

Abstract
Glioblastoma multiforme, due to its invasive nature, can be considered a disease of the entire brain. Despite recent advances in surgery, radiotherapy and chemotherapy, current treatment regimens have only a marginal impact on patient survival. A crucial challenge faced by cancer researchers is to effectively deliver drugs to invasive glioma cells residing in a sanctuary within the central nervous system. The blood–brain barrier (BBB) restricts delivery of many small and large molecules into the brain. Drug delivery to the brain is further restricted by active efflux transporters present at the BBB, which transport drugs out of the brain back into the blood. Current clinical assessment of drug delivery and hence efficacy is based on the measured drug levels in the bulk tumor mass that is usually removed by surgery. Mounting evidence suggests that the inevitable relapse and lethality of glioblastoma multiforme is due to a failure to effectively treat invasive glioma cells. These invasive cells hide in areas of the brain that are shielded by an intact BBB where they continue to grow and give rise to the recurrent tumor. Effective delivery of chemotherapeutics to the invasive glioma cells is therefore critical, and long-term efficacy will depend upon the ability of a molecularly targeted agent to penetrate an intact and functional BBB throughout the entire brain. This review highlights the various aspects of the BBB, and also the brain–tumor-cell barrier, a barrier due to expression of efflux transporters in tumor cells, that together can significantly influence drug response. It then discusses the special challenge of glioma as a disease of the whole brain, which lends particular emphasis to the need to effectively deliver drugs across the BBB to reach both the central tumor and the invasive glioma cells.
The past two decades have witnessed major advances in molecular and cellular biology that have substantially improved our understanding of human malignancies. Unfortunately, this period has also seen a significant rise in the incidence of malignant brain tumors along with only a modest increase in the survival rates associated with them, which are often poor (Ref. 1). Out of the approximately 22,020 new cases of primary malignant brain tumors that were estimated to be diagnosed in the USA in 2010, 80% were expected to be malignant gliomas (Refs 2, 3). Gliomas represent a group of highly malignant and lethal tumors of the brain that, despite all therapeutic advances, have an extremely poor prognosis. The median survival of patients with glioblastoma multiforme, the most common and most malignant subtype of glioma, is only 12-18 months (Ref. 4). The current standard of care in glioblastoma multiforme is treatment with the DNA-alkylating agent temozolomide combined with radiation, a treatment that has been proven to prolong patient survival by a few months (Ref. 4). Many new molecularly targeted agents that were developed to inhibit signaling pathways critical for glioma growth and proliferation have failed to elicit any clinical benefit (Ref. 5).
Compared with treatment of other types of tumors, targeting tumors of the central nervous system (CNS) is particularly challenging due to the location of the tumor in a pharmacological and immunological sanctuary within the CNS. The blood–brain barrier (BBB) presents a major obstacle to systemic chemotherapy and is capable of significantly limiting drug response (Ref. 6). Drug efflux transporters at the BBB restrict the passage of drugs into the brain and thus shield the tumor cells from exposure to cytotoxic chemotherapy. In addition to the BBB, the presence of similar drug efflux pumps within tumor cells (the brain–tumor-cell barrier; BTB) further protects them from chemotherapy. Systemically administered drugs thus have to cross these two sequential barriers to reach their intended molecular target.
This review focuses on the special challenge that these barriers pose to molecularly targeted and cytotoxic chemotherapeutic drugs. The aim is to provide an overview of the various molecular targets and target-directed chemotherapy for glioma. We review the most important ATP-driven transporters at the BBB and in tumor cells and their role in limiting the delivery and hence efficacy of systemic chemotherapy. Finally, we summarize how treatment of an infiltrative tumor like glioblastoma multiforme requires targeting the invasive tumor cells that often reside in areas away from the primary tumor – cells that are not removed by surgery and are shielded by multiple barriers, and therefore continue to grow and give rise to the recurrent tumor (Ref. 7).
Malignant Glioma
Malignant glioma represents one of the greatest challenges faced by the neuro-oncology community. Gliomas are tumors that are thought to arise from glial progenitor and glial cells and include astrocytoma, glioblastoma, oligodendroglioma, ependymoma, mixed glioma and a few other, rare histologies (Ref. 2). These tumors account for 32% of all primary brain tumors and, as stated above, 80% of all malignant primary brain tumors diagnosed in the USA (Ref. 2). The World Health Organization (WHO) classifies gliomas into four grades based on their histological features and malignancy. Grade I (pilocytic astrocytoma) and grade II (diffuse astrocytoma) tumors are slow growing and the least malignant forms of glioma, while grade III tumors (anaplastic astrocytoma) are more malignant and associated with poorer prognosis (Ref. 8). Grade IV is assigned to the most malignant and mitotically active tumors associated with extremely poor survival rates. Glioblastoma multiforme is a grade IV glioma and is characterized by uncontrolled cellular proliferation, diffuse infiltration, necrosis, angiogenesis and resistance to apoptosis. The name “multiforme” signifies the vast intratumoral heterogeneity seen in the disease. Glioblastoma multiforme is the most common subtype of glioma (accounting for ∼50% of gliomas); and glioblastoma multiforme and astrocytoma together account for ∼75% of gliomas. Survival rates for patients with malignant gliomas are the worst among all brain tumors: less than 5% of glioblastoma multiforme patients survive for 5 years post diagnosis (Refs 1, 2).
A majority of glioblastomas are primary tumors that develop de novo in the brain without any evidence of a precursor tumor; a relatively smaller fraction (∼10%) are secondary tumours that start as low-grade astrocytomas but subsequently progress to high-grade gliomas (Refs 9, 10). Progress in our understanding of the molecular pathogenesis of malignant gliomas has made it possible to distinguish between these two types of glioblastoma multiforme based on the genetic aberrations and deregulated growth-factor pathways presented by the tumor. Primary glioblastomas are characterized by amplification of the epidermal growth factor receptor (EGFR) and its mutant EGFR vIII, loss of heterozygosity of chromosome 10q, amplification/overexpression of the MDM2 gene (mouse double minute 2), deletion of the PTEN gene (phosphatase and tensin homologue) and alterations in the RB1 (retinoblastoma 1) and p53 (TP53) signaling pathways (Refs 9, 10). Secondary glioblastoma multiformes are characterized mainly by overexpression of the platelet derived growth factor receptor (PDGFR) and genetic mutations in the p53 and RB1 signaling pathways (Refs 9, 10). Despite the genetic differences, no differences in sensitivity to conventional chemotherapy between primary and secondary glioblastoma multiformes have been reported. The molecular and genetic aberrations in glioma have been extensively studied and show remarkable heterogeneity even within an individual tumor (Refs 11, 12). The enormous intratumoral variability combined with the complexity of the deregulated signaling pathways might be one of the reasons why most target-directed therapeutics are ineffective against the disease.
Despite aggressive treatment, essentially all malignant gliomas recur (Ref. 13), eventually leading to death. The median survival of a glioblastoma patient after recurrence is approximately 5-7 months (Ref. 5). Surgery remains one of the most effective treatments and almost all patients undergo surgery, unless the location of the tumor makes any degree of surgical debulking impossible (Ref. 14). Studies have shown a correlation between the extent of surgical debulking and increased patient survival (Refs 15, 16). Unfortunately, the grim reality is that regardless of the extent of resection, tumor recurrence and death is almost always inevitable. Radiotherapy is another treatment option for glioblastoma multiforme that has been proven to increase survival in patients post surgery (Ref. 4). Chemotherapy is fast assuming an increasingly important role in the treatment of malignant gliomas. Although many earlier studies failed to show benefit with adjuvant chemotherapy, the finding that temozolomide in combination with radiotherapy increases patient survival, dramatically changed chemotherapeutic treatment of glioma (Ref. 4). Temozolomide is now the standard of care in glioma, with almost every patient receiving the drug. However, reports of resistance to temozolomide have intensified the search for more effective target-directed therapies. A recent study showed that treatment with bevacizumab, a monoclonal antibody targeting vascular endothelial growth factor A (VEGFA), in combination with radiotherapy was well tolerated and resulted in better overall survival (Ref. 17). It is believed that such anti-angiogenic therapy can potentiate the effects of radiation mainly by normalizing tumor blood vessels and enhancing oxygen delivery (Ref. 18). Consequently, several ongoing clinical trials are evaluating the effects of concurrent chemotherapy with radiotherapy in glioma.
A potentially significant advancement in the treatment of gliomas is the development of molecularly targeted agents. There has been considerable progress in understanding the molecular pathogenesis of glioma and identification of key oncogenic pathways that can be targeted using small-molecule inhibitors. This has led to the development of several small-molecule agents that inhibit such deregulated signaling pathways in glioma. The recent success of such small-molecule inhibitors in other cancers has propelled rapid development of similar therapies for treatment of malignant gliomas.
Molecularly Targeted Therapy
Molecular abnormalities in signal transduction pathways are characteristic features of many brain tumors, including glioma, and result in uncontrolled tumor cell proliferation, survival and apoptotic resistance. The growth factor pathways that are commonly altered in malignant glioma the epidermal growth factor (EGF) (Refs 19-21), platelet derived growth factor (PDGF) (Refs 22-24) and the vascular endothelial growth factor (VEGF) (Refs 24-26). Deregulation in receptors of these pathways (EGFR, PDGFR, VEGFR) results in constitutive activation of downstream effectors that regulate gene transcription, ultimately leading to the phenotype in malignant glioma (Fig. 1). Thus, an attractive approach to inhibit the aberrant signaling pathways in glioma is to use small-molecule tyrosine kinase inhibitors (TKIs) that inhibit the activity of upstream receptors of these pathways.
Figure 1. Molecularly Targeted Therapy for Malignant Glioma.
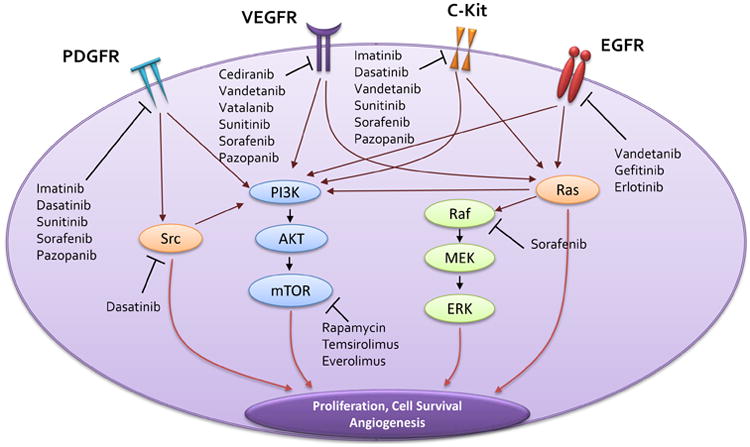
Several signaling pathways are aberrantly activated in glioma, the most common being signaling through EGFR, PDGFR, VEGFR and c-Kit (Ref. 24). These pathways can be deregulated due to one or more mechanisms such as auto-activation, aberrant expression, mutations and decreased activity of phosphatases that turn off the signal. Signaling through these pathways can be shut down by targeted therapies that inhibit these receptors, thereby preventing the downstream effects that ultimately lead to growth and proliferation of the tumor. Such molecularly targeted therapeutic agents have been shown in the figure near the targets that they inhibit. EGFR – epidermal growth factor receptor; PDGFR – platelet derived growth factor receptor; VEGFR – vascular endothelial growth factor receptor; mTOR – mammalian target of rapamycin; PI3K – phosphatidylinositol 3-kinase; ERK - extracellular signal-regulated kinase/mitogen activated protein kinase (MAPK); MEK –mitogen-activated protein kinase kinase; SRC - rous sarcoma oncogene cellular homolog; PI3K -phosphoinositide 3-kinase; AKT - AKT8 virus oncogene cellular homolog
Targeting EGFR and PDGFR
Aberrant signaling through the EGFR pathway is one of the most common genetic alterations seen in glioma (Refs 19, 27), and therefore several therapeutic strategies have used small-molecule TKIs to target EGFR in glioma. Gefitinib (Iressa, Astra Zeneca) and erlotinib (Tarceva, OSI Pharmaceuticals) were some of the first TKIs to show potent inhibitory effects on EGFR, prolonging survival in preclinical models of brain tumors (Refs 28-31). However, neither of these two promising drugs showed any significant survival benefit in glioblastoma multiforme patients (Refs 32-38). PDGFR is another attractive therapeutic target in glioma (Fig. 1) because it is commonly overexpressed in glioma and is thought to contribute to the aggressive phenotype of the tumor (Refs 22, 23).
Imatinib (Gleevec, Novartis), a potent inhibitor of the tyrosine kinases BCR–ABL, c-Kit (KIT) and PDGFR, was the first selective TKI to be approved for the treatment of cancer (Refs 39, 40). Imatinib showed encouraging antiglioma activity in preclinical studies, raising hopes in clinical trials that followed (Refs 41-43). However, the preclinical success did not translate into significant clinical benefit, with Phase II trials reporting insignificant antitumor effects in glioma patients (Refs 44, 45). Dasatinib (Sprycel, Bristol-Myers Squibb) is another PDGFR inhibitor with additional inhibitory effect on the Src family of kinases. It has also been shown that dasatinib can inhibit the growth and migration of glioma cells and induce cellular apoptosis, again warranting clinical investigation in glioma (Ref. 46); however, there is no published literature on the clinical efficacy of dasatinib in glioma (Table 1).
Table 1. Molecularly Targeted Agents for Tumors of the Central Nervous System.
| COMPOUND | MOLECULAR TARGET | RESULTS FROM CLINICAL TRIALS FOR GBM | NUMBER OF CLINCAL TRIALS |
|---|---|---|---|
| Cediranib | VEGFR 1,2,3 | Median OS in 16 patients of 211 days (Ref. 48) 6-month PFS 25.8 %, PR in 56.7 % (Ref. 49) |
6 |
| Dasatinib | Bcr-Abl, C-kit, PDGFR, Src | No Published Results | 4 |
| Erlotinib | EGFR | 6-month PFS 3.1% (Ref. 35) Median survival 19.3 months (Ref. 36) 6-month PFS of 3 %, 12-month survival of 57 % (Ref. 37) Median PFS 2.8 months, median OS 8.6 months (Ref. 38) |
20 |
| Everolimus | mTOR | stable disease in 36%, PR in 14 % of patients (Ref. 57) | 11 |
| Gefitinib | EGFR | Median EFS 8.1 weeks, median OS 39.4 weeks (Ref. 34) Median time to progression 8.4 weeks, 6-month PFS 14.3%, median OS 24.6 weeks (Ref. 33) |
4 |
| Imatinib | Bcr-Abl, C-kit, PDGFR | 6-month PFS 3% (Ref. 44) 6-month PFS 16% (Ref. 45) 6-month PFS 27%, median PFS 14.4 week (Ref. 185) 24% progression-free at 6 months (Ref. 186) |
7 |
| Lapatanib | EGFR2 | No Published Results | 3 |
| Pazopanib | C-kit, PDGFR, VEGFR 1,2,3 | No Published Results | 1 |
| Sirolimus | mTOR | 6-month PFS of 3.1% (Ref. 35) | 4 |
| Sorafenib | C-kit, PDGFR, Raf | Median PFS 6 months, median OS 16 months (Ref. 187) | 7 |
| Sunitinib | VEGFR 2,3, C-kit, FLT3, PDGFR | Median TTP 1.5 months and OS 3 months (Ref. 188) | 8 |
| Temsirolimus | mTOR | 6-month PFS 7.8 %, median OS 4.4 months (Ref. 58) | 9 |
| Vandetanib | EGFR, VEGFR | No Published Results | 10 |
| Vatalanib | C-kit, PDGFR, VEGFR1,2,3 | Partial Response in 29 % patients (Ref. 189) | 2 |
OS, overall survival; PFS, progression-free survival; PR, partial response; EFS, event-free survival; TTP, time to progression.
Number of clinical trials are determined from the clinical trials that have either been completed or are ongoing for therapy in glioma, searched on www.clinicaltrial.gov on 1st Dec 2010.
Targeting VEGFR
Angiogenesis, the process of vascular proliferation due to formation of new blood vessels, is a histopathological hallmark of malignant glioma. The angiogenic effect is mediated primarily through the VEGFR pathway, which is frequently upregulated in glioblastoma multiforme, making it a prime target for growth inhibition and therapeutic efficacy (Refs 25, 26). Numerous small-molecule VEGFR inhibitors such as cediranib, sunitinib, sorafenib, vatalanib and vandetanib, have shown promising results in preclinical glioma models.
Cediranib (Recentin, AstraZeneca), a pan inhibitor of the VEGFR tyrosine kinase, is one of the most exciting prospects for antiangiogenic therapy in glioma. It has demonstrated significant effects in mouse glioma models, decreasing oedema via vascular normalization in the tumor, leading to improvement in survival (Ref. 47). These preclinical effects have been mirrored in the clinic, where cediranib treatment results in normalization of tumor vessels, decreased vessel permeability and alleviation of vasogenic oedema (Ref. 48). Encouraging new data from a recently concluded Phase II trial suggest that cediranib therapy results in significant radiographic response and increases progression-free survival (Ref. 49).
Sunitinib (Sutent, Pfizer) and sorafenib (Nexavar, Bayer) are two multitargeted TKIs exhibiting both antiproliferative and antiangiogenic activity by simultaneously targeting VEGFR and PDGFR (Refs 50, 51). Separate studies have shown that both these compounds can increase survival in mouse glioma models at doses achievable in the clinic (Refs 52, 53). Several clinical trials are currently evaluating the efficacy of these two agents in human malignant glioma. Vandetanib (Zactima, AstraZeneca) is a novel small-molecule inhibitor that simultaneously targets VEGFR and EGFR (Ref. 54). It has demonstrated potent anti-glioma effects in clinically relevant glioblastoma multiforme models, suppressing tumor cell proliferation and angiogenesis while inducing apoptosis via inhibition of EGFR (Ref. 55). There are many ongoing clinical studies that are evaluating the efficacy and toxicity of vandetanib in glioma patients.
Targeting PI3K–AKT–mTOR
Other important molecularly targeted agents include inhibitors of the PI3K–AKT–mTOR pathway [comprising phosphoinositide 3-kinase, the serine/threonine protein kinase AKT and the mammalian target of rapamycin (MTOR)] (Fig. 1), which is thought to be highly activated in human glioblastomas, modulating key translational processes (Ref. 56). Rapamycin (sirolimus) and its analogues temsirolimus (CCI779) and everolimus (RAD001) are the three mTOR inhibitors that have undergone extensive preclinical and clinical evaluation for therapy in glioma. Clinical trials with mTOR inhibitors as a single agent in glioma have been largely unsuccessful, with no therapeutic benefits reported (Refs 35, 57, 58). However, several trials are currently evaluating mTOR inhibitors in combination with other TKIs with an aim to shutdown multiple signaling cascades feeding the tumor.
Improving efficacy of molecularly targeted agents
Most promising molecularly targeted agents have failed to provide any survival benefit in malignant gliomas (Table 1). Given the dismal prognosis of patients with glioma, the quest to find newer effective therapeutic options has gained precedence over the need to find the reasons behind the failure of these agents, although the two goals are closely linked. Some of the reasons for this lack of efficacy have been related to the genetic heterogeneity of gliomas and the complexity of signaling pathways, such as negative feedback mechanisms and upregulation of alternative pathways. However, all these hypotheses are reliant on the a priori assumption that there is adequate drug delivery to the target. The lack of drug delivery to the target is an often overlooked yet perfectly plausible explanation for a lack of efficacy. Would this delivery failure be detected in preclinical models that were used to justify the clinical trials? This would probably not be the case if the preclinical model used was not established in the brain (e.g. flank model), or if the assessment involved well-circumscribed brain tumors with a leaky BBB amidst no appreciable infiltrative growth to provide a pharmacological sanctuary (discussed further below). The latter well-circumscribed phenotype is the growth pattern of that majority of standard implanted models that are typically used for preclinical validation in the process of drug development (Ref. 59).
So the question germane to the efficacy of molecularly targeted agents in glioma is: are these drugs delivered to the tumor-infiltrated normal brain present after surgical removal of the bulk tumor mass at levels that are adequate to disrupt the function of their targets? Treatment of a brain tumor requires the drug to bypass several barriers and gain access to what is considered a ‘sanctuary’ in the CNS. The CNS is protected by a highly developed and well-regulated interface that separates it from the peripheral circulation and maintains homeostasis in the brain (Ref. 60). This interface also prevents most drugs and chemicals from entering the brain, thereby rendering them ineffective. Once inside the brain, the drug faces additional barriers that further limit its delivery to the ultimate target. It is critical to recognize that the intracellular targets in question are in the invasive glioma cell – that is, cells left behind after resection. Discussion of these barriers that limit drug delivery to tumor and hence their efficacy is the essence of this review.
Barriers Restricting Drug Delivery to Brain and Brain Tumor
The blood-brain barrier (BBB)
The BBB is a natural defence mechanism in the CNS that separates the brain from the peripheral circulation. The barrier is formed by a dense network of blood capillaries supplying the brain, wherein the endothelial cells are joined together by tight junctions such that most drugs and chemicals cannot readily cross into the brain parenchyma. The BBB thus shields the brain from exposure to circulating toxins and potentially harmful chemicals by preventing them from entering the brain. Besides the presence of tight junctions, a relative paucity of fenestrae and pinocytotic vesicles within the brain capillary endothelial cells along with the presence of the surrounding extracellular matrix, pericytes, and astrocyte foot processes further restrict brain uptake (Ref. 60). As a result of the tight junctions in the BBB, circulating molecules gain access to the brain only via (1) passive diffusion of small nonpolar molecules through the BBB or (2) active transport (Ref. 61).
Numerous studies have endeavored to correlate brain penetration and CNS activity of compounds to their physicochemical properties. These studies have used different approaches for predicting BBB permeability and reported that compounds that have activity within the CNS have high lipophilicity (log P ∼4), few hydrogen-bond donors (2-7), low polar surface area (PSA ∼40 Å) and low molecular weight (∼400 Da) (Refs 62-65). It is not surprising that all these properties impart greater membrane permeability to the drug molecule, resulting in enhanced transport to the brain (Ref. 65). However, several molecules with these favorable properties have been found to have a modest permeability into the brain, which is a result of active efflux transporters that further make the BBB impermeable (Refs 62, 66). The BBB is fortified by the presence of numerous drug transport proteins, many of which transport drugs out of the brain. It has been shown that ATP-dependent transporters can severely restrict brain penetration of therapeutic agents – even those molecules with favorable physicochemical properties that were predicted to cross the BBB with relative ease (Refs 62, 66). A majority of these transporters belong to two superfamilies: the ATP-binding cassette (ABC) and solute carrier (SLC) families. P-glycoprotein (P-gp, ABCB1), breast cancer resistance protein (BCRP, ABCG2) and multidrug-resistance-associated proteins (MRPs, ABCC) are important members of the ABC family. We limit our discussion in this review to P-gp, BCRP and MRPs. The reader is directed to several excellent reviews that cover other drug efflux transporters in greater detail than possible within the scope of this article (Refs 67-73).
P-glycoprotein
P-gp, the product of the ABCB1 gene (previously known as multidrug resistance 1 gene, MDR1), is by far the most extensively studied member of the ABC superfamily of transporters. It was originally discovered by Juliano et al. in 1976 while studying the mechanisms behind the resistance in tumor cell lines (Ref. 74). The group noticed that cell membranes of the resistant cells expressed a 170 kDa surface glycoprotein capable of altering permeability of drugs, and designated it as ‘permeability glycoprotein’ or ‘P glycoprotein’. A decade later, in 1986, the gene encoding the protein was discovered (Ref. 75) and the complete primary structure of P-gp was determined (Ref. 76). The existence of P-gp at the BBB was first reported in 1989 when Cordon-Cardo et al. detected P-gp expression in brain capillary endothelial cells and proposed that it played a role in regulating entry of drug molecules into the CNS (Ref. 76). Shortly thereafter, Theibaut and colleagues reported the expression of P-gp at the rat BBB (Ref. 77), which was followed by numerous studies showing the presence of P-gp in brain capillaries of other species such as mice, rats, bovine and porcine (Refs 78-80). However, it was a seminal study by Beaulieu et al. that reported the localization of P-gp on the luminal side of the capillary endothelial cells and bolstered theories that the transporter is involved in preventing drugs from entering the brain and in the development of multidrug resistance in cancer (Ref. 81).
The most compelling early evidence of the protective role of P-gp at the BBB was a chance discovery when mice deficient in the Abcb1a (Mdr1a) gene (P-gp knockout) were found to be 100-fold more sensitive to the neurotoxin ivermectin compared with the normal wild-type mice (Ref. 82). The study revealed elevated levels of ivermectin in the brains of the P-gp-knockout mice, a finding confirming that P-gp protects the CNS by preventing drugs and chemicals from crossing the BBB. P-gp has since been implicated in restricting CNS penetration of hundreds of drugs, including several chemotherapeutic agents in clinical practice. The development of the Abcb1a/1b(-/-) double knockout mice (Ref. 83) and Abcb1a/1b(-/-)Abcg2(-/-) triple knockout mice (Ref. 84) has provided researchers with powerful tools to examine the influence of P-gp in transport of drugs to the brain. Studies exploring the interaction of chemotherapeutic agents with P-gp have used these in vivo models to illustrate how potent anticancer drugs and many molecularly targeted TKIs are avid P-gp substrates and how this limits their distribution to the CNS (Table 2).
Table 2. Selected ABC Transporters at the Blood-Brain Barrier and Braintumor Cell Barrier and their Substrate Chemotherapeutic Agents.
| TRANSPORTER | GENE | LOCALIZATION AT THE BBB | PRESENCE IN GLIOMA CELLS | SELECTED SUBSTRATE CHEMOTHERAPEUTIC AGENTS |
|---|---|---|---|---|
| P-glycoprotein (P-9P) | ABCB1 (MDR1) | Luminal (Ref. 79) | Yes (Refs 124-126) | Vincristine, vinblastine, paclitaxel, docitaxel, doxorubicin, daunorubicin, mitoxantrone, etoposide, teniposide, methotrexate, topotecan, imatinib, dasatinib, lapatinib, gefitinib, sorafenib, erlotinib, tandutinib, |
| Breast Cancer Resistance Protein (BCRP) | ABCG2 (MXR) | Luminal (Ref. 101) | Yes (Refs 131-133) | doxorubicin, daunorubicin, mitoxantrone, methotrexate, topotecan, SN-38 (active metabolite of irinotecan), gimatecan, imatinib, dasatinib, lapatinib, gefitinib, sorafenib, erlotinib, tandutinib, |
| Multi-Drug Resistance Associated Protein 1 (MRP1) | ABCC1 (MRP1) | Luminal, Apical (Refs 87, 88) | Yes (Refs 127,128,136) | Etoposide, teniposide, vincristine, vinblastine, paclitaxel, docitaxel, doxorubicin, daunorubicin, mitoxantrone, topotecan, irinotecan, methotrexate |
| Multi-Drug Resistance Associated Protein 2 (MRP2) | ABCC2 (MRP2) | Luminal (Ref. 89) | ? | Cisplatin, etoposide, vincristine, vinblastine, doxorubicin, daunorubicin, topotecan, irinotecan, methotrexate, paclitaxel, docitaxel |
| Multi-Drug Resistance Associated Protein 3 (MRP3) | ABCC3 (MRP3) | ? | Yes (Refs 128,136, 138) | Etoposide, teniposide, vincristine, methotrexate |
| Multi-Drug Resistance Associated Protein 4 (MRP4) | ABCC4 (MRP4) | Luminal, Apical (Refs 87, 88) | Yes (Refs 128,135) | Methotrexate, topotecan, 6-mercaptopurine, thioguanine, cisplatin |
| Multi-Drug Resistance Associated Protein 5 (MRP5) | ABCC5 (MRP5) | Luminal, Apical (Refs 87, 88) | Yes (Refs 128, 135, 136) | 6-mercaptopurine, thioguanine, gemcitabine |
Multidrug-resistance-associated proteins
The discovery of P-gp as an efflux transporter capable of transporting drugs out of tumor cells led to an augmented interest among researchers to find other proteins involved in drug transport and resistance. In 1987, Cole and co-workers noticed that an adriamycin-selected lung cancer cell line was resistant to drugs such as colchicine, vinca alkaloids and anthracycline analogues (Ref. 85). These cells were known not to overexpress P-gp, leading researchers to believe that the observed resistance might be due to a transporter mediated mechanism that was similar to P-gp. Molecular analysis revealed the presence of a cDNA encoding a 190 kDa protein that was later confirmed to be present in several multidrug-resistance cell lines that did not express P-gp. This protein was called the multidrug-resistance-associated protein (Ref. 86), the first of 12 members of a subfamily of ABC transporters now designated as subfamily-C (ABCC). Cloning of MRP in 1992 resulted in a renewed enthusiasm for drug resistance investigations, which were now focused at identifying additional transporters capable of transporting drugs out of cells.
There is now evidence that nine of the twelve ABCC family members (MRP1–9) mediate some form of xenobiotic and/or drug resistance (Ref. 87). The discovery of MRPs also resulted in several studies investigating the localization and role of these transporters at the BBB. However, studies on the expression of MRP transporters at the BBB have been controversial and often contradictory. Huai-Yun et al. in 1998 demonstrated the functional expression of MRP1 in bovine brain microvessel endothelial cells (BBMEC) and suggested that the most likely localization of MRP1 at the BBB should be apical (Ref. 88). In 2004, Zhang et al. described the localization of various MRP analogues in bovine brain microvessel endothelial cells, showing that MRP1 and MRP5, which are predominantly localized basolaterally in various tissues, were highly expressed on the apical side, whereas MRP2 was not detected (Ref. 89). The group also reported equal localization of MRP4 on the apical and the basolateral plasma membranes in these cells. Nies and co-workers quantitatively studied the expression and localization of MRPs in several regions of adult human brain and showed the presence of MRP 1, 4 and 5 on the luminal side of the BBB, consistent with the findings in the bovine brain (Ref. 90). In contrast to these earlier studies that report absence of MRP2 at the BBB, some studies have shown MRP2 expression at the luminal membranes of the human (Ref. 91), rat and pig BBB (Ref. 92).
Although equivocal, expression of MRPs at the BBB has thus now been described in several studies; however, their exact localization and role at the BBB is still debated. There have been reports that demonstrate the influence of MRPs at the BBB, wherein absence or inhibition of the transporter(s) results in enhanced brain penetration of substrate drugs (Refs 93-97). Recently it was shown that brain transport of topotecan to the brain was enhanced when MRP4 was absent in the MRP4-knockout mice (Ref. 98). These studies strongly suggest that some of the MRPs act as an active drug efflux transporter at the BBB. However, further investigation is necessary to completely understand the function of these transporters at the BBB. The availability of newer tools such as knockout mice deficient in one or more of the MRPs can provide answers to questions that are still unanswered with respect to the protective role of MRPs at the BBB.
Breast Cancer Resistance Protein
Breast cancer resistance protein is another member of the ABC superfamily of transporters that confers drug resistance in cancer by virtue of its ability to translocate drugs out of cells. BCRP was originally identified independently and almost simultaneously by three different groups studying non-P-gp- and non-MRP-mediated drug resistance in cancer cell lines (Refs 99-101). In 1999, Doyle and colleagues observed an ATP-dependent reduction in the intracellular accumulation of anthracycline anticancer drugs in MCF-7 breast cancer cells and were not able to ascribe this to overexpression of known multidrug resistance transporters, P-gp or MRP. RNA fingerprinting identified overexpression of a mRNA that encoded a 655 amino acid protein in the resistant cells, a protein that they designated as the breast cancer resistance protein (Ref. 99). A similar study investigating the occurrence of mitoxantrone resistance in cancer cell lines isolated a novel cDNA that encoded for an ATP-dependent transporter that was named mitoxantrone resistance protein (MXR) (Ref. 100). Around the same time, Allikmets and co-workers identified a novel gene that was highly expressed in the human placenta. They showed that the gene encoded an ABC transporter protein, which they termed as the ABCP (ABC transporter in the placenta) (Ref. 101). When the sequences of genes from these three studies were eventually compared, they were recognized as essentially identical and belonging to a subfamily of ABC transporters not previously associated with drug resistance in humans (Ref. 102). Subsequently, the Human Genome Nomenclature Committee assigned this gene the name ABCG2. Following the cloning of BCRP, its role in the efflux of drugs from multidrug-resistant cells has been widely studied, and there are several reports on BCRP-mediated resistance to chemotherapeutic agents (Table 2).
The putative role of BCRP in the barrier function at the BBB has been controversial. Several studies have reported that BCRP is localized on the luminal side of the capillary endothelial cells in the human (Ref. 103) and rat brains (Ref. 104). Others have reported enriched presence of BCRP in brain capillaries of mice (Ref. 105) and pigs (Ref. 106). However, this presence of BCRP at the BBB has not been unequivocally correlated to the low brain penetration of all BCRP substrates. Lee et al. conducted in situ brain perfusion studies using dehydroepiandrosterone sulfate and mitoxantrone, two drugs that are efficiently transported by BCRP, and reported no enhancement in brain penetration of the two compounds in Abcg2(-/-) mice (Ref. 107). Similarly, another study showed that in vitro interaction of BCRP with substrate compounds rarely translates to visible effects at the BBB in vivo (Ref. 108). The authors from both studies concluded that BCRP plays a minor role in the efflux of drugs at the BBB. By contrast, there have been several studies that demonstrate the role of BCRP in efflux of drugs at the BBB. Cisternino and colleagues showed that BCRP-mediated efflux of prazosin and mitoxantrone at the BBB limits permeability of the brain to these prototypical substrates (Ref. 105). Likewise, Enokizono et al. showed that brain partitioning of drugs increased significantly when BCRP was absent in the Abcg2(-/-) mice (Ref. 109). Breedveld et al. showed that brain penetration of imatinib was restricted by BCRP (Ref. 110) and we recently reported that sorafenib transport to the brain was significantly increased in Abcg2(-/-) mice (Ref. 111). There has been a recent increase in the number of studies investigating the role of BCRP-mediated active efflux in the transport of drugs out of the brain. This surge has been driven by reports suggesting a possible cooperative role of P-gp and BCRP in keeping drugs out of the brain (Refs 111-118). Several studies have shown that there is a dramatic increase in the brain penetration of dual P-gp and BCRP substrates when these two transporters are absent simultaneously in the Abcb1(-/-) Abcg2(-/-) mice. First seen with topotecan (Ref. 112), this phenomenon has now been reported for several other compounds including important TKIs such as lapatinib (Ref. 113), dasatinib (Refs 114, 115), gefitinib (Ref. 116), erlotinib (Ref. 117) and sorafenib (Refs 111, 118). These findings, along with reports that there is extensive overlap in the expression pattern and substrate specificity of BCRP and P-gp (Ref. 119), suggest that P-gp and BCRP work together at the BBB to limit brain penetration of dual substrates.
Efflux Transport at BBB Restricts Drug Delivery
The BBB is a major bottleneck that limits drug delivery to the brain; a significant fraction of large and small molecules do not effectively cross the BBB (Ref. 6). It is clear that drug efflux transporters, a key component of this barrier, can significantly restricts passage of drugs into the brain, even those with favorable physiochemical properties to cross biological membranes. The fact that there are multiple drug transporters at the BBB, some of which might be working in concert with each other, further complicates the problem. Effective targeting of tumors in the brain will require novel strategies to inhibit these gatekeepers so that promising drug candidates are not rendered ineffective due to their inability to enter the brain.
Is the BBB Compromised in Glioma?
Recently, the role of the BBB in limiting treatment efficacy in glioma has been questioned based on studies that report high concentrations of chemotherapeutic agents in tumor resections (Ref. 120). These reports suggest that the BBB does not influence delivery in glioma. This has caused confusion in the clinical assessment of drug delivery when using drug concentrations in the tumor core (the resected tissue) as a guide for the adequacy of drug delivery. It is true that the BBB can be disrupted at or near the tumor since the central core of the tumor is highly angiogenic, containing new and leaky blood vessels (Ref. 121). While drug delivery might be greatly enhanced in such areas of the tumor, surgery almost completely removes the central core of the tumor in the brain (contrast-enhancing area). Therefore concentrations in these areas do not represent those in the brain areas that are not removed by surgery. Moreover, the BBB is intact at the growing edge of the tumor and early in the development of the vascular niche of invasive glioma cells (Ref. 122). The disruption of brain vasculature is directly related to tumor size, and the distance from the central core (Ref. 121). Invasive glioma cells that are not removed by surgery reside in areas of diffuse glioma invasion, which can be centimetres away from the main tumor (Ref. 123) and have an intact BBB capable of restricting drug levels. Given the diffusely infiltrating growth of glioma, it is not surprising that the tumor eventually recurs from areas of the tumor rim that are not resected (Ref. 13), where drug delivery is impaired due the BBB.
Effective delivery of chemotherapeutics to the invasive glioma cells is therefore critical, and long-term efficacy will depend upon the ability of a molecularly targeted agent to penetrate an intact and functional BBB throughout the entire brain. This idea of glioma as a disease of the whole brain lends particular credence to the need to use the systemic circulation to effectively deliver drug across the BBB to encompass the central tumor, growing edge of the tumor, and invasive glioma cells. We present this problem in Figure 2, where a hypothetical schematic of a brain tumor can be seen with a gradient of drug concentration around the tumor (Fig. 2a). The tumor core (the area with a disrupted BBB) can have high drug levels; however, areas immediately surrounding the core can receive significantly less drug owing to an intact BBB. The tumor core is usually removed after surgery, but glioma cells invade areas of restricted drug delivery away from the tumor (Fig. 2b). The goal of effective chemotherapy should be to effectively deliver drug in areas that can harbor the invasive glioma cells and not just the tumor core, the part of the tumor removed by surgery (Fig. 2c). This idea has been supported in a study by Fine et al., where the authors measured paclitaxel concentrations in resected tissue specimens from brain tumor patients and showed that concentrations in the normal brain surrounding the tumor were 10-fold lower than that in the tumor core (Ref. 124). Furthermore, Pitz et al. recently summarized clinical studies reporting anticancer drug concentrations in brain tumors and suggested that drug concentrations in contrast enhancing areas of tumor (tumor core) were relatively higher than that in noncontrast enhancing areas (tumor periphery/normal brain (Ref. 125)).
Figure 2. Hypothetical Schematic of Regional Drug Delivery in Glioma due to Invasive Nature of the Tumor.
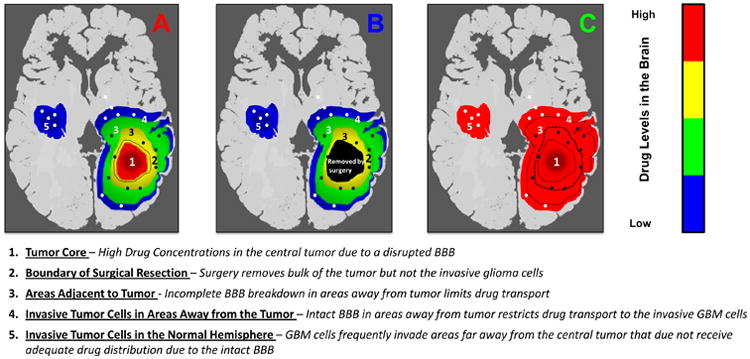
(A) Schematic of a brain tumor with a simulated gradient of drug concentration around the site of tumor. The tumor core, the area with a disrupted BBB, can have high drug levels (region ‘1’); however areas immediately surrounding the core can receive significantly less drug owing to an intact blood-brain barrier. (B) The tumor core is usually removed after surgery (up to boundary ‘2’); however, glioma cells invade areas of restricted drug delivery away from the tumor (regions ‘3-5’). These areas away from the tumor are the sites where the invasive glioma cells continue to grow and give rise to the recurrent tumor. (C) The goal of chemotherapy should be to effectively deliver drug in areas that can harbor the invasive glioma cells and not just the tumor core, the part of the tumor removed by surgery.
The brain–tumor-cell barrier (BTB)
In addition to the BBB, the BTB is another barrier that the drug has to cross to reach its intracellular target. The tumor cell membrane, which forms this barrier, regulates the transport of nutrients, growth factors, drugs and other substances into and out of the cell. A considerable amount of work has been done studying the expression, regulation and activity of ABC transporters in cells from various tumors, including glioblastoma multiforme. There is increasing evidence suggesting that drug efflux transporters on the tumor cells decrease intracellular drug uptake, resulting in the multidrug-resistant phenotype often observed in glioma cells.
P-gp is by far the most extensively studied efflux transporter in glial tumors and its presence has been confirmed by several studies. Fattori and co-workers used immunohistochemistry to show that P-gp was heterogeneously expressed in about 82% of glioblastomas (Ref. 126). Similarly, several other studies have reported enhanced expression of P-gp in tissue specimens from human gliomas (Refs 127, 128). However, there have also been several conflicting reports indicating the absence of P-gp in glioblastoma multiforme cells. These studies suggest that expression of P-gp in human glioma specimens is relatively low and rare (Ref. 129). Decleves et al. showed that P-gp was not expressed in human glioma cells at either the transcript or the protein level (Ref. 130). These widely differing results on the expression of P-gp have been attributed in part to the assay technique used for detection of P-gp (Ref. 131). Nevertheless, the recent reports mentioned above confirm the presence of P-gp in glioma cells and its effect on accumulation of anticancer drugs in these cells.
In contrast to P-gp, very few studies have investigated the expression of BCRP in tumor cells from glioma. Despite its original isolation from drug-resistant breast cancer cell lines, expression of BCRP in many solid tumors have been found to be negligible (Ref. 132). However, new evidence implicates this transporter with a special side-population of tumor cells that are believed to have stem-cell-like properties (Refs 133, 134). These precursor cells, responsible for driving tumor growth and proliferation, are thought to be drug resistant due to efflux by BCRP. In a mouse model of glioma, Bleau et al. recently demonstrated enhanced tumorigenicity of BCRP-enriched stem-like cells (Ref. 135). This evidence suggests a similar role of BCRP wherein the transporter confers resistance in glioma cells by virtue of its ability to pump drugs out of the cell.
Other than P-gp and BCRP, MRPs have also been found to be expressed in glioblastoma cells (Ref. 136). Transporters of this family have been found to be expressed at levels that are in some cases greater than that of P-gp (Ref. 129). Histochemical analysis of glioma specimens has revealed the presence of significant amounts of MRPs 1, 3, 4 and 5 (Refs 130, 137, 138). The influence of MRPs on chemoresistance in glioma has also been reported: nonspecific inhibition of MRPs enhanced the cytotoxic effects of anticancer agents in glioma cell lines (Ref. 139). In a recent study, Kuan and co-workers reported elevated expression of MRP3 in human glioblastoma multiforme in contrast to negligible presence in normal brain (Ref. 140) and suggested the potential use of MRP3 as a prognostic marker and molecular target for glioblastoma multiforme.
In summary, expression of ABC transporters in human glioma cells and their role in acquired drug resistance has been reported by several studies in the past few years. The findings have often been ambiguous and conflicting. While the genetic heterogeneity of the tumor in glioma can account for some of the variability in the reports, more research is clearly needed to elucidate the role of these transporters in tumor cells. Nonetheless, it is clear that the BTB can be a significant second barrier that has the ability to hamper drug delivery to the intracellular target.
BBB and BTB: Complex Barriers that Limit Delivery of TKIs to Glioma
The impact of the BBB and BTB on drug delivery to the target site can be significant, especially when the drug is a substrate for transporters present at both the barriers (Fig. 3). The recent surge in the development of molecularly targeted TKIs for CNS tumors has led to several investigations on their interaction with important efflux. The availability of tools in the form of transgenic mouse models and transporter overexpressing cell lines have made it possible to study drug–transporter interactions with an aim to modulate these and enhance drug transport to the target tissue.
Figure 3. Multiple Mechanisms and Barriers That Limit Drug Delivery to Glioma.
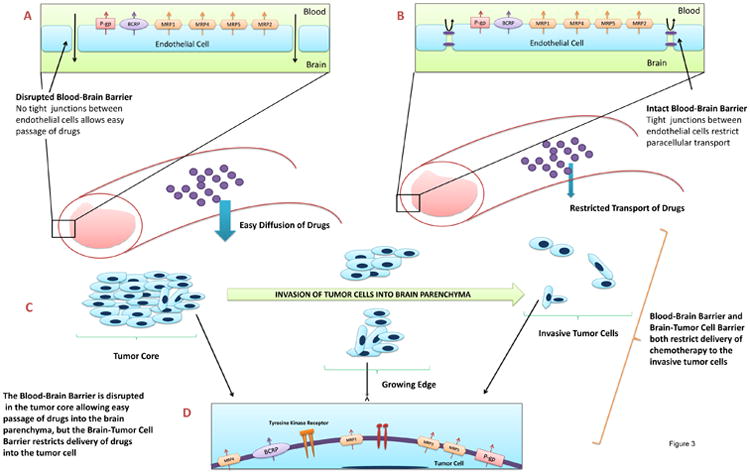
The blood-brain barrier and the brain-tumor cell barrier form sequential barriers that a systemically administered drug must cross to reach the tumor. A) The BBB is often disrupted at the site of the tumor allowing for easy diffusion of drugs and small molecules into the tumor. However this is also the part of the tumor that gets removed after surgery. B) The BBB however is intact in areas centimetres away from the tumor core. Drug delivery across this barrier is restricted by the presence of tight junctions between endothelial cells and more importantly by drug efflux transporters that pump drugs back into the blood. Drug that gets across this barrier and reaches the brain is usually a fraction of what reaches the tumor core. C) Invasion of glioma cells from the tumor core into the normal brain parenchyma. These small nests of tumor cells are protected by the intact BBB and receive only a small amount of drug. These cells eventually give rise to the recurrent tumor after surgery D) The brain-tumor cell barrier represents the barrier between the brain parenchyma and the tumor cell. Drug efflux transporters present in the tumor cell are a major component of this barrier and restrict intracellular drug uptake. This second barrier is especially important for molecularly targeted agents since they target intracellular domains of receptor tyrosine kinases. Pgp – p-glycoprotein; BCRP –breast cancer resistance protein; MRP – multidrug resistance associated protein.
Given that P-gp and BCRP are the two important transporters that limit drug delivery to the brain and tumor cells, most studies have investigated the interaction of TKIs with these two efflux pumps. Imatinib was the first TKI that was reported to be a substrate for drug-effluxing transporters, when it was discovered that distribution of imatinib to the brain was restricted by P-gp-mediated efflux (Ref. 141). This was followed by a number of studies that reported that imatinib was effluxed by both P-gp and BCRP at the BBB (Refs 110, 142). The finding that imatinib does not effectively cross the BBB was crucial in explaining its lack of efficacy against brain relapses in chronic myeloid leukemia (Ref. 143). Similarly, Polli et al. showed that the EGFR inhibitor lapatinib was a substrate for both P-gp and BCRP (Ref. 144) and then suggested that these two transporters work together at the BBB to limit brain penetration of dual substrates (Ref. 113). Thereafter, we showed that P-gp and BCRP work in concert to limit the brain penetration of several other TKIs, such as dasatinib, gefitinib and sorafenib (Refs 111, 114, 116). Subsequent studies have shown that this is true for tandutinib and erlotinib as well (Refs 145, 146). An extremely important finding in most of these studies is that pharmacological inhibition of the two transporters together significantly enhanced brain levels of the TKIs, over and above individual inhibition of one of the transporters. These preclinical studies used elacridar (GF120918), a dual inhibitor of P-gp and BCRP, and demonstrated that it increases the transport of the concurrently administered TKI to the brain. This suggests that co-administration of an inhibitor of P-gp and BCRP can be used as a strategy to enhance the delivery of these drugs to the brain.
Many promising TKIs are effective in treating non-CNS malignancies such as lung, breast and hepatic cancer. However, none of them shows any clinical efficacy against the metastatic disease in the brain or against primary brain tumors such as glioma (Table 1). The complication of delivery across an intact BBB has made it difficult to apply peripherally acting chemotherapeutic agents to invasive cancers of the brain. The problem is confounded by the fact that the BTB has the ability to further restrict intracellular delivery of drug into the invasive glioma cells. A more detailed understanding of these multiple barriers can help researchers devise strategies to overcome some of these barriers and thereby increase the effectiveness of these drugs against GBM.
Clinical Implications
Tyrosine Kinase Inhibitors in glioblastoma multiforme: Hopes and Disappointments
In May 2001, the first TKI, imatinib, was approved for treatment of a human malignancy (chronic myeloid leukemia), raising hopes within the oncology community of the promise of similar target-directed chemotherapeutics for treating other devastating cancers such as glioblastoma multiforme. However, to date, none of the TKIs has been able to show any clinical benefit against this disease. Imatinib's success in chronic myeloid leukemia started a wave of clinical trials that evaluated different TKIs either alone or in combination for therapy in glioma. The trials were backed by significant data showing efficacy of the compounds in preclinical models of glioblastoma multiforme, but most of them culminated in disappointing failures. The first clinical trial of a TKI in glioma tested gefitinib (Ref. 34), with the hope that inhibition of the highly deregulated EGFR signaling pathway would translate into improved patient survival. Its failure was soon followed by the clinical inefficacy of the other major EGFR inhibitor, erlotinib. Identification of newer targets led to introduction of newer targeted therapies: the clinical outcomes, however, did not change.
Studies explaining the failure of these trials have suggested that some reasons for this could be the heterogeneous molecular characteristics of individual gliomas and the complexity of signaling cascades that feed the tumor. However, several questions remain. (1) Does the drug cross the BBB? (2) What are drug concentrations in the brain? (3) What are the concentrations in the tumor? (4) Is the concentration sufficient to inhibit the target? Answers to these questions can help us gain an insight into the possible reasons behind the failure of these drugs. If therapeutic agents do not reach their intended molecular target, regardless of their potency, they cannot possibly be effective. It is well accepted that the BBB evolved to protect the brain and will be a barrier in the CNS delivery of most drugs. As discussed earlier, many of the TKIs are substrates for important transporters at the BBB and this significantly limits their concentrations in the brain. Whether these preclinical findings translate in humans and whether these transporters restrict penetration of drugs across a human BBB is still unknown. But there is no evidence to suggest otherwise and the inefficacy of these agents against brain tumors in humans adds further credence to the hypothesis.
Drug Concentrations in the Brain and the Tumor
Many of the questions raised above can be explored if drug levels in the brain could be measured in patients receiving chemotherapy. Unfortunately, very few studies have evaluated drug concentrations in brain tissues. This is due in part to the difficulty in sampling drug levels in the brain tissue and an uncertainty in the prediction of drug levels in brain from concentrations in surrogate tissues such as the cerebrospinal fluid (Ref. 147). However, recently Hofer and colleagues presented a few case reports where they investigated concentrations of chemotherapeutic agents in the brain and tumor. The group measured gefitinib concentrations in tissue specimens from seven glioblastoma multiforme patients and reported 10-fold higher concentrations in excised tumor tissue compared with plasma (Refs 120, 148). These findings were supported by preclinical reports describing gefitinib accumulation in tumor (Ref. 149). The groups concluded that delivery of drugs (gefitinib) to the tumor is not restricted in patients since the BBB is overcome by residual damage from radiotherapy and/or by the pathological infiltrative characteristics of glioblastoma multiforme that compromises the functional integrity of the BBB. In the 1980s, a few studies by Stewart and co-workers measured concentrations of cisplatin (Ref. 150), vinblastine (Ref. 151) and etoposide (Ref. 152) in brain tumors. All these studies reported high drug levels in the tumor similar to the above report. But the group also presented a very interesting finding. Drug concentrations in regions immediately adjacent to the tumor were surprisingly lower than that in the tumor, the concentrations decreasing with increasing distance from the tumor. In a similar study, Blakeley et al. used microdialysis to show that penetration of methotrexate was significantly lower in the brain areas adjacent to the tumor (Ref. 153). All these studies show significantly high drug levels in the tumor. So how does one explain the apparent contradiction that tumor distribution of drugs does not seem to be restricted by the BBB, yet at the same time their efficacy against the tumor is minimal and that the recurrence of tumor after surgery, centimetres away from the original tumor, is inevitable even with intensive radio- or chemotherapy?
Glioblastoma: A Whole-Brain Disease
Given its invasive and infiltrating nature, we consider glioma essentially a disease of the entire brain, and this idea can help understand the answers to some of the questions raised above. In addition to being one of the most malignant cancers, glioma is also one the most infiltrative tumors. Even complete surgical resection of the tumor-bearing hemisphere inevitably leads to recurrence and has been abandoned (Ref. 154). Historical reports show that more than 50 % of untreated brain tumors spread into the contralateral hemisphere (Ref. 155). Thus one of the most important hallmarks of malignant glioma is local invasion and has been described in studies as early as 1938. In a landmark study, Hans –Joachim Scherer described the diffuse invasion of glioblastomas by defining secondary patterns that reflected growth of tumor in neighboring brain tissue (Ref. 156). Thus glioblastoma multiforme is a disease of the whole brain. Tumor cells that migrate into the surrounding brain parenchyma escape surgical resection and are the putative source of the recurrent tumor (Figs 2, 3).
This pathological property of glioma can account for many of the pharmacokinetic findings mentioned above. First, the central core of the tumor is a highly necrotic mass and the BBB is most likely disrupted in this area. This allows systemically delivered chemotherapy to easily traverse the impaired barrier and reach the tumor, thus explaining the high concentrations seen in tumor by Hofer and Frei (Ref. 120). This is almost always true since the very ability of contemporary imaging techniques to detect a brain tumor relies on the ability of the contrast agent (gadolinium) to leak through a disrupted BBB and enhance the tumor core (Ref. 157). Nevertheless, this is also the part of the tumor that is removed by surgical debulking, rendering less relevant any correlations between drug concentrations in this area to eventual efficacy or lack thereof.
Second, disruption of the BBB becomes increasingly insignificant in areas away from the tumor. This is a valuable finding in the studies by Stewart et al. and Blakeley et al. (Refs 150-153). The fact that drug exposure in areas immediately adjacent to the tumor was an order of magnitude lower than the exposure in tumor confirms the presence of a functional BBB in these areas, capable of restricting passage of drugs into the brain. This has been elegantly demonstrated by Lockman et al., where the authors show that the BBB remains sufficiently intact in satellite legions of the metastatic tumor to significantly restrict drug delivery to the tumor cells (Ref. 158). This theory has also been supported by other recent studies that have shown that concentrations of paclitaxel (Ref. 124) and temozolomide (Ref. 159) in the tumor periphery were lower than that in the tumor core. A recent study by Pitz et al. summarizes findings from clinical studies and shows that concentrations of many anticancer drugs in contrast-enhancing areas of tumor were several fold higher than that in plasma (Ref. 125). More importantly, the study also reports that tissue-to-blood ratios were generally higher in contrast-enhancing regions than non-enhancing regions, and areas of brain distant to tumor (Ref. 125). Thus, in areas distant from the tumor core, where gadolinium does not cross the intact BBB, mechanisms that limit drug distribution (tight junctions and efflux transport) will still be operative and limit drug delivery. Consequently, less drug reaches these sites that harbor the infiltrated tumorigenic glioma cells, which continue to grow and ultimately reach a clinically significant size. Thus recurrence, an inevitable occurrence in glioma, might be due not only to tumor cells invading the adjoining brain areas but also to a lack of drug delivery in such areas.
Finally, there is a growing body of literature that suggests that a subset of these invasive cells have stem-like properties that allow them to repopulate the tumor (Ref. 160). The cancer stem cell hypothesis asserts that tumor development and maintenance in glioblastoma multiforme is controlled exclusively by these rare fractions of cells with unlimited proliferative and self-renewing capacities (Ref. 161). A basic tenet of this hypothesis is that these stem-like cells have an innate resistance to chemotherapy (Refs 162, 163), mainly due to the presence of drug transporters that efflux drugs out of the cells (Refs 164-167). This indicates that even if a drug crosses the BBB to reach brain parenchyma, its entry into an infiltrative tumor cell can be further restricted by drug efflux proteins present within such cells. These infiltrative cells, shielded by the BBB and the BTB, thus grow and eventually give rise to the recurrent tumor.
Thus, the two complex sequential barriers – the BBB and the BTB – are two important factors that govern the passage of drug from systemic circulation to the target site. The clinical failure of molecularly targeted therapy suggests two fundamental realities. One is that the BBB and the BTB can significantly limit drug delivery to the target site. The other reality is that regardless of how potent our targeted agents are, they will continue to be ineffective until strategies are devised to improve their delivery across the BBB and the BTB into the invasive glioma cells. An excellent depiction of this predicament is given by Berens et al., where the authors explain that the clinical course of glioma patients after surgery is determined by residual, invasive tumor cells – that is, “those left behind” (Ref. 7).
Outstanding Research Questions
The realization of the impact that the BBB and the BTB can have on chemotherapy in glioma has resulted in a renewed interest among researchers to pursue strategies that can overcome these barriers and increase the delivery of drug to the tumor targets. Several innovative methods have been developed and used to circumvent the BBB and improve drug delivery to the brain. These techniques can be divided into three broad categories; administration of chemotherapy directly into the brain parenchyma, osmotic disruption of the BBB, and inhibition of drug efflux.
Direct administration into the CNS is achieved via use of biodegradable polymers, convection-enhanced delivery or by intrathecal and intraventricular administration. In 2002, the US Food and Drug Administration (FDA) approved Gliadel® wafers for use as an adjunct to surgery in the treatment of malignant glioma. These are biogedradable polymeric wafers that slowly release the DNA-alkylating agent BCNU in the space remaining after surgical resection and have been shown to be well tolerated and offer a survival benefit in glioblastoma multiforme patients with concurrent chemotherapy (Refs 168, 169). However, new data indicate no survival benefit and significant adverse effects on treatment with these wafers (Ref. 170). Clinical evaluation of convection-enhanced delivery for enhancing tumoral delivery of chemotherapy has yielded similar results (Ref. 171). In a recent trial, convection-enhanced delivery afforded no survival benefit compared with Gliadel® (Ref. 172), while a separate clinical trial reported that treatment was associated with severe neurological complications (Ref. 173). Transient disruption of the BBB by intra-arterial infusion of a hyperosmotic solution of mannitol (Ref. 174) or the bradykinin analogue RMP-7 (Ref. 175) is a method used to enhance concentrations of chemotherapy in brain. Recent studies have shown that treatment with carboplatin, etoposide (Ref. 176) and bevacizumab (Ref. 177) after disruption of the BBB resulted in prolonged time to progression and reduction in tumor volume.
A primary drawback common to the above approaches is that these are complex techniques and are associated with a significant incidence of treatment-related complications. Modulation of drug transporters at the BBB may be an alternative possible method to improve delivery of chemotherapy to the brain. Compounds such as valspodar (PSC833), zosuquidar (LY335979) and elacridar (GF120918), which are potent inhibitors of drug transporters P-gp and/or BCRP, can significantly enhance systemic and brain concentrations of the concurrently administered chemotherapeutic agent (Refs 111, 112, 114-116). Consequently, several clinical trials have tested these chemical modulators with the aim to reverse multidrug resistance in hematological and solid tumors. The results from these clinical investigations have been disappointing, with many studies reporting no enhancement in drug efficacy and significant toxicities related to administration of the reversal agent. Treatment with valspodar has been associated with severe toxicities and no improvement in efficacy of concurrent chemotherapy (Refs 178, 179). The P-gp inhibitor zosuquidar has been reported to be relatively nontoxic but has again failed to show any improvement in treatment (Refs 180, 181). In contrast, coadministration of the dual P-gp and BCRP inhibitor elacridar resulted in significant enhancement in the oral bioavailability of topotecan and doxorubicin (Refs 182, 183). However, these effects were seen after high doses of elacridar, which were often toxic. While most of these failures were in trials for peripheral solid tumors, the scenario might be different in brain tumors where a moderate enhancement in drug delivery across the BBB can dramatically increase relative drug concentrations in the brain. Furthermore, in many of the studies, it was not clear if the observed toxicities were a result of the transport modulator or the simultaneously administered chemotherapeutic agent. Again, this may be different in brain tumors where the most common toxicity observed with the current TKIs is systemic and administration of an efflux inhibitor could even serve to reduce the chemotherapeutic dose if the desired brain concentrations were achieved at lower systemic doses. Clinical trials of modulation of multi-drug resistance have been limited by two major factors: inability to achieve adequate nontoxic levels of the modulators to reverse drug resistance in patients and the presence of multiple mechanisms of resistance (Ref. 184). Development of new, more potent inhibitors can help overcome some of these limitations. Further clinical studies are needed to better understand the benefit of increasing delivery of chemotherapeutic drugs to tumors in the brain. Additionally, preclinical studies that will be used to justify these clinical trials must employ intracranial models that exhibit appreciable tumor-infiltrated normal brain protected by the BBB in order to be most informative.
Conclusion
The BBB and the BTB are two important obstacles that restrict the passage of molecularly targeted agents to the tumor. Increase in our understanding of the molecular biology of glioma has resulted in new potent compounds that intervene in various signaling pathways that drive the tumor growth. However, regardless of their potency, if the therapeutic agents do not reach their intended molecular target, they cannot possibly be effective. Numerous strategies have been devised to circumvent some of these barriers and improve delivery of drug to the tumor cells in the brain. Although some of these strategies have shown promising results in the preclinical setting, results in patients have thus far been poor. The molecular heterogeneity in glioma calls for the use of multitargeted agents – ‘dirty drugs’ that can inhibit multiple signaling pathways simultaneously. However, we also need ‘sharp needles’ that can effectively deliver such drugs to the site of the invasive tumor. The next generation of clinical trials is exploring the use of multitargeted TKIs or combinations of single-targeting TKIs. Further research investigating delivery of chemotherapeutics to the tumor will ensure that these clinical trials do not follow the same pattern as that of the previous trials. Approaching the treatment of glioma by assuming that the tumor is localized in the contrast-enhancing area (hence resection) will lead to continued failure. The dismal prognosis in glioma may remain unchanged until measures are taken to ensure that promising anticancer treatments are delivered effectively to the invasive glioma cells, those hiding behind an intact BBB. We must effectively treat “those left behind”.
Acknowledgments
Funding: This work was supported by grants from the Children's Cancer Research Fund at the University of Minnesota and the National Institutes of Health-National Cancer Institute [CA138437] to W.F.E. and J.R.O. Funding for S.A. was provided by the Doctoral Dissertation Fellowship by the University of Minnesota. We thank the reviewers for their thoughtful comments and suggestions that helped further improve this article.
References
- 1.Altekruse SF, Kosary CL, Krapcho M, et al. National Cancer Institute; Bethesda, MD: 2010. SEER Cancer Statistics Review, 1975-2007. http://seer.cancer.gov/csr/1975_2007/, based on November 2009 SEER data submission, posted to the SEER web site, 2010. [Google Scholar]
- 2.CBTRUS. Central Brain Tumor Registry of the United States. Hinsdale, IL: 2010. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2004-2006. website: www.cbtrus.org. [Google Scholar]
- 3.Cancer Facts & Figures 2010. Atlanta: American Cancer Society; 2010. [Google Scholar]
- 4.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 5.Wen PY, Brandes AA. Treatment of recurrent high-grade gliomas. Curr Opin Neurol. 2009;22:657–664. doi: 10.1097/WCO.0b013e32833229e3. [DOI] [PubMed] [Google Scholar]
- 6.Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berens ME, Giese A. “…those left behind.” Biology and oncology of invasive glioma cells. Neoplasia. 1999;1:208–219. doi: 10.1038/sj.neo.7900034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170:1445–1453. doi: 10.2353/ajpath.2007.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maher EA, Brennan C, Wen PY, et al. Marked genomic differences characterize primary and secondary glioblastoma subtypes and identify two distinct molecular and clinical secondary glioblastoma entities. Cancer Res. 2006;66:11502–11513. doi: 10.1158/0008-5472.CAN-06-2072. [DOI] [PubMed] [Google Scholar]
- 11.Liang Y, Diehn M, Watson N, et al. Gene expression profiling reveals molecularly and clinically distinct subtypes of glioblastoma multiforme. Proc Natl Acad Sci U S A. 2005;102:5814–5819. doi: 10.1073/pnas.0402870102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 14.Kreisl TN. Chemotherapy for malignant gliomas. Semin Radiat Oncol. 2009;19:150–154. doi: 10.1016/j.semradonc.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 15.McGirt MJ, Chaichana KL, Gathinji M, et al. Independent association of extent of resection with survival in patients with malignant brain astrocytoma. J Neurosurg. 2009;110:156–162. doi: 10.3171/2008.4.17536. [DOI] [PubMed] [Google Scholar]
- 16.Lacroix M, Abi-Said D, Fourney DR, et al. A multivariate analysis of 416 patients with glioblastoma multiforme: prognosis, extent of resection, and survival. J Neurosurg. 2001;95:190–198. doi: 10.3171/jns.2001.95.2.0190. [DOI] [PubMed] [Google Scholar]
- 17.Niyazi M, Ganswindt U, Schwarz SB, et al. Irradiation and Bevacizumab in High-Grade Glioma Retreatment Settings. Int J Radiat Oncol Biol Phys. 2010 doi: 10.1016/j.ijrobp.2010.09.002. Oct 27. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 18.Lee CG, Heijn M, di Tomaso E, et al. Anti-Vascular endothelial growth factor treatment augments tumor radiation response under normoxic or hypoxic conditions. Cancer Res. 2000;60:5565–5570. [PubMed] [Google Scholar]
- 19.Libermann TA, Nusbaum HR, Razon N, et al. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature. 1985;313:144–147. doi: 10.1038/313144a0. [DOI] [PubMed] [Google Scholar]
- 20.Rich JN, Bigner DD. Development of novel targeted therapies in the treatment of malignant glioma. Nat Rev Drug Discov. 2004;3:430–446. doi: 10.1038/nrd1380. [DOI] [PubMed] [Google Scholar]
- 21.Lund-Johansen M, Bjerkvig R, Humphrey PA, Bigner SH, Bigner DD, Laerum OD. Effect of epidermal growth factor on glioma cell growth, migration, and invasion in vitro. Cancer Res. 1990;50:6039–6044. [PubMed] [Google Scholar]
- 22.Nister M, Claesson-Welsh L, Eriksson A, Heldin CH, Westermark B. Differential expression of platelet-derived growth factor receptors in human malignant glioma cell lines. J Biol Chem. 1991;266:16755–16763. [PubMed] [Google Scholar]
- 23.Guha A, Dashner K, Black PM, Wagner JA, Stiles CD. Expression of PDGF and PDGF receptors in human astrocytoma operation specimens supports the existence of an autocrine loop. Int J Cancer. 1995;60:168–173. doi: 10.1002/ijc.2910600206. [DOI] [PubMed] [Google Scholar]
- 24.Maher EA, Furnari FB, Bachoo RM, et al. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001;15:1311–1333. doi: 10.1101/gad.891601. [DOI] [PubMed] [Google Scholar]
- 25.Schmidt NO, Westphal M, Hagel C, et al. Levels of vascular endothelial growth factor, hepatocyte growth factor/scatter factor and basic fibroblast growth factor in human gliomas and their relation to angiogenesis. Int J Cancer. 1999;84:10–18. doi: 10.1002/(sici)1097-0215(19990219)84:1<10::aid-ijc3>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 26.Plate KH, Breier G, Weich HA, Risau W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature. 1992;359:845–848. doi: 10.1038/359845a0. [DOI] [PubMed] [Google Scholar]
- 27.Mukasa A, Wykosky J, Ligon KL, Chin L, Cavenee WK, Furnari F. Mutant EGFR is required for maintenance of glioma growth in vivo, and its ablation leads to escape from receptor dependence. Proc Natl Acad Sci U S A. 2010;107:2616–2621. doi: 10.1073/pnas.0914356107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heimberger AB, Learn CA, Archer GE, et al. Brain tumors in mice are susceptible to blockade of epidermal growth factor receptor (EGFR) with the oral, specific, EGFR-tyrosine kinase inhibitor ZD1839 (iressa) Clin Cancer Res. 2002;8:3496–3502. [PubMed] [Google Scholar]
- 29.Ciardiello F, Caputo R, Bianco R, et al. Inhibition of growth factor production and angiogenesis in human cancer cells by ZD1839 (Iressa), a selective epidermal growth factor receptor tyrosine kinase inhibitor. Clin Cancer Res. 2001;7:1459–1465. [PubMed] [Google Scholar]
- 30.Griffero F, Daga A, Marubbi D, et al. Different response of human glioma tumor-initiating cells to epidermal growth factor receptor kinase inhibitors. J Biol Chem. 2009;284:7138–7148. doi: 10.1074/jbc.M807111200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halatsch ME, Gehrke EE, Vougioukas VI, et al. Inverse correlation of epidermal growth factor receptor messenger RNA induction and suppression of anchorage-independent growth by OSI-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in glioblastoma multiforme cell lines. J Neurosurg. 2004;100:523–533. doi: 10.3171/jns.2004.100.3.0523. [DOI] [PubMed] [Google Scholar]
- 32.Preusser M, Gelpi E, Rottenfusser A, et al. Epithelial Growth Factor Receptor Inhibitors for treatment of recurrent or progressive high grade glioma: an exploratory study. J Neurooncol. 2008;89:211–218. doi: 10.1007/s11060-008-9608-3. [DOI] [PubMed] [Google Scholar]
- 33.Franceschi E, Cavallo G, Lonardi S, et al. Gefitinib in patients with progressive high-grade gliomas: a multicentre phase II study by Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO) Br J Cancer. 2007;96:1047–1051. doi: 10.1038/sj.bjc.6603669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rich JN, Reardon DA, Peery T, et al. Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol. 2004;22:133–142. doi: 10.1200/JCO.2004.08.110. [DOI] [PubMed] [Google Scholar]
- 35.Reardon DA, Desjardins A, Vredenburgh JJ, et al. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J Neurooncol. 2009;96:219–230. doi: 10.1007/s11060-009-9950-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prados MD, Chang SM, Butowski N, et al. Phase II study of erlotinib plus temozolomide during and after radiation therapy in patients with newly diagnosed glioblastoma multiforme or gliosarcoma. J Clin Oncol. 2009;27:579–584. doi: 10.1200/JCO.2008.18.9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raizer JJ, Abrey LE, Lassman AB, et al. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro Oncol. 2010;12:95–103. doi: 10.1093/neuonc/nop015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peereboom DM, Shepard DR, Ahluwalia MS, et al. Phase II trial of erlotinib with temozolomide and radiation in patients with newly diagnosed glioblastoma multiforme. J Neurooncol. 2009;98:93–99. doi: 10.1007/s11060-009-0067-2. [DOI] [PubMed] [Google Scholar]
- 39.Buchdunger E, Cioffi CL, Law N, et al. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther. 2000;295:139–145. [PubMed] [Google Scholar]
- 40.Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov. 2002;1:493–502. doi: 10.1038/nrd839. [DOI] [PubMed] [Google Scholar]
- 41.Ranza E, Mazzini G, Facoetti A, Nano R. In-vitro effects of the tyrosine kinase inhibitor imatinib on glioblastoma cell proliferation. J Neurooncol. 2009;96:349–357. doi: 10.1007/s11060-009-9975-4. [DOI] [PubMed] [Google Scholar]
- 42.Geng L, Shinohara ET, Kim D, et al. STI571 (Gleevec) improves tumor growth delay and survival in irradiated mouse models of glioblastoma. Int J Radiat Oncol Biol Phys. 2006;64:263–271. doi: 10.1016/j.ijrobp.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 43.Kilic T, Alberta JA, Zdunek PR, et al. Intracranial inhibition of platelet-derived growth factor-mediated glioblastoma cell growth by an orally active kinase inhibitor of the 2-phenylaminopyrimidine class. Cancer Res. 2000;60:5143–5150. [PubMed] [Google Scholar]
- 44.Wen PY, Yung WK, Lamborn KR, et al. Phase I/II study of imatinib mesylate for recurrent malignant gliomas: North American Brain Tumor Consortium Study 99-08. Clin Cancer Res. 2006;12:4899–4907. doi: 10.1158/1078-0432.CCR-06-0773. [DOI] [PubMed] [Google Scholar]
- 45.Raymond E, Brandes AA, Dittrich C, et al. Phase II study of imatinib in patients with recurrent gliomas of various histologies: a European Organisation for Research and Treatment of Cancer Brain Tumor Group Study. J Clin Oncol. 2008;26:4659–4665. doi: 10.1200/JCO.2008.16.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Premkumar DR, Jane EP, Agostino NR, Scialabba JL, Pollack IF. Dasatinib synergizes with JSI-124 to inhibit growth and migration and induce apoptosis of malignant human glioma cells. J Carcinog. 2010;9:7. doi: 10.4103/1477-3163.65448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamoun WS, Ley CD, Farrar CT, et al. Edema control by cediranib, a vascular endothelial growth factor receptor-targeted kinase inhibitor, prolongs survival despite persistent brain tumor growth in mice. J Clin Oncol. 2009;27:2542–2552. doi: 10.1200/JCO.2008.19.9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Batchelor TT, Sorensen AG, di Tomaso E, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11:83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Batchelor TT, Duda DG, di Tomaso E, et al. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol. 2010;28:2817–2823. doi: 10.1200/JCO.2009.26.3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008;7:3129–3140. doi: 10.1158/1535-7163.MCT-08-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O'Farrell AM, Abrams TJ, Yuen HA, et al. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood. 2003;101:3597–3605. doi: 10.1182/blood-2002-07-2307. [DOI] [PubMed] [Google Scholar]
- 52.de Bouard S, Herlin P, Christensen JG, et al. Antiangiogenic and anti-invasive effects of sunitinib on experimental human glioblastoma. Neuro Oncol. 2007;9:412–423. doi: 10.1215/15228517-2007-024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siegelin MD, Raskett CM, Gilbert CA, Ross AH, Altieri DC. Sorafenib exerts anti-glioma activity in vitro and in vivo. Neurosci Lett. 2010;478:165–170. doi: 10.1016/j.neulet.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rich JN, Sathornsumetee S, Keir ST, et al. ZD6474, a novel tyrosine kinase inhibitor of vascular endothelial growth factor receptor and epidermal growth factor receptor, inhibits tumor growth of multiple nervous system tumors. Clin Cancer Res. 2005;11:8145–8157. doi: 10.1158/1078-0432.CCR-05-0319. [DOI] [PubMed] [Google Scholar]
- 55.Yiin JJ, Hu B, Schornack PA, et al. ZD6474, a multitargeted inhibitor for receptor tyrosine kinases, suppresses growth of gliomas expressing an epidermal growth factor receptor mutant, EGFRvIII, in the brain. Mol Cancer Ther. 2010;9:929–941. doi: 10.1158/1535-7163.MCT-09-0953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu X, Pandolfi PP, Li Y, Koutcher JA, Rosenblum M, Holland EC. mTOR promotes survival and astrocytic characteristics induced by Pten/AKT signaling in glioblastoma. Neoplasia. 2005;7:356–368. doi: 10.1593/neo.04595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kreisl TN, Lassman AB, Mischel PS, et al. A pilot study of everolimus and gefitinib in the treatment of recurrent glioblastoma (GBM) J Neurooncol. 2009;92:99–105. doi: 10.1007/s11060-008-9741-z. [DOI] [PubMed] [Google Scholar]
- 58.Galanis E, Buckner JC, Maurer MJ, et al. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol. 2005;23:5294–5304. doi: 10.1200/JCO.2005.23.622. [DOI] [PubMed] [Google Scholar]
- 59.Lee J, Kotliarova S, Kotliarov Y, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 60.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 61.Pardridge WM. Blood-brain barrier biology and methodology. J Neurovirol. 1999;5:556–569. doi: 10.3109/13550289909021285. [DOI] [PubMed] [Google Scholar]
- 62.Pardridge WM. CNS drug design based on principles of blood-brain barrier transport. J Neurochem. 1998;70:1781–1792. doi: 10.1046/j.1471-4159.1998.70051781.x. [DOI] [PubMed] [Google Scholar]
- 63.Ajay, Bemis GW, Murcko MA. Designing libraries with CNS activity. J Med Chem. 1999;42:4942–4951. doi: 10.1021/jm990017w. [DOI] [PubMed] [Google Scholar]
- 64.Vilar S, Chakrabarti M, Costanzi S. Prediction of passive blood-brain partitioning: straightforward and effective classification models based on in silico derived physicochemical descriptors. J Mol Graph Model. 2010;28:899–903. doi: 10.1016/j.jmgm.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mahar Doan KM, Humphreys JE, Webster LO, et al. Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J Pharmacol Exp Ther. 2002;303:1029–1037. doi: 10.1124/jpet.102.039255. [DOI] [PubMed] [Google Scholar]
- 66.Golden PL, Pollack GM. Blood-brain barrier efflux transport. J Pharm Sci. 2003;92:1739–1753. doi: 10.1002/jps.10424. [DOI] [PubMed] [Google Scholar]
- 67.Kusuhara H, Sugiyama Y. Active efflux across the blood-brain barrier: role of the solute carrier family. NeuroRx. 2005;2:73–85. doi: 10.1602/neurorx.2.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sun H, Dai H, Shaik N, Elmquist WF. Drug efflux transporters in the CNS. Adv Drug Deliv Rev. 2003;55:83–105. doi: 10.1016/s0169-409x(02)00172-2. [DOI] [PubMed] [Google Scholar]
- 69.Nicolazzo JA, Katneni K. Drug transport across the blood-brain barrier and the impact of breast cancer resistance protein (ABCG2) Curr Top Med Chem. 2009;9:130–147. doi: 10.2174/156802609787521580. [DOI] [PubMed] [Google Scholar]
- 70.Borst P, Evers R, Kool M, Wijnholds J. A family of drug transporters: the multidrug resistance-associated proteins. J Natl Cancer Inst. 2000;92:1295–1302. doi: 10.1093/jnci/92.16.1295. [DOI] [PubMed] [Google Scholar]
- 71.Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev. 2003;55:3–29. doi: 10.1016/s0169-409x(02)00169-2. [DOI] [PubMed] [Google Scholar]
- 72.Schinkel AH. P-Glycoprotein, a gatekeeper in the blood-brain barrier. Adv Drug Deliv Rev. 1999;36:179–194. doi: 10.1016/s0169-409x(98)00085-4. [DOI] [PubMed] [Google Scholar]
- 73.Loscher W, Potschka H. Role of drug efflux transporters in the brain for drug disposition and treatment of brain diseases. Prog Neurobiol. 2005;76:22–76. doi: 10.1016/j.pneurobio.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 74.Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455:152–162. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- 75.Ueda K, Cornwell MM, Gottesman MM, et al. The mdr1 gene, responsible for multidrug-resistance, codes for P-glycoprotein. Biochem Biophys Res Commun. 1986;141:956–962. doi: 10.1016/s0006-291x(86)80136-x. [DOI] [PubMed] [Google Scholar]
- 76.Cordon-Cardo C, O'Brien JP, Casals D, et al. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc Natl Acad Sci U S A. 1989;86:695–698. doi: 10.1073/pnas.86.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. Immunohistochemical localization in normal tissues of different epitopes in the multidrug transport protein P170: evidence for localization in brain capillaries and crossreactivity of one antibody with a muscle protein. J Histochem Cytochem. 1989;37:159–164. doi: 10.1177/37.2.2463300. [DOI] [PubMed] [Google Scholar]
- 78.Barrand MA, Robertson KJ, von Weikersthal SF. Comparisons of P-glycoprotein expression in isolated rat brain microvessels and in primary cultures of endothelial cells derived from microvasculature of rat brain, epididymal fat pad and from aorta. FEBS Lett. 1995;374:179–183. doi: 10.1016/0014-5793(95)01104-m. [DOI] [PubMed] [Google Scholar]
- 79.Hegmann EJ, Bauer HC, Kerbel RS. Expression and functional activity of P-glycoprotein in cultured cerebral capillary endothelial cells. Cancer Res. 1992;52:6969–6975. [PubMed] [Google Scholar]
- 80.Tsuji A, Terasaki T, Takabatake Y, et al. P-glycoprotein as the drug efflux pump in primary cultured bovine brain capillary endothelial cells. Life Sci. 1992;51:1427–1437. doi: 10.1016/0024-3205(92)90537-y. [DOI] [PubMed] [Google Scholar]
- 81.Beaulieu E, Demeule M, Ghitescu L, Beliveau R. P-glycoprotein is strongly expressed in the luminal membranes of the endothelium of blood vessels in the brain. Biochem J. 1997;326(Pt 2):539–544. doi: 10.1042/bj3260539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schinkel AH, Smit JJ, van Tellingen O, et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. 1994;77:491–502. doi: 10.1016/0092-8674(94)90212-7. [DOI] [PubMed] [Google Scholar]
- 83.Schinkel AH, Mayer U, Wagenaar E, et al. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc Natl Acad Sci U S A. 1997;94:4028–4033. doi: 10.1073/pnas.94.8.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jonker JW, Freeman J, Bolscher E, et al. Contribution of the ABC transporters Bcrp1 and Mdr1a/1b to the side population phenotype in mammary gland and bone marrow of mice. Stem Cells. 2005;23:1059–1065. doi: 10.1634/stemcells.2005-0150. [DOI] [PubMed] [Google Scholar]
- 85.Mirski SE, Gerlach JH, Cole SP. Multidrug resistance in a human small cell lung cancer cell line selected in adriamycin. Cancer Res. 1987;47:2594–2598. [PubMed] [Google Scholar]
- 86.Cole SP, Bhardwaj G, Gerlach JH, et al. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science. 1992;258:1650–1654. doi: 10.1126/science.1360704. [DOI] [PubMed] [Google Scholar]
- 87.Haimeur A, Conseil G, Deeley RG, Cole SP. The MRP-related and BCRP/ABCG2 multidrug resistance proteins: biology, substrate specificity and regulation. Curr Drug Metab. 2004;5:21–53. doi: 10.2174/1389200043489199. [DOI] [PubMed] [Google Scholar]
- 88.Huai-Yun H, Secrest DT, Mark KS, et al. Expression of multidrug resistance-associated protein (MRP) in brain microvessel endothelial cells. Biochem Biophys Res Commun. 1998;243:816–820. doi: 10.1006/bbrc.1997.8132. [DOI] [PubMed] [Google Scholar]
- 89.Zhang Y, Schuetz JD, Elmquist WF, Miller DW. Plasma membrane localization of multidrug resistance-associated protein homologs in brain capillary endothelial cells. J Pharmacol Exp Ther. 2004;311:449–455. doi: 10.1124/jpet.104.068528. [DOI] [PubMed] [Google Scholar]
- 90.Nies AT, Jedlitschky G, Konig J, et al. Expression and immunolocalization of the multidrug resistance proteins, MRP1-MRP6 (ABCC1-ABCC6), in human brain. Neuroscience. 2004;129:349–360. doi: 10.1016/j.neuroscience.2004.07.051. [DOI] [PubMed] [Google Scholar]
- 91.Dombrowski SM, Desai SY, Marroni M, et al. Overexpression of multiple drug resistance genes in endothelial cells from patients with refractory epilepsy. Epilepsia. 2001;42:1501–1506. doi: 10.1046/j.1528-1157.2001.12301.x. [DOI] [PubMed] [Google Scholar]
- 92.Miller DS, Nobmann SN, Gutmann H, Toeroek M, Drewe J, Fricker G. Xenobiotic transport across isolated brain microvessels studied by confocal microscopy. Mol Pharmacol. 2000;58:1357–1367. doi: 10.1124/mol.58.6.1357. [DOI] [PubMed] [Google Scholar]
- 93.Potschka H, Fedrowitz M, Loscher W. Multidrug resistance protein MRP2 contributes to blood-brain barrier function and restricts antiepileptic drug activity. J Pharmacol Exp Ther. 2003;306:124–131. doi: 10.1124/jpet.103.049858. [DOI] [PubMed] [Google Scholar]
- 94.Potschka H, Loscher W. Multidrug resistance-associated protein is involved in the regulation of extracellular levels of phenytoin in the brain. Neuroreport. 2001;12:2387–2389. doi: 10.1097/00001756-200108080-00020. [DOI] [PubMed] [Google Scholar]
- 95.Potschka H, Fedrowitz M, Loscher W. P-glycoprotein and multidrug resistance-associated protein are involved in the regulation of extracellular levels of the major antiepileptic drug carbamazepine in the brain. Neuroreport. 2001;12:3557–3560. doi: 10.1097/00001756-200111160-00037. [DOI] [PubMed] [Google Scholar]
- 96.Loscher W, Potschka H. Role of multidrug transporters in pharmacoresistance to antiepileptic drugs. J Pharmacol Exp Ther. 2002;301:7–14. doi: 10.1124/jpet.301.1.7. [DOI] [PubMed] [Google Scholar]
- 97.Sun H, Johnson DR, Finch RA, Sartorelli AC, Miller DW, Elmquist WF. Transport of fluorescein in MDCKII-MRP1 transfected cells and mrp1-knockout mice. Biochem Biophys Res Commun. 2001;284:863–869. doi: 10.1006/bbrc.2001.5062. [DOI] [PubMed] [Google Scholar]
- 98.Leggas M, Adachi M, Scheffer GL, et al. Mrp4 confers resistance to topotecan and protects the brain from chemotherapy. Mol Cell Biol. 2004;24:7612–7621. doi: 10.1128/MCB.24.17.7612-7621.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Doyle LA, Yang W, Abruzzo LV, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci U S A. 1998;95:15665–15670. doi: 10.1073/pnas.95.26.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Miyake K, Mickley L, Litman T, et al. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: demonstration of homology to ABC transport genes. Cancer Res. 1999;59:8–13. [PubMed] [Google Scholar]
- 101.Allikmets R, Schriml LM, Hutchinson A, Romano-Spica V, Dean M. A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res. 1998;58:5337–5339. [PubMed] [Google Scholar]
- 102.Robey RW, Polgar O, Deeken J, To KW, Bates SE. ABCG2: determining its relevance in clinical drug resistance. Cancer Metastasis Rev. 2007;26:39–57. doi: 10.1007/s10555-007-9042-6. [DOI] [PubMed] [Google Scholar]
- 103.Cooray HC, Blackmore CG, Maskell L, Barrand MA. Localisation of breast cancer resistance protein in microvessel endothelium of human brain. Neuroreport. 2002;13:2059–2063. doi: 10.1097/00001756-200211150-00014. [DOI] [PubMed] [Google Scholar]
- 104.Hori S, Ohtsuki S, Tachikawa M, et al. Functional expression of rat ABCG2 on the luminal side of brain capillaries and its enhancement by astrocyte-derived soluble factor(s) J Neurochem. 2004;90:526–536. doi: 10.1111/j.1471-4159.2004.02537.x. [DOI] [PubMed] [Google Scholar]
- 105.Cisternino S, Mercier C, Bourasset F, Roux F, Scherrmann JM. Expression, up-regulation, and transport activity of the multidrug-resistance protein Abcg2 at the mouse blood-brain barrier. Cancer Res. 2004;64:3296–3301. doi: 10.1158/0008-5472.can-03-2033. [DOI] [PubMed] [Google Scholar]
- 106.Eisenblatter T, Galla HJ. A new multidrug resistance protein at the blood-brain barrier. Biochem Biophys Res Commun. 2002;293:1273–1278. doi: 10.1016/S0006-291X(02)00376-5. [DOI] [PubMed] [Google Scholar]
- 107.Lee YJ, Kusuhara H, Jonker JW, Schinkel AH, Sugiyama Y. Investigation of efflux transport of dehydroepiandrosterone sulfate and mitoxantrone at the mouse blood-brain barrier: a minor role of breast cancer resistance protein. J Pharmacol Exp Ther. 2005;312:44–52. doi: 10.1124/jpet.104.073320. [DOI] [PubMed] [Google Scholar]
- 108.Zhao R, Raub TJ, Sawada GA, et al. Breast cancer resistance protein interacts with various compounds in vitro, but plays a minor role in substrate efflux at the blood-brain barrier. Drug Metab Dispos. 2009;37:1251–1258. doi: 10.1124/dmd.108.025064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Enokizono J, Kusuhara H, Ose A, Schinkel AH, Sugiyama Y. Quantitative investigation of the role of breast cancer resistance protein (Bcrp/Abcg2) in limiting brain and testis penetration of xenobiotic compounds. Drug Metab Dispos. 2008;36:995–1002. doi: 10.1124/dmd.107.019257. [DOI] [PubMed] [Google Scholar]
- 110.Breedveld P, Pluim D, Cipriani G, et al. The effect of Bcrp1 (Abcg2) on the in vivo pharmacokinetics and brain penetration of imatinib mesylate (Gleevec): implications for the use of breast cancer resistance protein and P-glycoprotein inhibitors to enable the brain penetration of imatinib in patients. Cancer Res. 2005;65:2577–2582. doi: 10.1158/0008-5472.CAN-04-2416. [DOI] [PubMed] [Google Scholar]
- 111.Agarwal S, Sane R, Ohlfest JR, Elmquist WF. Role of Breast Cancer Resistance Protein (ABCG2/BCRP) in the Distribution of Sorafenib to the Brain. J Pharmacol Exp Ther. 2010;336:223–233. doi: 10.1124/jpet.110.175034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.de Vries NA, Zhao J, Kroon E, Buckle T, Beijnen JH, van Tellingen O. P-glycoprotein and breast cancer resistance protein: two dominant transporters working together in limiting the brain penetration of topotecan. Clin Cancer Res. 2007;13:6440–6449. doi: 10.1158/1078-0432.CCR-07-1335. [DOI] [PubMed] [Google Scholar]
- 113.Polli JW, Olson KL, Chism JP, et al. An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethy l]amino}methyl)-2-furyl]-4-quinazolinamine; GW572016) Drug Metab Dispos. 2009;37:439–442. doi: 10.1124/dmd.108.024646. [DOI] [PubMed] [Google Scholar]
- 114.Chen Y, Agarwal S, Shaik NM, Chen C, Yang Z, Elmquist WF. P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. J Pharmacol Exp Ther. 2009;330:956–963. doi: 10.1124/jpet.109.154781. [DOI] [PubMed] [Google Scholar]
- 115.Lagas JS, van Waterschoot RA, van Tilburg VA, et al. Brain accumulation of dasatinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by elacridar treatment. Clin Cancer Res. 2009;15:2344–2351. doi: 10.1158/1078-0432.CCR-08-2253. [DOI] [PubMed] [Google Scholar]
- 116.Agarwal S, Sane R, Gallardo JL, Ohlfest JR, Elmquist WF. Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J Pharmacol Exp Ther. 2010;334:147–155. doi: 10.1124/jpet.110.167601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kodaira H, Kusuhara H, Ushiki J, Fuse E, Sugiyama Y. Kinetic analysis of the cooperation of P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. J Pharmacol Exp Ther. 2010;333:788–796. doi: 10.1124/jpet.109.162321. [DOI] [PubMed] [Google Scholar]
- 118.Lagas JS, van Waterschoot RA, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Breast cancer resistance protein and P-glycoprotein limit sorafenib brain accumulation. Mol Cancer Ther. 2010;9:319–326. doi: 10.1158/1535-7163.MCT-09-0663. [DOI] [PubMed] [Google Scholar]
- 119.Tanaka Y, Slitt AL, Leazer TM, Maher JM, Klaassen CD. Tissue distribution and hormonal regulation of the breast cancer resistance protein (Bcrp/Abcg2) in rats and mice. Biochem Biophys Res Commun. 2005;326:181–187. doi: 10.1016/j.bbrc.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 120.Hofer S, Frei K. Gefitinib concentrations in human glioblastoma tissue. J Neurooncol. 2007;82:175–176. doi: 10.1007/s11060-006-9257-3. [DOI] [PubMed] [Google Scholar]
- 121.Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8:610–622. doi: 10.1038/nrn2175. [DOI] [PubMed] [Google Scholar]
- 122.Gilbertson RJ, Rich JN. Making a tumour's bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer. 2007;7:733–736. doi: 10.1038/nrc2246. [DOI] [PubMed] [Google Scholar]
- 123.Silbergeld DL, Chicoine MR. Isolation and characterization of human malignant glioma cells from histologically normal brain. J Neurosurg. 1997;86:525–531. doi: 10.3171/jns.1997.86.3.0525. [DOI] [PubMed] [Google Scholar]
- 124.Fine RL, Chen J, Balmaceda C, et al. Randomized study of paclitaxel and tamoxifen deposition into human brain tumors: implications for the treatment of metastatic brain tumors. Clin Cancer Res. 2006;12:5770–5776. doi: 10.1158/1078-0432.CCR-05-2356. [DOI] [PubMed] [Google Scholar]
- 125.Pitz MW, Desai A, Grossman SA, Blakeley JO. Tissue concentration of systemically administered antineoplastic agents in human brain tumors. J Neurooncol. 2011:1–10. doi: 10.1007/s11060-011-0564-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fattori S, Becherini F, Cianfriglia M, Parenti G, Romanini A, Castagna M. Human brain tumors: multidrug-resistance P-glycoprotein expression in tumor cells and intratumoral capillary endothelial cells. Virchows Arch. 2007;451:81–87. doi: 10.1007/s00428-007-0401-z. [DOI] [PubMed] [Google Scholar]
- 127.Demeule M, Shedid D, Beaulieu E, et al. Expression of multidrug-resistance P-glycoprotein (MDR1) in human brain tumors. Int J Cancer. 2001;93:62–66. doi: 10.1002/ijc.1306. [DOI] [PubMed] [Google Scholar]
- 128.Nabors MW, Griffin CA, Zehnbauer BA, et al. Multidrug resistance gene (MDR1) expression in human brain tumors. J Neurosurg. 1991;75:941–946. doi: 10.3171/jns.1991.75.6.0941. [DOI] [PubMed] [Google Scholar]
- 129.Spiegl-Kreinecker S, Buchroithner J, Elbling L, et al. Expression and functional activity of the ABC-transporter proteins P-glycoprotein and multidrug-resistance protein 1 in human brain tumor cells and astrocytes. J Neurooncol. 2002;57:27–36. doi: 10.1023/a:1015735815111. [DOI] [PubMed] [Google Scholar]
- 130.Decleves X, Fajac A, Lehmann-Che J, et al. Molecular and functional MDR1-Pgp and MRPs expression in human glioblastoma multiforme cell lines. Int J Cancer. 2002;98:173–180. doi: 10.1002/ijc.10135. [DOI] [PubMed] [Google Scholar]
- 131.Ashmore SM, Thomas DG, Darling JL. Does P-glycoprotein play a role in clinical resistance of malignant astrocytoma? Anticancer Drugs. 1999;10:861–872. doi: 10.1097/00001813-199911000-00001. [DOI] [PubMed] [Google Scholar]
- 132.Doyle LA, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2) Oncogene. 2003;22:7340–7358. doi: 10.1038/sj.onc.1206938. [DOI] [PubMed] [Google Scholar]
- 133.Zhou S, Schuetz JD, Bunting KD, et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001;7:1028–1034. doi: 10.1038/nm0901-1028. [DOI] [PubMed] [Google Scholar]
- 134.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 135.Bleau AM, Hambardzumyan D, Ozawa T, et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4:226–235. doi: 10.1016/j.stem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mohri M, Nitta H, Yamashita J. Expression of multidrug resistance-associated protein (MRP) in human gliomas. J Neurooncol. 2000;49:105–115. doi: 10.1023/a:1026528926482. [DOI] [PubMed] [Google Scholar]
- 137.Bronger H, Konig J, Kopplow K, et al. ABCC drug efflux pumps and organic anion uptake transporters in human gliomas and the blood-tumor barrier. Cancer Res. 2005;65:11419–11428. doi: 10.1158/0008-5472.CAN-05-1271. [DOI] [PubMed] [Google Scholar]
- 138.Calatozzolo C, Gelati M, Ciusani E, et al. Expression of drug resistance proteins Pgp, MRP1, MRP3, MRP5 and GST-pi in human glioma. J Neurooncol. 2005;74:113–121. doi: 10.1007/s11060-004-6152-7. [DOI] [PubMed] [Google Scholar]
- 139.Bahr O, Rieger J, Duffner F, Meyermann R, Weller M, Wick W. P-glycoprotein and multidrug resistance-associated protein mediate specific patterns of multidrug resistance in malignant glioma cell lines, but not in primary glioma cells. Brain Pathol. 2003;13:482–494. doi: 10.1111/j.1750-3639.2003.tb00479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kuan CT, Wakiya K, Herndon JE, 2nd, et al. MRP3: a molecular target for human glioblastoma multiforme immunotherapy. BMC Cancer. 2010;10:468. doi: 10.1186/1471-2407-10-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dai H, Marbach P, Lemaire M, Hayes M, Elmquist WF. Distribution of STI-571 to the brain is limited by P-glycoprotein-mediated efflux. J Pharmacol Exp Ther. 2003;304:1085–1092. doi: 10.1124/jpet.102.045260. [DOI] [PubMed] [Google Scholar]
- 142.Bihorel S, Camenisch G, Lemaire M, Scherrmann JM. Influence of breast cancer resistance protein (Abcg2) and p-glycoprotein (Abcb1a) on the transport of imatinib mesylate (Gleevec) across the mouse blood-brain barrier. J Neurochem. 2007;102:1749–1757. doi: 10.1111/j.1471-4159.2007.04808.x. [DOI] [PubMed] [Google Scholar]
- 143.Leis JF, Stepan DE, Curtin PT, et al. Central nervous system failure in patients with chronic myelogenous leukemia lymphoid blast crisis and Philadelphia chromosome positive acute lymphoblastic leukemia treated with imatinib (STI-571) Leuk Lymphoma. 2004;45:695–698. doi: 10.1080/10428190310001625728. [DOI] [PubMed] [Google Scholar]
- 144.Polli JW, Humphreys JE, Harmon KA, et al. The role of efflux and uptake transporters in [N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethy l]amino}methyl)-2-furyl]-4-quinazolinamine (GW572016, lapatinib) disposition and drug interactions. Drug Metab Dispos. 2008;36:695–701. doi: 10.1124/dmd.107.018374. [DOI] [PubMed] [Google Scholar]
- 145.Yang JJ, Milton MN, Yu S, et al. P-glycoprotein and Breast Cancer Resistance Protein Affect Disposition of Tandutinib, A Tyrosine Kinase Inhibitor. Drug Metab Lett. 2010;4:201–212. doi: 10.2174/187231210792928279. [DOI] [PubMed] [Google Scholar]
- 146.de Vries NA, Buckle T, Zhao J, Beijnen JH, Schellens JH, van Tellingen O. Restricted brain penetration of the tyrosine kinase inhibitor erlotinib due to the drug transporters P-gp and BCRP. Invest New Drugs. 2010:1–7. doi: 10.1007/s10637-010-9569-1. [DOI] [PubMed] [Google Scholar]
- 147.de Lange EC, Danhof M. Considerations in the use of cerebrospinal fluid pharmacokinetics to predict brain target concentrations in the clinical setting: implications of the barriers between blood and brain. Clin Pharmacokinet. 2002;41:691–703. doi: 10.2165/00003088-200241100-00001. [DOI] [PubMed] [Google Scholar]
- 148.Hofer S, Frei K, Rutz HP. Gefitinib accumulation in glioblastoma tissue. Cancer Biol Ther. 2006;5:483–484. doi: 10.4161/cbt.5.5.2653. [DOI] [PubMed] [Google Scholar]
- 149.McKillop D, Partridge EA, Kemp JV, et al. Tumor penetration of gefitinib (Iressa), an epidermal growth factor receptor tyrosine kinase inhibitor. Mol Cancer Ther. 2005;4:641–649. doi: 10.1158/1535-7163.MCT-04-0329. [DOI] [PubMed] [Google Scholar]
- 150.Stewart DJ, Leavens M, Maor M, et al. Human central nervous system distribution of cis-diamminedichloroplatinum and use as a radiosensitizer in malignant brain tumors. Cancer Res. 1982;42:2474–2479. [PubMed] [Google Scholar]
- 151.Stewart DJ, Lu K, Benjamin RS, et al. Concentration of vinblastine in human intracerebral tumor and other tissues. J Neurooncol. 1983;1:139–144. doi: 10.1007/BF00182959. [DOI] [PubMed] [Google Scholar]
- 152.Stewart DJ, Richard MT, Hugenholtz H, et al. Penetration of VP-16 (etoposide) into human intracerebral and extracerebral tumors. J Neurooncol. 1984;2:133–139. doi: 10.1007/BF00177899. [DOI] [PubMed] [Google Scholar]
- 153.Blakeley JO, Olson J, Grossman SA, He X, Weingart J, Supko JG. Effect of blood brain barrier permeability in recurrent high grade gliomas on the intratumoral pharmacokinetics of methotrexate: a microdialysis study. J Neurooncol. 2009;91:51–58. doi: 10.1007/s11060-008-9678-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bell E, Jr, Karnosh LJ. Cerebral hemispherectomy; report of a case 10 years after operation. J Neurosurg. 1949;6:285–293. doi: 10.3171/jns.1949.6.4.0285. [DOI] [PubMed] [Google Scholar]
- 155.Matsukado Y, Maccarty CS, Kernohan JW. The growth of glioblastoma multiforme (astrocytomas, grades 3 and 4) in neurosurgical practice. J Neurosurg. 1961;18:636–644. doi: 10.3171/jns.1961.18.5.0636. [DOI] [PubMed] [Google Scholar]
- 156.Scherer HJ. Structural development in gliomas. Am J Cancer. 1938;34:334–351. [Google Scholar]
- 157.Hesselink JR, Press GA. MR contrast enhancement of intracranial lesions with Gd-DTPA. Radiol Clin North Am. 1988;26:873–887. [PubMed] [Google Scholar]
- 158.Lockman PR, Mittapalli RK, Taskar KS, et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin Cancer Res. 2010;16:5664–5678. doi: 10.1158/1078-0432.CCR-10-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rosso L, Brock CS, Gallo JM, et al. A new model for prediction of drug distribution in tumor and normal tissues: pharmacokinetics of temozolomide in glioma patients. Cancer Res. 2009;69:120–127. doi: 10.1158/0008-5472.CAN-08-2356. [DOI] [PubMed] [Google Scholar]
- 160.Molina JR, Hayashi Y, Stephens C, Georgescu MM. Invasive glioblastoma cells acquire stemness and increased Akt activation. Neoplasia. 2010;12:453–463. doi: 10.1593/neo.10126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 162.Liu G, Yuan X, Zeng Z, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 164.Bleau AM, Huse JT, Holland EC. The ABCG2 resistance network of glioblastoma. Cell Cycle. 2009;8:2936–2944. [PubMed] [Google Scholar]
- 165.Nakai E, Park K, Yawata T, et al. Enhanced MDR1 Expression and Chemoresistance of Cancer Stem Cells Derived from Glioblastoma. Cancer Invest. 2009;27:901–908. doi: 10.3109/07357900801946679. [DOI] [PubMed] [Google Scholar]
- 166.Chu L, Huang Q, Zhai DZ, et al. Expression and significance of ABCG2 in human malignant glioma. Ai Zheng. 2007;26:1090–1094. [PubMed] [Google Scholar]
- 167.Shervington A, Lu C. Expression of multidrug resistance genes in normal and cancer stem cells. Cancer Invest. 2008;26:535–542. doi: 10.1080/07357900801904140. [DOI] [PubMed] [Google Scholar]
- 168.Westphal M, Hilt DC, Bortey E, et al. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro Oncol. 2003;5:79–88. doi: 10.1215/S1522-8517-02-00023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Attenello FJ, Mukherjee D, Datoo G, et al. Use of Gliadel (BCNU) wafer in the surgical treatment of malignant glioma: a 10-year institutional experience. Ann Surg Oncol. 2008;15:2887–2893. doi: 10.1245/s10434-008-0048-2. [DOI] [PubMed] [Google Scholar]
- 170.Bock HC, Puchner MJ, Lohmann F, et al. First-line treatment of malignant glioma with carmustine implants followed by concomitant radiochemotherapy: a multicenter experience. Neurosurg Rev. 2010;33:441–449. doi: 10.1007/s10143-010-0280-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bidros DS, Liu JK, Vogelbaum MA. Future of convection-enhanced delivery in the treatment of brain tumors. Future Oncol. 2009;6:117–125. doi: 10.2217/fon.09.135. [DOI] [PubMed] [Google Scholar]
- 172.Kunwar S, Chang S, Westphal M, et al. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro Oncol. 2010;12:871–881. doi: 10.1093/neuonc/nop054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lidar Z, Mardor Y, Jonas T, et al. Convection-enhanced delivery of paclitaxel for the treatment of recurrent malignant glioma: a phase I/II clinical study. J Neurosurg. 2004;100:472–479. doi: 10.3171/jns.2004.100.3.0472. [DOI] [PubMed] [Google Scholar]
- 174.Kroll RA, Neuwelt EA. Outwitting the blood-brain barrier for therapeutic purposes: osmotic opening and other means. Neurosurgery. 1998;42:1083–1099. doi: 10.1097/00006123-199805000-00082. discussion 1099-1100. [DOI] [PubMed] [Google Scholar]
- 175.Nomura T, Inamura T, Black KL. Intracarotid infusion of bradykinin selectively increases blood-tumor permeability in 9L and C6 brain tumors. Brain Res. 1994;659:62–66. doi: 10.1016/0006-8993(94)90863-x. [DOI] [PubMed] [Google Scholar]
- 176.Hall WA, Doolittle ND, Daman M, et al. Osmotic blood-brain barrier disruption chemotherapy for diffuse pontine gliomas. J Neurooncol. 2006;77:279–284. doi: 10.1007/s11060-005-9038-4. [DOI] [PubMed] [Google Scholar]
- 177.Boockvar JA, Tsiouris AJ, Hofstetter CP, et al. Safety and maximum tolerated dose of superselective intraarterial cerebral infusion of bevacizumab after osmotic blood-brain barrier disruption for recurrent malignant glioma. J Neurosurg. 2010;114:624–632. doi: 10.3171/2010.9.JNS101223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lhomme C, Joly F, Walker JL, et al. Phase III study of valspodar (PSC 833) combined with paclitaxel and carboplatin compared with paclitaxel and carboplatin alone in patients with stage IV or suboptimally debulked stage III epithelial ovarian cancer or primary peritoneal cancer. J Clin Oncol. 2008;26:2674–2682. doi: 10.1200/JCO.2007.14.9807. [DOI] [PubMed] [Google Scholar]
- 179.Carlson RW, O'Neill AM, Goldstein LJ, et al. A pilot phase II trial of valspodar modulation of multidrug resistance to paclitaxel in the treatment of metastatic carcinoma of the breast (E1195): a trial of the Eastern Cooperative Oncology Group. Cancer Invest. 2006;24:677–681. doi: 10.1080/07357900600981349. [DOI] [PubMed] [Google Scholar]
- 180.Ruff P, Vorobiof DA, Jordaan JP, et al. A randomized, placebo-controlled, double-blind phase 2 study of docetaxel compared to docetaxel plus zosuquidar (LY335979) in women with metastatic or locally recurrent breast cancer who have received one prior chemotherapy regimen. Cancer Chemother Pharmacol. 2009;64:763–768. doi: 10.1007/s00280-009-0925-9. [DOI] [PubMed] [Google Scholar]
- 181.Cripe LD, Uno H, Paietta EM, et al. Zosuquidar, a novel modulator of P-glycoprotein, does not improve the outcome of older patients with newly diagnosed acute myeloid leukemia: a randomized, placebo-controlled trial of the Eastern Cooperative Oncology Group 3999. Blood. 2010;116:4077–4085. doi: 10.1182/blood-2010-04-277269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kruijtzer CM, Beijnen JH, Rosing H, et al. Increased oral bioavailability of topotecan in combination with the breast cancer resistance protein and P-glycoprotein inhibitor GF120918. J Clin Oncol. 2002;20:2943–2950. doi: 10.1200/JCO.2002.12.116. [DOI] [PubMed] [Google Scholar]
- 183.Planting AS, Sonneveld P, van der Gaast A, et al. A phase I and pharmacologic study of the MDR converter GF120918 in combination with doxorubicin in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2005;55:91–99. doi: 10.1007/s00280-004-0854-6. [DOI] [PubMed] [Google Scholar]
- 184.Sikic BI. Pharmacologic approaches to reversing multidrug resistance. Semin Hematol. 1997;34:40–47. [PubMed] [Google Scholar]
- 185.Reardon DA, Egorin MJ, Quinn JA, et al. Phase II study of imatinib mesylate plus hydroxyurea in adults with recurrent glioblastoma multiforme. J Clin Oncol. 2005;23:9359–9368. doi: 10.1200/JCO.2005.03.2185. [DOI] [PubMed] [Google Scholar]
- 186.Desjardins A, Quinn JA, Vredenburgh JJ, et al. Phase II study of imatinib mesylate and hydroxyurea for recurrent grade III malignant gliomas. J Neurooncol. 2007;83:53–60. doi: 10.1007/s11060-006-9302-2. [DOI] [PubMed] [Google Scholar]
- 187.Lamar RE, Spigel DR, Burris HA, et al. Phase II trial of radiation therapy/temozolomide followed by temozolomide/sorafenib in the first-line treatment of glioblastoma multiforme (GBM) ASCO Meeting Abstracts. 2009;27:2018. [Google Scholar]
- 188.Grisanti S, Pedersini R, Ferrari VD, et al. Phase II study of sunitinib and irinotecan in patients with recurrent high-grade glioma (HGG) ASCO Meeting Abstracts. 2010;28:e12537. [Google Scholar]
- 189.Sathornsumetee S, Rich JN, Vredenburgh JJ, et al. Phase I trial of imatinib mesylate, hydroxyurea and vatalanib for patients with recurrent glioblastoma multiforme (GBM) 2007 ASCO Annual Meeting Proceedings (Post-Meeting Edition) 2007;25:2027. [Google Scholar]
Future Reading, Resources and Contacts
- 1.Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin. 2010;60:166–193. doi: 10.3322/caac.20069. Reviews the latest developments in therapeutic options for glioma and highlights the current and future direction of clinical trials. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berens ME, Giese A. “…those left behind.” Biology and oncology of invasive glioma cells. Neoplasia. 1999;1(3):208–19. doi: 10.1038/sj.neo.7900034. An excellent report on the invasive nature of glioma and the need to target the invasive glioma cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lagas JS, Vlaming ML, Schinkel AH. Pharmacokinetic assessment of multiple ATP-binding cassette transporters: the power of combination knockout mice. Mol Interv. 2009;9(3):136–45. doi: 10.1124/mi.9.3.7. A review on the role of ABC transporters at the blood-brain barrier and the availability of newer research tools in the form of transgenic mice. [DOI] [PubMed] [Google Scholar]