

Summary
Nucleolar disassembly occurs during mitosis and nucleolar stress, releasing several MDM2-interactive proteins residing in the nucleolus that share the common activity of p53 stabilization. Here, we demonstrated that mobilization of nucleostemin (NS), a cancer and stem cell-enriched nucleolar protein, plays the opposite role by stabilizing MDM2 and suppressing p53 functions. Our results showed that NS increases the protein stability and nucleoplasmic retention of MDM2, and competes with L23 for MDM2 binding. These activities are significantly elevated when NS is released into the nucleoplasm by mutations that abolish its nucleolar localization or by chemotherapeutic agents that disassemble the nucleoli. NS depletion decreases MDM2 protein, increases the transcriptional activities without changing the protein level of p53, and triggers G2/M arrest and cell death in U2OS but not in H1299 cells. This work reveals that nucleoplasmic relocation of NS during nucleolar disassembly safeguards the G2/M transit and survival of continuously dividing cells by MDM2 stabilization and p53 inhibition.
Keywords: nucleostemin, MDM2, cancer, ubiquitylation, p53
Introduction
Multiple stress signals activate p53 and trigger cell-cycle arrest, apoptosis, and DNA repair mechanisms. Although the p53’s function is essential for safeguarding genome integrity and preventing tumor formation, it needs to be harnessed in continuously dividing cells to avoid premature cell-cycle exit or death. A primary regulator of p53 is MDM2 (mouse double minute 2), which suppresses the activity of p53 by working as an E3 ubiquitin ligase to promote its protein degradation (Haupt et al., 1997; Kubbutat et al., 1997) or by binding to the N-terminal domain of p53 to inhibit its transcriptional activity (Momand et al., 1992; Oliner et al., 1993).
Nucleostemin (NS) was isolated as a gene enriched in neural stem cells (NSCs) but not in their differentiated progeny (Tsai and McKay, 2002). It encodes a nucleolar GTP-binding protein abundantly expressed by cancer and stem cells, and is required for maintaining the proliferation of embryonic NSCs and human cancer cells in vitro, as well as for early embryogenesis (Beekman et al., 2006; Zhu et al., 2006). The mechanism underlying the NS activity is not completely understood, but is indicated by its ability to bind and regulate p53 (Tsai and McKay, 2002) and telomeric repeat-binding factor 1 (Zhu et al., 2006). The molecular basis of a nucleolar-related p53 regulation began to emerge when several nucleolar proteins were shown to exhibit the ability to bind MDM2 and stabilize p53. ARF (alternative reading frame), PML (promyelocytic leukemic protein), B23, L5, L11, and L23 all enhance p53 stability by inhibiting or sequestering MDM2 in the nucleolus (Bernardi et al., 2004; Dai et al., 2004; Jin et al., 2004; Kurki et al., 2004; Tao and Levine, 1999; Zhang et al., 2003).
A number of studies investigated the relationship between NS and p53, and showed that knocking down the expression of NS increased the level of p53 (Ma and Pederson, 2007) and that the early embryonic lethal phenotype of NS-null mice cannot be rescued by p53 deletion (Beekman et al., 2006). Questions remain regarding how nucleolar NS and nucleoplasmic p53 come in contact with each other and what the molecular connection between these two proteins in tumor cells is. The association of NS and p53 in living cells can be envisioned in several ways. First, NS shuttles between the nucleolus and nucleoplasm on a GTP-driven cycle, thus allowing individual NS protein the opportunity to interact with proteins residing in the nucleoplasm (Tsai and McKay, 2005). p53 has also been found in the active site of transcription within the nucleolus (Rubbi and Milner, 2000). In addition, NS can be relocated to the nucleoplasm upon nucleolar disassembly during mitosis or induced by drugs that block the RNA polymerase activity or de novo GTP synthesis. Finally, the interaction between NS and p53 may be mediated by other yet unidentified proteins.
While investigating the role of NS in p53 regulation, we discovered that the association between NS and p53 is mediated by MDM2, and began to explore the mechanistic and biological relevance of the NS-MDM2 interaction. At the completion of this work, another study came out that reported the same interaction between NS and MDM2 (Dai et al., 2008), but showed that both overexpression and knockdown of NS lead to the same phenotypes of p53 activation, MDM2 upregulation, and G1/S cell-cycle arrest, and that these findings depended on the L5 and/or L11 interaction with MDM2. In this study, we showed that the NS-MDM2 interaction occurs mainly when nucleolar NS is mobilized into the nucleoplasm in living cells. Nucleoplasmic relocation of NS increases its MDM2-binding and the nucleoplasmic retention of MDM2. Contrary to the effect of other MDM2-interactive nucleolar proteins, NS exhibits the unique ability to stabilize MDM2 by preventing its ubiquitylation, compete with L23 for MDM2 binding, and lower the transcriptional activity of p53. Further analyses reveal a role of NS in promoting the G2/M transit and cell survival in U2OS cells.
Results
MDM2 binds NS independently of p53, and mediates association of NS and p53
To define the interaction between NS, MDM2, and p53, HEK293 cells were triple-transfected with HA-tagged NS, FLAG-tagged MDM2, and/or Myc-tagged p53 expression plasmids, and immunoprecipitated with anti-tag antibodies. While all three proteins showed up in the same protein complexes in the triple-transfected cells (Fig. 1A1), the binding between NS and p53 in the double-transfected cells was significantly reduced (Fig. 1A2). By contrast, the NS-MDM2 and MDM2-p53 interactions were unaffected by the coexpression of p53 or NS, respectively (Fig. S1A and Fig. 1A1). We confirmed the in vivo binding of NS and MDM2 by showing that the endogenous NS and MDM2 coexisted in the same protein complexes in U2OS cells (Fig. 1B). These results demonstrate that MDM2 mediates part of the binding between NS and p53.
Figure 1.
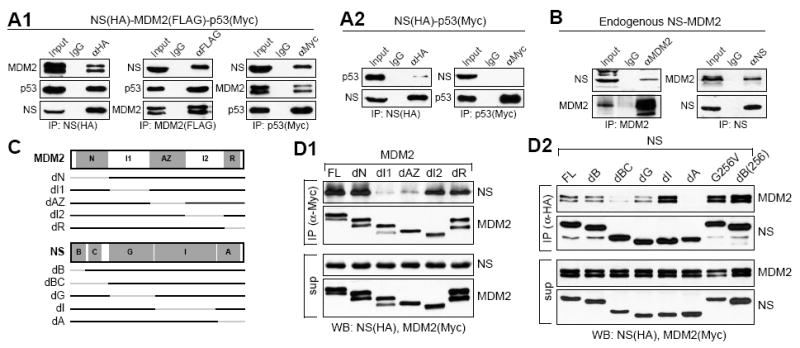
MDM2 mediates association between nucleostemin (NS) and p53 via the central domain of MDM2 and the coiled-coil and acidic domains of NS.
(A1) Triple-coIP assays of HA-tagged NS, FLAG-tagged MDM2, and Myc-tagged p53 showed that NS, MDM2, and p53 coexisted in the same protein complex. (A2) Binding between NS and p53 was significantly reduced without MDM2 coexpression. (B) In vivo binding of endogenous NS and MDM2 was confirmed by coIP assays, which immunoprecipitated MDM2 or NS complexes from U2OS cells. (C) Diagram shows MDM2 (top) and NS (bottom) deletion mutants, with grey lines indicating the deleted regions. Abbreviations: N, p53-binding; I1 and I2, intermediate-1 and -2; AZ, acidic/zinc finger; R, RING-finger; B, basic; C, coiled-coil; G, GTP-binding; I, intermediate; and A, acidic domains. CoIP assays showed that NS failed to bind MDM2 mutants deleted of the I1- or AZ-domain (D1), and that deleting the A-domain or the BC-domain of NS abolished its ability to bind MDM2 (D2). Sup, supernatant.
Binding of MDM2 and NS requires the central domain of MDM2 and the coiled-coil and acidic domains of NS
To map the NS-binding domains of MDM2, non-overlapping deletions were made on MDM2 that correspond to its p53-binding (N, a.a. 1-108), intermediate-1 (I1, a.a. 109-222), acidic-zinc finger (a.a. 223-322), intermediate-2 (I2, a.a. 323-434), and RING-finger domains (R, a.a. 435-491) (Fig. 1C, top). CoIP assays of Myc-tagged MDM2 mutants and HA-tagged NS showed that deleting the I1-domain (dI1) or the AZ-domain (dAZ) of MDM2 reduced its ability to bind NS (Fig. 1D1). To define the MDM2-interactive domain of NS, NS mutants deleted of the basic (B, a.a. 1-46), basic-coiled-coil (BC, a.a. 1-115), GTP-binding (G, a.a. 116-283), intermediate (I, a.a. 284-464), or acidic (A, a.a. 465-549) domain, as well as a single-residue mutant (G256V) lacking the GTP-binding and nucleolus-targeting capabilities, were generated (Fig. 1C, bottom). CoIP assays of Myc-tagged MDM2 and HA-tagged NS mutants by anti-HA (Fig. 1D2) or anti-Myc antibody (Fig. S1B) both demonstrated that deleting either the BC domain or the A-domain of NS reduced its ability to bind MDM2, while the B-domain alone deletion (dB) did not. These findings indicate that the NS-MDM2 binding requires the central region (a.a. 109-322) of MDM2 and the C-domain (a.a. 47-115) and the A-domain of NS.
NS binds MDM2 in the nucleoplasm and increases the nucleoplasmic retention of MDM2
Next, we used the BiFC (bimolecular fluorescence complementation) assay to show the actual complexing of NS and MDM2 in living cells. BiFC involves coexpression of two potentially interacting proteins fused individually to the N-terminal (VN173, Yn) or the C-terminal domain (VC155, Yc) of the Venus variant of yellow fluorescent protein (YFP), and measures the reconstitution of a functional YFP complex when the interactive protein pairs bring the Yn and Yc fragments into close proximity (Fig. 2A1). In our experiments, HeLa cells were cotransfected with plasmids encoding the Yn- and Yc-fused proteins and a nucleolar localization signal (NoLS)-tagged cyan fluorescent protein (noCFP). The BiFC efficiencies were measured by counting the percentages of YFP+ cells in the CFP+ population by fluorescence-activated cell sorting (FACS) analyses. While an 48.7% BiFC efficiency was observed between wild-type MDM2 and NS, the NS mutant lacking the BC- and A-domains (NS-GI) displayed only a 27.2% BiFC efficiency with the wild-type MDM2. The BiFC efficiencies between the wild-type NS and the MDM2 mutants lacking the AZ-domain (dAZ) or the I1- and AZ-domains (dIAZ) were reduced to 15% and 9.4%, respectively (Fig. 2A2). Western blots showed that the expression levels of wild-type and mutant FLAG-tagged MDM2-Yn (or Myc-tagged NS-Yc) were the same (Fig. S2A), excluding the possibility that the observed findings were caused by different expression levels of the fusion proteins.
Figure 2.
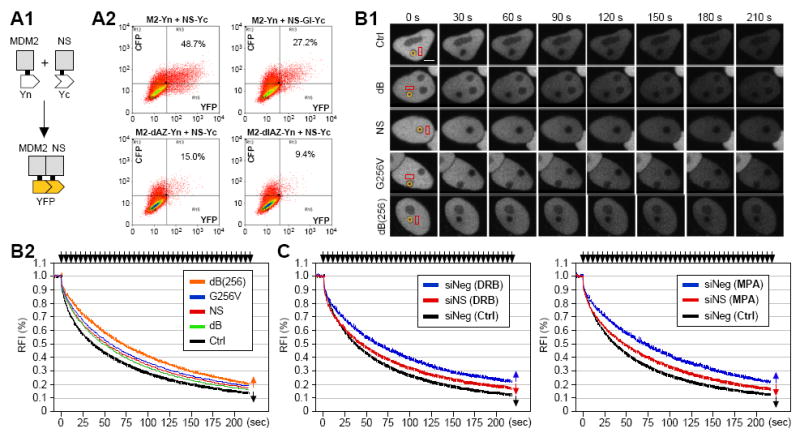
NS binds and retains MDM2 in the nucleoplasm of living cells under nucleolar stress.
(A1) In vivo interaction between NS and MDM2 was shown by the bimolecular fluorescence complementation (BiFC) approach. The FLAG-tagged N-terminal (Yn) and Myc-tagged C-terminal (Yc) fragments of Venus YFP were fused to MDM2 and NS, respectively. (A2) Yn-fused MDM2 (wild-type or mutant) and Yc-fused NS (wild-type or mutant) were coexpressed in HeLa cells with a nucleolar CFP (noCFP) marker. The percentages of YFP+ cells in the CFP+ population were measured by FACS and indicated in the histogram. NS-GI-Yc was deleted of the BC and A domains of NS. M2-dAZ-Yn and M2-dIAZ-Yn were deleted of the AZ domain and the I1 and AZ domains of MDM2, respectively. (B1) The nucleoplasmic retention time of MDM2 was measured by FLIP in HeLa cells, in which the nucleolus is bleached and the nucleoplasmic intensity is was measured. Time-sequenced images with labels indicating the bleached areas in the nucleolus (yellow circles), the measured areas in the nucleoplasm (red rectangles), and intervals between image acquisition and the first bleaching pulse (in seconds) were shown. Scale bar, 5um. (B2) The average FLIP rates of MDM2 were calculated from 20 cells of 2-3 independent experiments. Coexpression of wild-type or nucleoplasmic mutants of NS increased the nucleoplasmic retention time of MDM2 (p < 0.0001, by Repeated Measures ANOVA). Error bars represent standard error mean (s.e.m.) and are shown on one side (indicated by arrows) of the control and dB(256) curves. Y-axis represents the relative fluorescence index (RFI), and top arrows indicate bleaching pulses. (C) The role of endogenous NS in regulating the dynamic distribution of MDM2 is revealed by doxorubicin (ADR) and mycophenolic (MPA) treatments, which mobilize NS from the nucleolus to the nucleoplasm. When exposed to ADR (2uM, 4h) (left panel) or MPA (40uM, 4h) (right panel), the retention time of MDM2 in the nucleoplasm was increased (blue) compared to the mock-treated cells (Ctrl, black, p < 0.0001). Knocking down the endogenous expression of NS (siNS) was able to reverse a significant portion of the drug-induced retention of MDM2 in the nucleoplasm (red, p < 0.0001).
Because the BiFC binding is irreversible, we applied the FLIP (fluorescence loss in photobleaching) approach to determine the dynamic interaction between NS and MDM2 in living cells. The FLIP paradigm was set up where the rate of fluorescence loss in the nucleoplasm was measured while bleaching one nucleolus with repetitive bleaching pulses. The validity of using the C-terminally GFP-fused MDM2 to track the distribution of endogenous MDM2 protein was verified by the results showing that the C-terminally GFP-fused MDM2 was able to reduce p53 protein as did the wild-type MDM2 (Fig. S2B), and that its dynamic property is the same as that of the N-terminally GFP-fused MDM2 (Fig. S2C, p = 0.95, Repeated Measures ANOVA). FLIP analyses demonstrated that coexpression of wild-type NS (mean decay half-time (T1/2) = 51.5 seconds), dB (T1/2 = 47.4s), G256V (T1/2 = 56.0s), or dB(256) mutant (T1/2 = 69.5s) all increased the nucleoplasmic retention time of MDM2 compared to the control-transfected cells (T1/2 = 37.0s) (Fig. 2B, p < 0.0001 for all). Among them, the dB(256) mutant had the most ability to retain MDM2 in the nucleoplasm (p < 0.0001 when compared with wild-type NS and dB; p < 0.01 when compared with G256V).
To demonstrate that this MDM2-retaining effect by overexpressing wild-type and mutant NS proteins can also be seen with the native NS protein, we used doxorubicin (ADR, 2uM for 4 h) and mycophenolic acid (MPA, 40uM for 4 h) to mobilize the endogenous NS from the nucleolus to the nucleoplasm. The activities of ADR and MPA are to trigger nucleolar stress by inactivating the transcriptional activity and to block de novo GTP synthesis, respectively. Our FLIP results showed that when cells were exposed to ADR or MPA, their nucleoplasmic retention time of MDM2 was significantly prolonged (blue traces; T1/2 = 59.4s and 68.0s for ADR- and MPA-treated cells, respectively) compared to that of mock-treated samples (black traces; T1/2 = 37.2s) (p < 0.0001 for both drugs) (Fig. 2C, left panel for ADR treatment and right panel for MPA treatment). To determine how much of this drug-induced increase of MDM2 nucleoplasmic retention is mediated by NS translocation, we compared the drug effects between control (siScr, blue traces) and NS-knockdown (siNS, red traces) cells. Our results showed that knocking down the endogenous NS reverses a major portion of this drug-induced MDM2 retention (T1/2 = 44.7s and 47.7s for ADR and MPA-treated cells, respectively) (p < 0.0001 for both). By contrast, NS knockdown in mock-treated cells did not affect the nucleoplasmic retention time of MDM2 significantly (Fig. S2D, p = 0.97). These results demonstrate that the interaction between NS and MDM2 occurs when the endogenous NS is released from the nucleolus to the nucleoplasm, and that NS binding increases the nucleoplasmic residence of MDM2.
NS increases MDM2 protein by decreasing its degradation and ubiquitylation
To address the functional importance of NS-MDM2 binding, we first asked how does NS affect the protein level of MDM2. H1299 cells were cotransfected with the same amount of MDM2 and increasing amounts of NS-expression plasmids. MDM2 protein levels were compared between different samples after normalization to a coexpressed GFP control. Western blots showed that MDM2 protein was increased by coexpression of wild-type NS in a dose-dependent manner (Fig. 3A). The ability to increase MDM2 protein was abolished by deleting the MDM2-binding domains of NS (Fig. S3A) but preserved in the nucleoplasmic mutants (Fig. S3B). To confirm that the MDM2 protein level is also regulated by the endogenous NS, two micro-RNA-adapted short hairpin RNA constructs (shRNAmir) were created that exhibited a 54% (shNS-1) and 84% (shNS-2) knockdown efficiency of NS protein (Fig. 3B, bottom panel). Compared to the sample treated with a scrambled shRNAmir construct (shScr), cells transfected with the shNS-1 or shNS-2 construct had reduced amounts of exogenous MDM2 protein at levels comparable to their NS knockdown efficiencies (Fig. 3B, top panel). NS depletion also showed the same effect on the endogenous MDM2 (Fig. 5A). Since the expressions of exogenous MDM2 and GFP were both driven by the same EF1α promoter, we reasoned that the NS effect on MDM2 protein must occur post-transcriptionally, and tested this idea by measuring the protein stability of MDM2 in the NS-perturbed cells. For overexpression experiments, H1299 cells were transfected with MDM2 and with or without NS expression plasmid. Thirty-six hours (h) after transfection, cells were treated with cycloheximide (CHX, 100ug/ml), and lysates were collected at 0.5h-to-1h intervals. Western analyses showed that MDM2 proteins in the NS-overexpression cells were degraded much slower than that in the control cells (Fig. 3C, p < 0.0001 by Repeated Measures ANOVA). To confirm these findings by the knockdown approach, the protein stability of MDM2 was measured in the shNS-2 and shScr-transfected cells, and shown to be decreased by NS depletion (Fig. 3D, p = 0.04).
Figure 3.
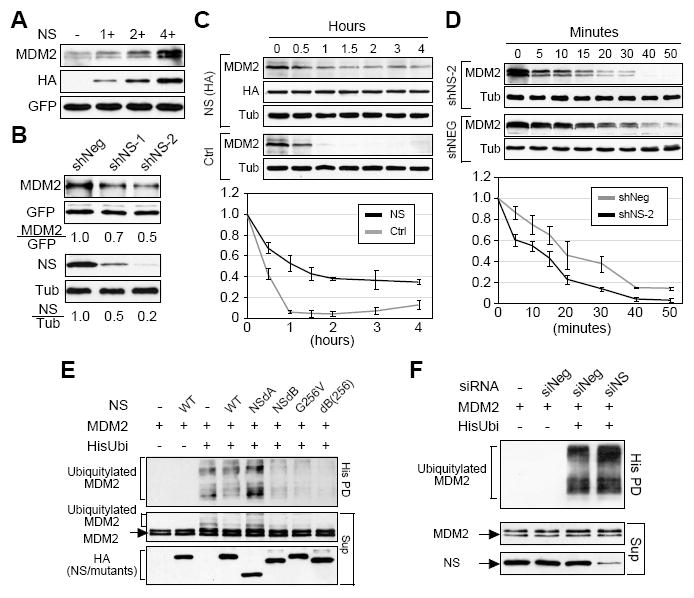
NS increases the protein level of MDM2 by preventing its degradation and ubiquitylation.
(A) Coexpression of NS (HA-tagged) increased the protein levels of exogenously expressed MDM2 in a dose-dependent manner. (B) Knocking down the expression of NS by the NS-targeting shRNAmir constructs (shNS-1 and shNS-2) decreased the amount of MDM2 protein. (C) MDM2 protein stability, with or without NS overexpression (NS v.s. Ctrl), was measured in H1299 cells. After cycloheximide treatment, cell lysates were collected from 0 to 4 hours (h) at 0.5-1h intervals. The MDM2 protein amounts at every time point were measured from three independent experiments, adjusted based on their α-tubulin amounts, and expressed as percentages of the MDM2 protein amount at the 0h time-point. (D) NS depletion by shNS-2 cotransfection increased the protein degradation of MDM2. Protein degradation assays were performed over a 50-minute (m) window. (E) HEK293 cells were transfected with (His)6-tagged ubiquitin, MDM2, and/or NS (wild-type or mutant) plasmids as specified. Ubiquitylated MDM2 products were pulled down by Ni2+ sepharose (His PD) and detected by anti-MDM2 (SMP14) antibody. Overexpression of wild-type NS slightly decreased the polyubiquitylation of MDM2 compared to the control sample. Overexpression of the nucleoplasmic mutants of NS (dB, G256V, and dB(256)) significantly decreased the ubiquitylated products of MDM2. Sup, supernatant. (F) Conversely, depleting the endogenous NS by the NS-targeting siRNA (siNS) increased the ubiquitylation of MDM2 compared to cells treated with a scrambled siRNA (siScr).
Figure 5.
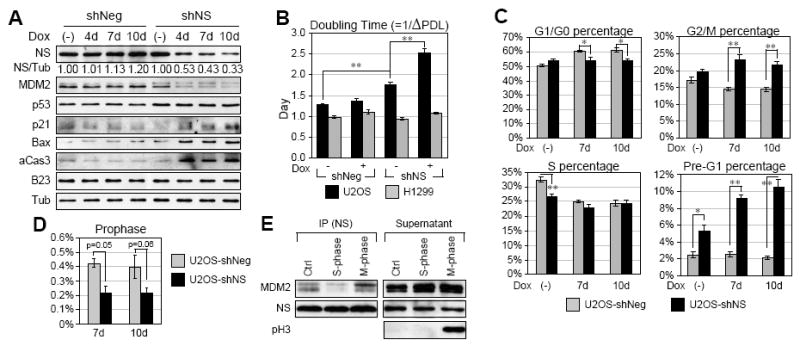
NS depletion triggers G2/M arrest and apoptosis.
Doxycycline (Dox)-inducible NS-knockdown (shNS) U2OS and H1299 cells and their respective controls (shScr) were created (refer to Fig. S4). (A) U2OS-shNS cells displayed a 47%, 57%, and 67% loss of NS protein after 4d, 7d, and 10d of Dox (20ug/ml) treatment, respectively. NS depletion reduced MDM2 protein without changing the protein level of p53. The p53 transcriptional activity, as determined by the protein levels of two of its transcriptional targets, p21 and Bax, and cleaved caspase-3 (aCas3) were increased. Tub, α-tubulin. (B) Population doubling levels (PDL) and time ((=1/ΔPDL in days) were measured over a six-day period. The doubling time of Dox-treated U2OS-shNS cells was significantly prolonged compared to the shScr cells and the untreated shNS cells (black bars). The doubling time of H1299 cells was unchanged by NS knockdown (grey bars). (C) Cell-cycle analyses showed decreased G1/G0 and increased G2/M and sub-G1 cell percentages in the NS-knockdown U2OS cells. (D) The percentages of anti-phospho-Histone3 (pH3)-labeled prophase cells were reduced by NS knockdown. (E) Binding between the endogenous NS and MDM2 increased in the M-phase-synchronized cells.
MDM2 protein is degraded by the ubiquitin-proteasome-mediated mechanism. To determine how NS influences the ubiquitylation of MDM2, in vivo ubiquitylation assays were conducted, in which HEK293 cells were transfected with (His)6-tagged ubiquitin, MDM2, and NS (wild-type or mutant) expression plasmids. Ubiquitylated proteins were captured from protein extracts by Ni2+-chelating sepharose. Anti-MDM2 western blots showed that overexpression of wild-type NS, but not the non-MDM2-binding dA mutant, slightly and consistently reduced the amount of ubiquitylated MDM2 in the pulldown fraction (Fig. 3E). Notably, this activity of NS was significantly enhanced in the nucleoplasmic mutants of NS, i.e. NSdB, G256V, and dB(256). Confirming these findings, knocking down the endogenous NS by a NS-targeting siRNA duplex (siNS) (Tsai and McKay, 2002) increased the ubiquitylation of MDM2 (Fig. 3F). These data demonstrate that NS stabilizes MDM2 protein by reducing its ubiquitylation, and such activity is more evident with the nucleoplasmic mutants of NS than with the wild-type NS.
Nucleoplasmic NS competes against ribosomal protein L23 for MDM2 binding
The NS-interactive domain of MDM2 overlaps with its binding sites for L5, L11, and L23. Here, we used L23 as an example to determine the MDM2-binding relationship between NS and this group of proteins. HEK293 cells were triple-transfected with HA-tagged NS, Myc-tagged MDM2, and FLAG-tagged L23 plasmids. Protein complexes were immunoprecipitated by anti-tag antibodies. Compared to the samples expressing only NS and MDM2 (Fig. S1A1), the interaction between NS and MDM2 was significantly reduced when L23 was coexpressed (Fig. 4A1). Binding between MDM2 and L23 did not require coexpression of wild-type NS (Fig. 4A2), and no direct interaction was detected between NS and L23 (Fig. 4A3), indicating that in the normal growing cells, more MDM2 proteins were bound by L23 than by NS. Because most NS proteins are localized in the nucleolus in the interphase cells and only binds MDM2 when it is translocated into the nucleoplasm, we next examined the abilities of the three nucleoplasmic mutants of NS (dB, G256V, and dB(256)) to compete with L23 for MDM2 binding. Triple-coIP experiments revealed that dB(256) has the strongest activity to bind MDM2 in the presence of L23 (Fig. 4B), consistent with its stronger ability to change the nucleoplasmic retention and ubiquitylation of MDM2 than that of wild-type NS, dB, and G256V. To determine if dB(256) can compete against L23 for MDM2 binding, MDM2 protein complexes were immunoprecipitated by anti-MDM2 antibody from cells expressing the same amount of L23 but different levels of dB(256). The coIP results showed that increased binding of dB(256) to MDM2 reduced the amount of L23 bound by MDM2 (Fig. 4C).
Figure 4.
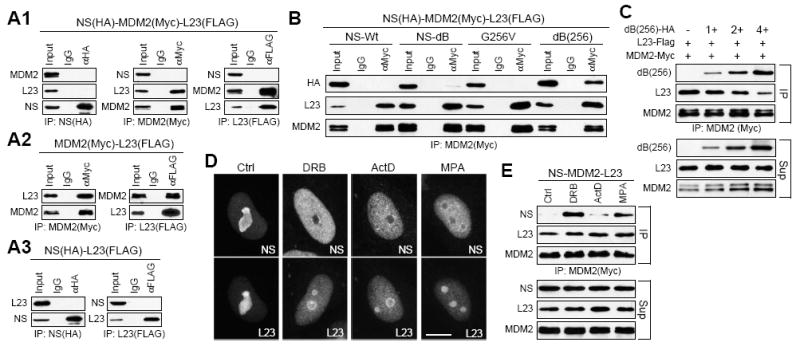
Nucleoplasmic NS competes against L23 for MDM2 binding.
(A1) Triple coIP of NS (HA), MDM2 (Myc), and L23 (FLAG) by the indicated antibodies (IP) showed that L23 coexpression reduces the interaction between NS and MDM2. Double coIP showed that L23 interacts with MDM2 (A2) but not with NS (A3). (B) The ability of NS to bind MDM2 in the presence of L23 overexpression was significantly increased by the combined mutations of dB and G256V (dB(256)). (C) The dB(256) mutant was capable of competing against L23 for MDM2 binding in the triple-coIP experiments. The MDM2 protein complexes (top panel) were immunoprecipitated from lysates (bottom panel) containing the same amount of L23 and increasing amounts of dB(256). (D) Both NS (HA) and L23 (FLAG) reside primarily in the nucleolus under normal growth conditions (Ctrl). When exposed to ADR (2uM), actinomycin-D (ActD, 0.05ug/ml) and MPA (40uM) for 4h, L23 was redistributed to the nucleoplasm as NS but to a less extent. Distributions of NS and L23 were shown in different ADR-treated cells due to the autofluorescent property of ADR. Scale bar shows 10um. (E) Compared to mock-treated cells, ADR and MPA significantly increased the binding between NS and MDM2, and ActD had a smaller effect. (F) Proteins were extracted from cells coexpressing HA-tagged NS, Myc-tagged MDM2, and FLAG-tagged L5, L11, or L23, and immunoprecipitated by anti-Myc antibody. Western blots showed that L23 competes against NS for MDM2 binding better than L5 and L11 do.
To confirm that the increased MDM2 binding by dB(256) can also be seen with the native NS protein, we used ADR (2uM, 4h), actinomycin-D (ActD, 0.05ug/ml, 4h) and MPA (40uM, 4h) to mobilize the endogenous NS from the nucleolus to the nucleoplasm based on the previously described rationale, and measured the coIP efficiency between NS and MDM2. Cofocal analyses showed that these drugs trigger nucleoplasmic translocation of both NS and L23. Notably, the nucleoplasmic relocation of NS was more sensitive to these treatments than that of L23 (Fig. 4D). To test the drug effects on NS-MDM2 binding, coIP assays were performed in cells triple-transfected with MDM2, NS, and L23 plasmids, treated with ADR, ActD, or MPA, and immunoprecipitated by anti-Myc antibody for MDM2. Western blots showed that the coIP efficiency between NS and MDM2 was increased by these drugs even in the presence of L23, and that this effect was more significant in the ADR and MPA-treated samples than in the ActD-treated sample (Fig. 4E). To determine if L5 and L11 show the same activity as L23 in competing with NS for MDM2 binding, triple-coIP experiments were performed and showed that L23 competes against NS for MDM2 binding better than L5 and L11 do (Fig. 4F). This result indicates that the relationships between NS, MDM2 and these three ribosomal proteins are not exactly the same.
NS depletion reduces MDM2 protein and increases p53 transcriptional activity without changing the p53 protein level
To address how NS may affect the protein level and transcriptional activity of p53, U2OS and H1299 stable cell lines with doxycycline (Dox)-inducible NS knockdown capabilities were established (Fig. S4A), both of which displayed comparable knockdown efficiencies of NS proteins after Dox treatment (Fig. 5A, S4B). Cell lysates were collected from control (shScr) and NS-knockdown (shNS) U2OS cells, receiving no treatment or Dox (20ug/ml) treatment for 4d, 7d, and 10d. Compared to the non-treated cells, the Dox-treated U2OS-shNS cells showed a time-dependent reduction of NS protein along with a decrease in MDM2 protein, whereas the U2OS-shScr cells did not (Fig. 5A). Although the p53 protein level was unchanged, its transcriptional activity, as assessed by two of its transcriptional targets (p21 and Bax), was upregulated. The increase in Bax expression was paralleled by elevated protein levels of cleaved caspase-3, the convergent point of both the intrinsic and extrinsic cell death pathways. These results demonstrate that NS depletion decreases MDM2 protein and enhances the transcriptional activity without changing the protein level of p53.
NS promotes cell survival and G2/M transit during nucleolar stress
To determine the biological functions of NS, we measured the cell proliferation rates of NS-depleted U2OS (p53-wild-type) and H1299 (p53-null) cells. The population doubling levels (PDLs) were measured daily over a 6-day period by the formula: ΔPDL = log(nf/n0)/log2, where n0 is the initial number of cells and nf is the final number of cells. The time (in days) for one population doubling was calculated as 1/ΔPDL. Our results showed that NS-depleted U2OS cells had a longer doubling time compared to the shScr cells and the non-treated shNS cells (Fig. 5B, black bars). The doubling time of non-treated U2OS-shNS cells was slightly longer than that of U2OS-shScr cells, indicating a possible leakage expression of the shNS-2 construct before Dox treatment. In contrast to the inhibitory effect of NS knockdown on the proliferation of U2OS cells, NS depletion did not slow down the proliferation rate of H1299 cells (Fig. 5B, grey bars), suggesting that the NS’s ability to promote cell proliferation may be partially mediated by a p53-dependent mechanism in human cancer cells.
A reduced population-doubling rate can be caused by an increase in cell death, cell-cycle arrest, or cell-cycle length elongation. These possibilities were addressed by propidium iodide (PI)-labeled cell-cycle analyses of control (shScr) and NS-knockdown (shNS) U2OS cells (Fig. 5C). Before Dox treatment, the S-phase cell percentage of the U2OS-shNS culture was less and its sub-G1-cell percentage was more than that of the U2OS-shScr culture, consistent with a low-level expression of shNS-2 before Dox induction. After Dox treatment for 7d or 10d, the NS-knockdown cells displayed lower G1/G0 cell percentages (p < 0.01) and higher G2/M cell percentages (p < 0.001) compared to the time-matched shScr cells, indicating cell-cycle arrest at the G2/M stage. Most significantly, NS depletion increased the percentage of sub-G1 (apoptotic) cells (p < 0.001). To determine if the G2/M arrest occurs before, during, or after mitosis, prophase cells with condensed chromatin and anti-phospho-Histone H3 (pH3) labeling were measured in the 7d- and 10d-Dox-treated shScr and shNS cultures (Fig. 5D). We found that the NS-knockdown culture contained more G2/M-phase cells but less prophase cells than did the control culture, suggesting that the G2/M arrest occurs before the mitotic entry. These results pointed out that NS depletion blocks mitotic entry and triggers apoptosis, which were not seen in the NS-knockdown H1299 cells (Fig. S4C).
One notable change in the nucleoli during cell-cycle progression is that they dissemble during the prophase and reform at the late stage of mitosis, suggesting a possibility that, in normal dividing cells, NS mainly interacts with MDM2 during mitosis when NS is released from the nucleolus. To test this idea, bindings of endogenous MDM2 and NS were examined by coIP experiments in the S-phase or M-phase-synchronized U2OS cells. CoIP results showed that the interaction between NS and MDM2 is increased in the mitotic cells compared to the S-phase cells or the nonsynchronized cells (Fig. 5E), supporting the idea that the NS-mediated MDM2 binding and stabilization mainly occur during mitosis in non-stressed cells. Nucleoplasmic relocation of NS also occurs during nucleolar stress induced by ADR and ActD or during MPA-triggered GTP depletion. To demonstrate that cells with more NS will be better protected from the drug-induced cell death or cell-cycle arrest than cells with less NS, NS-overexpression U2OS stable cells (NS#12, Fig. 6, black bars) (Zhu et al., 2006) and vector-transfected U2OS stable cells (grey bars) were exposed to ActD (0.05ug/ml) or MPA (40uM) for 18 hours and analyzed for their cell-cycle profiles. Here, we did not use ADR because the autofluorescence of ADR overlapped with that of PI. In mock-treated cells, ActD treatment triggered cell-cycle arrest within the S-phase, and both ActD and MPA increase the apoptotic cell percentages (sub-G1). In non-treated cultures, NS overexpression arrested cells at the G1/S stage. Notably, overexpression of NS reduced the ActD or MPA-induced cell death and reversed the ActD-dependent S-phase delay. These findings show that NS-overexpressing cells are better protected from cell death induced by drugs that trigger nucleolar stress or GTP depletion.
Figure 6.
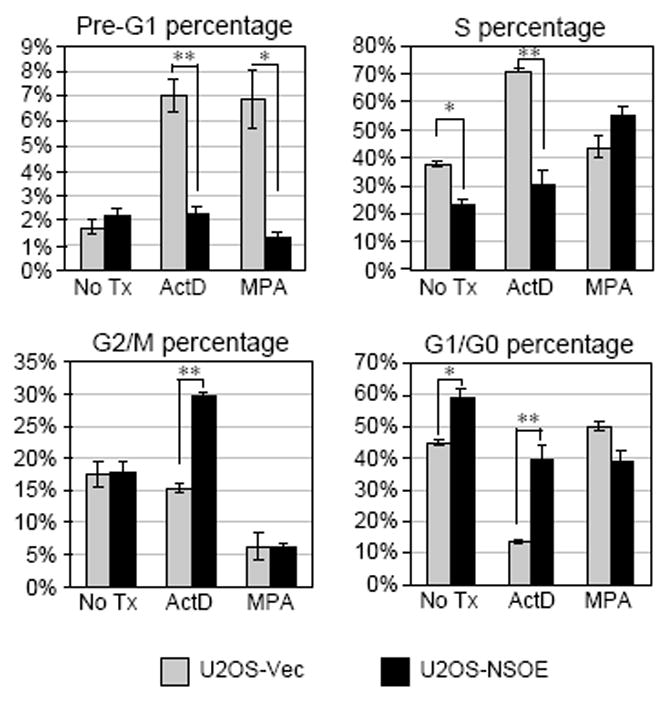
NS protects against drug-induced death and cell-cycle arrest.
Cell-cycle analyses showed that NS overexpression (NSOE, black bars) has a significant effect in protecting against ActD and MPA-induced cell death (sub-G1) and reducing the S-phase block triggered by ActD. Bars, average of three independent duplicate experiments (n=6); error bars, s.e.m.; *, p < 0.01; **, p < 0.001.
Discussion
This work identifies MDM2 as a NS-binding protein responsible for most of the NS-p53 interaction, and assigns their protein-binding sites to the central acidic-zinc finger domain of MDM2 and the coiled-coil and acidic domains of NS. Our findings on the interactive domains of these two proteins are largely consistent with the results described by the recent study on NS-MDM2 interaction (Dai et al., 2008), except for the involvement of the acidic domain of NS. We show further evidence for the association between NS and MDM2 in living cells and determine that their binding occurs in the nucleoplasm. As a result of this interaction, MDM2 protein degradation and ubiquitylation are reduced. A major role of MDM2 is to suppress p53 function by either increasing its protein degradation or directly inhibiting its transcriptional activity. Our data demonstrate that loss of NS enhances the transcriptional activity without changing the protein level of p53 in vivo and triggers G2/M arrest and cell death in U2OS cells.
Mobilized NS regulates MDM2 protein stability and p53 activity
In the interphase cells, MDM2 is localized in the nucleoplasm and NS resides in the nucleolus. Nucleolar sequestration of MDM2 has been proposed as a potential mechanism that controls its activity via association with several nucleolar proteins, including ARF, L5, L11, and L23. Our findings showed that MDM2 binding of NS does not require the nucleolar distribution of NS. In fact, the nucleoplasmic mutants of NS show stronger activities in binding and retaining MDM2 in the nucleoplasm (Fig. 2B, 4B) and inhibiting MDM2 ubiquitylation (Fig. 3E) than wild-type NS does. In addition, overexpression of NS does not promote the nucleolar accumulation of MDM2 (data not shown). Based on these reasons, we conclude that the NS-mediated regulation of MDM2 occurs when NS is mobilized from the nucleolus, which happens during mitosis or nucleolar stress. Under normal growth condition, the majority of cells are in the interphase, and, therefore, their NS proteins are inactive in stabilizing MDM2. This may account for why overexpression of wild-type NS or knockdown of endogenous NS showed only a mild but reproducible effect on MDM2 ubiquitylation in non-synchronized cultures.
NS depletion increases the transcriptional activity of p53 but not its protein level, indicating that this NS-mediated MDM2 stabilization may regulate p53’s functions by a direct inhibition rather than the ubiquitylation-dependent mechanism. Because MDM2 serves as an E3 ubiquitin ligase for itself and for p53, the activity of NS to regulate the ubiquitylation of MDM2 may at the same time affect the ability of MDM2 to ubiquitylate p53, which may explain why MDM2 decrease by NS knockdown does not lead to an increase of p53 protein. An alternative mechanism for the NS-mediated p53 inhibition is by neutralizing the p53-stabilizing activity of L23. In the normal growing cultures, more MDM2 proteins were bound by L23 than by NS. When exposed to stimuli that release NS from the nucleolus to the nucleoplasm, the overall binding between NS and MDM2 increases even though some L23 proteins also relocate to the nucleoplasm in the same process. Such findings may be caused by the differential sensitivities of NS and L23 to drug-induced nucleoplasmic translocation (Fig. 4D). Another explanation is that the MDM2 binding of NS is highly regulated by both its GTP-binding status and nucleolar distribution, whereas a good amount of MDM2-L23 interaction already occurs at the baseline level in the nucleoplasm and cytoplasm (Dai et al., 2004; Jin et al., 2004). Finally, MDM2 is known to interact and affect (or be affected by) a number of other proteins that take part in the p53 regulation. These proteins include L5, L11, PML, and ARF, and not all of them are expressed in the same cell type or at the same level as NS. Although the whole picture of this NS-mediated MDM2 regulation requires further investigation, this work provides a molecular basis to begin to address these questions.
Biological roles of NS in safeguarding G2/M progression and preventing drug-induced cell death
Consistent with what has been reported by Dai et al, we observed a G1/S arrest effect associated with NS overexpression. While Dai et al described an increase of MDM2 protein and G1/S arrest by NS knockdown, our results showed that NS depletion leads to MDM2 decrease and G2/M arrest. The first finding is supported by both gain- and loss-of-function experiments in this study. The latter finding was consistently observed and is also supported by our previous FACS analyses of NS+/- MEF cells (Zhu et al., 2006). Although the p53-mediated cell cycle arrest was initially thought to occur mainly at the G1/S phase of the cell cycle, there is now ample evidence supporting the role of p53 in controlling G2/M entry. The mechanism by which p53 delays the G2/M transition is mediated by Cdc2 inhibition via three transcriptional targets of p53, p21, 14-3-3δ, and Gadd45. p21 can directly inhibit Cdc2 (Bunz et al., 1998; Taylor and Stark, 2001). 14-3-3δ anchors Cdc25C in the cytoplasm where it cannot activate Cdc2 and induce mitosis (Peng et al., 1997). Gadd45 dissociates Cdc2 from Cyclin B1 (Zhan et al., 1999). The p53’s effect on the G2/M transition in response to genotoxic stress is cell type-dependent. Therefore, the NS-regulated G2/M transition may be context-dependent and mediated by multiple p53 target genes collectively.
Based on our data, we predict the following model. In normal interphase cells, NS is localized in the nucleolus and does not interact with MDM2 (Fig. 7A). When exposed to stress signals or chemotherapeutic agents, the nucleoli are disassembled and NS protein is mobilized from the nucleolus to the nucleoplasm. In the NS-enriched cells, nucleoplasmic relocation of NS increases the binding and nucleoplasmic retention of MDM2, which stabilizes MDM2 on one hand and competes against L23 binding of MDM2 on the other hand. Both events suppress p53 activities and prevent cell-cycle arrest and cell death (Fig. 7B, left panel). In cells expressing little or no NS, MDM2 is either sequestered by the remaining L23 in the nucleolus (grey circles) or ubiquitylated and degraded. As a result, p53 is activated and triggers cell-cycle arrest and apoptosis (Fig. 7B, right panel). The nucleoli also undergo a process of disassembly and reformation during mitosis. During this cell-cycle window, NS and other nucleolar proteins are temporarily released into the nucleoplasm/cytoplasm, allowing their interaction with nucleoplasmic proteins and potentially setting up a counting mechanism that counts the number of cell division by the loss of MDM2 protein during mitosis and signals cell-cycle exit when MDM2 protein falls below a threshold level. Here, the role of NS is to inactivate this counting mechanism to safeguard the proliferative status of continuously dividing cells (Fig. 7C). Because the early embryonic lethality of NS-null mice cannot be rescued by p53 deletion (Beekman et al., 2006) and the early embryonic lethality of mdm2-null mice is due to the missing p53 ubiquitylation by MDM2 (Itahana et al., 2007), the MDM2-p53 pathway may be the principal mediator of the NS activity only in cancer cells but not in early embryos.
Figure 7.
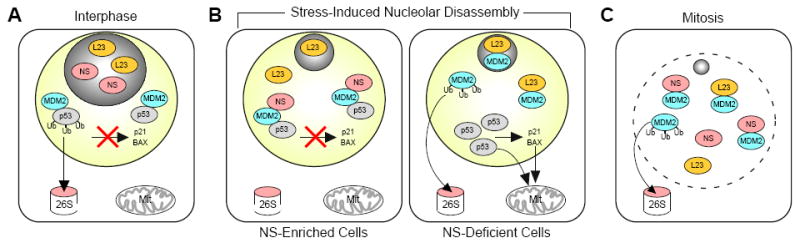
Nucleoplasmic mobilization of NS stabilizes MDM2 and promotes G2/M transition and cell survival.
(A) In dividing interphase cells, NS is localized in the nucleolus (grey circle), while MDM2 resides in the nucleoplasm (yellow circle) and blocks the activities of p53 by ubiquitylation (Ub) and transcriptional inhibition. (B) The nucleoli are disassembled when exposed to drugs that trigger nucleolar stress or GTP depletion. In the NS-enriched cells (left panel), nucleoplasmic translocation of NS inhibits the p53’s activity by stabilizing MDM2 and by competing against L23 for MDM2 binding. In the NS-deficient cells (right panel), MDM2 is either sequestered in the nucleolus by L23 or degraded, leading to G2/M arrest and cell death. (C) Nucleolar disassembly during mitosis releases NS into the nucleoplasm/cytoplasm, allowing NS to bind and stabilize MDM2. Stabilized MDM2 inhibits p53’s functions and safeguards the proliferation and survival of continuously dividing cells. Mit, mitochondria; 26S, 26S-proteasome.
In conclusion, this study shows that NS is a unique MDM2-interactive nucleolar protein that stabilizes MDM2, inhibits p53 functions, and promotes cell proliferation and survival. It does so by binding and retaining the MDM2 protein in the nucleoplasm during mitosis and nucleolar stress.
Materials and Method
Epitope-tagged full-length, deletion, and point-mutation cDNA constructs
Deletions and point mutations were introduced by stitching PCR reactions as described previously (Tsai and McKay, 2002; Tsai and McKay, 2005). cDNAs were subcloned into pCIS expression vectors containing Myc, hemagglutinin (HA), or FLAG-epitopes at the N- or C-terminus. The N- and C-terminally GFP-fused MDM2 constructs were created in the pCIS and pEGFP-N1 vectors, respectively.
Cell culture, transfection, and western blot
Cells culture and plasmid transfection procedures were described previously (Meng et al., 2007). Primary antibodies used in western analyses include anti-HA (HA.11), anti-Myc (9E10), anti-FLAG (Sigma), anti-MDM2 (SMP14), anti-p53 (DO-1), anti-p21 (Santa Cruz), anti-Bax (Santa Cruz), anti-cleaved caspase-3 (Cell Signaling), anti-B23 (Zymed), and anti-NS antibodies raised in chicken (Ab2438) or rabbits (Ab138).
Short hairpin RNA, siRNA duplex, and inducible NS-knockdown cells
Transient knockdown experiments were performed by transfection of shRNAmir constructs or siRNA duplexes. shRNAmir constructs were generated in the pShag Magic vector (pSM2c) based on a mir-30 hairpin design that targets 21-bp sequences of NS, capped by mir-5' and mir-3' sequences and driven by a U6 promoter. Two shRNAmir constructs were tested for their NS knockdown efficiencies. The targeted sequences for NS are: 5’-GCT GTA CTG CCA AGA ACT TAA-3’ (shNS-1) and 5’-CCT GAT ATT AAG CCA TCA AAT-3’ (shNS-2). The shScr construct targets a scrambled sequence of 5’-TCT CGC TTG GGC GAG AGT AAG-3’. siRNA duplexes for NS and control knockdown were described in (Tsai and McKay, 2002). Creation of stable lines with inducible NS-knockdown capabilities was described in Fig. S4.
Protein degradation and in vivo ubiquitination assays
Protein degradation assays were performed in cycloheximide-treated H1299 cells as described before (Zhu et al., 2006). For in vivo ubiquitination assays, His-tagged ubiquitin and MDM2-expression plasmids were coexpressed with or without NS or shNS-2 in HEK293 cells. Two days after transfection, cells were treated with MG132 (10uM) for 6 hours before protein extraction in 6M guanidinium buffer. Ubiquitylated proteins were pulled down by Ni2+-chelating sepharose.
Cell-cycle profile and synchronization
Cell-cycle profiles were analyzed by counting the PI-labeled cells with a COULTER EPICS XL flow cytometer and the XL System II software (Zhu et al., 2006). Each cell-cycle profile was compiled from 2×104 gated events, and analyzed using the Multi Cycle AV software. Early S-phase synchronization was achieved by incubation with 2mM thymidine for 20h, and mitotic arrest was achieved by incubation with 0.5uM nocodazole for 20h.
Coimmunoprecipitation
Cells were harvested in NTEN buffer (20mM Tris pH8.0, 150mM NaCl, 1mM EDTA, 0.5% NP40, 0.1mM DTT, supplemented with 1mM PMSF, 1ug/ml leupeptin, 0.5ug/ml aprotinin, 0.7ug/ml pepstatin A, and 1uM E64). Lysates were incubated with primary antibody for 1 hour at 4C, followed by incubation with protein G sepharose beads (Pharmacia) for an additional 4 hours at 4C. Immunoprecipitates were washed 5 times with RIPA buffer (1X PBS, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP40, 1mM PMSF, 1ug/ml leupeptin, 0.5ug/ml aprotinin, 0.7ug/ml pepstatin A, and 1uM E64), fractionated by 10% SDS-PAGE, and detected by western blots.
Fluorescence loss in photobleaching (FLIP)
Bleaching experiments were performed on HeLa cells grown on Nalgene Lab Tek II chamber slides by using a Zeiss LSM510 confocal microscope equipped with a 63X plan-apochromat oil objective as described previously (Meng et al., 2007). The nucleoplasmic retention time was measured by the rate of fluorescence loss in the nucleoplasm while bleaching a 2-um circular region within one nucleolus with repetitive bleaching pulses of 150-ms duration and 0.59-s interval. The relative fluorescence index (RFI) in the nucleoplasm of bleached cells was normalized to the nucleoplasmic intensity of neighboring non-bleached cells after background subtraction by the following calculation: RFI=(It/I0)*(C0/Ct), where It and I0 are the background-subtracted intensities of the nucleoplasm in the bleached cell at time-point t and before photobleaching, respectively. Ct and C0 are the background-subtracted intensities of the nucleoplasm in the neighboring control cell at time-point t and before photobleaching, respectively.
Bimolecular fluorescence complementation (BiFC)
Protein pairs were individually fused to a FLAG-tagged Venus YFP N-terminal fragment (amino acids 1-173, Yn) and a Myc-tagged YFP C-terminal fragment (amino acids 156-239, Yc), and coexpressed with a nucleolar localization signal-tagged CFP (noCFP) in HeLa cells grown on Nalgene Lab Tek II chamber slides. After a 24-hour incubation at 37C and a 15-hour incubation at 30C, cells were collected for fluorescence-activated cell sorting (FACS) analyses. Live cell images were recorded on a Zeiss Axiovert 200 fluorescence microscope, equipped with a 63X oil objective (NA 1.4), a Zeiss AxioCam MRm CCD camera, and filter sets described as below: YFP (excitation, BP 500/20; emission, BP 535/30), CFP (excitation, BP 436/20, emission, BP 480/40).
Supplementary Material
Acknowledgments
We gratefully acknowledge Karen Vousden for providing the MDM2 cDNA and Chang-Deng Hu for the BiFC constructs. This work is supported by NCI-PHS grant R01 CA113750 to R.Y. Tsai.
References
- Beekman C, Nichane M, De Clercq S, Maetens M, Floss T, Wurst W, Bellefroid E, Marine JC. Evolutionarily Conserved Role of Nucleostemin: Controlling Proliferation of Stem/Progenitor Cells during Early Vertebrate Development. Mol Cell Biol. 2006;26:9291–301. doi: 10.1128/MCB.01183-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi R, Scaglioni PP, Bergmann S, Horn HF, Vousden KH, Pandolfi PP. PML regulates p53 stability by sequestering Mdm2 to the nucleolus. Nat Cell Biol. 2004;6:665–72. doi: 10.1038/ncb1147. [DOI] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Dai MS, Sun XX, Lu H. Aberrant expression of nucleostemin activates p53 and induces cell cycle arrest via inhibition of MDM2. Mol Cell Biol. 2008;28:4365–76. doi: 10.1128/MCB.01662-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai MS, Zeng SX, Jin Y, Sun XX, David L, Lu H. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol Cell Biol. 2004;24:7654–68. doi: 10.1128/MCB.24.17.7654-7668.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–9. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Itahana K, Mao H, Jin A, Itahana Y, Clegg HV, Lindstrom MS, Bhat KP, Godfrey VL, Evan GI, Zhang Y. Targeted inactivation of Mdm2 RING finger E3 ubiquitin ligase activity in the mouse reveals mechanistic insights into p53 regulation. Cancer Cell. 2007;12:355–66. doi: 10.1016/j.ccr.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Jin A, Itahana K, O’Keefe K, Zhang Y. Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol Cell Biol. 2004;24:7669–80. doi: 10.1128/MCB.24.17.7669-7680.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Kurki S, Peltonen K, Latonen L, Kiviharju TM, Ojala PM, Meek D, Laiho M. Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell. 2004;5:465–75. doi: 10.1016/s1535-6108(04)00110-2. [DOI] [PubMed] [Google Scholar]
- Ma H, Pederson T. Depletion of the Nucleolar Protein Nucleostemin Causes G1 Cell Cycle Arrest via the p53 Pathway. Mol Biol Cell. 2007;18:2630–5. doi: 10.1091/mbc.E07-03-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L, Zhu Q, Tsai RY. Nucleolar trafficking of nucleostemin family proteins: common versus protein-specific mechanisms. Mol Cell Biol. 2007;27:8670–82. doi: 10.1128/MCB.00635-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–45. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993;362:857–60. doi: 10.1038/362857a0. [DOI] [PubMed] [Google Scholar]
- Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277:1501–5. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- Rubbi CP, Milner J. Non-activated p53 co-localizes with sites of transcription within both the nucleoplasm and the nucleolus. Oncogene. 2000;19:85–96. doi: 10.1038/sj.onc.1203378. [DOI] [PubMed] [Google Scholar]
- Tao W, Levine AJ. P19(ARF) stabilizes p53 by blocking nucleo-cytoplasmic shuttling of Mdm2. Proc Natl Acad Sci U S A. 1999;96:6937–41. doi: 10.1073/pnas.96.12.6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene. 2001;20:1803–15. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- Tsai RY, McKay RD. A nucleolar mechanism controlling cell proliferation in stem cells and cancer cells. Genes Dev. 2002;16:2991–3003. doi: 10.1101/gad.55671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai RY, McKay RD. A multistep, GTP-driven mechanism controlling the dynamic cycling of nucleostemin. J Cell Biol. 2005;168:179–84. doi: 10.1083/jcb.200409053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Q, Antinore MJ, Wang XW, Carrier F, Smith ML, Harris CC, Fornace AJ., Jr Association with Cdc2 and inhibition of Cdc2/Cyclin B1 kinase activity by the p53-regulated protein Gadd45. Oncogene. 1999;18:2892–900. doi: 10.1038/sj.onc.1202667. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wolf GW, Bhat K, Jin A, Allio T, Burkhart WA, Xiong Y. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol Cell Biol. 2003;23:8902–12. doi: 10.1128/MCB.23.23.8902-8912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Yasumoto H, Tsai RY. Nucleostemin Delays Cellular Senescence and Negatively Regulates TRF1 Protein Stability. Mol Cell Biol. 2006;26:9279–90. doi: 10.1128/MCB.00724-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.