

ABSTRACT
Specific immunotherapy for acute leukemia remains a great unmet need. Native unmodified monoclonal antibody therapies, while promising, are inadequately effective for these malignancies, and multiple mechanisms for failure have been described. Antibody-dependent cellular cytotoxicity or phagocytosis is the primary modality of mAb-mediated cell killing in vivo, but ultimately leads to relapse of the leukemias, in model systems and in humans. By use of a T-cell receptor mimic mAb ESKM, derived against a WT1 peptide expressed in complex with HLA-A*02:01, whose only mechanism of therapeutic action is ADCC, we evaluated the mechanisms of leukemic relapse from its potent therapeutic action in mouse xenograft models of human leukemia. Leukemia escape was not associated with loss of the antigenic target, downregulation of cell surface HLA, antibody pharmacokinetic or biodistribution issues, or development of leukemia cell-intrinsic resistance to ADCC. Interestingly, the rapidity of leukemic growth determined whether leukemia was able to evade cytotoxicity independent of the presence of sufficient effector cells. By engineering leukemia cells with upregulated p27Kip1 and slower cell cycling times, we show that relapse was inversely correlated with growth rates resulting in the eventual inadequacy of effector to target ratio. Moreover, lack of migration of effector cells into lymphomatous pockets of ALL also allowed local escape. Successful leukemia therapy with mAb might therefore be improved in similar situations by combination with measures to reduce burden and slow leukemia cell growth.
KEYWORDS: ADCC, antibody, escape mechanisms, Leukemia, T-cell receptor mimicking
Introduction
Monoclonal antibody (mAb) therapies are not currently approved for the treatment of acute or chronic myeloid leukemia (AML or CML) or as first-line treatment of acute lymphoid leukemia (ALL) because of poor efficacy, lack of leukemia-specific targets, and toxicity.1,2 The main therapeutic mechanism in vivo of unmodified mAb is antibody-dependent cellular cytotoxicity or phagocytosis (ADCC/ADCP). These functions may be improved by altering the monoclonal IgG Fc glycosylation.3 Even with this enhancement that improves activity in chronic lymphoid leukemia,4 previous studies show an inadequate effect on overall survival in acute leukemias.5 Failure has been attributed to target downregulation or heterogeneity, as well as lack of potency.6 FDA-approved strategies enhancing mAb therapy potency include the attachment of potent small molecule drugs or β-emitting radioactive elements to mAb, and the creation of truncated Ig bi-specific constructs (BiTEs) for T-cell recruitment.7-10 These hybrid molecules have alternative modes of action to ADCC/ADCP, and while more potent, have significantly increased toxicity, result in off-target organ damage, and display difficult pharmacokinetics. Understanding the mechanisms by which cancer cells resist ADCC and developing strategies to defeat these hurdles is therefore crucial to improving efficacy of native mAb therapy.
ESK1 and ESKM (an afucosylated Fc form of ESK1) are human T-cell receptor mimic (TCRm) monoclonal IgG1 generated against a 9-mer peptide derived from the oncogenic transcription factor Wilm's Tumor 1 (WT1). This Db126 peptide (RMFPNAPYL) is expressed in the context of HLA-A*02:01 molecules on the surface of cancer cells.11 HLA-A*02:01 is found in approximately 40% of the US and European population, and a lower fraction in the rest of the world. The Fc portion of ESKM has altered glycosylation, enhancing its affinity for the Fc receptors, resulting in more potent and effective ADCC.12 ESKM has not been shown to kill via complement-dependent cytotoxicity (CDC), nor does its binding to the cell have any direct effects on cell growth or viability. Several TCRm antibodies have been developed to various leukemia antigens and are in preclinical development.13-15
WT1 encodes for a zinc finger transcription factor found in the embryonic development of multiple organ systems.16-18 Importantly, multiple cancers have been identified with significantly increased expression of the WT1 product, including nearly all high-risk MDS,19 CML, acute leukemias (AML, ALL) and their CD34+ stem cells,20-23 and many solid tumors.24-26 It is suppressed in nearly all normal cells after birth.27 Therefore, WT1 a useful tumor marker and a desirable antigenic target for therapy.28
ESK1 and ESKM have demonstrated selective HLA-restricted killing against leukemias overexpressing WT1 in patient cells and cell lines without observed off-target toxicity, in vitro and in xenograft NSG mouse models.29 However, though there are profound anti-leukemia effects, mice are not cured by continued dosing of the mAb alone in these model systems. Unless additional anti-leukemic drugs, such as dasatinib, were used concurrently, the leukemias invariably relapsed within weeks even while on ESKM therapy. Because ESKM works solely by ADCC/ADCP,14 it provides an elegant in vivo model system to explore previously undescribed causes of resistance to ADCC/ADCP. Elucidation of possible mechanisms of mAb treatment failure should be helpful in designing better strategies for overcoming mAb immunotherapy resistance for the large number of mAb in development. The data here show that cell kinetics and effector to target ratios are the most important determinants of treatment failure and provide a guide to the most effective treatment regimens to benefit from mAb therapies in leukemia, in setting where this may be an obstacle.
Results
In vivo leukemia growth patterns
The simplest form of acquired resistance to ADCC/ADCP would be loss of target antigen from leukemia cell surface by selective pressure. However, we previously reported that BV173 ALL cells on ESKM therapy harvested from mouse BM did not demonstrate downregulation of surface HLA-A*02:01 or ESKM mAb binding.29 Thus, antigen loss did not account for treatment failure in this model. We therefore hypothesized that resistance to ADCC could be secondary to mechanisms intrinsic to the target cells, extrinsic factors such as those affecting the pharmacokinetics or biodistribution of the mAb hindering its delivery to the target, or properties of the effector cells or their function in the host.
To better understand the system features that may play a role in the extrinsic escape mechanisms, we first carefully characterized the growth patterns of leukemia models in NSG mice and their relationship to the host effector cells. BV173 is a Philadelphia positive ALL cell line, whose growth and response to therapy with both TCRm and tyrosine kinase inhibitors (TKIs) was previously described.29 Lymphomatous relapse, in addition to bone marrow (BM) relapse, was frequently observed with this cell line after mAb therapy in NSG mouse models. Histologic analysis with immunohistochemical staining (IHC) of this leukemia confirmed that BV173 grows in clusters (Figs. 1A and B). The clearance of this leukemia in the liver with ESKM therapy can be most easily explained by the infiltration of macrophages within these clusters, which perform ADCC/ADCP and cell-mediated clearance (Fig. 1C). NK cells are reduced in function in these mice and therefore do not substantially contribute to the killing in this organ.
Figure 1.
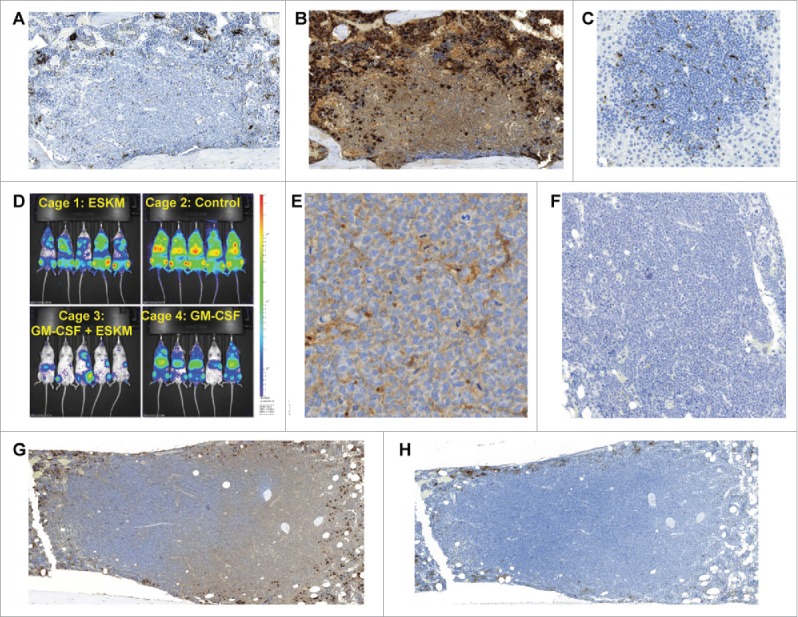
BV173 engrafted xenograft NSG mouse model. All histology analyses were performed on mice 3 weeks after leukemia injection. For mice receiving ESKM therapy, treatment was started on day 6 after injection, with mice receiving 2 weeks of therapy. (A) Untreated mouse bone marrow. IHC with mac-2 for macrophage staining. Large lymphomatous BV173 cell cluster evident, with no macrophage infiltration. (B) Same section of bone marrow as in (A), with myeloperoxidase IHC for neutrophil (including precursor) evaluation. As in (A), minimal effector cell penetration into lymphomatous BV173 cluster. (C) Mouse liver from non-treated mouse, IHC for mac-2, demonstrating mouse Kupffer cells (macrophages) infiltrating BV173 lymphomatous cluster. (D) BLI, exponential scale, of BV173 growth 34 d after injection (4 weeks of therapy, starting on day 6). (E) IHC for human IgG, 1 d after treatment dose of ESKM. Diffuse penetration of ESKM is seen throughout lymphoma. (F) Control IHC for human IgG in untreated mouse. No human IgG is seen, demonstrating that the IHC in Fig. 1E can only be ESKM. (G) Large BM BV173 lymphoma, IHC mac-2 staining, showing no macrophage infiltration. (H) Same as BM as (E) with myeloperoxidase staining showing no neutrophil infiltration into lymphomatous BV173 tumor.
We postulated that if effector cell insufficiency was responsible for the treatment failures, then increasing the number and activity of myeloid cell populations within the host should reduce relapses. Increasing and stimulating effectors with murine GM-CSF30 slowed BV173 engraftment and growth in mouse BM but ultimately did not prevent leukemic outgrowth (Fig. 1D). The addition of large numbers of NK cells to the mice improved outcomes in previous work.14 Thus, insufficient effector cell number alone was not a sufficient explanation for escape. Furthermore, the antibody itself displayed excellent penetration in the lymphomatous nests (Figs. 1E and F), suggesting that resistance based on pharmacokinetics of the mAb does not play a major role. Without effector cell penetration, such as neutrophilic or monocytic infiltration of the lymphoma, ESKM has no efficacy (Figs. 1G and H). Leukemia that follows such lymphomatous growth patterns appears to be minimally susceptible to ADCC/ADCP. While this may describe one mechanism of escape, it does not explain the observed systemic and BM escape.
In the second model, the AML cell line SET2 grows as discrete tumor cells in the BM (Figs. 2A and B). Initial treatments of SET2 with ESKM resulted in modest, short-lived therapeutic effects (Fig. 2C). Increasing E:T ratio by decreasing the number of injected SET2 cells from 3 × 106 to 0.5 × 106 per mouse improved initial cytotoxicity, but ultimately did not prevent rapid leukemic overgrowth. Increasing E:T further with GM-CSF did not increase ADCC/ADCP potency in this model (Fig. 2D).
Figure 2.
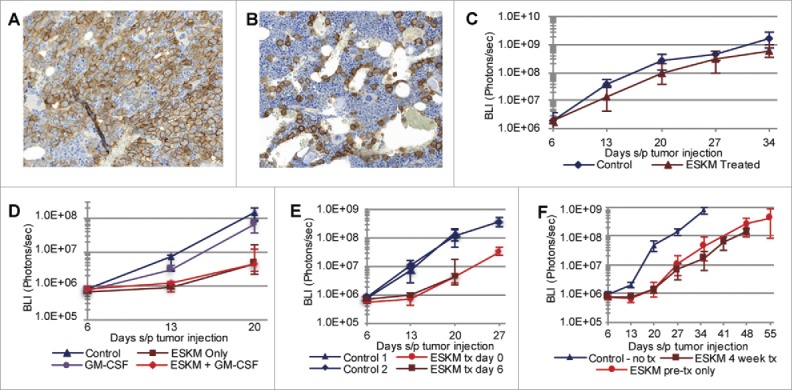
SET2 cell line engrafted xenograft NSG mouse model growth patterns. Error bars: 5th to 95th percentile. All BLI imaging and growth curves use exponential scale. (A) IHC for CD33, outlining discreet SET2 cells in mouse BM with no therapy, 3 weeks after injection. (B) CD33 IHC of SET2 cells in mouse BM 3 weeks after injection on 2 weeks of ESKM therapy (started on day 6). (C) In vivo SET2 growth quantification by BLI over time, with 3 × 106 cells injected on day 0, with and without ESKM therapy. (D) In vivo SET2 growth quantification by BLI over time with and without ESKM therapy. Effector to target ratios are increased from (C) with only 5 × 105 cells injected on day 0 and the addition of GM-CSF. (E) SET2 growth by BLI quantification, comparing starting ESKM after leukemic BM engraftment (on day 6) to leukemic growth in ESKM pre-treated mice (ESKM injected 2 h prior to injection of SET2). (F) SET2 growth by BLI quantification comparing 4 weeks of continuous (bi-weekly) ESKM therapy to only 1 week of treatment.
Pre-treatment of mice with ESKM prior to injection of leukemia, which should provide a situation of maximum effector to target ratio, did not change outcome when compared to therapy on already engrafted SET2 cells in the mouse BM (Fig. 2E). Furthermore, after the first week of ESKM therapy, additional mAb doses had minimal efficacy of slowing leukemic growth (Fig. 2F), implying the outgrowth of a resistant clone or the failure of therapy after leukemic overgrowth past a certain tumor burden.
Selection of germline or epigenetic durable resistance in vivo and in vitro
As we previously reported, relapsing BV173 ALL cells did not demonstrate HLA loss or antigen downregulation.29 This was confirmed by repeated BV173 harvests after 3 to 5 weeks of ESKM therapy. SET2 cells harvested from mouse BM likewise did not down regulate the target antigen as measured by flow cytometry (Figs. 3A and B).
Figure 3.
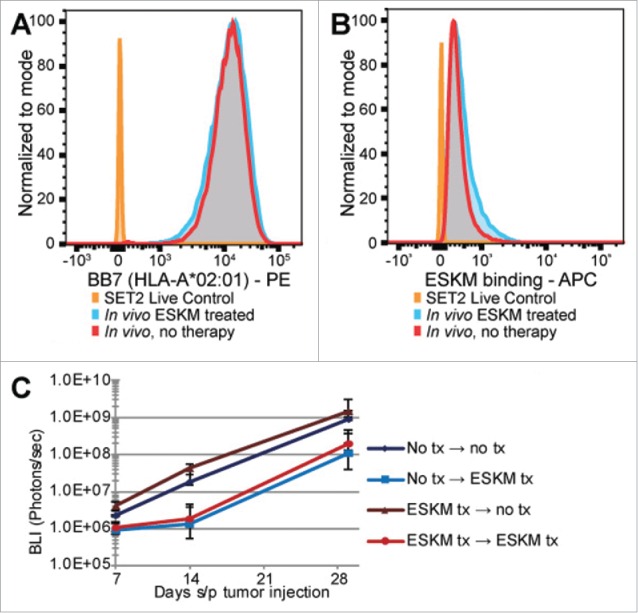
SET2 cell evaluation status-post harvest from murine BM. (A) Flow cytometry for surface HLA-A*02:01 expression, showing no difference between SET2 cells harvested from control mice and those treated with ESKM. (B) Same cells as in (A), evaluating for ESKM binding, showing no difference between cells extracted from ESKM treated and untreated mice. (C) SET2 cells from ESKM treated and untreated mice were harvested and passaged into a secondary group of mice. Ten initial mice (5 control, 5 treated) had BM harvested, and the SET2 cells extracted by Ficoll from each mouse were injected into two mice, (a control mouse and an ESKM pre-treated mouse). Error bars: 5th to 95th percentile. BLI growth curve utilizing exponential scale.
However, these data do not rule out other forms of intrinsic resistance such as the outgrowth of an ADCC/ADCP resistant clone. Therefore, SET2 cells were extracted from the BM of ESKM treated and untreated mice, and then passaged in naive NSG mice. The resistant tumor cells extracted from the ESKM treated mice showed no durable resistance to renewed ESKM therapy compared to their untreated counterparts (Fig. 3C).
We also explored whether resistance to ADCC could be selected for in vitro, using healthy volunteer PBMCs as effectors and SET2 target cells. A 3 d ADCC assay with E:T 25:1 was set up, with flow cytometry used to evaluate cytotoxicity and HLA expression. No plateau in killing of SET2 was observed, and HLA-A*02 expression actually increased, likely due to cytokine release by the effector cells (Figs. 4A and B).31
Figure 4.
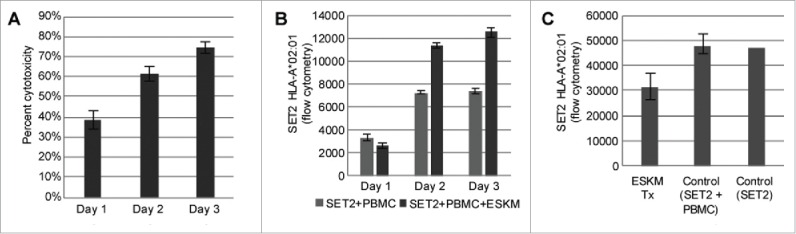
Lack of outgrowth of resistant SET2 cells in vitro. Error bars: 5th to 95th percentile. (A) 3 d ADCC: daily cytotoxicity as measured by flow cytometry. Effector to target ratio set at 25:1, with normal donor PBMCs used as effectors. Cytotoxicity measured as ratio of live SET2 cells with:without ESKM in media. (B) Median HLA-A*02:01 cell surface expression of cells in (A) on days 1–3 of ADCC by flow cytometry. HLA-A*02:01 increases in cells exposed to PBMC with ESKM compared to SET2 cells with PBMC without ESKM likely due to inflammatory cytokines. (C) Median surface HLA-A*02:01 expression by flow cytometry of passaged SET2 cells, status-post 2nd round of ADCC.
Increasing E:T to 50:1 greatly improved cytotoxicity overnight to 93.1% (± 0.9%, 5%–95% confidence). To definitively assess whether a resistant SET2 clone existed in this model, SET2 cells were exposed to 2 d of ADCC with E:T 50:1. The reaction was then quenched by killing the effector cells with puromycin (to which the SET2 target cells were made resistant). The remaining cells then underwent a second 18 h ADCC with E:T 50:1, and results were compared to SET2 with PBMCs without ESKM. The pre-treated SET2 cells remained susceptible to ADCC with 97.8% cytotoxicity, and no evidence of target loss (Fig. 4C). These results make treatment escape via selection and outgrowth of an intrinsically resistant SET2 clone very unlikely. Rather, a kinetic escape mechanism is strongly suggested.
Slowing leukemia growth kinetics improved therapeutic efficacy
The evaluation of the kinetic escape hypothesis required the controlled slowing of leukemic growth without direct toxicity to the cell. This has been previously done in another model by inducing the overexpression of the cyclin-dependent kinase inhibitor p27Kip1 inducing cell cycle arrest without apoptosis.32 We transduced SET2 with a doxycycline inducible vector coding for CDKN1B gene (coding for p27Kip1 protein). The SET2 p27Kip1 expressing cell line (SET2-S) responded to doxycycline mediated induction of p27Kip1 with dose-dependent growth inhibition (Figs. 5A–C). In the NSG mouse model, ESKM-mediated ADCC was dramatically improved by slowing SET2-S growth by p27Kip1 overexpression (Figs. 5D and E). p27Kip1 overexpression alone slowed growth but was not therapeutically effective. A comparison of the doxycycline treated SET2-S growth in vivo with and without ESKM therapy demonstrates that even a modest slowing of leukemic growth results in dramatic increase in therapeutic effect (Fig. 5E). Evaluation of this p27Kip1 cell line by flow cytometry revealed no downregulation of ESKM binding with the overexpression of p27Kip1 (Fig. 5F). In vitro ADCC performed by chromium assay showed minimal differences in ESKM-mediated toxicity of SET2-S between normal and high p27Kip1 expressing cells (Fig. 5G). The highly improved potency of ADCC/ADCP in these experiments in vivo appears to result from slower tumor growth, and therefore supports the hypothesis of kinetic escape mechanism of ADCC resistance.
Figure 5.
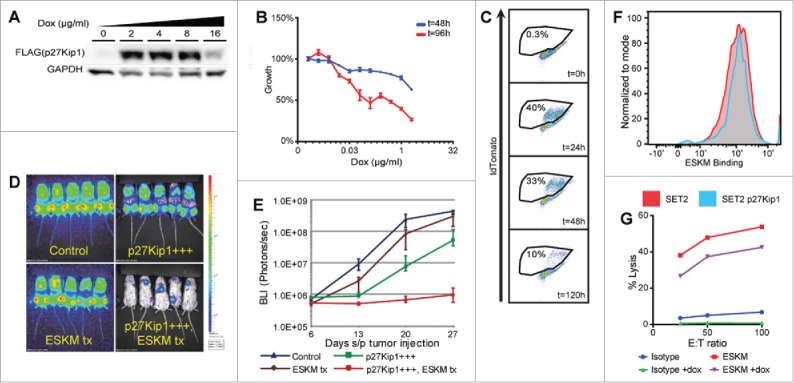
SET2-S cell evaluation in vitro and in vivo. Error bars: 5th to 95th percentile. BLI imaging and growth curves use exponential scale. (A) Western blot evaluating the protein expression of p27Kip1 measured using a FLAG specific antibody. (B) Dose-dependent growth inhibition of SET2-S cells in vitro with increasing doxycycline concentrations. (C) Competition assay demonstrating decrease in population of SET2-S cells when compared to SET2 cells. SET2 and SET2-S cells were mixed 50:50 at t = 0 h and 2 µg/mL doxycycline was added to the media. Expression of tdTomato, marking SET-S cells, was measured over time. (D) BLI at the end of 3 weeks of therapy in NSG mouse model of SET2-S leukemia (day 28 status-post leukemic injection). Controls had injections of SET2-S cells and no therapy. ESKM treated mice received the antibody only on day 6 onwards. The other two groups got doxycycline alone or doxycycline with ESKM. (E) A plot on log scale of the mice treated in panel D over one month. SET2-S in vivo growth, by BLI, evaluating ESKM ADCC with and without p27Kip1 overexpression, demonstrating clear superiority of ADCC treatment on slower growing SET2-S cells compared to wild type controls. (F) Flow cytometry of SET2-S cells with and without doxycycline exposure, evaluating ESKM binding. (G) ADCC of SET2-S with and without upregulation of p27Kip1, activated by doxycycline exposure. The overexpression of p27Kip1 appears to stabilize SET2-S, decreasing the chromium release of both control and ESKM treated cells.
Slower growth and better effector cells improved therapeutic efficacy
We investigated therapeutic activity in the Rag2−/− model system. These mice have higher macrophage and NK numbers with increased function than NSG mice. The BV173 rate of growth in this model was about half as fast as the rates in NSG mice (1 log increase of BLI in 2 weeks vs. 1 log increase BLI in 1 week in the other models described above.) After treatment, BLI measured demonstrated substantial reduction in tumor volume (Figs. 6A and B), even when tumors were treated this late from engraftment. In addition, the version of the ESK mAb used did not have an Fc region enhanced for better ADCC. Radiance measurements out to day 105 demonstrated that at least two of the five mice treated with ESK1 lacked detectable signal over background, suggesting that cure might have been achieved in these mice.
Figure 6.
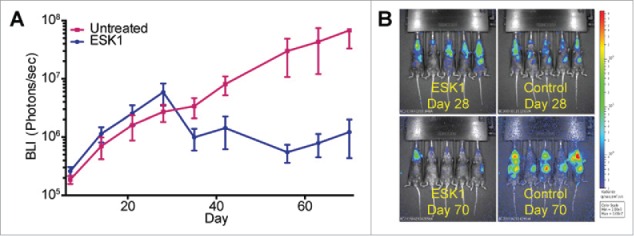
Therapy in a Rag2 mouse knock out model. Rag2−/− mice injected with BV173 leukemia xenograft (day 0) after cytoreduction with radiation and anti-NK mAb. Mice were treated with 3 weeks of biweekly ESK1 starting day 28. BLI was measured per mouse in each group (mean +/− SEM). (A) BV173 tumor growth by BLI comparing ESK1 treated mice with their control (untreated) counterparts. (B) BLI of ESK1 and untreated groups on day 28, at the start of therapy, and day 70, 3 weeks after the final week of therapy.
Discussion
The work described here demonstrates new potential mechanisms of resistance to a mAb targeting a WT1 derived epitope. While many FDA approved antibodies have been shown to have activity by blocking signaling pathways, or via complement, the TCRm mAb ESKM has activity solely through ADCC. Unlike other mAb systems such as rituximab or trastuzumab, which have multiple mechanisms of action, we could use this mAb-antigen system to probe for new avenues of escape to ADCC or ADCP. We expect that future antibodies targeting similar tumor antigens presented on MHC-I will also rely completely on ADCC for activity. Surprisingly, in this system target downregulation or intrinsic resistance to ADCC did not contribute to failure. Instead, cell growth kinetics, E:T ratio and growth patterns, were found to be the main factors in treatment failure.
Cancer therapies with unmodified mAb generally result in small, but significant additive therapeutic effect, and require combination with other modalities of treatment, such as chemotherapy, to achieve optimal results.33-35 Other than mAbs that mediate the upregulation of T-cell activity via checkpoint blockade, a mechanism of action completely independent of ADCC, most native mAb alone do not result in complete remissions or long-term survivals. Even more complex constructs, such as brentuximab vedontin, are used in combination or sequenced with chemotherapy.36 Other effective mAb, such as rituximab and trastuzumab, rely on additional mechanisms of action to achieve improved results. The lack of mAb ADCC/ADCP potency is a significant limitation for this class of drugs, given the relative ease of manufacturing and the specificity of such therapy. While combining antibody therapy with chemotherapy has improved outcomes, these two treatments may not synergize well in many instances, since chemotherapeutic agents can be naturally toxic to the effector cells required for ADCC.
The data here are derived from experiments that utilize the antibody ESKM, whose sole method of cytotoxicity is ADCC/ADCP, designed to elucidate the in vitro and in vivo factors, which result in leukemic escape from TCRm antibody therapy. The data do not support downregulation of target epitope, inadequate pharmacokinetics of the mAb, the inhibition of effector cells, suboptimal effector to target ratio in the BM, or the microenvironment as important mechanisms of mAb therapy failure in this system. Instead, we observed that growth kinetics of the leukemia, and the quality and number of effectors, was critical to leukemic relapse. This was demonstrated by the superior efficacy of ESKM in mice on slower growing clones of AML cells, which differed from the parental line only by the introduction of an inducible cyclin-dependent kinase inhibitor p27Kip1 to slow the AML cell cycling time and by superior efficacy in Rag−/− mice in which tumors grew half as fast and which have more active NK and macrophage compartments. These results indicate that leukemia may be able to evade this TCRm mAb by outgrowing effectors. These findings are especially relevant for the use of mAb in the setting of rapidly growing population of leukemia cells, with a limited number of effectors whose activity may be suppressed by chemotherapy or the disease itself. For mAb that act via other mechanisms not requiring effectors, such as the direct killing demonstrated by rituximab or trastuzumab, or other mAb that work by blocking signaling, these issues may be less important.
The concept of outgrowing antibody-mediated destruction in humans has been documented in another disease. In idiopathic thrombocytopenia purpura (ITP), the antibody-mediated destruction of platelets can be treated by thrombopoietic agents, which are very effective at increasing platelet numbers simply by increasing platelet production.37,38 The converse therefore is feasible, and documented by our data: slowing down target cell growth increases susceptibility to the effector mechanisms. Interestingly, the escape mechanism does not appear to be simply a matter of inadequate E:T ratios as the initiation of therapy at time zero or addition of more effectors, either by infusion or cytokine induction, did not prevent relapse.
These findings may be highly relevant currently, given the rapid development and approval of many cellular growth pathway inhibitors, each targeting specific overactive signaling pathways in cancer cells, many of which are cytostatic. With the ever-increasing ability to detect cell growth drivers and an expanding library of small molecules, such as TKIs, including BRAF inhibitors, MEK inhibitors, and JAK inhibitors, it will soon be possible to slow the growth of many tumor cells safely in vivo. Indeed, in our previous study we showed that in mouse models of Ph+ ALL, ESKM plus imatinib was more than additive, and ESKM plus dasatinib was sometimes curative.29 Combining cytostatic therapy with targeted immune therapy, such as TCRm mAbs, may therefore result in synergistic and possibly curative outcomes without resorting to cytotoxic chemotherapy. Furthermore, chemotherapy may contribute to the effector cell dysfunction. Of course, chemotherapy may still be required to lower tumor burden for more optimal E:T ratios.
Materials and methods
In vitro ADCC
Use of human cells was done under IRB approved protocols. Six hour ADCC in vitro was performed via chromium (51Cr) release assay, as previously described.29 One to three day ADCC assay was performed by incubating healthy donor PBMCs isolated by Ficoll with target leukemia cells at E:T 50:1 and 25:1 in triplicate. Treatment groups had ESKM added to media at 3 µg/mL, while control groups had no antibody added. The SET2 cells used in these assays were GFP tagged, and flow cytometry was utilized to measure the concentration of live (DAPI negative) GFP positive cells in solution compared to total number of cells. Constitutively on GFP was transduced with a lentiviral GFP vector specifically for the purpose of easily identifying these cells in the in vitro ADCC assays.
Flow cytometry
Fortessa flow cytometer (BD Biosciences) was utilized for all flow cytometry measurements. APC- or PE-tagged BB7 antibody (BioLegend) was utilized to measure HLA-A02 expression on cell surface for both in vitro and in vivo studies. After mouse BM resection in xenograft models, APC-tagged ESK1 antibody (Innova Biosciences – Lightning-Link® Allophycocyanin) was used to measure RMF expression on cell surface. Xenografted SET2 cells were identified by CD33-BV711, and BV173 by CD19-PE (BioLegend). Xenografted SET2 and BV173 leukemia cells were both dimly GFP positive, and GFP was therefore not used for flow cytometry.
In vivo mouse xenograft models
Animal studies were done on IACUC approved protocols. NOD SCID IL-2γ knockout mice (NSG) from Jackson Labs were used for all mouse models.39 Luciferase-tagged SET2, BV173, and SET2-S cell lines were used in all xenograft models, and leukemia growth was measured by bioluminescent imaging (BLI) using the IVIS Spectrum, with intraperitoneal luciferin injections 5 min prior to imaging. Day 0 in studies was defined as the day of leukemia injection by tail vein. Because no differences were noted in the therapeutic effect between biweekly ESKM dosing of 100 µg and 50 µg per dose, 50 µg ESKM biweekly IP were used in these studies. No significant differences in BLI were seen previously between isotype control therapy and no IgG therapy, and control mice were therefore not treated with isotype IgG.12,14 GM-CSF was dosed at 1 µg, administered daily IP. For SET2-S cell line experiments, mice were administered doxycycline via food pellets (625 mg/kg) (Harlan–Teklad) starting on day 6 and continued until the end of the experiment.
Rag2−/− mice underwent whole body irradiation with either 500cGy (ESK1 treated group) or 600cGy (untreated group) on Day 20. On Day 1, the mice were NK depleted using the anti-NK1.1 antibody (clone PK136) IP. 1 × 107 BV173 cells per mouse were injected via tail vein on Day 0. ESK1 therapy (100 µg retro-orbital) began on Day 28 and continued twice a week for 3 weeks until Day 47. BLI was measured weekly until 3 weeks after the final week of therapy.
Cloning, vector construction, and purification of SET2-S
The 3rd generation lentiviral vector pLV was obtained from VectorBuilder (Santa Clara, CA, USA) and the human CDKN1B (coding for p27Kip1 protein) cDNA was cloned under the TRE3G inducible promoter with a C-terminal FLAG tag and IRES (for expression of tdTomato). The pLV vector also had constitutive expression of rtTA3 under the CMV promoter. HEK293T cells were used to produce lentiviral particles using the pRSV (Addgene #12253), pMDL (Addgene #12251), and pMD2.G (Addgene #12259) vectors along with the CDKN1B gene. Viral supernatant was harvested from HEK293T cells and used to transduce SET2 GFP-luciferase expressing cells using spinfection at 1 × 106 cells in 8 µg/mL polybrene with viral supernatant at 2000x g, 2 h, for two rounds on transduction. A clonogenic assay was set up to insure 100% purity of transduced SET2-S cells. Colonies positive for tdTomato after 2 µg/mL doxycycline induction were pooled and expanded for downstream use.
Flag-tag specific antibody used in the Western blot assay were purchased from Cell Signaling Technologies (#14793). Doxycycline was obtained from Sigma. SET2 cells stably transduced with CDKN1B were exposed to increasing doses of doxycycline for 48 h and 96 h. Cell-Titer Glo (Promega) was performed at indicated time points to assess viability. Flow cytometry studies for tdTomato expression were performed on the Accuri C6 flow cytometer (BD Biosciences). Flow cytometry for ESKM binding was performed using ESK conjugated with APC fluorophore. Cells were stained with 3 µg/mL ESK-APC for 45 min on ice. DAPI was used for viability assessment. Cells were analyzed using the Fortessa flow cytometer (BD Biosciences).
Disclosure of potential conflicts of interest
D.A.S., T.D., and L.D. are inventors of technology described in this paper licensed by Sloan Kettering to Novartis.
Acknowledgments
We thank Tanya Korontsvit and Victoriya Zakhaleva for their assistance.
Author Contributions
L.D., E.J.B., T.D., C.E.L., E. C., and D.A.S. designed the study. D.A.S. coordinated the study. L.D., D.P., E.C., and E.J.B. conducted experiments and analysis with assistance from D.P and E.J.B. SET2-S cell line was synthesized by E.J.B. ESKM was synthesized and supplied by C.E.L. L.D. drafted the manuscript, which was subsequently revised by all co-authors.
Funding
This study was supported by the Leukemia and Lymphoma Society, NIH R01CA55349, P01CA23766, P30CA 008748 T32CA62948-18, MSTP-GM07739, Lymphoma Foundation, Tudor and Glades funds, the MSKCC Technology Development Fund and the Experimental Therapeutics Center.
References
- 1.Bhojwani D, Pui CH. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol 2013; 14(6):e205-17; PMID:23639321; http://dx.doi.org/ 10.1016/S1470-2045(12)70580-6 [DOI] [PubMed] [Google Scholar]
- 2.Tasian SK, Pollard JA, Aplenc R. Molecular therapeutic approaches for pediatric acute myeloid leukemia. Front Oncol 2014; 4:55; PMID:24672775; http://dx.doi.org/ 10.3389/fonc.2014.00055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jefferis R. Antibody therapeutics: isotype and glycoform selection. Expert Opin Biol Ther 2007; 7(9):1401-13; PMID:17727329; http://dx.doi.org/ 10.1517/14712598.7.9.1401 [DOI] [PubMed] [Google Scholar]
- 4.Shah A. New developments in the treatment of chronic lymphocytic leukemia: role of obinutuzumab. Ther Clin Risk Manag 2015; 11:1113-22; PMID:26251607; http://dx.doi.org/ 10.2147/TCRM.S71839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jabbour E, O'Brien S, Ravandi F, Kantarjian H. Monoclonal antibodies in acute lymphoblastic leukemia. Blood 2015; 125(26):4010-16; PMID:25999456; http://dx.doi.org/ 10.1182/blood-2014-08-596403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Czuczman MS, Olejniczak S, Gowda A, Kotowski A, Binder A, Kaur H, Knight J, Starostik P, Deans J, Hernandez-Ilizaliturri FJ. Acquirement of rituximab resistance in lymphoma cell lines is associated with both global CD20 gene and protein down-regulation regulated at the pretranscriptional and posttranscriptional levels. Clin Cancer Res 2008; 14(5):1561-70; PMID:18316581; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-1254 [DOI] [PubMed] [Google Scholar]
- 7.Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, Noppeney R, Viardot A, Hess G, Schuler M et al.. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008; 321(5891):974-77; PMID:18703743; http://dx.doi.org/ 10.1126/science.1158545 [DOI] [PubMed] [Google Scholar]
- 8.Younes A, Bartlett NL, Leonard JP, Kennedy DA, Lynch CM, Sievers EL, Forero-Torres A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med 2010; 363(19):1812-21; PMID:21047225; http://dx.doi.org/ 10.1056/NEJMoa1002965 [DOI] [PubMed] [Google Scholar]
- 9.Daver N, O'Brien S. Novel therapeutic strategies in adult acute lymphoblastic leukemia–a focus on emerging monoclonal antibodies. Curr Hematol Malig Rep 2013; 8(2):123-31; PMID:23539383; http://dx.doi.org/ 10.1007/s11899-013-0160-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milenic DE, Brady ED, Brechbiel MW. Antibody-targeted radiation cancer therapy. Nat Rev Drug Discov 2004; 3(6):488-99; PMID:15173838; http://dx.doi.org/ 10.1038/nrd1413 [DOI] [PubMed] [Google Scholar]
- 11.Maslak PG, Dao T, Krug LM, Chanel S, Korontsvit T, Zakhaleva V, Zhang R, Wolchok JD, Yuan J, Pinilla-Ibarz J et al.. Vaccination with synthetic analog peptides derived from WT1 oncoprotein induces T-cell responses in patients with complete remission from acute myeloid leukemia. Blood 2010; 116(2):171-9; PMID:20400682; http://dx.doi.org/ 10.1182/blood-2009-10-250993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Veomett N, Dao T, Liu H, Xiang J, Pankov D, Dubrovsky L, Whitten JA, Park SM, Korontsvit T, Zakhaleva V et al.. Therapeutic efficacy of an Fc-enhanced TCR-like antibody to the intracellular WT1 oncoprotein. Clin Cancer Res 2014; 20(15):4036-46; PMID:24850840; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alatrash G, Mittendorf EA, Sergeeva A, Sukhumalchandra P, Qiao N, Zhang M, St John LS, Ruisaard K, Haugen CE, Al-Atrache Z et al.. Broad cross-presentation of the hematopoietically derived PR1 antigen on solid tumors leads to susceptibility to PR1-targeted immunotherapy. J Immunol 2012; 189(11):5476-84; PMID:23105141; http://dx.doi.org/ 10.4049/jimmunol.1201221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dao T, Yan S, Veomett N, Pankov D, Zhou L, Korontsvit T, Scott A, Whitten J, Maslak P, Casey E et al.. Targeting the intracellular WT1 oncogene product with a therapeutic human antibody. Sci Transl Med 2013; 5(176):176ra133; PMID:23486779; http://dx.doi.org/26942058 10.1126/scitranslmed.3005661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dubrovsky L, Dao T, Gejman RS, Brea EJ, Chang AY, Oh CY, Casey E, Pankov D, Scheinberg DA. T cell receptor mimic antibodies for cancer therapy. Oncoimmunology 2016; 5(1):e1049803; PMID:26942058; http://dx.doi.org/ 10.1080/2162402X.2015.1049803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nurmemmedov E, Thunnissen M. Expression, purification, and characterization of the 4 zinc finger region of human tumor suppressor WT1. Protein Expr Purif 2006; 46(2):379-89; PMID:16343939; http://dx.doi.org/ 10.1016/j.pep.2005.10.029 [DOI] [PubMed] [Google Scholar]
- 17.Pritchard-Jones K, Fleming S, Davidson D, Bickmore W, Porteous D, Gosden C, Bard J, Buckler A, Pelletier J, Housman D et al.. The candidate Wilms' tumour gene is involved in genitourinary development. Nature 1990; 346(6280):194-7; PMID:2164159; http://dx.doi.org/ 10.1038/346194a0 [DOI] [PubMed] [Google Scholar]
- 18.Wang ZY, Qiu QQ, Deuel TF. The Wilms' tumor gene product WT1 activates or suppresses transcription through separate functional domains. J Biol Chem 1993; 268(13):9172-5; PMID:8486616 [PubMed] [Google Scholar]
- 19.Tamaki H, Ogawa H, Ohyashiki K, Ohyashiki JH, Iwama H, Inoue K, Soma T, Oka Y, Tatekawa T, Oji Y et al.. The Wilms' tumor gene WT1 is a good marker for diagnosis of disease progression of myelodysplastic syndromes. Leukemia 1999; 13(3):393-9; PMID:10086730; http://dx.doi.org/ 10.1038/sj.leu.2401341 [DOI] [PubMed] [Google Scholar]
- 20.Im HJ, Kong G, Lee H. Expression of Wilms tumor gene (WT1) in children with acute leukemia. Pediatr Hematol Oncol 1999; 16(2):109-18; PMID:10100271; http://dx.doi.org/ 10.1080/088800199277434 [DOI] [PubMed] [Google Scholar]
- 21.Niegemann E, Wehner S, Kornhuber B, Schwabe D, Ebener U. wt1 gene expression in childhood leukemias. Acta Haematol 1999; 102(2):72-6; PMID:10529509; http://dx.doi.org/ 10.1159/000040973 [DOI] [PubMed] [Google Scholar]
- 22.Menssen HD, Renkl HJ, Rodeck U, Maurer J, Notter M, Schwartz S, Reinhardt R, Thiel E. Presence of Wilms' tumor gene (wt1) transcripts and the WT1 nuclear protein in the majority of human acute leukemias. Leukemia 1995; 9(6):1060-7; PMID:7596170 [PubMed] [Google Scholar]
- 23.Gerber JM, Qin L, Kowalski J, Smith BD, Griffin CA, Vala MS, Collector MI, Perkins B, Zahurak M, Matsui W et al.. Characterization of chronic myeloid leukemia stem cells. Am J Hematol 2011; 86(1):31-7; PMID:21132730; http://dx.doi.org/ 10.1002/ajh.21915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oji Y, Ogawa H, Tamaki H, Oka Y, Tsuboi A, Kim EH, Soma T, Tatekawa T, Kawakami M, Asada M et al.. Expression of the Wilms' tumor gene WT1 in solid tumors and its involvement in tumor cell growth. Jpn J Cancer Res 1999; 90(2):194-204; PMID:10189890; http://dx.doi.org/ 10.1111/j.1349-7006.1999.tb00733.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amin KM, Litzky LA, Smythe WR, Mooney AM, Morris JM, Mews DJ, Pass HI, Kari C, Rodeck U, Rauscher FJ 3rd et al.. Wilms' tumor 1 susceptibility (WT1) gene products are selectively expressed in malignant mesothelioma. Am J Pathol 1995; 146(2):344-56; PMID:7856747 [PMC free article] [PubMed] [Google Scholar]
- 26.Schittenhelm J, Beschorner R, Simon P, Tabatabai G, Herrmann C, Schlaszus H, Capper D, Weller M, Meyermann R, Mittelbronn M. Diagnostic value of WT1 in neuroepithelial tumours. Neuropathol Appl Neurobiol 2009; 35(1):69-81; PMID:18466223; http://dx.doi.org/ 10.1111/j.1365-2990.2008.00957.x [DOI] [PubMed] [Google Scholar]
- 27.Fanni D, Fanos V, Monga G, Gerosa C, Locci A, Nemolato S, Van Eyken P, Faa G. Expression of WT1 during normal human kidney development. J Matern Fetal Neonatal Med 2011; 24(Suppl 2):44-7; PMID:21888469; http://dx.doi.org/ 10.3109/14767058.2011.606619 [DOI] [PubMed] [Google Scholar]
- 28.Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL, Weiner LM et al.. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res 2009; 15(17):5323-37; PMID:19723653; http://dx.doi.org/ 10.1158/1078-0432.CCR-09-0737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dubrovsky L, Pankov D, Brea EJ, Dao T, Scott A, Yan S, O'Reilly RJ, Liu C, Scheinberg DA. A TCR-mimic antibody to WT1 bypasses tyrosine kinase inhibitor resistance in human BCR-ABL+ leukemias. Blood 2014; 123(21):3296-304; PMID:24723681; http://dx.doi.org/ 10.1182/blood-2014-01-549022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pennline KJ, Pellerito F, DaFonseca M, Monahan P, Siegel MI, Smith SR. Flow cytometric analysis of recombinant murine GM-CSF (rmuGM-CSF) induced changes in the distribution of specific cell populations in vivo. Cytometry 1990; 11(2):283-91; PMID:2180653; http://dx.doi.org/ 10.1002/cyto.990110209 [DOI] [PubMed] [Google Scholar]
- 31.Keskinen P, Ronni T, Matikainen S, Lehtonen A, Julkunen I. Regulation of HLA class I and II expression by interferons and influenza A virus in human peripheral blood mononuclear cells. Immunology 1997; 91(3):421-9; PMID:9301532; http://dx.doi.org/ 10.1046/j.1365-2567.1997.00258.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drexler HC, Pebler S. Inducible p27(Kip1) expression inhibits proliferation of K562 cells and protects against apoptosis induction by proteasome inhibitors. Cell Death Differ 2003; 10(3):290-301; PMID:12700629; http://dx.doi.org/ 10.1038/sj.cdd.4401159 [DOI] [PubMed] [Google Scholar]
- 33.Kushner BH, Kramer K, Cheung NK. Phase II trial of the anti-G(D2) monoclonal antibody 3F8 and granulocyte-macrophage colony-stimulating factor for neuroblastoma. J Clin Oncol 2001; 19(22):4189-94; PMID:11709561 [DOI] [PubMed] [Google Scholar]
- 34.Hiddemann W, Kneba M, Dreyling M, Schmitz N, Lengfelder E, Schmits R, Reiser M, Metzner B, Harder H, Hegewisch-Becker S et al.. Frontline therapy with rituximab added to the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) significantly improves the outcome for patients with advanced-stage follicular lymphoma compared with therapy with CHOP alone: results of a prospective randomized study of the German low-grade lymphoma study group. Blood 2005; 106(12):3725-32; PMID:16123223; http://dx.doi.org/ 10.1182/blood-2005-01-0016 [DOI] [PubMed] [Google Scholar]
- 35.Habermann TM, Weller EA, Morrison VA, Gascoyne RD, Cassileth PA, Cohn JB, Dakhil SR, Woda B, Fisher RI, Peterson BA et al.. Rituximab-CHOP versus CHOP alone or with maintenance rituximab in older patients with diffuse large B-cell lymphoma. J Clin Oncol 2006; 24(19):3121-7; PMID:16754935; http://dx.doi.org/ 10.1200/JCO.2005.05.1003 [DOI] [PubMed] [Google Scholar]
- 36.Moskowitz CH, Nademanee A, Masszi T, Agura E, Holowiecki J, Abidi MH, Chen AI, Stiff P, Gianni AM, Carella A et al.. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin's lymphoma at risk of relapse or progression (AETHERA): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015; 385(9980):1853-62; PMID:25796459; http://dx.doi.org/ 10.1016/S0140-6736(15)60165-9 [DOI] [PubMed] [Google Scholar]
- 37.Frampton JE, Lyseng-Williamson KA. Romiplostim. Drugs 2009; 69(3):307-17; PMID:19275274; http://dx.doi.org/ 10.2165/00003495-20-0969030-00006 [DOI] [PubMed] [Google Scholar]
- 38.Garnock-Jones KP, Keam SJ. Eltrombopag. Drugs 2009; 69(5):567-76; PMID:19368418; http://dx.doi.org/ 10.2165/00003495-200969050-00005 [DOI] [PubMed] [Google Scholar]
- 39.Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J, Greiner DL et al.. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol 2005; 174(10):6477-89; PMID:15879151; http://dx.doi.org/ 10.4049/jimmunol.174.10.6477 [DOI] [PubMed] [Google Scholar]