetics.74 One advantage to the use of NPs as drug delivery vehicles is that they can be formulated to contain more than one therapeutic agent. For example, the encapsulation of doxorubicin and cisplatin into a single nanocarrier was found to improve therapeutic efficacy compared to a treatment regimen of either drug alone, or co-administration of the two free drugs.74 Other studies have also shown that the delivery of NPs containing platinum drugs and another anticancer drug results in a synergistic antitumor effect.74,75 For an extensive review of polymer-based platinum combination therapy delivery systems, see ref. 74. Given the unique delivery challenges of CNS tumors, and the difficulty of effectively administering combination therapies, NPs present a means to deliver combination therapies consisting of a platinum drug and another anticancer drug. NPs can be designed to delivery platinum agents in combination with a tumor antigen, adjuvant, or other immunostimulatory factor in order to foster an antitumor immune response.23 NPs have been dual loaded with a TGF-β inhibitor and IL-2, providing a sustained local delivery of the drug combination, resulting in a synergistic antitumor effect in melanoma models.76 Platinum drugs may be dual loaded with other immunomodulatory therapeutics into NPs for a synergistic and sustained antitumor immunomodulation.
The immunomodulatory effects of platinum drugs may be enhanced or exploited for the treatment of GBM by loading NPs with a platinum drug and one or more other therapeutic agents. For example, NP formulations of chemokines are under investigation for anticancer therapy.77 The dual loading of chemokines and platinum-chemotherapeutics may offer synergistic anti-glioma effects; specifically, platinum drugs may generate a more permissive environment for immune cells to enter the tumor in response to chemokine co-treatment. Alternatively, dual-loading of dendritic cell stimulatory molecules such as cytosine-phosphate-guanine with platinum drugs may help generate a strong effector cell anti-glioma response.78 Furthermore, as some platinum drugs are capable of causing immunogenic cell death, it may be beneficial to co-deliver therapeutics to reinforce the resulting antitumor adaptive immune response. Although platinum drugs alone have multiple immunomodulatory effects, the co-delivery of platinum drugs and other immunomodulatory or cytotoxic therapeutics offers a way to strengthen or complement these multi-modal effects.
Conclusions
Many therapeutics, including platinum agents, have been investigated extensively as potential therapies for GBM patients. The limitations of these agents for GBM treatment have become increasingly clear as evidenced by the minimal improvement in patient survival and/or treatment-related toxicities. However, emerging evidence suggests there may be previously unrecognized and heretofore inaccessible therapeutic potential of such treatments, specifically platinum-based drugs, if the delivery and dosing can be controlled carefully. The principles of nanomedicine and formulation chemistry offer new opportunities to re-align the therapeutic ratio of otherwise toxic chemotherapeutics, especially in GBM because NPs can overcome some of the drug delivery challenges presented by the BBB and brain tissue, provide a sustained drug release, targeted to GBM cells, and reduce toxicity. Included in the newly recognized therapeutic effects appears to be immunomodulation, which may have broad application in future combination therapies for GBM.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the National Institutes of Health under Grant K12NS080223 (GW), National Institutes of Health under Grant K25EB018370 (AK), National Institutes of Health under Grant K08NS09043 (GW), Institutional Research Grant of American Cancer Society under Grant IRG-97-153-10 (AK), American Cancer Society under Grant RSG-16-012-01-CDD (GW), Passano Foundation Physician Scientist Award (GW), Elsa U. Pardee Foundation Research Grant (AK and JW), PhRMA Foundation Research Starter Grant in Pharmaceutics (AK), AAPS Foundation New Investigator Grant Award (AJK), and Dean's Challenge Award to Accelerate Innovation and Discovery in Medicine (JW and GW).
References
- 1.Wilson TA, Karajannis MA, Harter DH. Glioblastoma multiforme: state of the art and future therapeutics. Surg Neurol Int 2014; 5:64; PMID:24991467; http://dx.doi.org/ 10.4103/2152-7806.137196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg B, VanCamp L, Trosko JE, Mansour VH. Platinum compounds: a new class of potent antitumour agents. Nature 1969; 222:385-6; PMID:5782119 [DOI] [PubMed] [Google Scholar]
- 3.Oberoi HS, Nukolova NV, Kabanov AV, Bronich TK. Nanocarriers for delivery of platinum anticancer drugs. Adv Drug Deliv Rev 2013; 65:1667-85; PMID:24113520; http://dx.doi.org/ 10.1016/j.addr.2013.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basu A, Krishnamurthy S. Cellular responses to cisplatin-induced DNA damage. J Nucleic Acids 2010; 2010:16; PMID:20811617; http://dx.doi.org/ 10.4061/2010/201367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charest G, Sanche L, Fortin D, Mathieu D, Paquette B. Optimization of the route of platinum drugs administration to optimize the concomitant treatment with radiotherapy for glioblastoma implanted in the Fischer rat brain. J Neurooncol 2013; 115:365-73; PMID:24026531; http://dx.doi.org/ 10.1007/s11060-013-1238-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buckner JC, Ballman KV, Michalak JC, Burton GV, Cascino TL, Schomberg PJ, Hawkins RB, Scheithauer BW, Sandler HM, Marks RS, et al. Phase III trial of carmustine and cisplatin compared with carmustine alone and standard radiation therapy or accelerated radiation therapy in patients with glioblastoma multiforme: North Central Cancer Treatment Group 93-72-52 and Southwest Oncology Group 9503 Trials. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2006; 24:3871–9; PMID:16921039 [DOI] [PubMed] [Google Scholar]
- 7.Eiseman JL, Beumer JH, Rigatti LH, Strychor S, Meyers K, Dienel S, Horn CC. Plasma pharmacokinetics and tissue and brain distribution of cisplatin in musk shrews. Cancer Chemother Pharmacol 2015; 75:143-52; PMID:25398697; http://dx.doi.org/ 10.1007/s00280-014-2623-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeremic B, Grujicic D, Jevremovic S, Stanisavljevic B, Milojevic L, Djuric L, Mijatovic L. Carboplatin and etoposide chemotherapy regimen for recurrent malignant glioma: a phase II study. J Clin Oncol 1992; 10:1074-7; PMID:1318951 [DOI] [PubMed] [Google Scholar]
- 9.Peterson K, Harsh GT, Fisher PG, Adler J, Le Q. Daily low-dose carboplatin as a radiation sensitizer for newly diagnosed malignant glioma. J Neurooncol 2001; 53:27-32; PMID:11678427; http://dx.doi.org/ 10.1023/A:1011891209900 [DOI] [PubMed] [Google Scholar]
- 10.Brandes AA, Rigon A, Zampieri P, Ermani M, Carollo C, Altavilla G, Turazzi S, Chierichetti F, Florentino MV. Carboplatin and teniposide concurrent with radiotherapy in patients with glioblastoma multiforme: a phase II study. Cancer 1998; 82:355-61; PMID:9445194; http://dx.doi.org/ 10.1002/(SICI)1097-0142(19980115)82:2%3c362::AID-CNCR17%3e3.0.CO;2-X [DOI] [PubMed] [Google Scholar]
- 11.Jeremic B, Shibamoto Y, Acimovic L, Milicic B, Milisavljevic S, Nikolic N, Dagovic A, Aleksandrovic J, Radosavljevic-Asic G. Hyperfractionated radiation therapy and concurrent low-dose, daily carboplatin/etoposide with or without weekend carboplatin/etoposide chemotherapy in stage III non-small-cell lung cancer: a randomized trial. Int J Radiat Oncol Biol Phys 2001; 50:19-25; PMID:11316542; http://dx.doi.org/ 10.1016/S0360-3016(00)01546-7 [DOI] [PubMed] [Google Scholar]
- 12.Fortin D, McAllister LD, Nesbit G, Doolittle ND, Miner M, Hanson EJ, Neuwelt EA. Unusual cervical spinal cord toxicity associated with intra-arterial carboplatin, intra-arterial or intravenous etoposide phosphate, and intravenous cyclophosphamide in conjunction with osmotic blood brain-barrier disruption in the vertebral artery. Am J Neuroradiol 1999; 20:1794-802; PMID:10588099 [PMC free article] [PubMed] [Google Scholar]
- 13.Newton HB. Intra-arterial chemotherapy of primary brain tumors. Curr Treatment Options Oncol 2005; 6:519-30; PMID:16242056; http://dx.doi.org/ 10.1007/s11864-005-0030-1 [DOI] [PubMed] [Google Scholar]
- 14.Chintala S, Ali-Osman F, Mohanam S, Rayford A, Go Y, Gokaslan ZL, Gagercas E, Vekaiah B, Sawaya R, Nicolson G et al.. Effect of cisplatin and BCNU on MMP-2 levels in human glioblastoma cell lines in vitro. Clin Exp Metastasis 1997; 14:361-7; PMID:9219724; http://dx.doi.org/10890917 10.1023/A:1018442003163 [DOI] [PubMed] [Google Scholar]
- 15.Mishima K, Mazar AP, Gown A, Skelly M, Ji XD, Wang XD, Jones TR, Cavenee WK, Huang HJ. A peptide derived from the non-receptor-binding region of urokinase plasminogen activator inhibits glioblastoma growth and angiogenesis in vivo in combination with cisplatin. Proc Natl Acad Sci U S A 2000; 97:8484-9; PMID:10890917; http://dx.doi.org/ 10.1073/pnas.150239497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spiro TP, Liu L, Majka S, Haaga J, Willson JKV, Gerson SL. Temozolomide: the effect of once- and twice-a-day dosing on tumor tissue levels of the DNA repair protein O6-alkylguanine-DNA-alkyltransferase. Clin Cancer Res 2001; 7:2309-17; PMID:11489806 [PubMed] [Google Scholar]
- 17.Zheng Y, Hunting DJ, Ayotte P, Sanche L. Role of secondary low-energy electrons in the concomitant chemoradiation therapy of cancer. Phys Rev Lett 2008; 100:198101; PMID:18518490; http://dx.doi.org/ 10.1103/PhysRevLett.100.198101 [DOI] [PubMed] [Google Scholar]
- 18.Rolle CE, Sengupta S, Lesniak MS. Mechanisms of immune evasion by gliomas. Adv Exp Med Biol 2012; 746:53-76; PMID:22639159; http://dx.doi.org/ 10.1007/978-1-4614-3146-6_5 [DOI] [PubMed] [Google Scholar]
- 19.Nduom EK, Weller M, Heimberger AB. Immunosuppressive mechanisms in glioblastoma. Neuro Oncol 2015; 17 Suppl 7:vii9-14; PMID:26516226; http://dx.doi.org/ 10.1093/neuonc/nov151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patel MA, Pardoll DM. Concepts of immunotherapy for glioma. J Neurooncol 2015; 123:323-30; PMID:26070552; http://dx.doi.org/ 10.1007/s11060-015-1810-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson RC, Elder JB, Brown MD, Mandigo CE, Parsa AT, Kim PD, Senatus P, Anderson DE, Bruce JN. Changes in the immunologic phenotype of human malignant glioma cells after passaging in vitro. Clin Immunol 2002; 102:84-95; PMID:11781071; http://dx.doi.org/ 10.1006/clim.2001.5152 [DOI] [PubMed] [Google Scholar]
- 22.Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wohrer A, Dieckmann K, Filipits M, Brandstetter A, Weller M et al.. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol 2015; 17:1064-75; PMID:25355681; http://dx.doi.org/ 10.1093/neuonc/nou307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC et al.. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med 2007; 13:84-8; PMID:17159987; http://dx.doi.org/ 10.1038/nm1517 [DOI] [PubMed] [Google Scholar]
- 24.Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin Cancer Res 2013; 19:3165-75; PMID:23613317; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-3314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brantley EC, Nabors LB, Gillespie GY, Choi YH, Palmer CA, Harrison K, Roarty K, Benveniste EN. Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: implications for STAT-3 activation and gene expression. Clin Cancer Res 2008; 14:4694-704; PMID:18676737; http://dx.doi.org/ 10.1158/1078-0432.CCR-08-0618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goenka S, Kaplan MH. Transcriptional regulation by STAT6. Immunol Res 2011; 50:87-96; PMID:21442426; http://dx.doi.org/ 10.1007/s12026-011-8205-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Merk BC, Owens JL, Lopes MB, Silva CM, Hussaini IM. STAT6 expression in glioblastoma promotes invasive growth. BMC Cancer 2011; 11:184; PMID:21595984; http://dx.doi.org/ 10.1186/1471-2407-11-184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell 2015; 28:690-714; PMID:26678337; http://dx.doi.org/ 10.1016/j.ccell.2015.10.012 [DOI] [PubMed] [Google Scholar]
- 29.Kyi C, Postow MA. Checkpoint blocking antibodies in cancer immunotherapy. FEBS Lett 2014; 588:368-76; PMID:24161671; http://dx.doi.org/ 10.1016/j.febslet.2013.10.015 [DOI] [PubMed] [Google Scholar]
- 30.Barbee MS, Ogunniyi A, Horvat TZ, Dang TO. Current status and future directions of the immune checkpoint inhibitors ipilimumab, pembrolizumab, and nivolumab in oncology. Ann Pharmacother 2015; 49:907-37; PMID:25991832; http://dx.doi.org/ 10.1177/1060028015586218 [DOI] [PubMed] [Google Scholar]
- 31.Hato SV, Khong A, de Vries IJ, Lesterhuis WJ. Molecular pathways: the immunogenic effects of platinum-based chemotherapeutics. Clin Cancer Res 2014; 20:2831-7; PMID:24879823; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-3141 [DOI] [PubMed] [Google Scholar]
- 32.Bergmann-Leitner ES, Abrams SI. Treatment of human colon carcinoma cell lines with anti-neoplastic agents enhances their lytic sensitivity to antigenspecific CD8+ cytotoxic T lymphocytes. Cancer immunology, immunotherapy: CII 2001; 50:445–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lesterhuis WJ, Punt CJ, Hato SV, Eleveld-Trancikova D, Jansen BJ, Nierkens S, Schreibelt G, de Boer A, Van Herpen CM, Kaanders JH et al.. Platinum-based drugs disrupt STAT6-mediated suppression of immune responses against cancer in humans and mice. J Clin Investig 2011; 121:3100-8; PMID:21765211; http://dx.doi.org/ 10.1172/JCI43656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu WM, Fowler DW, Smith P, Dalgleish AG. Pre-treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br J Cancer 2009; 102:115-23; PMID:19997099; http://dx.doi.org/ 10.1038/sj.bjc.6605465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fletcher S, Turkson J, Gunning PT. Molecular approaches towards the inhibition of the signal transducer and activator of transcription 3 (STAT3) protein. ChemMedChem 2008; 3:1159-68; PMID:18683176; http://dx.doi.org/ 10.1002/cmdc.200800123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanterman J, Sade-Feldman M, Biton M, Ish-Shalom E, Lasry A, Goldshtein A, Hubert A, Baniyash M. Adverse immunoregulatory effects of 5FU and CPT11 chemotherapy on myeloid-derived suppressor cells and colorectal cancer outcomes. Cancer Res 2014; 74:6022-35; PMID:25209187; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-0657 [DOI] [PubMed] [Google Scholar]
- 37.Roselli M, Cereda V, di Bari MG, Formica V, Spila A, Jochems C, Farsaci B, Donahue R, Gulley JL, Schlom J et al.. Effects of conventional therapeutic interventions on the number and function of regulatory T cells. Oncoimmunology 2013; 2:e27025; PMID:24353914; http://dx.doi.org/ 10.4161/onci.27025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramakrishnan R, Assudani D, Nagaraj S, Hunter T, Cho HI, Antonia S, Altiok S, Celis E, Gabrilovich DI. Chemotherapy enhances tumor cell susceptibility to CTL-mediated killing during cancer immunotherapy in mice. J Clin Investig 2010; 120:1111-24; PMID:20234093; http://dx.doi.org/ 10.1172/JCI40269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bezu L, Gomes-de-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R, Galluzzi L, Kepp O, Kroemer G. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol 2015; 6:187; PMID:25964783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alcindor T, Beauger N. Oxaliplatin: a review in the era of molecularly targeted therapy. Current Oncol 2011; 18:18-25; PMID:21331278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kadowaki H, Nishitoh H. Signaling pathways from the endoplasmic reticulum and their roles in disease. Genes 2013; 4:306-33; PMID:24705207; http://dx.doi.org/ 10.3390/genes4030306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Tellingen O, Yetkin-Arik B, de Gooijer MC, Wesseling P, Wurdinger T, de Vries HE. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist Updat 2015; 19:1-12; PMID:25791797; http://dx.doi.org/ 10.1016/j.drup.2015.02.002 [DOI] [PubMed] [Google Scholar]
- 43.Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med 2013; 19:1584-96; PMID:24309662; http://dx.doi.org/ 10.1038/nm.3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx 2005; 2:3-14; PMID:15717053; http://dx.doi.org/ 10.1602/neurorx.2.1.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prados MD, Schold SC, Fine HA, Jaeckle K, Hochberg F, Mechtler L, Fetell MR, Phuphanich S, Feun L, Janus TJ et al.. A randomized, double-blind, placebo-controlled, phase 2 study of RMP-7 in combination with carboplatin administered intravenously for the treatment of recurrent malignant glioma. Neuro Oncol 2003; 5:96-103; PMID:12672281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sheleg S, Korotkevich E, Zhavrid E, Muravskaya G, Smeyanovich A, Shanko Y, Yurkshtovich T, Bychkovsky P, Belyaev S. Local chemotherapy with cisplatin-depot for glioblastoma multiforme. J Neurooncol 2002; 60:53-9; PMID:12416546; http://dx.doi.org/ 10.1023/A:1020288015457 [DOI] [PubMed] [Google Scholar]
- 47.White E, Bienemann A, Taylor H, Hopkins K, Cameron A, Gill S. A phase I trial of carboplatin administered by convection-enhanced delivery to patients with recurrent/progressive glioblastoma multiforme. Contemporary Clin Trials 33:320-31; PMID:22101221; http://dx.doi.org/ 10.1016/j.cct.2011.10.010 [DOI] [PubMed] [Google Scholar]
- 48.Schneider CS, Perez JG, Cheng E, Zhang C, Mastorakos P, Hanes J, Winkles JA, Woodworth GF, Kim AJ. Minimizing the non-specific binding of nanoparticles to the brain enables active targeting of Fn14-positive glioblastoma cells. Biomaterials 2015; 42:42-51; PMID:25542792; http://dx.doi.org/ 10.1016/j.biomaterials.2014.11.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Charest G, Paquette B, Fortin D, Mathieu D, Sanche L. Concomitant treatment of F98 glioma cells with new liposomal platinum compounds and ionizing radiation. J Neurooncol 2010; 97:187-93; PMID:19760366; http://dx.doi.org/ 10.1007/s11060-009-0011-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peng XH, Wang Y, Huang D, Wang Y, Shin HJ, Chen Z, Spewak MB, Mao H, Wang X, Wang Y et al.. Targeted delivery of cisplatin to lung cancer using ScFvEGFR-heparin-cisplatin nanoparticles. ACS Nano 2011; 5:9480-93; PMID:22032622; http://dx.doi.org/ 10.1021/nn202410f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller MA, Zheng YR, Gadde S, Pfirschke C, Zope H, Engblom C, Kohler RH, Iwamoto Y, Yang KS, Askevold B et al.. Tumour-associated macrophages act as a slow-release reservoir of nano-therapeutic Pt(IV) pro-drug. Nat Commun 2015; 6:8692; PMID:26503691; http://dx.doi.org/ 10.1038/ncomms9692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muldoon LL, Soussain C, Jahnke K, Johanson C, Siegal T, Smith QR, Hall WA, Hynynen K, Senter PD, Peereboom DM et al.. Chemotherapy delivery issues in central nervous system malignancy: a reality check. J Clin Oncol 2007; 25:2295-305; PMID:17538176; http://dx.doi.org/ 10.1200/JCO.2006.09.9861 [DOI] [PubMed] [Google Scholar]
- 53.Sykova E, Nicholson C. Diffusion in brain extracellular space. Physiol Rev 2008; 88:1277-340; PMID:18923183; http://dx.doi.org/ 10.1152/physrev.00027.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kreuter J. Drug delivery to the central nervous system by polymeric nanoparticles: what do we know? Adv Drug Deliv Rev 2014; 71:2-14; PMID:23981489; http://dx.doi.org/ 10.1016/j.addr.2013.08.008 [DOI] [PubMed] [Google Scholar]
- 55.Karanam V, Marslin G, Krishnamoorthy B, Chellan V, Siram K, Natarajan T, Bhaskar B, Franklin G. Poly (varepsilon-caprolactone) nanoparticles of carboplatin: preparation, characterization and in vitro cytotoxicity evaluation in U-87 MG cell lines. Colloids Surf B: Biointerfaces 2015; 130:48-52; PMID:25899843; http://dx.doi.org/ 10.1016/j.colsurfb.2015.04.005 [DOI] [PubMed] [Google Scholar]
- 56.Arshad A, Yang B, Bienemann AS, Barua NU, Wyatt MJ, Woolley M, Johnson DE, Edler KJ, Gill SS. Convection-enhanced delivery of carboplatin PLGA nanoparticles for the treatment of glioblastoma. PLoS One 2015; 10:e0132266; PMID:26186224; http://dx.doi.org/ 10.1371/journal.pone.0132266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saida Y, Watanabe S, Tanaka T, Baba J, Sato K, Shoji S, Igarashi N, Kondo R, Okajima M, Koshio J et al.. Critical roles of chemoresistant effector and regulatory T cells in antitumor immunity after lymphodepleting chemotherapy. J Immunol 2015; 195:726-35; PMID:26041539; http://dx.doi.org/ 10.4049/jimmunol.1401468 [DOI] [PubMed] [Google Scholar]
- 58.Duan X, He C, Kron SJ, Lin W. Nanoparticle formulations of cisplatin for cancer therapy Wiley Interdiscip Rev: Nanomed Nanobiotechnol 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boulikas T. Low toxicity and anticancer activity of a novel liposomal cisplatin (Lipoplatin) in mouse xenografts. Oncol Rep 2004; 12:3-12; PMID:15201951 [PubMed] [Google Scholar]
- 60.Stathopoulos GP, Antoniou D, Dimitroulis J, Stathopoulos J, Marosis K, Michalopoulou P. Comparison of liposomal cisplatin versus cisplatin in non-squamous cell non-small-cell lung cancer. Cancer Chemother Pharmacol 2011; 68:945-50; PMID:21301848; http://dx.doi.org/ 10.1007/s00280-011-1572-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stathopoulos GP, Boulikas T, Kourvetaris A, Stathopoulos J. Liposomal oxaliplatin in the treatment of advanced cancer: a phase I study. Anticancer Res 2006; 26:1489-93; PMID:16619562 [PubMed] [Google Scholar]
- 62.Shi M, Fortin D, Paquette B, Sanche L. Convection-enhancement delivery of liposomal formulation of oxaliplatin shows less toxicity than oxaliplatin yet maintains a similar median survival time in F98 glioma-bearing rat model. Invest New Drugs 2016; 34:269-76; PMID:26961906; http://dx.doi.org/ 10.1007/s10637-016-0340-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Plummer R, Wilson RH, Calvert H, Boddy AV, Griffin M, Sludden J, Tilby MJ, Eatock M, Pearson DG, Ottley CJ et al.. A phase I clinical study of cisplatin-incorporated polymeric micelles (NC-6004) in patients with solid tumours. Br J Cancer 2011; 104:593-8; PMID:21285987; http://dx.doi.org/ 10.1038/bjc.2011.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maeda H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv Drug Deliv Rev 2015; 91:3-6; PMID:25579058; http://dx.doi.org/ 10.1016/j.addr.2015.01.002 [DOI] [PubMed] [Google Scholar]
- 65.Nukolova NV, Baklaushev VP, Abakumova TO, Mel'nikov PA, Abakumov MA, Yusubalieva GM, Bychkov DA, Kabanov AV, Chekhonin VP. Targeted delivery of cisplatin by connexin 43 vector nanogels to the focus of experimental glioma C6. Bull Exp Biol Med 2014; 157:524-9; PMID:25110098; http://dx.doi.org/ 10.1007/s10517-014-2606-x [DOI] [PubMed] [Google Scholar]
- 66.Marrache S, Pathak RK, Dhar S. Detouring of cisplatin to access mitochondrial genome for overcoming resistance. Proc Natl Acad Sci USA 2014; 111:10444-9; PMID:25002500; http://dx.doi.org/26234400 10.1073/pnas.1405244111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Feldhaeusser B, Platt SR, Marrache S, Kolishetti N, Pathak RK, Montgomery DJ, Reno LR, Howerth E, Dhar S. Evaluation of nanoparticle delivered cisplatin in beagles. Nanoscale 2015; 7:13822-30; PMID:26234400; http://dx.doi.org/ 10.1039/C5NR03447G [DOI] [PubMed] [Google Scholar]
- 68.Perez JG, Tran NL, Rosenblum MG, Schneider CS, Connolly NP, Kim AJ, Woodworth GF, Winkles JA. The TWEAK receptor Fn14 is a potential cell surface portal for targeted delivery of glioblastoma therapeutics. Oncogene 2015; 35(17):2145-55; PMID:26300004; http://dx.doi.org/ 10.1038/onc.2015.310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sadhukha T, Prabha S. Encapsulation in nanoparticles improves anti-cancer efficacy of carboplatin. AAPS PharmSciTech 2014; 15:1029-38; PMID:24831091; http://dx.doi.org/ 10.1208/s12249-014-0139-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nance EA, Woodworth GF, Sailor KA, Shih TY, Xu Q, Swaminathan G, Xiang D, Eberhart C, Hanes J. A dense poly(ethylene glycol) coating improves penetration of large polymeric nanoparticles within brain tissue. Sci Transl Med 2012; 4:149ra119; PMID:22932224; http://dx.doi.org/26546464 10.1126/scitranslmed.3003594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.He C, Tang Z, Tian H, Chen X. Co-delivery of chemotherapeutics and proteins for synergistic therapy. Adv Drug Deliv Rev 2016; 98:64-76; PMID:26546464; http://dx.doi.org/ 10.1016/j.addr.2015.10.021 [DOI] [PubMed] [Google Scholar]
- 72.Jang B, Kwon H, Katila P, Lee SJ, Lee H. Dual delivery of biological therapeutics for multimodal and synergistic cancer therapies. Adv Drug Deliv Rev 2016; 98:113-33; PMID:26654747; http://dx.doi.org/ 10.1016/j.addr.2015.10.023 [DOI] [PubMed] [Google Scholar]
- 73.Piccolo MT, Menale C, Crispi S. Combined anticancer therapies: an overview of the latest applications. Anti-cancer Agents Med Chem 2015; 15:408-22; PMID:25584691; http://dx.doi.org/ 10.2174/1871520615666150113123039 [DOI] [PubMed] [Google Scholar]
- 74.Kang L, Gao Z, Huang W, Jin M, Wang Q. Nanocarrier-mediated co-delivery of chemotherapeutic drugs and gene agents for cancer treatment. Acta Pharm Sin B 2015; 5:169-75; PMID:26579443; http://dx.doi.org/ 10.1016/j.apsb.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang YT, Shi Y, Jay M, Di Pasqua AJ. Enhanced toxicity of cisplatin with chemosensitizer phenethyl isothiocyanate toward non-small cell lung cancer cells when delivered in liposomal nanoparticles. Chem Res Toxicol 2014; 27:946-8; PMID:24836554; http://dx.doi.org/ 10.1021/tx5001128 [DOI] [PubMed] [Google Scholar]
- 76.Brinker CJ. Nanoparticle immunotherapy: Combo combat. Nat Mater 2012; 11:831-2; PMID:23001226; http://dx.doi.org/ 10.1038/nmat3434 [DOI] [PubMed] [Google Scholar]
- 77.Lin Y, Sharma S, John MS. CCL21 cancer immunotherapy. Cancers 2014; 6:1098-110; PMID:24810425; http://dx.doi.org/ 10.3390/cancers6021098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shao K, Singha S, Clemente-Casares X, Tsai S, Yang Y, Santamaria P. Nanoparticle-based immunotherapy for cancer. ACS Nano 2015; 9:16-30; PMID:25469470; http://dx.doi.org/ 10.1021/nn5062029 [DOI] [PubMed] [Google Scholar]