

ABSTRACT
The beneficial effects of checkpoint blockade in tumor immunotherapy are limited to patients with increased tumor-infiltrating lymphocytes (TILs). Delineation of the regulatory networks that orchestrate the presence of TILs holds great promise for the design of effective immunotherapies. Podoplanin/gp38 (PDPN)-expressing lymph node stromal cells (LNSCs) are present in tumor stroma; however, their effect in the regulation of TILs remains elusive. Herein we demonstrate that intratumor injection of ex-vivo-isolated PDPN+ LNSCs into melanoma-bearing mice induces elimination of TILs and promotes tumor growth. In support, PDPN+ LNSCs exert their function through direct inhibition of CD4+ T cell proliferation in a cell-to-cell contact independent fashion. Mechanistically, we demonstrate that PDPN+ LNSCs mediate T cell growth arrest and induction of apoptosis to activated CD69+CD4+ T cells. Importantly, LTbR-Ig-mediated blockade of PDPN+ LNSCs expansion and function significantly attenuates melanoma tumor growth and enhances the infiltration and proliferation of CD4+ TILs. Overall, our findings decipher a novel role of PDPN-expressing LNSCs in the elimination of CD4+ TILs and propose a new target for tumor immunotherapy.
KEYWORDS: CD4+ tumor infiltrating lymphocytes, lymph node stroma cells, lymphocytes, melanoma, tumor
Abbreviations
- PDPN
podoplanin/gp38
- LNSCs
lymph node stroma cells
- LNs
lymph nodes
- TILs
tumor-infiltrating lymphocytes
- ELS
ectopic lymphoid-like structures
- LTbR
lymphotoxin beta receptor
- Tregs
T regulatory cells
- tdLNs
tumor draining lymph nodes
Introduction
The contexture of tumor microenvironment plays a crucial role in tumor development as well as progression and importantly dictates response to therapies.1 Although the advent of checkpoint blockade immunotherapies has attained remarkable benefit in solid tumors, their clinical effectiveness is limited in a small proportion of patients and is associated with the presence of tumor-infiltrating lymphocytes (TILs).2 Diverse regulatory pathways have been proposed to participate in elimination of TILs from the tumor milieu. To this end, chemokines that facilitate lymphocyte trafficking, immunomodulatory enzymes, and regulatory cells subsets such as Foxp3+ T regulatory cells (Tregs) and myeloid-derived suppressor cells (MDSCs) have all been demonstrated to impair the accumulation and function of TILs.3 However, targeting of these pathways has been shown to partially restore TIL presence and the generation of antitumor response.4,5 Therefore, a better understanding of the molecules and pathways that orchestrate the elimination of TILs is an important step toward the development of more efficacious and enduring cancer immunotherapies.
Lymph node stromal cells (LNSCs) play a pivotal role in the induction of peripheral tolerance under steady state but also in the course of an immune response. Thus, podoplanin/gp38 (PDPN)-expressing LNSCs inhibit the in vitro polyclonal and antigen-specific proliferation of both CD8+ and CD4+ T cells,6-8 and abrogation of CD8+ T cell/PDPN+ LNSCs interaction in vivo enhances the proliferation of CD8+ T cells.9 In addition, reduction of fibroblastic reticular cell (FRC), a subset of PDPN+ LNSCs, impaired the generation of anti-viral CD4+ and CD8+ T cell responses,10,11 whereas transplantation of FRCs in septic mice demonstrated a therapeutic effect.12 A potential role of the LNSC compartment in antitumor immune responses is emerging. To this end, ectopic lymphoid-like structures (ELSs) formed by LNSCs are found in solid tumors but their contribution to disease remains controversial.13 Thus, the presence of ELSs has been associated with better overall survival and favorable clinical outcome in several tumor types,14,15 whereas other studies demonstrate ELSs niches to promote the survival and growth of Tregs or tumor progenitor cells resulting in enhanced tumor growth in breast and hepatic cancer models.16-18 In support, PDPN+ LNSC subtypes, such as lymphatic endothelial cells (LECs), have been shown to enhance tumor growth by promoting the proliferation of tumor cells,19 or by presenting tumor antigens and leading to apoptosis of antitumor specific CD8+ T cells, thus accelerating metastasis.20 Collectively, although presence of PDPN+ LNSCs in solid tumors is well documented, their functional properties as well as the underlying mechanism via which PDPN+ LNSCs shape the antitumor immune response remains elusive.
Here, we demonstrate that PDPN+ LNSCs act in favor of tumor growth by inhibiting antitumor specific CD4+ T cell proliferation and by inducing death to activated CD4+ T cells. Importantly, depletion of PDPN+ LNSCs during melanoma development enhances the frequency and proliferation of CD4+ TILs and significantly reduces tumor growth.
Results
PDPN+ LNSCs infiltrate melanoma tumor and inhibit tumor growth
Stroma cells infiltrate solid tumors and orchestrate the formation of ELSs.13 But their role in antitumor immune responses remains controversial. Herein we focused on the PDPN-expressing stroma cells as the major subset of LNSCs21 that have been implicated in peripheral tolerance induction.22 Interestingly, immunohistological assessment of B16/F10 melanoma solid tumors revealed a significant degree of infiltration of PDPN+ ER-TR7+ stroma cells that belong to FRCs and LYVE-1+ PDPN+ cells characteristic of LECs (Fig. 1A). To dissect their role in tumor growth, PDPN+ cells were sorted in high purity (>98%) from lymph nodes (LNs) isolated from naive mice (Fig. 1B) and co-injected with B16/F10 tumor cells in syngeneic recipients. A repetitive injection of PDPN+ LNSCs was performed intratumoraly (i.t.) on day 10 after inoculation that tumors were palpable (Fig. 1C). Notably, PDPN+ LNSCs-treated animals demonstrated significantly increased tumor volume compared to PBS-injected mice (Fig. 1D). Analysis of TILs revealed decreased numbers of both CD4+ and CD8+ T cells compared to control animals (Fig. 1E). PDPN+ LNSCs retained their functional properties upon isolation as demonstrated by the increased expression of IL-7 and CCL21 and the enhanced survival of naive CD4+ T cells in vitro (Fig. S1A and B). Overall, these results provided evidence that PDPN+ LNSCs in B16/F10 melanoma solid tumors promoted tumor growth and dampened antitumor immune responses.
Figure 1.

Enhanced tumor growth and reduced TILs in mice i.t. injected with PDPN+ LNSCs. (A) Immunohistochemical LYVE-1, ER-TR7, PDPN, and DAPI staining of day 14 tumor sections is shown. (B) Gating strategy for the isolation of PDPN+ LNSCs from LNs of naive mice is presented. (C) Experimental outline for B16/F10 and PDPN+ LNSCs in vivo administration. Mice were injected s.c. on day 0 with 3 × 105 B16/F10 and 5–10 × 104 PDPN+ LNSCs sorted from LNs of naïve mice. On day 10 mice received an i.t. injection of 5–10 × 104 PDPN+ LNSCs. On day 15 mice were sacrificed and their TILs were analyzed. (D) Mean and standard deviation of tumor volume of mice treated as in (B) 15 d after tumor inoculation are denoted. (E) Gating strategy, mean, and standard deviation of CD4+ and CD8+ T cell numbers per 5 × 105 total tumor cells are shown. Means were calculated from three independent experiments. Numbers on FACS plots denote frequency of gated population. *p < 0.05, **p < 0.005.
PDPNC+ LNSCs infiltrate melanoma tumor and promote tumor growth
The emerging role of CD4+ T cells in antitumor immune responses23-25 prompted us to elucidate the mechanism of action of PDPN+ LNSCs on CD4+ T cells. To this end, sorted PDPN+ LNSCs from tumors or tumor-draining lymph nodes (tdLNs) were co-cultured with naive CD4+CD25−CD69− T cells in the presence of aCD3/IL-2. PDPN+ cells from both tumors and tdLNs significantly inhibited the proliferation of CD4+ T cells (Fig. 2A). Interestingly, PDPN+ LNSCs isolated from skin-draining LNs of naive mice also inhibited the proliferation of CD4+ T cells in vitro, as determined by the decreased percentage of dividing cells (Fig. 2B). Thus, PDPN+ LNSCs retained their suppressive properties in tumor microenvironment and attenuated the proliferation of CD4+ T cells. To gain insights into the inhibition of CD4+ T cell proliferation by PDPN+ LNSCs, we performed a transwell experiment. As shown in Fig. 2C even in the absence of cell-to-cell contact PDPN+ LNSCs diminished the number of dividing CD4+ T cells and the percentage of CD4+ T cells that have underwent four or more rounds of proliferation. In support, addition of supernatants from PDPN+ LNSCs-CD4+ T cells co-cultures significantly decreased the number of cell divisions (Fig. 2D). These data suggest that PDPN+ LNSC-mediated suppression of CD4+ T cell proliferation was cell contact independent.
Figure 2.
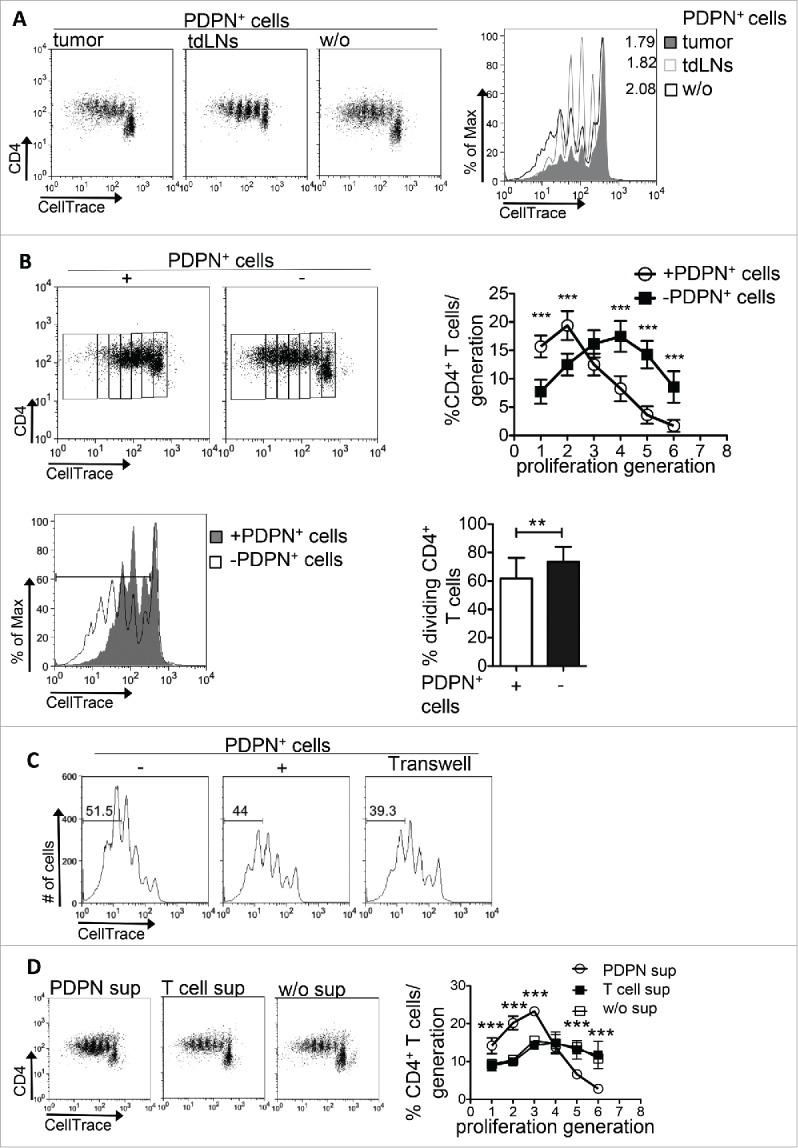
PDPN+ LNSCs inhibit CD4+ T cell proliferation in vitro. (A) CellTrace-labeled CD4+CD25−CD69−CD11c− (15 × 104) T cells sorted from splenocytes of naive C57BL/6 mice were activated with aCD3 (0.2 μg/mL) and IL-2 (20 ng/mL) for 3.5 d in the presence or absence of PDPN+ LNSCs (5 × 104) sorted from tumors and tdLNs on day 14 after tumor inoculation. (B) T cells isolated and activated as in A were co-cultured with PDPN+ LNSCs sorted from naive mice. (C) PDPN+ LNSCs were separated from T cells using a 0.4 μm transwell insert. Proliferation was assessed by flow cytometry based on CellTrace dilution. Graphs denote the mean percentages and standard deviations of CD4+ T cells in each proliferation generation and of dividing CD4+ T cells. (D) Mean and standard deviation of the percentage of CD4+ T cells per generation of proliferation in cultures of CellTrace-labeled CD4+CD25−CD69−CD11c− (15 × 104) T cells with aCD3 (0.2 μg/mL) and IL-2 (20 ng/mL) in the absence (w/o sup) or presence of 3.5 d supernatants from co-cultures of PDPN+ LNSCs with CD4+ T cells (PDPN sup) or from cultures of CD4+ T cells alone (T cell sup) are presented. Means have been calculated from 15 to 19 independent experiments. Numbers on FACS plots denote frequency of gated population or proliferation index (p.i.). **p < 0.005, ***p < 0.0005.
PDPN+ LNSCs block TCR signaling and activation of CD4+ T cells
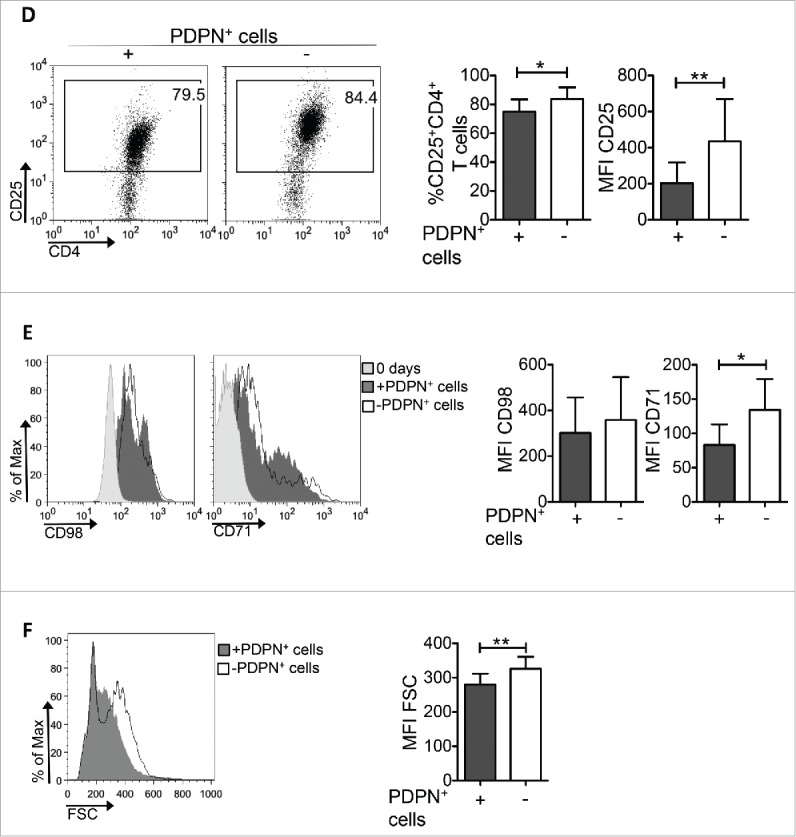
The duration and strength of TCR triggering determines the magnitude of activation and proliferation of CD4+ T cells.26,27 Our results show that activated CD4+ T cells presented the same levels of TCRβ internalization during the first 18 h (0.5 d) in the presence or absence of PDPN+ LNSCs (Fig. 3A) whereas higher levels of TCRβ were expressed 1.5 d of incubation with PDPN+ LNSCs (Fig. 3A). In line with this, phosphorylation levels of ZAP70 and SYK, two downstream molecules of TCR triggering, were decreased in CD4+ T cells activated in the presence of PDPN+ LNSCs (Fig. 3B). Furthermore, analysis of CD4+ T cells 1.5 d after stimulation revealed a significant difference in CD69 expression in PDPN+ LNSC-cultured CD4+ T cells but not the first hours of culture (0.5 d) (Fig. 3C). In addition, expression of CD25 by CD4+ T cells 3.5 d after stimulation was also significantly attenuated in the presence of PDPN+ LNSCs (Fig. 3D). To determine whether cell growth was affected, we examined the expression of amino-acid transporter CD98 and the transferring receptor CD71, responsible for iron uptake. To this end, surface level of CD71 was downregulated in CD4+CD69+ T cells activated with PDPN+ LNSCs compared to those cultured without PDPN+ LNSCs (Fig. 3E). Consistent with this result, the cell growth of CD4+ T cells was hindered by PDPN+ LNSCs compared to that of CD4+ T cells cultured alone (Fig. 3F). Collectively, these results demonstrated a dominant suppressive effect of PDPN+ LNSCs on cell growth and activation of CD4+ T cells.
Figure 3.

PDPN+ LNSCs impair TCR signaling, CD4+ T cell activation and growth. CD4+CD25−CD69−CD11c− (15 × 104) T cells sorted from splenocytes of naive C57BL/6 mice were activated with aCD3 (0.2 μg/mL) and IL-2 (20 ng/mL) in the presence or absence of PDPN+ LNSCs (5 × 104) sorted from LNs of naive mice. (A) Geometric mean (Geo Mean) and standard deviation of TCRβ surface expression on days 0.5 and 1.5 are shown. (B) Histogram represents phosphorylation of ZAP70 and SYK(Y319/Y352) on day 0.5. (C) Mean and standard deviation of the percentage of CD69+CD4+ T cells on days 0.5 and 1.5 are presented. (D) On day 3.5 mean and standard deviation of the percentage of CD25+CD4+ T cells and of CD25 mean fluorescent intensity (MFI) were calculated and are denoted. (E) MFI and standard deviation of CD98 and CD71 cell surface expression on CD4+CD69+ T cells on day 0 and 1.5 are shown. (F) Histogram and graph represent the MFI of forward side scatter (FSC) of CD4+ T cells 3.5 d after the initiation of the culture. Means and standard deviation were calculated from 4 (A, B, E) and 20 (C, D, F) replicates. Numbers on FACS plots denote frequency of gated population. *p < 0.05, **p < 0.005, ***p < 0.0005.
Figure 3.
(Continued)
PDPN+ LNSCs promote apoptosis of activated CD4+ T cells
Next, we sought to delineate the mechanism via which PDPN+ LNSCs exert their suppressive function on activated CD4+ T cells. To examine whether PDPN+ LNSCs induced anergy, PDPN+ LNSC-cultured CD4+ T cells were re-activated with aCD3/aCD28 but no significant differences in proliferation were observed (Fig. S2A) excluding the possibility of anergy induction.
It was recently shown that LNSCs could induce Foxp3+ Tregs.28 Thus, we assessed Foxp3 expression using CD4+Foxp3− T cells from Foxp3gfp reporter mice. Culture of CD4+Foxp3− T cells with PDPN+ LNSC did not cause upregulation of Foxp3 expression or intracellular expression of IL-10 (Fig. S2B and C). Therefore, our results indicated that PDPN+ LNSCs directly inhibited CD4+ T cell proliferation in vitro without any contribution from Tregs.
Deletion of activated T cells could also be accounted for tolerance induction. We analyzed the frequency of 7AAD+ cells in proliferating (CFSElow) and non-proliferating (CFSEhigh) CD4+ T cells cultured with PDPN+ LNSCs. PDPN+ LNSCs induced death of proliferating CD4+ T cells only (Fig. 4A, data not shown), and this was accompanied by significantly increased expression of total (Fig. 4B) as well as activated 3, 7 executive caspases (Fig. S3A) on activated CD69+ CD4+ T cells. In support, a greater percentage of CD69+ CD4+ T cells stained positive for histone γH2Ax (Fig. 4C) and cleaved caspase 3 (Fig. S3B) in the co-cultures of T cells with PDPN+ LNSCs than in cultures of T cells alone as shown by confocal microscopy. Finally, addition of PDPN+ LNSCs on day 1.5 after activation of CD4+ T cells resulted in enhanced percentages of apoptotic cells compared to the culture of CD4+ T cells alone (Fig. 4D), indicating that PDPN+ LNSCs induced death to activated CD4+ T cells. In conclusion, we demonstrated that PDPN+ LNSCs induced apoptosis of activated CD4+ T cells.
Figure 4.
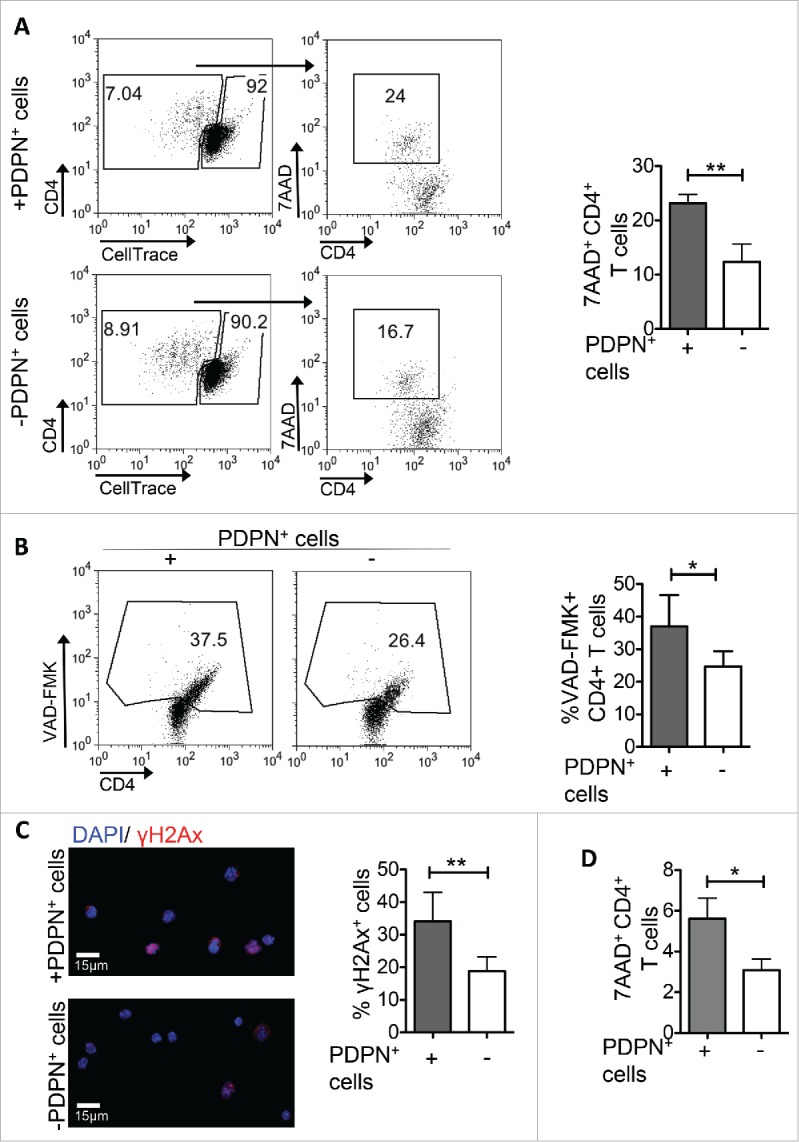
PDPN+ LNSCs induce apoptosis to activated CD4+ T cells. CellTrace-labeled CD4+CD25−CD69−CD11c− (15 × 104) T cells sorted from splenocytes of naive C57BL/6 mice were activated with aCD3 (0.2 μg/mL) and IL-2 (20 ng/mL) and either at day 0 (A, B, C) or at day 1.5 (D) PDPN+ LNSCs (5 × 104) sorted from LNs of naive mice were added. (A, D) Mean percentages and standard deviation of 7AAD+ cells in the gated populations of proliferating (CellTrace low) CD4+ T cells on day 3.5 are presented. Gating strategy is denoted in the FACS plots. (B) After 1.5 d of incubation the mean percentage and standard deviation of VAD-FMK+ cells in the gated population of CD4+CD69+ T cells are presented. (C) 1.5 d following the initiation of the co-culture CD4+CD69+ T cells were sorted, fixed with PFA, permeabilized with Triton, and stained with anti-γH2Ax. Mean percentage and standard deviation of γH2Ax positive cells were calculated from a total of 100 cells. Means were calculated from four independent experiments. Numbers on FACS plots denote frequency of gated population. *p < 0.05, **p < 0.005.
FRCs promote cell growth arrest and LECs induce apoptosis to CD4+ T cells
The major subsets of PDPN+ LNSCs are FRCs (PDPN+CD31−) and LECs (PDPN+CD31+).21 To examine whether the impaired CD4+ T cell proliferation is attributed to FRCs or LECs, CD4+ T cells were activated with aCD3/IL-2 stimuli in the presence or absence of PDPN+ LNSCs subsets (Fig. 5A). Both FRCs and LECs inhibited the proliferation of CD4+ T cells as indicated by the decreased percentages of dividing cells compared to CD4+ T cells cultured alone (Fig. 5B). Notably, FRCs impeded the activation of CD4+ T cells as indicated by the decreased expression of CD25 as well as their cell growth, whereas LECs were found to induce apoptosis to activated CD4+ T cells (Fig. 5 C and D). Overall, these results strongly suggested that FRCs dampened CD4+ T cell proliferation by inhibiting CD4+ T cell activation whereas LECs by inducing death to activated CD4+ T cells.
Figure 5.
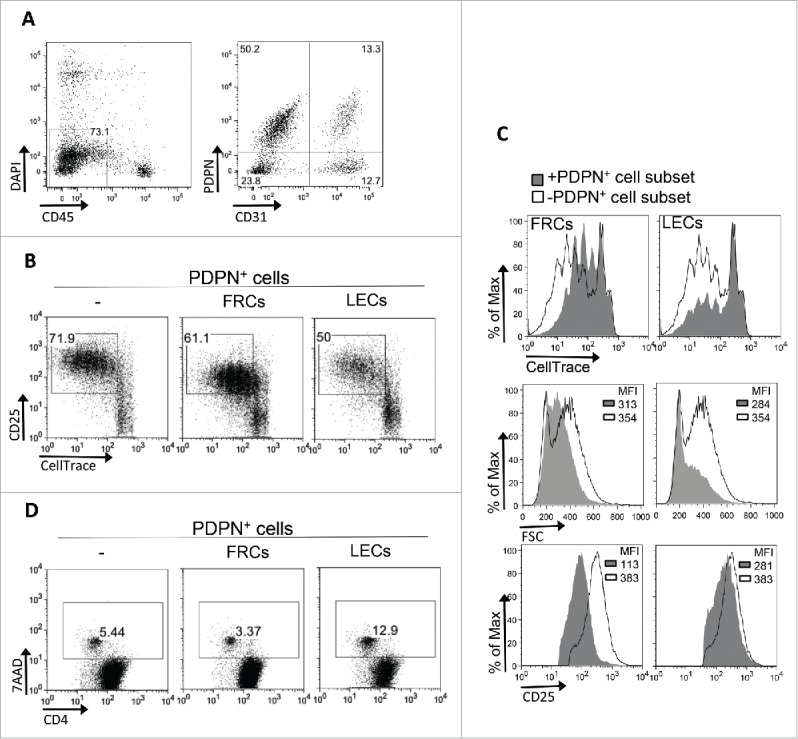
FRCs and LECs PDPN+ LNSC subsets inhibit CD4+ T cell proliferation. (A) Gating strategy for the sorting of FRCs and LECs from CD45+ cells-enriched LNs of naive mice is shown. (B–D) CD4+CD25−CD69−CD11c− (15 × 104) T cells sorted from splenocytes of naive C57BL/6 mice were activated with aCD3 (0.2 μg/mL) and IL-2 (20 ng/mL) in the presence or absence of FRCs or LECs (5 × 104) sorted from LNs of naive mice. (B) Proliferation and activation was assessed by flow cytometry based on CellTrace dilution and CD25 expression, respectively. Numbers on FACS plots represent percentages of dividing cells. (C) Overlays of CellTrace and FSC of cells gated on CD4+ T cells and of CD25 gated on CD25+CD4+ T cells are presented. (D) Percentages of 7AAD+ cells gated on CellTrace low CD4+ T cells (as in Fig. 4A) are denoted on the gated populations of the FACS plots. Results are representative of three independent experiments.
Increased numbers and proliferation of CD4+ T cells and reduced tumor growth in LTbR-Ig-treated tumor-bearing mice
Next, we prompted to examine whether targeting PDPN+ LNSCs during tumor growth could have a therapeutic effect. Lymphotoxin beta receptor (LTbR) signaling on LNSCs is crucial for their function and expansion during inflammation.13,29,30 Thus, we dissected the therapeutic value of an LTbR immunoglobulin fusion protein (LTbR-Ig) that blocks LTbR signaling. To this end, mice were inoculated with B16/F10 melanoma cells and treated i.p. with LTbR-Ig every 3 d. Treated mice presented a significant reduction of the PDPN+ cells in tumor microenvironment, as compared to isotype-treated controls (Fig. 6A). Histological observation was confirmed by quantitative RT-PCR performed on frozen sections from the tumor explants (Fig. 6B). Interestingly, LTbR-Ig-treated mice demonstrated significantly decreased tumor volume compared to control-treated animals (Fig. 6C). Using PET/CT with 18F-FDG, we confirmed that the markedly reduced tumor volume in LTbR-Ig-treated group was accompanied by reduced metabolic activity as determined by decreased total lesion glycolysis (TLG) (Isotype 10930 ± 1826 and LTbR-Ig 3769 ± 1481) (Fig. 6D). Importantly, inhibition of tumor growth in LTbR-Ig-treated mice was accompanied by significantly increased frequency of CD4+ T cells in the tdLNs and in TILs, whereas the frequency of CD4+Foxp3+ Tregs was significantly decreased (Fig. 6E). Indeed, we found that the percentage of CD4+ T cells expressing the proliferation marker Ki67 was significantly elevated in the tdLNs of B16/F10-inoculated mice treated with LTbR-Ig, compared to mice treated with the control Ig (Fig. 7A). Moreover, the frequency of apoptotic CD4+ T cells in the tdLNs of mice treated with LTbR-Ig was significantly reduced compared to control-treated mice (Fig. 7B). Furthermore, upon adoptive transfer of CellTrace-labeled Ovabumin (OVA)-specific OTII Va2+ CD4+ T cells into B16-OVA tumor bearing mice treated either with LTbR-Ig or control, we demonstrated that the total number of transferred cells was significantly enhanced (Fig. 7C) and exhibited increased proliferation (Fig. 7C) in the spleen of LTbR-Ig-treated mice compared to mice treated with isotype control. Finally, i.t. administration of LTbR-Ig, when the tumor was palpable, demonstrated a greater percentage of CD3+CD4+ TILs compared to isotype-treated mice whereas the percentages of CD4+ T cells in tdLNs had no difference (Fig. S4), suggesting a local PDPN+ LNSC-T cell interaction. In conclusion, LTbR-Ig-treatment during melanoma growth elicited potent antitumor immune responses and significantly suppressed tumor burden associated with a decrease in PDPN+ cells in the tumor microenvironment.
Figure 6.
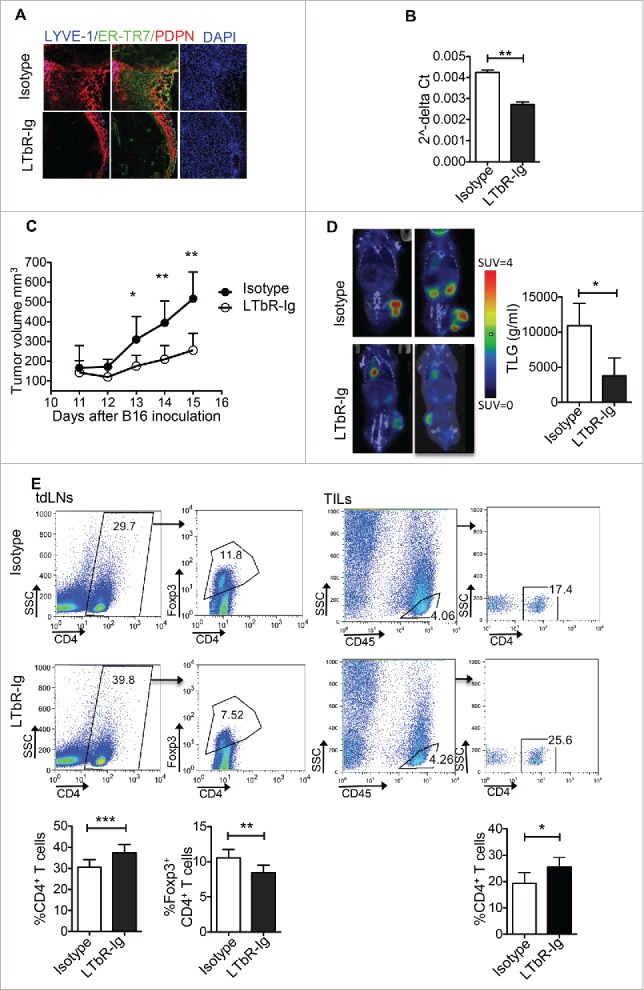
Reduced tumor growth and increased numbers of CD4+ T cells in tumor-bearing mice treated with LTbR-Ig. Mice inoculated s.c. with 3 × 105 B16/F10 melanoma cells were treated i.p. every three days with LTbR-Ig (100 μg/mouse) or isotype control. (A) Immunohistochemical LYVE-1, ER-TR7, PDPN, and DAPI staining of day 14 tumor sections is presented. (B) Quantitative RT-PCR analysis of PDPN mRNA obtained from tumor tissues at day 14. (C) Mean tumor volume (mm3) and standard deviation are presented. (D) Representative PET/CT images, mean and standard deviation of total lesion glycolysis (TLG) of mice 13–14 d following tumor inoculation are denoted. SUV stands for standardized uptake value. (E) FACS plots and graphs represent gating strategy, mean percentages, and standard deviation of CD4+ T cells and CD4+ Foxp3+ T cells in tdLNs and CD45+CD4+ T cells in TILs 14–15 d after tumor inoculation. Data are representative of three independent experiments with 5–8 replicates each. Numbers on FACS plots denote frequency of gated population. *p < 0.05, **p < 0.005, ***p < 0.0005.
Figure 7.
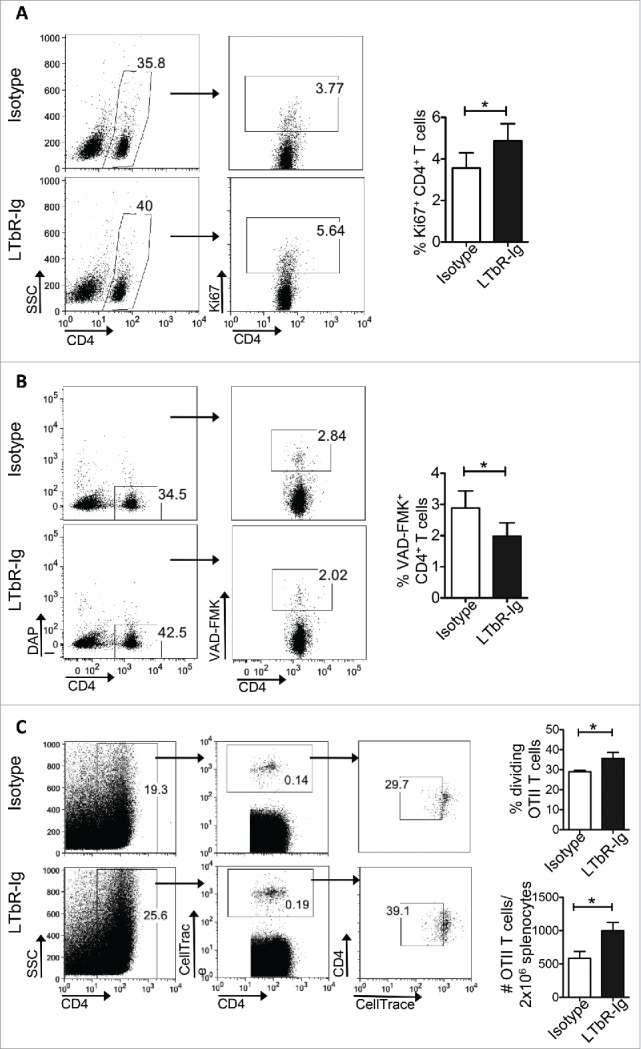
PDPN+ LNSCs inhibit the proliferation and induce apoptosis to antitumor CD4+ T cells in vivo. (A, B) Graphs represent gating strategy, mean, and standard deviation of the percentage of Ki67+ (A) and VAD-FMK+ (B) CD4+ T cells in the tdLNs of mice inoculated s.c. at the back with B16-F10 melanoma cells (3 × 105), treated i.p. every three days with LTbR-Ig (100 μg/mouse) or isotype and analyzed 15 d later. (C) CellTrace-labeled OTII Va2+ CD4+ T cells adoptively transferred (1 × 106/mouse) i.v. at day 8 into B16-OVA inoculated mice treated i.p. every three days with LTbR-Ig (100 μg/mouse) or isotype. Graphs denote gating strategy, mean, and standard deviation of total spleen numbers and percentages of dividing of OTII Va2+ CD4+ T cells. Means and standard deviations were calculated from one to two experiments with five replicates each. *p < 0.05, **p < 0.005.
Discussion
The effectiveness of antitumor immune responses is critically depended on infiltration of tumors by activated effector T cells. Presence of TILs is considered to possess a prognostic benefit in solid tumors and is associated with clinical response to current immunotherapies.31 Therefore, understanding of the mechanisms that impede antitumor immune responses will facilitate the design of more effective therapeutic strategies aiming to enhance the antitumor T cell-mediated immune responses. Toward this, our findings demonstrate a novel role of PDPN-expressing LNSCs in tumor evasion, through suppression of antitumor CD4+ T cell responses. Contraction of the PDPN+ LNSC compartment obtained through treatment of melanoma-inoculated mice with LTbR-Ig results in inhibition of tumor growth and increased proliferation of CD4+ T cells in the tdLNs.
Besides the pivotal role of LNSCs in the structural organization of secondary lymphoid organs and the orchestration of immune responses, accumulating evidence underlies a crucial involvement of LNSCs in the regulation of immunity and induction of peripheral tolerance. The notion of LNSCs as critical regulators of inflammatory responses has been strengthened by the identification of ELS in a variety of inflamed tissues;13,32-35 however, their contribution to tumor development and progression as well as response to therapy is not known. In this context, our data suggest that presence of PDPN+ LNSCs in the tumor promotes tumor growth and impede antitumor responses. The underlying mechanism responsible for the recruitment of PDPN+ LNSCs in the tumor milieu remains elusive. It is possible that chemokines secreted by tumor cells or other stromal populations might form a cue to direct the recruitment of PDPN+ LNSCs. This is supported by a seminal study where over-expression of the chemoattractant CCL21 by melanoma cells resulted in enhanced recruitment of CCR7+ lymphoid tissue inducer (LTi) cells and induction of ELS at the tumor site that promoted tumor evasion.36 During embryogenesis expression of lymphotoxin alpha and beta by LTi cells is required for the maturation of resident messenchymal cells in LNSCs via upregulation of adhesion molecules and PDPN.37,38 To this end, assessment of Lta and Ltb gene expression by tumor cells as well as tumor-infiltrating cells demonstrated that CD3+ TILs express high levels of both genes suggesting that TILs contribute to LNSCs recruitment into tumor milieu (A.K. and V.P. unpublished data). Accordingly, here we demonstrate that blocking of LTbR signaling during melanoma growth affects the formation of PDPN+ stromal cell network and overall decreases the PDPN transcript in the tumor microenvironment. This in turn results in an enhancement of the antitumor immune responses and promotion of tumor rejection suggesting the PDPN+ LNSCs as a potent cell population in cancer immunotherapy.
Diverse mechanisms have been attributed to LNSC-mediated induction of peripheral tolerance. Ectopic expression of peripheral tissue antigens (PTAs) by LNSCs was shown to drive clonal deletion of self-reactive CD8+ T cells22,39-44 in a nitric oxide synthetase (NOS2)-dependent manner.6 In line with this, tumor-antigen uptake and cross-presentation by LECs led to CD8+ T cell apoptosis.20 Furthermore, suppression of proliferation and subsequent apoptosis of CD4+ T cells was induced by LNSCs upon acquisition of peptide–MHC II complexes through DC-derived exosomes45 or through induction of Tregs.28 Finally, splenic stromal cells induced tolerance via induction of tolerogenic dendritic cells.46,47 Our findings extend the current knowledge on the tolerogenic properties of LNSCs since we demonstrate for the first time that within the tumor microenvironment, the expanded population of PDPN+ cells mediates the elimination of CD4+ TILs in a cell contact independent fashion. The delineation of the precise molecular pathways that facilitate the PDPN+ LNSC-mediated contraction of CD4+ TILs in melanoma tumor is under investigation. Of interest, expression of PDPN has been associated with tumor cell spreading, migration, and invasion in several types of cancer48 but the respective mechanism remains elusive. Accumulating evidence suggests, however, that PDPN-expressing cancer-associated fibroblasts (CAFs) promote tumor cell invasion in vitro49 with the serine residues in the intracellular domain of PDPN to accommodate CAF–tumor cell interactions.50 Although CAFs have been extensively shown to promote tumor growth,51 whether they constitute a different cell type of PDPN+ LNSCs described in this study remains to be seen. For example, in line with our results, co-injection of CAFs with B16-melanoma cells enhanced tumor growth in vivo.52 However, only the FRC subset of LNSCs could resemble CAFs whereas our data demonstrate that both FRCs and LECs impede CD4+ T cell proliferation.
Tumor-reactive effector CD4+ T cells could not only orchestrate an effective antitumor CD8+ T cell response but they could also themselves provide protection against tumor. In support of this, CD4+ T cells have been shown to enhance and sustain the accumulation of CD8+ cytotoxic T cells within tumors.23,24 Moreover, tumor-specific Th17 cells were shown to be sufficient to diminish B16 melanoma growth53 via recruitment of dendritic cells into the tumor and development of potent antitumor cytotoxic T cell response.54 Finally, adoptive transfer of tumor-specific CD4+ T cells into tumor-bearing lymphopenic hosts resulted in complete tumor eradication mediated by a potent cytotoxic activity of the CD4+ T cells.25 Our findings support a PDPN+ LNSC-mediated apoptosis and inhibition of proliferation on activated CD4+ T cells raising the idea for a dominant effect of PDPN-expressing LNSCs in tumor evasion mechanisms.
Delineation of the pathways that deliver negative regulatory signals to TILs leading to their elimination or to the induction of tolerance holds great promise for the design of better and more potent therapeutic protocols. Toward this, combination of immunotherapies has been proposed to produce durable antitumor responses in patients that did not respond to monotherapies.55,56 However, the choice of the therapeutic regimens that should be combined is not an obvious task. Several parameters, including the immune contexture of the tumor, should be taken into account for the right integration of combined immunotherapy. Toward this, our findings describe a mechanism that promotes elimination of TILs upon encountering PDPN+ stroma cells and propose that targeting of this cell subset could increase the lymphocytic immune infiltrate and thus might enhance the clinical benefit of immunotherapy. The more knowledge acquired on the diverse mechanism that impede antitumor immunity and promote tumor evasion not only would facilitate the design of more efficient therapeutic agents but also could help in biomarker development and patient selection who will benefit from the respective immunotherapies.
Materials and methods
Animals – cell lines
C57BL/6 were maintained in the animal facility of Biomedical Research Foundation of Academy of Athens, Foxp3gfp.KI mice (C57BL/6 background), kindly provided by Dr. Alexander Rudensky (Department of Immunology, Memorial Sloan-Kettering Cancer Center, New York, USA), OTII TCR CD45.1 Tg mice kindly provided by Dr. Federica Sallusto (Institute of Research in Biomedicine, Universita’ della Svizzera italiana, Italy). All procedures were in accordance to institutional guidelines and were approved by the Greek Federal Veterinary Office. All mice used in the experiments were female 8–10 weeks old.
The cancer cell line B16/F10 was kindly provided by Dr. Aris Eliopoulos (School of Medicine, University of Crete, Greece) and the B16 cell line stably expressing ovalbumin with gfp (B16-OVA) was kindly provided by Dr. Caetano Reis e Sousa (The Francis Crick Institute, London, UK).
Antibodies and reagents
The following antibodies targeting: CD4+ (clone RM4-4, GK1.5), I-Ab (AF6-120.1), CD44 (IM7), Foxp3 (150D), Ki67 (16A8), CD45 (30-F11), PDPN (8.1.1), CD31 (390), CD11c (117308), CD25 (3C7), CD69 (H1.2F3), CD98 (RL388), CD71 (RI7217), TCRβ (H57-597), IL-10 (JES5-1688), TCR Va2 (B20.1) were purchased from Biolegend and pZAP70/pSYK (n3kobu5) was purchased from eBioscience.
Functional grade purified anti-CD3e (aCD3) (clone 145-2C11) was from Biolegend and anti-CD28 (aCD28) (clone 37.51) was from eBioscience. 7-amino-actinomycin D (7AAD) (Biolegend) and 4′,6-diamidino-2-phenylindole (DAPI) were from Sigma-Aldrich. Recombinant murine IL-2 and recombinant human TGFβ were from Peprotech, and CellTrace Violet Cell Proliferation Kit was from Invitrogen.
Tumor experiments
C57BL/6 mice were implanted subcutaneously (s.c.) on the back with a mixture of 3 × 105 B16/F10 melanoma cells with 5–10 × 104 PDPN+ LNSCs sorted from LNs of naive mice. Ten days later mice received intratumoraly (i.t.) 5–10 × 104 PDPN+ LNSCs sorted from LNs of naive mice. For the therapeutic protocol, C57BL/6 mice were implanted subcutaneously (s.c.) on the back with 3 × 105/100 μL B16/F10 or B16-OVA melanoma cells. One hundred microgram of mLTbR-mIgG1 (LTbR-Ig) and of a control murine monoclonal IgG1 antibody MOPC-21 (prepared by Biogen/Idec) were administered intraperitoneally (i.p.) every 3 d following the melanoma cells injection. Mice inoculated with B16-OVA melanoma cells received OTII T cells 8 d after the inoculation. In more detail, CD4+ Va2+ T cells were sorted from LNs and splenocytes of naive OTII mice, stained with CellTrace (50 μM) and adoptively transferred intravenous (i.v.) (1 × 106/mouse) into tumor bearing mice. Tumor growth was monitored by measurement of two perpendicular diameters (d) of the tumor by caliper every day from day nine to day fourteen following melanoma cells inoculation. The total tumor volume was calculated using the equation .
Histology and immunofluorescence
For the immunofluorescence staining of tumors, 7 μm sections of tumor tissue were cut by cryosectioning and fixed for 20 min in ice-cold acetone. Then, sections were incubated for 60 min with the following primary antibodies in PBS (1%BSA); PDPN clone 8.1.1 (Biolegend) and ER-TR7 (AbD Serotech). PDPN was detected using goat anti-hamster biotin (Cambridge Bioscience) and then streptavidin-Alexa Fluor 555 (Molecular Probes). ER-TR7 was detected with goat anti-rat FITC (Southern Biotech). DAPI (Molecular Probes) was used for nuclear staining. All secondary antibodies were incubated for 30 min. Sections were mounted using Prolong Gold Antifade reagent (Invitrogen Life Technologies). Images were acquired on an LSM 780 laser scanning confocal microscope. Confocal micrographs were stored as digital arrays of 2048×2048 pixels with 8-bit sensitivity; detectors were routinely set so that intensities in each channel spanned the 0–255 scale optimally. The ZEN 2012 imaging software was used to process the images. For the staining of sorted CD69+ CD4+ T cells 10 × 104–10 × 105 cells were incubated for 15 min on poly-lysine-treated coverslips and fixed with 4% PFA for 15 min at RT. For γH2Ax staining cells were permeabilized with Triton X 0.2% for 30 min and incubated for another 30 min with blocking buffer (1% BSA with 0.1% Triton X). For cleaved caspase-3 staining cells were incubated with blocking buffer 5% FBS and 0.1% Triton X. A 60 min incubation with the anti-mouse γH2Ax (MABE205, Merck Millipore) (1/200) or anti-cleaved caspase-3 (Cell Signaling) (1/500) diluted in blocking buffer followed. Anti-γH2Ax was detected with Alexa fluor® 555 anti-mouse IgG (1:500, Molecular Probes) and anti-cleaved caspase-3 with Alexa Fluor® 488 anti-Rabbit IgG (H+L) (1:200, Invitrogen). Again DAPI was used for nuclear staining and cells were mounted using Mowiol (Sigma). Images were acquired using an inverted confocal live cell imaging system Leica SP5.
LNSCs isolation
LNSCs were isolated as described.21 Briefly, inguinal, branchial, and axillary LNs of naive C57BL/6 mice and tumors or tdLNs of C57BL/6 mice inoculated with B16/F10 cells were cut into smaller pieces and incubated in digestion medium (RPMI medium (Gibco) containing 0.1 mg/mL DNaseI (Invitrogen), 0.2 mg/mL collagenase P (Roche) and 0.8 mg/mL dispase (Gibco)) for 25 min at 37°C and the cell fraction was collected. The same procedure was repeated for the remaining undigested LN fractions for 20 min. The single-cell suspensions were filtered (100 μm). For the cell-sorting of PDPN+ LNSCs, the single-cell suspension was enriched for CD45− cells with MACS cell separation technology (Miltenyi) and stained with antibodies.
Flow cytometry
For analysis of TILs, tumors were dissected and incubated for 45 min at 37°C in RPMI medium containing 0.1 mg/mL DNaseI, 0.2 mg/mL collagenase P and 0.8 mg/mL dispase then was homogenized and strained through a nylon filter with a pore size of 40 μm (BD Falcon). For analysis of tdLNs and spleen, LN cell suspension and splenocytes cell suspension were prepared by passing through a nylon strainer with a pore size of 40 μm. For analysis of T cells co-cultured with PDPN+ LNSCs, cells were collected from the wells using PBS and stained as follows.
For staining of extracellular markers, cell suspensions were incubated with antibodies for 20 min at 4°C. For caspases staining, FITC-VAD-FMK (eBioscience) or FAM-DEVD-FMK (Flica3,7) (ImmunoChemistry Technologies) in complete medium were incubated with the cell suspensions for 1 h at 37°C and 5% CO2, washed, and stained for the extracellular markers, according to the vendors instructions. For Ki67 staining, cells were stained for extracellular markers, fixed for 1 h with 96% ethanol at −20°C and then stained for Ki67 for 30 min at RT. For Foxp3 intracellular staining, cells were stained for the extracellular markers and then fixed and stained using the Foxp3 Staining Set (eBioscience) according to the vendor instructions. For pZAP/SYK intracellular staining, cells were stained for extracellular markers and then fixed and stained using the BD Cytofix/Cytoperm Plus Fixation/Permeabilization kit (BD). For IL-10 intracellular staining, cells were incubated with 50 ng/mL PMA (Sigma-Aldrich), 2 μg/mL Ionomycin (Sigma-Aldrich) and Golgi plug (1/1000) (BD) for 6 h at 37°C and 5% CO2, stained for extracellular markers, fixed and stained for IL-10 using the BD Cytofix/Cytoperm Plus Fixation/Permeabilization kit (BD), according to vendor instructions. All samples were analyzed with ARIA III (BD).
Flow cytometry data were analyzed with FlowJo 8.7 software. The proliferation index (p.i.) shows the average number of divisions the responding cells have undergone.
PET/CT imaging
C57BL/6 mice (n = 8, with average weight 16.3 ± 0 .44 g) were fasted but with a free access to water, for a period of 12 h prior to imaging. PET studies were performed under anesthesia using 2% isoflurane in 0.8 L/min O2. Vital signs were monitored throughout the anesthesia period. Mice were placed on a warmed bed so that temperature was maintained at 36°C throughout the study. 18F-FDG (10.53 ± 3 .81 MBq, in a mean volume of 132.86 ± 33 .52 μL) was injected in the lateral tail vein, except from one animal, where due to vein canulation difficulties, an injection was given in the intraperitonial cavity. List mode acquisition over 20 min was employed preceded by 60 min uptake time. Imaging was performed using a Mediso scanner (Mediso nanoScan PC, with 8 detector modules) with an axial field of view of 98.6 mm and a spatial resolution of 0.8 mm at the center of the scanner (with Tera-Tomo 3D PET iterative reconstruction). PET imaging was preceded by CT, for attenuation correction and anatomical localization of the PET images. X-ray beam energy of 50 kVp, exposure time 300 msec and current 670 μA were employed. The number of projections per rotation was 480 and the slice thickness was 250 μm. For the CT image reconstruction, a modified version of the Feldkamp algorithm and the Ram-Lak filter were used. PET and CT data were reconstructed using Nucline software version 2.01 (Build 011.0005). The PET image reconstruction was performed using a version of 3D OSEM algorithm (Tera-Tomo 3D PET iterative reconstruction) and voxel dimensions of 0.4 × 0.4 × 0.4 mm3 (image matrix size 87 × 75 × 246). Corrections for dead time, decay, scatter, attenuation and axial sensitivity as well as normalization were applied to the PET data. Image analysis was performed with the Mediso InterView Fusion software package version 3.00.039.0000 and total lesion glycolysis (TLG), a parameter that reflects viable cell density in the whole tumor was obtained by drawing volumes of interests around the tumor.
Co-cultures of CD4+ T cells and PDPN+ LNSCs
Cell-sorted CD4+CD25−CD69−CD11c− T cells from splenocytes of naive C57BL/6 mice or CD4+CD25−CD69−CD11c−Foxp3− T cells from splenocytes of naive Foxp3gfp reporter mice were stained with CellTrace (10 μM) according to vendor's instructions, cultured in 96-well round-bottomed plates (Sarstedt), and activated or not with soluble aCD3 (0.2 μg/mL) and IL-2 (20 ng/mL) (Peprotech). Either simultaneously or 1.5 d after the initiation of the culture cell-sorted PDPN+ LNSCs were added in the culture in a ratio of T cells/PDPN+ LNSCs 3/1 (15 × 104 T cells and 5 × 104 PDPN+ LNSCs). CD4+ T cells were analyzed in different time points from 4 h to 3.5 d after the initiation of the culture. For some experiments, a Transwell insert with a pore size of 0.4 μm (Corning HTS Transwell) was included and CD4+ T cells were cultured in the plate whereas PDPN+ LNSCs were cultured in the transwell.
In some experiments, cell-sorted CellTrace labeled CD4+CD25−CD69−CD11c− T cells (15 × 104) from splenocytes of naive C57BL/6 mice were activated with soluble aCD3 (0.2 μg/mL) and IL-2 (20 ng/mL) and the culture medium was supplemented with 20% of 3.5 d supernatant of PDPN+ LNSCs and CD4+ T cells co-cultures stimulated with aCD3 (0.2 μg/mL) and IL-2 (20 ng/mL) or of CD4+ T cells cultured alone and stimulated with aCD3 (0.2 μg/mL) and IL-2 (20 ng/mL). After 3.5 d proliferation of CD4+ T cells was analyzed.
Restimulation of CD4+ T cells activated in the presence of PDPN+ LNSCs
Cell‐sorted CellTrace labeled CD4+CD25−CD69−CD11c− T cells (15 × 104) from splenocytes of naive C57BL/6 mice were stimulated with soluble aCD3 (0.2 μg/mL) and IL‐2 (20 ng/mL) in the presence or absence of cell‐sorted PDPN+ LNSCs (5 × 104) from LNs of naive C57BL/6 mice. After 1.5 d CD4+7AAD− T cells (5 × 104) were sorted from the cultures and restimulated with plate bound aCD3 (10 μg/mL) and soluble aCD28 (2 μg/mL) for another 2.5 d.
Quantitative RT‐PCR
Cell‐sorted PDPN+ LNSCs, PDPN− LNSCs, and CD45+ cells from LNs of naive mice as well as 14 d tumors were lysed in RLT buffer (Qiagen). There RNA was extracted using Qiagen RNeasy mini kit or micro kit according to manufacturer's guidelines. First‐strand cDNA synthesis was performed using Superscript II reverse transcriptase (Invitrogen). RT‐PCR was carried out in 20 μL reactions using the iTaq Universal SYBR Green Supermix (BioRad). The program used was: 95°C for 30 sec, 95°C for 10 sec and 60°C for 30 sec for 40 cycles. Relative expression of target genes was calculated by comparing them to the expression of the house‐keeping genes HPRT and β‐actin. The following primers were used:
HPRT: Forward 5′-GTGAAACTGGAAAAGCCAAA‐3′, Reverse 5′‐GGACGCAGCAACTGACAT‐3′, IL‐7: Forward 5′‐CGCAGACCATGTTCCAT‐3′, Reverse 5′‐AATGTGGCACTCAGATGATGT‐3′, CCL21: Forward 5′‐CAAGGCAGTGATGGAGG‐3′, Reverse 5′‐GGGGTGAGAACAGGATTG‐3′ and PDPN from Applied Biosystems.
Data analysis and statistics
For statistical analysis, Prism 5 (GraphPad Software) was used and data were compared with an unpaired two-tailed t-test, with 95% confidence intervals (Student's t-test).
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors wish to thank Themis Alissafi, Aggelos Banos and Nikos Paschalidis for their excellent cooperation and assistance, Paulos Alexakos for its technical assistant on animal handling and maintenance, Anastasia Apostolidou and Arianna Gavriil for their technical assistant on flow cytometry.
Funding
A.H. was supported by a fellowship from the Greek Foundation of Public Fellowships (IKY). This work was supported by a grant from the Greek General Secretariat of Research and Technology (Aristeia II 3468 to P.V.).
References
- 1.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013; 19:1423-37; PMID:24202395; http://dx.doi.org/ 10.1038/nm.3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 2013; 14:1014-1022; PMID:24048123; http://dx.doi.org/ 10.1038/ni.2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 2005; 5:263-74; PMID:15776005; http://dx.doi.org/ 10.1038/nrc1586 [DOI] [PubMed] [Google Scholar]
- 4.Byrne WL, Mills KH, Lederer JA, O'Sullivan GC. Targeting regulatory T cells in cancer. Cancer Res 2011; 71:6915-20; PMID:22068034; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Najjar YG, Finke JH. Clinical perspectives on targeting of myeloid derived suppressor cells in the treatment of cancer. Front Oncol 2013; 3:49; PMID:23508517; http://dx.doi.org/ 10.3389/fonc.2013.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lukacs-Kornek V, Malhotra D, Fletcher AL, Acton SE, Elpek KG, Tayalia P, Collier AR, Turley SJ. Regulated release of nitric oxide by nonhematopoietic stroma controls expansion of the activated T cell pool in lymph nodes. Nat Immunol 2011; 12:1096-104; PMID:21926986; http://dx.doi.org/ 10.1038/ni.2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siegert S, Huang HY, Yang CY, Scarpellino L, Carrie L, Essex S, Nelson PJ, Heikenwalder M, Acha-Orbea H, Buckley CD et al.. Fibroblastic reticular cells from lymph nodes attenuate T cell expansion by producing nitric oxide. PLoS One 2011; 6:e27618; PMID:22110693; http://dx.doi.org/ 10.1371/journal.pone.0027618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ng CT, Nayak BP, Schmedt C, Oldstone MB. Immortalized clones of fibroblastic reticular cells activate virus-specific T cells during virus infection. Proc Natl Acad Sci USA 2012; 109:7823-8; PMID:22550183; http://dx.doi.org/ 10.1073/pnas.1205850109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Astarita JL, Cremasco V, Fu J, Darnell MC, Peck JR, Nieves-Bonilla JM, Song K, Kondo Y, Woodruff MC, Gogineni A et al.. The CLEC-2-podoplanin axis controls the contractility of fibroblastic reticular cells and lymph node microarchitecture. Nat Immunol 2015; 16:75-84; PMID:25347465; http://dx.doi.org/ 10.1038/ni.3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denton AE, Roberts EW, Linterman MA, Fearon DT. Fibroblastic reticular cells of the lymph node are required for retention of resting but not activated CD8+ T cells. Proc Natl Acad Sci USA 2014; 111:12139-44; PMID:25092322; http://dx.doi.org/ 10.1073/pnas.1412910111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cremasco V, Woodruff MC, Onder L, Cupovic J, Nieves-Bonilla JM, Schildberg FA, Chang J, Cremasco F, Harvey CJ, Wucherpfennig K et al.. B cell homeostasis and follicle confines are governed by fibroblastic reticular cells. Nat Immunol 2014; 15:973-981; PMID:25151489; http://dx.doi.org/ 10.1038/ni.2965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fletcher AL, Elman JS, Astarita J, Murray R, Saeidi N, D'Rozario J, Knoblich K, Brown FD, Schildberg FA, Nieves JM et al.. Lymph node fibroblastic reticular cell transplants show robust therapeutic efficacy in high-mortality murine sepsis. Sci Transl Med 2014; 6:249ra109; PMID:25122637; http://dx.doi.org/ 10.1126/scitranslmed.3009377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol 2014; 14:447-62; PMID:24948366; http://dx.doi.org/ 10.1038/nri3700 [DOI] [PubMed] [Google Scholar]
- 14.Messina JL, Fenstermacher DA, Eschrich S, Qu X, Berglund AE, Lloyd MC, Schell MJ, Sondak VK, Weber JS, Mule JJ. 12-Chemokine gene signature identifies lymph node-like structures in melanoma: potential for patient selection for immunotherapy? Sci Rep 2012; 2:765; PMID:23097687; http://dx.doi.org/ 10.1038/srep00765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Behr DS, Peitsch WK, Hametner C, Lasitschka F, Houben R, Schonhaar K, Michel J, Dollt C, Goebeler M, Marx A et al.. Prognostic value of immune cell infiltration, tertiary lymphoid structures and PD-L1 expression in Merkel cell carcinomas. Int J Clin Exp Pathol 2014; 7:7610-21; PMID:25550797 [PMC free article] [PubMed] [Google Scholar]
- 16.Sautes-Fridman C, Fridman WH. TLS in Tumors: What Lies Within. Trends Immunol 2016; 37:1-2; PMID:26706045; http://dx.doi.org/ 10.1016/j.it.2015.12.001 [DOI] [PubMed] [Google Scholar]
- 17.Finkin S, Yuan D, Stein I, Taniguchi K, Weber A, Unger K, Browning JL, Goossens N, Nakagawa S, Gunasekaran G et al.. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat Immunol 2015; 16:1235-44; PMID:26502405; http://dx.doi.org/ 10.1038/ni.3290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joshi NS, Akama-Garren EH, Lu Y, Lee DY, Chang GP, Li A, DuPage M, Tammela T, Kerper NR, Farago AF et al.. Regulatory T cells in tumor-associated tertiary lymphoid structures suppress anti-tumor T cell responses. Immunity 2015; 43:579-90; PMID:26341400; http://dx.doi.org/ 10.1016/j.immuni.2015.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee E, Pandey NB, Popel AS. Lymphatic endothelial cells support tumor growth in breast cancer. Sci Rep 2014; 4:5853; PMID:25068296; http://dx.doi.org/ 10.1038/srep05853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lund AW, Duraes FV, Hirosue S, Raghavan VR, Nembrini C, Thomas SN, Issa A, Hugues S, Swartz MA. VEGF-C promotes immune tolerance in B16 melanomas and cross-presentation of tumor antigen by lymph node lymphatics. Cell Rep 2012; 1:191-9; PMID:22832193; http://dx.doi.org/ 10.1016/j.celrep.2012.01.005 [DOI] [PubMed] [Google Scholar]
- 21.Fletcher AL, Malhotra D, Acton SE, Lukacs-Kornek V, Bellemare-Pelletier A, Curry M, Armant M, Turley SJ. Reproducible isolation of lymph node stromal cells reveals site-dependent differences in fibroblastic reticular cells. Front Immunol 2011; 2:35; PMID:22566825; http://dx.doi.org/ 10.3389/fimmu.2011.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fletcher AL, Lukacs-Kornek V, Reynoso ED, Pinner SE, Bellemare-Pelletier A, Curry MS, Collier AR, Boyd RL, Turley SJ. Lymph node fibroblastic reticular cells directly present peripheral tissue antigen under steady-state and inflammatory conditions. J Exp Med 2010; 207:689-97; PMID:20308362; http://dx.doi.org/ 10.1084/jem.20092642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marzo AL, Kinnear BF, Lake RA, Frelinger JJ, Collins EJ, Robinson BW, Scott B. Tumor-specific CD4+ T cells have a major “post-licensing” role in CTL mediated anti-tumor immunity. J Immunol 2000; 165:6047-55; PMID:11086036; http://dx.doi.org/ 10.4049/jimmunol.165.11.6047 [DOI] [PubMed] [Google Scholar]
- 24.Bos R, Sherman LA. CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res 2010; 70:8368-77; PMID:20940398; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, Blasberg R, Yagita H, Muranski P, Antony PA et al.. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med 2010; 207:637-650; PMID:20156971; http://dx.doi.org/ 10.1084/jem.20091918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huppa JB, Gleimer M, Sumen C, Davis MM. Continuous T cell receptor signaling required for synapse maintenance and full effector potential. Nat Immunol 2003; 4:749-55; PMID:12858171; http://dx.doi.org/ 10.1038/ni951 [DOI] [PubMed] [Google Scholar]
- 27.Schrum AG, Palmer E, Turka LA. Distinct temporal programming of naive CD4+ T cells for cell division versus TCR-dependent death susceptibility by antigen-presenting macrophages. Eur J Immunol 2005; 35:449-59; PMID:15682456; http://dx.doi.org/ 10.1002/eji.200425635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baptista AP, Roozendaal R, Reijmers RM, Koning JJ, Unger WW, Greuter M, Keuning ED, Molenaar R, Goverse G, Sneeboer MM et al.. Lymph node stromal cells constrain immunity via MHC class II self-antigen presentation. Elife 2014; 3:1-18; PMID:25407678; http://dx.doi.org/ 10.7554/eLife.04433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Randall TD, Carragher DM, Rangel-Moreno J. Development of secondary lymphoid organs. Annu Rev Immunol 2008; 26:627-50; PMID:18370924; http://dx.doi.org/ 10.1146/annurev.immunol.26.021607.090257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chai Q, Onder L, Scandella E, Gil-Cruz C, Perez-Shibayama C, Cupovic J, Danuser R, Sparwasser T, Luther SA, Thiel V et al.. Maturation of lymph node fibroblastic reticular cells from myofibroblastic precursors is critical for antiviral immunity. Immunity 2013; 38:1013-24; PMID:23623380; http://dx.doi.org/ 10.1016/j.immuni.2013.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melero I, Rouzaut A, Motz GT, Coukos G. T-cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov 2014; 4:522-6; PMID:24795012; http://dx.doi.org/ 10.1158/2159-8290.CD-13-0985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bergomas F, Grizzi F, Doni A, Pesce S, Laghi L, Allavena P, Mantovani A, Marchesi F. Tertiary intratumor lymphoid tissue in colo-rectal cancer. Cancers (Basel) 2011; 4:1-10; PMID:24213222; http://dx.doi.org/ 10.3390/cancers4010001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Chaisemartin L, Goc J, Damotte D, Validire P, Magdeleinat P, Alifano M, Cremer I, Fridman WH, Sautes-Fridman C, Dieu-Nosjean MC. Characterization of chemokines and adhesion molecules associated with T cell presence in tertiary lymphoid structures in human lung cancer. Cancer Res 2011; 71:6391-9; PMID:21900403; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-0952 [DOI] [PubMed] [Google Scholar]
- 34.Dieu-Nosjean MC, Antoine M, Danel C, Heudes D, Wislez M, Poulot V, Rabbe N, Laurans L, Tartour E, de Chaisemartin L et al.. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J Clin Oncol 2008; 26:4410-7; PMID:18802153; http://dx.doi.org/ 10.1200/JCO.2007.15.0284 [DOI] [PubMed] [Google Scholar]
- 35.Goc J, Fridman WH, Sautes-Fridman C, Dieu-Nosjean MC. Characteristics of tertiary lymphoid structures in primary cancers. Oncoimmunology 2013; 2:e26836; PMID:24498556; http://dx.doi.org/ 10.4161/onci.26836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science 2010; 328:749-52; PMID:20339029; http://dx.doi.org/ 10.1126/science.1185837 [DOI] [PubMed] [Google Scholar]
- 37.Benezech C, Mader E, Desanti G, Khan M, Nakamura K, White A, Ware CF, Anderson G, Caamano JH. Lymphotoxin-beta receptor signaling through NF-kappaB2-RelB pathway reprograms adipocyte precursors as lymph node stromal cells. Immunity 2012; 37:721-734; PMID:22940098; http://dx.doi.org/ 10.1016/j.immuni.2012.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol 2010; 10:664-74; PMID:20706277; http://dx.doi.org/ 10.1038/nri2832 [DOI] [PubMed] [Google Scholar]
- 39.Lee JW, Epardaud M, Sun J, Becker JE, Cheng AC, Yonekura AR, Heath JK, Turley SJ. Peripheral antigen display by lymph node stroma promotes T cell tolerance to intestinal self. Nat Immunol 2007; 8:181-90; PMID:17195844; http://dx.doi.org/ 10.1038/ni1427 [DOI] [PubMed] [Google Scholar]
- 40.Nichols LA, Chen Y, Colella TA, Bennett CL, Clausen BE, Engelhard VH. Deletional self-tolerance to a melanocyte/melanoma antigen derived from tyrosinase is mediated by a radio-resistant cell in peripheral and mesenteric lymph nodes. J Immunol 2007; 179:993-1003; PMID:17617591; http://dx.doi.org/ 10.4049/jimmunol.179.2.993 [DOI] [PubMed] [Google Scholar]
- 41.Gardner JM, Devoss JJ, Friedman RS, Wong DJ, Tan YX, Zhou X, Johannes KP, Su MA, Chang HY, Krummel MF et al.. Deletional tolerance mediated by extrathymic Aire-expressing cells. Science 2008; 321:843-7; PMID:18687966; http://dx.doi.org/ 10.1126/science.1159407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Magnusson FC, Liblau RS, von Boehmer H, Pittet MJ, Lee JW, Turley SJ, Khazaie K. Direct presentation of antigen by lymph node stromal cells protects against CD8 T-cell-mediated intestinal autoimmunity. Gastroenterology 2008; 134:1028-37; PMID:18395084; http://dx.doi.org/ 10.1053/j.gastro.2008.01.070 [DOI] [PubMed] [Google Scholar]
- 43.Yip L, Su L, Sheng D, Chang P, Atkinson M, Czesak M, Albert PR, Collier AR, Turley SJ, Fathman CG et al.. Deaf1 isoforms control the expression of genes encoding peripheral tissue antigens in the pancreatic lymph nodes during type 1 diabetes. Nat Immunol 2009; 10:1026-33; PMID:19668219; http://dx.doi.org/ 10.1038/ni.1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cohen JN, Guidi CJ, Tewalt EF, Qiao H, Rouhani SJ, Ruddell A, Farr AG, Tung KS, Engelhard VH. Lymph node-resident lymphatic endothelial cells mediate peripheral tolerance via Aire-independent direct antigen presentation. J Exp Med 2010; 207:681-8; PMID:20308365; http://dx.doi.org/ 10.1084/jem.20092465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dubrot J, Duraes FV, Potin L, Capotosti F, Brighouse D, Suter T, LeibundGut-Landmann S, Garbi N, Reith W, Swartz MA et al.. Lymph node stromal cells acquire peptide-MHCII complexes from dendritic cells and induce antigen-specific CD4(+) T cell tolerance. J Exp Med 2014; 211:1153-66; PMID:24842370; http://dx.doi.org/ 10.1084/jem.20132000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu X, Yi H, Guo Z, Qian C, Xia S, Yao Y, Cao X. Splenic stroma-educated regulatory dendritic cells induce apoptosis of activated CD4 T cells via Fas ligand-enhanced IFN-gamma and nitric oxide. J Immunol 2012; 188:1168-77; PMID:22205032; http://dx.doi.org/ 10.4049/jimmunol.1101696 [DOI] [PubMed] [Google Scholar]
- 47.Zhang M, Tang H, Guo Z, An H, Zhu X, Song W, Guo J, Huang X, Chen T, Wang J et al.. Splenic stroma drives mature dendritic cells to differentiate into regulatory dendritic cells. Nat Immunol 2004; 5:1124-33; PMID:15475957; http://dx.doi.org/ 10.1038/ni1130 [DOI] [PubMed] [Google Scholar]
- 48.Wicki A, Lehembre F, Wick N, Hantusch B, Kerjaschki D, Christofori G. Tumor invasion in the absence of epithelial-mesenchymal transition: podoplanin-mediated remodeling of the actin cytoskeleton. Cancer Cell 2006; 9:261-72; PMID:16616332; http://dx.doi.org/ 10.1016/j.ccr.2006.03.010 [DOI] [PubMed] [Google Scholar]
- 49.Neri S, Ishii G, Hashimoto H, Kuwata T, Nagai K, Date H, Ochiai A. Podoplanin-expressing cancer-associated fibroblasts lead and enhance the local invasion of cancer cells in lung adenocarcinoma. Int J Cancer 2015; 137:784-96; PMID:25648219; http://dx.doi.org/ 10.1002/ijc.29464 [DOI] [PubMed] [Google Scholar]
- 50.Krishnan H, Ochoa-Alvarez JA, Shen Y, Nevel E, Lakshminarayanan M, Williams MC, Ramirez MI, Miller WT, Goldberg GS. Serines in the intracellular tail of podoplanin (PDPN) regulate cell motility. J Biol Chem 2013; 288:12215-21; PMID:23530051; http://dx.doi.org/ 10.1074/jbc.C112.446823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer 2006; 6:392-401; PMID:16572188; http://dx.doi.org/ 10.1038/nrc1877 [DOI] [PubMed] [Google Scholar]
- 52.Kinugasa Y, Matsui T, Takakura N. CD44 expressed on cancer-associated fibroblasts is a functional molecule supporting the stemness and drug resistance of malignant cancer cells in the tumor microenvironment. Stem Cells 2014; 32:145-56; PMID:24395741; http://dx.doi.org/ 10.1002/stem.1556 [DOI] [PubMed] [Google Scholar]
- 53.Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, Paulos CM, Palmer DC, Touloukian CE, Ptak K et al.. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood 2008; 112:362-73; PMID:18354038; http://dx.doi.org/ 10.1182/blood-2007-11-120998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, Hwu P, Restifo NP, Overwijk WW, Dong C. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity 2009; 31:787-98; PMID:19879162; http://dx.doi.org/ 10.1016/j.immuni.2009.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell 2015; 28:690-714; PMID:26678337; http://dx.doi.org/ 10.1016/j.ccell.2015.10.012 [DOI] [PubMed] [Google Scholar]
- 56.Kroemer G, Galluzzi L. Combinatorial immunotherapy with checkpoint blockers solves the problem of metastatic melanoma-An exclamation sign with a question mark. Oncoimmunology 2015; 4:e1058037; PMID:26140249; http://dx.doi.org/ 10.1080/2162402X.2015.1058037 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.