

Abstract
We have recently described a novel form of hyperalgesic priming (type II) induced by agonists at two clinically important Gi-protein coupled receptors (Gi-GPCRs), mu-opioid and A1-adenosine. Like mu-opioids, the anti-migraine triptans, which act at 5-HT1B/D Gi-GPCRs have been implicated in pain chronification. We determined if sumatriptan, a prototypical 5-HT1B/D agonist produces type II priming. Characteristic of hyperalgesic priming, intradermal injection of sumatriptan (10 ng) induced a change in nociceptor function such that a subsequent injection of prostaglandin-E2 (PGE2) induces prolonged mechanical hyperalgesia. However, onset to priming was delayed 3 days, characteristic of type I priming. Also characteristic of type I priming, a protein kinase Cε (PKCε), but not a PKA inhibitor attenuated the prolongation phase of PGE2 hyperalgesia. The prolongation of PGE2 hyperalgesia was also permanently reversed by intradermal injection of cordycepin, a protein translation inhibitor. Also, hyperalgesic priming did not occur in animals pre-treated with pertussis toxin or IB4-positive nociceptor toxin, IB4-saporin. Finally, as observed for other agonists that induce type I priming, sumatriptan did not induce priming in female rats. The prolongation of PGE2 hyperalgesia induced by sumatriptan was partially prevented by co-injection of antagonists for the 5-HT1B and 5-HT1D, but not 5-HT7, serotonin receptors, and completely prevented by co-administration of a combination of the 5-HT1B and 5-HT1D antagonists. Moreover, the injection of selective agonists, for 5-HT1B and 5-HT1D receptors, also induced hyperalgesic priming. Our results suggest that sumatriptan, which signals via Gi-GPCRs, induces type I hyperalgesic priming, unlike agonists at other Gi-GPCRs, which induce type II priming.
Keywords: hyperalgesic priming, hyperalgesia, 5-HT1B receptor, 5-HT1D receptor, Gi-protein coupled receptors (GPCRs), chronic pain, triptans
Introduction
While the mechanism underlying the transition from acute to chronic pain remains poorly understood, it has been suggested that some analgesics can produce pain chronification. Thus, opioids, which are used clinically to treat moderate-to-severe pain, especially agonists at the mu-opioid receptor (MOR), may come to exacerbate or actually produce pain during their chronic administration, a phenomenon referred to as opioid-induced hyperalgesia (OIH) [6; 60; 63]. Similarly, the triptan class of drugs, used to treat migraine [25; 35; 76], also facilitates pain chronification and/or contributes to migraine chronification, a condition referred to as medication overuse headache (MOH; [9; 11; 26; 28; 38; 41; 45; 77]). The target for the therapeutic effect of triptans is thought to be 5-HT1B and 5-HT1D receptors that, like MOR are inhibitory G-protein coupled receptors (Gi-GPCRs) [37; 42; 64; 86; 95; 96]. Therefore, in the present study we explored the possibility that, like MOR and A1-adenosine receptor agonists [7; 8], triptans would also induce type II hyperalgesic priming. In addition, we explored the 5-HT receptor subtypes at which triptans act (5-HT1B, 5-HT1D and 5-HT7) to induce priming. We report that while sumatriptan, a prototypical 5-HT1B/D receptor agonist induces hyperalgesic priming, this priming meets the criteria for type I rather than type II priming.
Methods
Animals
Experiments were performed on 230–280 g male and female Sprague–Dawley rats (Charles River Laboratories, Hollister, CA, USA). Rats were housed in a controlled environment in the animal care facility at the University of California, San Francisco, under a 12-h light/dark cycle. Food and water were available ad libitum. The experimental protocols were approved by the Institutional Animal Care and Use Committee at UCSF and adhered to the guidelines of the American Association of Laboratory Animal Care, the National Institutes of Health (NIH), and the Committee for Research and Ethical Issues of the International Association for the Study of Pain (IASP), for the use of animals in research. In the design of experiments, all efforts were made to minimize the number of animals used and their suffering.
Mechanical nociceptive threshold testing
Mechanical nociceptive threshold was quantified using an Ugo Basile Analgesymeter® (Randall-Selitto paw-withdrawal test; Stoelting, Chicago, IL), which applies a linearly increasing mechanical force to the dorsum of the rat's hind paw, as previously described [32; 78; 80]. Nociceptive threshold was defined as the force in grams at which the animal withdrew its paw; baseline paw-pressure nociceptive mechanical threshold was defined as the mean of the 3 readings taken just before a test agent was injected. Each paw was treated as an independent measure and each experiment performed on a separate group of rats. Data are presented as mean change from baseline mechanical nociceptive threshold.
Drugs and their administration
The chemicals used in this study were: cordycepin 5′-triphosphate sodium salt (protein translation inhibitor; [7; 8; 34]), prostaglandin E2 (PGE2; a hyperalgesic agent that acts at receptors on nociceptors to sensitizes them), sumatriptan succinate (a 5-HT1B and 5-HT1D receptor agonist), CP-93129 dihydrochloride hydrate (a 5-HT1B receptor agonist), pertussis toxin (Gi-protein inhibitor) and SB-269970 (a 5-HT7 receptor antagonist), all from Sigma-Aldrich (St. Louis, MO, USA); PKCεV1–2 (PKCε-I, a PKCε-specific translocation inhibitor peptide; [7; 8; 48; 58]), from Calbiochem (La Jolla, CA, USA); H-89 dihydrochloride (inhibitor of protein kinase A [PKA]; [7; 8]) from Santa Cruz Biotechnology (Dallas, TX, USA); L-694,247 (a 5-HT1D receptor agonist), BRL 15572 (a 5-HT1D receptor antagonist) and NAS-181 (a 5-HT1B receptor antagonist), all from Tocris Bioscience (Avonmouth, Bristol, UK).
The selection of doses was based on previous studies that showed their effectiveness when injected intradermally on the dorsum of the hind paw [3; 7; 8; 32; 52]. The stock solution of PGE2 (1 μg/μL) was prepared in 10% ethanol and additional dilutions made with physiological saline (0.9% NaCl), yielding a final ethanol concentration of less than 1%. Cordycepin, sumatriptan, CP-93129, pertussis toxin, PKCεV1–2 and SB-269970 were dissolved in saline. All other drugs were dissolved in 100% DMSO (Sigma-Aldrich) and further diluted in saline containing 2% Tween 80 (Sigma-Aldrich). The final concentration of DMSO and Tween 80 was 2%. All drugs were injected intradermally on the dorsum of the hind paw, in a volume of 5 μL, using a 30-gauge hypodermic needle adapted to a 50 μl Hamilton syringe (Reno, NV, USA). The injection of cordycepin, H-89, PKCεV1–2 and pertussis toxin, was preceded by a hypotonic shock (2 μL of distilled water, separated by a bubble of air to avoid mixing in the same syringe), to facilitate the entry of compounds into the nerve terminal [14; 16].
Intrathecal administration of IB4-saporin or SSP-saporin
IB4-saporin
Isolectin B4 (IB4)-saporin, an IB4-positive nociceptor neurotoxin (Advanced Targeting Systems, San Diego, CA), was diluted in saline, and a dose of 3.2 μg, in a volume of 20 μL was administered intrathecally, 14 days prior to the priming experiments. The dose of IB4-saporin and time protocol were chosen based on previous reports from us and other groups [7; 8; 51; 52; 66; 89].
SSP-saporin
The neurotoxin [Sar9,Met(O2)11]-substance P-saporin (SSP-Saporin, Advanced Targeting Systems, San Diego, CA) was diluted in saline, and a dose of 100 ng, in a volume of 20 μL was administered intrathecally, 14 days prior to the priming experiments. The addition of [Sar9Met(O2)11] to substance P conjugated to saporin makes the agent more stable and potent than when substance P is bound to saporin. The dose and pre-treatment interval that we used were based on previous reports [22; 93], that demonstrated the effectiveness of this agent to deplete substance P-containing fibers with no loss of lumbar dorsal horn neurons expressing the neurokinin 1 (NK1) receptor in deeper laminae and prominent loss of NK1 receptor in laminae I [56; 59; 87; 92].
Rats were briefly anesthetized with 2.5% isoflurane (Phoenix Pharmaceuticals, St. Joseph, MO, USA) in 97.5% O2. Then, a 30-gauge hypodermic needle was inserted, on the midline, into the subarachnoid space, between the L4 and L5 vertebrae. The control treatment consisted of intrathecal injection of saline (vehicle; 20 μL). Animals regained consciousness approximately 1 min after removal from the anesthetic chamber. There was no effect of IB4-saporin or SSP-saporin on the mechanical nociceptive threshold per se.
Sumatriptan-induced changes in nociceptor function
Our group has previously shown that a single intradermal injection of a selective 5-HT1 receptor agonist produced mechanical hyperalgesia (Fig. 1A; [2; 79; 81]). To investigate the changes in nociceptor function produced by the injection of sumatriptan (a 5-HT1B/1D receptor agonist, 10 ng), 5-HT1B receptor agonist (CP-93129, 10 or 100 ng) or 5-HT1D receptor agonist (L-694,247, 10 or 100 ng) — measured as prolonged response to a hyperalgesic mediator, at a point in time when the mechanical nociceptive threshold was not different from pre-sumatriptan baseline-PGE2 (100 ng) was injected at the same site, and the change in nociceptive threshold evaluated 30 min and 4 h later. Presence of hyperalgesia at the 4th h is characteristic of priming [4; 7; 8; 31]. To evaluate the intracellular signaling pathways that play a role in priming induced by sumatriptan, and to investigate the mechanisms that play a role in the induction of the changes in nociceptor function produced by the activation of the 5-HT1B/D receptor, pharmacological agents were injected before sumatriptan (prevention protocol). To investigate the second messengers involved in the expression of the neuroplasticity, inhibitors were administered before the injection of PGE2 in the already primed paw (inhibition protocol), at a time when mechanical nociceptive threshold was not different from pre-sumatriptan levels.
Figure 1. Sumatriptan induces mechanical hyperalgesia and hyperalgesic priming in male rats.

A. Rats were treated with a single intradermal injection of vehicle (5 μL; black bar) or sumatriptan (10 ng in 5 μL; white bar) and 30 min later, mechanical nociceptive threshold was evaluated by the Randall-Sellitto paw withdrawal test. The group treated with sumatriptan showed significant mechanical hyperalgesia, when compared with the group treated with vehicle (***p < 0.0001; Unpaired Student's t-test), indicating that sumatriptan produces a pronociceptive effect. Average mechanical nociceptive threshold before the injection was 126.2 ± 2.7 g, for vehicle, and 118.5 ± 1.5 g, for sumatriptan group. B. Twenty-four hours later, when the mechanical nociceptive threshold was no longer different from pre-injection baseline (t5 = 1.667; p = 0.1942, for the vehicle group; t5 = 0.2116; p = 0.8460, for the sumatriptan group; paired Student's t-test), PGE2 (100 ng) was injected intradermally at the same site on the dorsum of the hind paw, and mechanical nociceptive threshold evaluated 30 min and 4 h later. In both groups PGE2 induced significant hyperalgesia at 30 min, which was no longer present at the 4th h, in the both groups (NS, p > 0.05, when both groups are compared; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that hyperalgesic priming was not present 24 hours after the injection of vehicle or sumatriptan. However, 72 hours later (C) and 30 days later (D) when PGE2 (100 ng) was again injected, at the same site, hyperalgesia induced by PGE2 was present 30 min after injection in the group previously treated with sumatriptan, and was still present at the 4th h (F1,20 = 72.97, ***p < 0.0001, when both groups are compared at the 4th h; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). These data demonstrate that a single injection of sumatriptan, 72 hours or 30 days prior, produced long-term changes in nociceptor function characteristic of hyperalgesic priming. (N = 6 paws per group)
Statistics
In all experiments, the dependent variable was mechanical paw-withdrawal threshold, expressed as percentage change from baseline. The average paw withdrawal thresholds before the injection of sumatriptan and before the tests with PGE2 (3 or more days, depending on the experiment) were 123.0 ± 1.05 g and 123.3 ± 1.12 g, respectively; paired Student's t-test showed no significant difference between these values (t125 = 0.4285, p = 0.6753). The total number of paws used in this study was 126. To compare the mechanical hyperalgesia induced by injection of the neuroplasticity-inducing agents (i.e., sumatriptan, 5-HT1B [CP-93129] or 5-HT1D [L-694,247] receptor agonists) and to compare the effect of PGE2 in different groups, in the presence or absence of inhibitors of signaling pathways, two-way repeated-measures ANOVA, followed by Bonferroni post hoc test, was performed. Graph Pad Prism 5.0 (GraphPad Software, Inc., San Diego, CA, USA) was used to plot graphs and to perform statistical analyses; a p-value less than 0.05 was considered statistically significant. Data are presented as mean ± SEM.
Results
Sumatriptan-induced hyperalgesic priming
One of the defining features of both type I and II priming is the prolongation of hyperalgesia induced by pronociceptive mediators, prototypically prostaglandin E2 (PGE2) [4; 12; 33; 57; 69; 73]. To determine if sumatriptan induces hyperalgesic priming we injected PGE2 intradermally, at the site of nociceptive testing. We report that a single low dose of sumatriptan (10 ng) induces mechanical hyperalgesia (Fig. 1A). This differs from the pronociceptive effects of MOR and A1-adenosine agonists that took repeated administration before they induced hyperalgesia [7; 8]. Therefore, in the following experiments, we tested for priming after a single administration of sumatriptan. Unlike type II priming, in which the prolongation of PGE2 hyperalgesia is observed immediately after the repeated injections of mu-opioid or A1-adenosine receptor agonists, no prolongation of PGE2 hyperalgesia was observed when PGE2 was administered 24 hours post sumatriptan (Fig. 1B). However, when it was injected 72 hours (Fig. 1C) or 30 days (Fig. 1D) after sumatriptan, we observed prolonged hyperalgesia. This demonstrates the time course for the onset of priming to be similar to that observed for type I hyperalgesic priming [4; 12].
Role of 5-HT1B and 5-HT1D receptors in sumatriptan-induced priming
Since triptans are agonists at two Gi-GPCRs, 5-HT1B and 5-HT1D [5; 24; 35; 39; 43; 82; 84] we evaluated the ability of antagonists at these two receptors to prevent sumatriptan-induced hyperalgesic priming. We found that, while alone the 5-HT1B and 5-HT1D antagonists (NAS-181 and BRL 15572, respectively) each partially attenuate the magnitude of the prolongation of PGE2 hyperalgesia (Fig. 2, gray and white bars), the co-administration of the two antagonists completely prevented sumatriptan-induced prolongation of PGE2 hyperalgesia (Fig. 2, dotted bars). Since, it has been suggested that triptans also produce some of their effects by action at the 5-HT7 receptor [71; 75; 88; 90; 91], we also determined if a 5-HT7 receptor antagonist (SB-269970) would attenuate sumatriptan-induced priming. We found that the SB-269970 did not attenuate sumatriptan-induced priming (Fig. 2, horizontally striped bars).
Figure 2. Role of 5-HT1B and 5-HT1D, but not 5-HT7, receptors in sumatriptan-induced prolongation of PGE2-induced hyperalgesia.

Male rats received vehicle (5 μL; black bars), NAS-181 (1 μg; 5-HT1B antagonist; gray bars), BRL 15572 (1 μg; 5-HT1D antagonist; white bars), the combination of NAS-181 (1 μg)/BRL 15572 (1 μg; dotted bars) or SB-269970 (1 μg; 5-HT7 antagonist; horizontally striped bars) on the dorsum of the hind paw. Ten minutes later, sumatriptan (10 ng) was injected at the same site. Four days later, when the mechanical nociceptive thresholds were not different from pre sumatriptan-injection control baseline (t5 = 0.8165; p = 0.4740, for the vehicle group; t5 = 1.260; p = 0.2967, for the NAS-181 group; t5 = 0.3015; p = 0.7827, for the BRL 15572 group; t5 = 1.000; p = 0.3910, for the NAS-181/BRL 15572 group; t5 = 1.667; p = 0.1942, for the SB-269970 group; paired Student's t-test), PGE2 (100 ng) was injected at the same site, and the mechanical nociceptive threshold evaluated. In all groups of rats evaluated 30 min after its injection PGE2 induced significant hyperalgesia. However, in the groups previously treated with NAS-181 (gray bars) or BRL 15572 (white bars), the prolongation of PGE2-induced hyperalgesia was significantly attenuated, and completely eliminated in the group previously treated with the combination of NAS-181/BRL 15572 (dotted bars; F2,30 = 507.75; ***p < 0.0001, when NAS-181, BRL 15572 and NAS-181/BRL 15572 groups are compared with the vehicle-treated group; two-way repeated measures ANOVA followed by Bonferroni post hoc test), indicating the participation of both 5-HT1B and 5-HT1D, but not 5-HT7, receptors in the induction of hyperalgesic priming by sumatriptan. (N = 6 paws per group)
Direct activation of 5-HT1B and 5-HT1D receptors induces hyperalgesic priming
We found that the prolongation of PGE2-induced hyperalgesia by injection of sumatriptan (10 ng) is dependent of activation of both 5-HT1B and 5-HT1D receptors (Fig. 2). Therefore, we next tested the effect of selective agonists for 5-HT1B (CP-93129; Fig. 3, gray bars) and 5-HT1D (L-694,247; Fig. 3, white bars). We observed that the dose of 100 ng, but not 10 ng, of both agonists, induced priming as manifest by prolongation of PGE2-induced hyperalgesia (Fig. 3).
Figure 3. Agonists for 5-HT1B and 5-HT1D receptors induced hyperalgesic priming.

Male rats were injected intradermally with agonists for 5-HT1B (CP-93129; 10 or 100 ng; gray bars) or 5-HT1D (L-694,247; 10 or 100 ng; white bars) receptors. Four days later, PGE2 (100 ng) was injected at the same site and the mechanical nociceptive threshold was evaluated 30 min and 4 h after its injection. In all groups of rats PGE2 induced significant hyperalgesia, evaluated 30 min after injection. Also, we observed prolongation of PGE2-induced hyperalgesia in the groups previously treated with 100 ng of agonist for 5-HT1B (F1,20 = 94.06, ***p < 0.0001) and 5-HT1D (F1,20 = 127.07, ***p < 0.0001; two-way repeated-measure ANOVA followed by Bonferroni post hoc test showed) receptor, when the 4th h of the groups were compared. BL: baseline. (N = 6 paws per group)
PKCε dependence in expression of sumatriptan-induced priming
Another mechanism that distinguishes type I from type II priming is the second messengers mediating the prolongation of PGE2 hyperalgesia, PKCε for type I priming [4; 70] and PKA for type II [7; 8]. Compatible with the above findings, which support the suggestion that sumatriptan induces type I priming, we observed that the prolongation of PGE2 hyperalgesia induced by sumatriptan was attenuated by a PKCε (Fig. 4A), but not by a PKA (Fig. 4B) inhibitor.
Figure 4. PKCε but not PKA plays a role in the expression of hyperalgesic priming induced by sumatriptan.
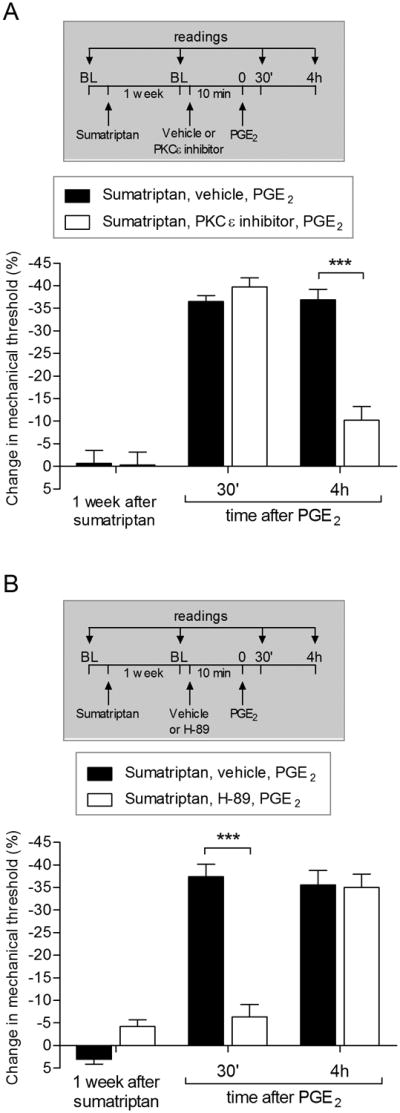
A. Male rats received a single injection of sumatriptan (10 ng) on the dorsum of the hind paw. One week later, when the mechanical nociceptive thresholds were not different from pre sumatriptan-injection baseline (t5 = 0.3203; p = 0.7697, for black bars; t5 = 0.2089; p = 0.8479, for white bars; paired Student's t-test), PGE2 (100 ng) was injected at the same site, in the presence of vehicle (control, black bars) or PKCε inhibitor (1 μg, white bars). The mechanical nociceptive threshold was then evaluated 30 min and 4 h later, by Randall-Sellitto paw withdrawal test. In both groups PGE2 induced significant hyperalgesia, evaluated 30 min after injection. However, we observed significant attenuation of PGE2-induced prolongation of hyperalgesia in the group previously treated with PKCε inhibitor (F1,15 = 132.58, ***p < 0.0001, when the vehicle and PKCε inhibitor group were compared; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). B. A different group of male rats were primed with an intradermal injection of sumatriptan (10 ng) and, one week later, received at the same site, vehicle (5 μL) or a PKA inhibitor (H-89; 1 μg). Ten min later, PGE2 (100 ng) was injected on the dorsum of the hind paw and the mechanical nociceptive thresholds were evaluated 30 min and 4 h later. While PGE2-induced hyperalgesia was still present after 30 min, in the group that received vehicle, in the group treated with H-89 it was significantly attenuated. However, it was present at the 4th h (F2,12 = 132.95, ***p < 0.0001, when vehicle and H-89 groups are compared at 30 min; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that the expression of priming induced by previous injection of sumatriptan is not dependent on PKA. BL: baseline. (N = 6 paws per group)
Peripheral protein translation dependence of sumatriptan-induced priming
A third distinction between type I and type II priming is the dependence of type I [30] but not type II [7; 8] priming, on protein translation in the peripheral terminal of the primary afferent nociceptor. We report that intradermal injection of cordycepin, an inhibitor of protein translation, permanently reversed sumatriptan-induced priming (Fig. 5), further suggesting that sumatriptan induces type I priming.
Figure 5. Hyperalgesic priming induced by sumatriptan is dependent on local protein translation.

Male rats that were treated with intradermal injection of sumatriptan (10 ng) on the dorsum of the hind paw received, three days later, PGE2 (100 ng) injected at the same site, in the presence of vehicle (5 μL, black bars) or the inhibitor of protein translation, cordycepin (1 μg, white bars), administered 15 min before. Mechanical nociceptive threshold was evaluated 30 min and 4 h after injection of PGE2. While the hyperalgesia induced by PGE2 was present 30 min after injection in the group previously treated with cordycepin, it was significantly inhibited at the 4th hour (F1,15 = 104.94, ***p < 0.0001, when both groups are compared at the 4th h; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). BL: baseline. (N = 6 paws per group)
Role of the inhibitory G-protein αi subunit in sumatriptan-induced priming
Since the 5-HT1B and 5-HT1D receptors are Gi-GPCRs [37; 42; 64; 86; 95; 96], to determine the signaling pathway downstream of the 5-HT1B/D receptor that contributes to the prolongation of PGE2-induced hyperalgesia, we tested the effect of the treatment with the G-protein αi subunit inhibitor, pertussis toxin (PTX), injected before sumatriptan. We found that the prolongation of PGE2-induced hyperalgesia was inhibited by PTX (Fig. 6). Of note, the ability of PTX to inhibit the induction of sumatriptan-induced priming (Fig. 6), the expression of type II priming induced by A1-adenosine receptor agonist CPA [8] and also type I hyperalgesic priming [29; 33; 57] contrasts with mu-opioid receptor agonist DAMGO-induced type II priming where PTX failed to inhibit the prolongation of PGE2-induced hyperalgesia induced by DAMGO [7].
Figure 6. Role of inhibitory G-protein αi subunit in sumatriptan-induced priming.

Male rats were treated with vehicle (5 μL, black bars) or pertussis toxin (PTX; 1 μg, white bars) by intradermal injection and, 30 min later, sumatriptan (10 ng) was injected at the same side. Five days later, PGE2 (100 ng) was injected intradermally at the same site on the dorsum of the hind paw, and the mechanical hyperalgesia was evaluated 30 min and 4 h later, by Randall-Sellitto paw withdrawal test. In both groups PGE2 induced significant hyperalgesia, evaluated 30 min after injection. However, we observed significant attenuation of PGE2-induced prolongation of hyperalgesia in the group previously treated with PTX (F2,12 = 169.04, ***p < 0.0001, when the vehicle and PTX group were compared; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that the αi subunit plays a role in the induction of sumatriptan-induce priming. BL: baseline. (N = 6 paws per group)
IB4-positive nociceptor dependence of sumatriptan-induced priming
We have previously demonstrated that the induction of type I priming is prevented by spinal intrathecal treatment with IB4-saporin, a neurotoxin that eliminates IB4-positive nociceptors [7; 8; 51; 52; 66; 89], whereas type II priming induction by the MOR agonist DAMGO is prevented by intrathecal administration of a stabilized form of substance P, conjugated to saporin (SSP-saporin; unpublished results), a specific neurotoxin that destroys neurons with processes containing NK1 receptors in the superficial but not the deeper laminae of the dorsal horn [22; 59; 62; 93], by eliminating substance P (SP) containing sensory neurons. In the present experiments we found that, in rats pretreated with IB4-saporin, but not SSP-saporin, the injection of sumatriptan did not induce prolongation of PGE2 hyperalgesia, evaluated 4 days later (Fig. 7).
Figure 7. Type I priming induced by sumatriptan is dependent on IB4-positive neurons.

Male rats were treated with vehicle (5 μL, black bars), IB4-saporin (3.2 μg/20 μL; white bars) or SSP-saporin (100 ng/20 μL; gray bars) by intrathecal injection. Fourteen days later, sumatriptan (10 ng) was injected on the dorsum of the hind paw. Four days later, when mechanical thresholds were not different from pre-sumatriptan baseline, (t5 = 1.663; p = 0.2010, for the vehicle group; t5 = 1.667; p = 0.1942, for the IB4-saporin group; t5 = 0.6547; p = 0.5799, for the SSP-saporin group; paired Student's t-test), PGE2 (100 ng) was injected intradermally at the same site on the dorsum of the hind paw, and the mechanical hyperalgesia evaluated 30 min and 4 h later. Two-way repeated-measures ANOVA followed by Bonferroni post hoc test showed PGE2-induced hyperalgesia at 30 min in all groups, that was still present at the 4th h, in vehicle (black bars) and SSP-saporin (gray bars)-treated groups, but not in IB4-saporin (white bars) treated-group, which at the 4th hour, the PGE2-induced hyperalgesia was significant blocked (F1,15 = 26.95, ***p = 0.0006, when vehicle-treated groups are compared at the 4th h with IB4-saporin-treated group; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that the prolonged hyperalgesia induced by PGE2 observed in the priming induced by sumatriptan occurs in IB4-positive neurons. BL: baseline. (N = 6 paws per group)
Sexual dimorphism in sumatriptan-induced priming
We have previously shown that the administration of agonists for cell surface receptors (such as TNF-α, MCP-1 and IL-6), induce type I priming only in male rats [53]. On the other hand, induction of type II priming by Gi-GPCR agonists is observed in females as well as males [7; 8]. In the present study we observed that sumatriptan did not induce priming in female rats (Fig. 8), another characteristic feature of type I hyperalgesic priming [53].
Figure 8. Sumatriptan did not induce prolongation of PGE2 hyperalgesia in female rats.

Female rats received intradermal injection of sumatriptan (10 ng, white bars) or vehicle (black bars) on the dorsum of the hind paw. Three days later, when the mechanical thresholds were not different from pre-vehicle or pre-sumatriptan baseline levels (t5 = 0.7559; p = 0.4838, for the vehicle group; t5 = 2.150; p = 0.0842, for the sumatriptan group, paired Student's t-test), PGE2 (100 ng) was injected at the same site, and the mechanical hyperalgesia was evaluated 30 min and 4 h later. Two way ANOVA followed by Bonferroni post hoc test showed that PGE2 induced significant hyperalgesia at 30 min in both groups, that was not present at the 4th h after its injection (F1,15 = 0.83; p = 0.3779, when compared both groups). BL: baseline. (N = 6 paws per group)
Discussion
The triptans, agonists at serotonin 5-HT1B and 5-HT1D receptors, are an important class of drugs used in the treatment of migraine [1; 35; 36; 40; 47; 55; 84]. One limitation in their clinical use is that they can contribute to migraine chronification and more frequent episodes of headache, a condition referred to as medication overuse headache (MOH), recently characterized as a global epidemic [9; 11; 27; 28; 38; 41; 45; 77]. The triptan receptors are both Gi-protein coupled, which they share with other classes of analgesics, including those that act at mu-opioid or CB cannabinoid receptors [67; 74]. Importantly, opioids can also produce exacerbation of the pain for which they are clinically administered [21; 23; 44; 60; 65], a phenomenon referred to as opioid-induced hyperalgesia (OIH). We have studied opioid-induced hyperalgesia, using a model in the primary afferent nociceptor, hyperalgesic priming (type II) [7; 8]. Therefore, in the present experiments we determined if sumatriptan, a clinically used triptan [35; 36; 49; 83] would also induce type II priming.
In the present experiments we observed that intradermal injection of sumatriptan produces mechanical hyperalgesia at the injection site, as has been reported in patients being treated for migraine [17; 18; 68; 85]. Also, the frequent use of triptans can lead to transition to MOH or chronic migraine [9; 10; 27; 28; 38; 41; 68; 77; 85]. We observed that sumatriptan produces a neuroplastic change in nociceptor function such that PGE2-induced mechanical hyperalgesia is markedly prolonged, a characteristic feature of both type I [4; 29; 33; 50; 57; 69; 73] and type II [7; 8] hyperalgesic priming. However, the mechanism underlying priming induced by sumatriptan resembled type I rather than type II priming. Thus, unlike type II priming, which develops within a matter of a few hours [7; 8], that induced by sumatriptan required 3 days to develop, characteristic of type I priming [4; 12]. A second difference was that, unlike type II priming, in which the prolongation of PGE2 hyperalgesia is PKA dependent, that induced by sumatriptan was PKCε dependent, another characteristic feature of type I priming [4; 70]. Furthermore, characteristic of type I [30], but not type II priming [7; 8], that induced by sumatriptan was reversed by administration of a protein translation inhibitor to the peripheral terminal of the primed nociceptor. Moreover, the ability of pertussis toxin to inhibit the induction of sumatriptan-induce priming is also characteristic of type I priming [33; 52; 57] and type II priming induced by CPA [8], but not type II priming induced by DAMGO [7]. In addition, sumatriptan is unable to produce priming in rats in which IB4-positive (non-peptidergic), but not IB4-negative (peptidergic), nociceptors have been destroyed, a further feature of type I [29] but not type II priming [7; 8]. Finally, unlike the type II priming induced by mu-opioid or A1-adenosine receptor agonists [7; 8], which occurs in female as well as male rats, sumatriptan induced priming only in males. Thus, we conclude that while the triptans act at Gi-protein coupled receptors, which are involved in pain chronification as well as analgesia, when tested in a preclinical model, unlike other Gi-protein coupled receptor agonists, sumatriptan induces type I priming. However, the downstream signaling pathway through which an agonist at a Gi-protein coupled receptor induces type I priming remains to be established. In Fig. 9 the signaling pathways that participate in sumatriptan-induced type I hyperalgesic priming are illustrated.
Figure 9. Schematic representation of the signaling pathways involved in sumatriptan-induced type I hyperalgesic priming.

As shown in “A”, the administration of sumatriptan (a 5-HT1B/D receptor agonist), applied at the terminal of the IB4-positive nociceptor, triggers the events that will lead to mechanical hyperalgesia and the development of type I hyperalgesic priming. Activation of 5-HT1B and 5-HT1D receptors by administration of sumatriptan and the following activation of G-protein αi subunit (Gαi), ultimately producing neuroplastic changes that are expressed as prolongation of the PGE2-induced hyperalgesia. B. PGE2-induced hyperalgesia, which is dependent only on PKA in the normal state [34], in the primed state is prolonged due to activation of an additional pathway involving PKCε and protein translation. Abbreviations: 1B, 5-HT1B receptor subtype; 1D, 5-HT1D receptor subtype; PKCε, protein kinase C epsilon; PGE2, prostaglandin-E2.
Since the triptans act at two serotonin receptors, 5-HT1B and 5-HT1D, both of which are GPCRs, we evaluated the role that each subtype of receptor plays in the induction of hyperalgesic priming. We observed that while alone the antagonists of 5-HT1B and 5-HT1D partially attenuate the prolongation of PGE2 hyperalgesia, their co-administration completely prevented sumatriptan-induced prolongation of PGE2 hyperalgesia. Furthermore, when agonists for each of these two serotonin receptors were injected, alone, each induced prolongation of PGE2-induced hyperalgesia.
Why agonists at 5-HT1B and 5-HT1D induce type I priming, while agonists at other Gi-protein coupled receptors, namely mu-opioid and A1-adenosine, induce type II priming remains to be explained. One possible way to address the underlying differences would be to determine if repeated administration of agonists at mu- or A1-receptors induce a state whereby triptan agonists will now produce prolonged hyperalgesia, or vice versa. In preliminary experiments we observed that while the repeated administration of the A1-adenosine receptor agonist CPA induces a state whereby the mu-agonist DAMGO induces hyperalgesia, the repeated administration of the mu-opioid receptor agonist DAMGO did not induce a state whereby the CPA induced hyperalgesia (unpublished results). Given this initial complexity, even for receptors whose agonists both induce type II priming, these experiments are beyond the scope of the present analysis.
In conclusion, in a model of pain chronification, induced by agonists at the triptan receptors 5-HT1B and 5-HT1D, key to the treatment of migraine, we have demonstrated that a clinically used member of this class of agonists, sumatriptan, induces both mechanical hyperalgesia at the site of injection and type I hyperalgesic priming, in nociceptors innervating the cutaneous injection site. While our studies were executed outside of the trigeminal system, the site of migraine, the basic neurovascular unit at both spinal and trigeminal levels is innervated by nociceptors that contain 5-HT1B and 5-HT1D [13; 15; 46; 61; 71; 72; 94]. Moreover, support for shared mechanisms in spinal and trigeminal distributions comes from both the presence of a migraine variant, abdominal migraine [20], which has been reported to be treatable with triptans in some patients [54], as well as the observation that migraineurs do have lowered threshold in extra-cranial sites [19].
Acknowledgments
This study was funded by a grant from the National Institutes of Health (NIH), NS084545.
Footnotes
Conflict of Interest: The authors declare no competing financial interests.
Contributor Information
Dioneia Araldi, Email: Dioneia.Araldi@ucsf.edu, Departments of Medicine and Oral Surgery, and Division of Neuroscience, University of California at San Francisco, 513 Parnassus Avenue, San Francisco, CA 94143, USA.
Luiz F. Ferrari, Email: Luiz.Ferrari@ucsf.edu, Departments of Medicine and Oral Surgery, and Division of Neuroscience, University of California at San Francisco, 513 Parnassus Avenue, San Francisco, CA 94143, USA.
Jon D. Levine, Departments of Medicine and Oral Surgery, and Division of Neuroscience, University of California at San Francisco, 513 Parnassus Avenue, San Francisco, CA 94143, USA
References
- 1.Ahn AH, Basbaum AI. Where do triptans act in the treatment of migraine? Pain. 2005;115(1-2):1–4. doi: 10.1016/j.pain.2005.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aley KO, Levine JD. Role of protein kinase A in the maintenance of inflammatory pain. J Neurosci. 1999;19(6):2181–2186. doi: 10.1523/JNEUROSCI.19-06-02181.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aley KO, Martin A, McMahon T, Mok J, Levine JD, Messing RO. Nociceptor sensitization by extracellular signal-regulated kinases. J Neurosci. 2001;21(17):6933–6939. doi: 10.1523/JNEUROSCI.21-17-06933.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aley KO, Messing RO, Mochly-Rosen D, Levine JD. Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci. 2000;20(12):4680–4685. doi: 10.1523/JNEUROSCI.20-12-04680.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amrutkar DV, Ploug KB, Hay-Schmidt A, Porreca F, Olesen J, Jansen-Olesen I. mRNA expression of 5-hydroxytryptamine 1B, 1D, and 1F receptors and their role in controlling the release of calcitonin gene-related peptide in the rat trigeminovascular system. Pain. 2012;153(4):830–838. doi: 10.1016/j.pain.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 6.Angst MS, Clark JD. Opioid-induced hyperalgesia: a qualitative systematic review. Anesthesiology. 2006;104(3):570–587. doi: 10.1097/00000542-200603000-00025. [DOI] [PubMed] [Google Scholar]
- 7.Araldi D, Ferrari LF, Levine JD. Repeated Mu-Opioid Exposure Induces a Novel Form of the Hyperalgesic Priming Model for Transition to Chronic Pain. J Neurosci. 2015;35(36):12502–12517. doi: 10.1523/JNEUROSCI.1673-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Araldi D, Ferrari LF, Levine JD. Adenosine-A1 receptor agonist induced hyperalgesic priming type II. Pain. 2016;157(3):698–709. doi: 10.1097/j.pain.0000000000000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Becerra L, Bishop J, Barmettler G, Xie Y, Navratilova E, Porreca F, Borsook D. Triptans disrupt brain networks and promote stress-induced CSD-like responses in cortical and subcortical areas. J Neurophysiol. 2016;115(1):208–217. doi: 10.1152/jn.00632.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bigal ME, Lipton RB. Excessive acute migraine medication use and migraine progression. Neurology. 2008;71(22):1821–1828. doi: 10.1212/01.wnl.0000335946.53860.1d. [DOI] [PubMed] [Google Scholar]
- 11.Bigal ME, Lipton RB. Overuse of acute migraine medications and migraine chronification. Curr Pain Headache Rep. 2009;13(4):301–307. doi: 10.1007/s11916-009-0048-3. [DOI] [PubMed] [Google Scholar]
- 12.Bogen O, Alessandri-Haber N, Chu C, Gear RW, Levine JD. Generation of a pain memory in the primary afferent nociceptor triggered by PKCepsilon activation of CPEB. J Neurosci. 2012;32(6):2018–2026. doi: 10.1523/JNEUROSCI.5138-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonaventure P, Voorn P, Luyten WH, Leysen JE. 5HT1B and 5HT1D receptor mRNA differential co-localization with peptide mRNA in the guinea pig trigeminal ganglion. Neuroreport. 1998;9(4):641–645. doi: 10.1097/00001756-199803090-00015. [DOI] [PubMed] [Google Scholar]
- 14.Borle AB, Snowdowne KW. Measurement of intracellular free calcium in monkey kidney cells with aequorin. Science. 1982;217(4556):252–254. doi: 10.1126/science.6806904. [DOI] [PubMed] [Google Scholar]
- 15.Bruinvels AT, Landwehrmeyer B, Gustafson EL, Durkin MM, Mengod G, Branchek TA, Hoyer D, Palacios JM. Localization of 5-HT1B, 5-HT1D alpha, 5-HT1E and 5-HT1F receptor messenger RNA in rodent and primate brain. Neuropharmacology. 1994;33(3-4):367–386. doi: 10.1016/0028-3908(94)90067-1. [DOI] [PubMed] [Google Scholar]
- 16.Burch RM, Axelrod J. Dissociation of bradykinin-induced prostaglandin formation from phosphatidylinositol turnover in Swiss 3T3 fibroblasts: evidence for G protein regulation of phospholipase A2. Proc Natl Acad Sci U S A. 1987;84(18):6374–6378. doi: 10.1073/pnas.84.18.6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burstein R, Jakubowski M. Analgesic triptan action in an animal model of intracranial pain: a race against the development of central sensitization. Ann Neurol. 2004;55(1):27–36. doi: 10.1002/ana.10785. [DOI] [PubMed] [Google Scholar]
- 18.Burstein R, Levy D, Jakubowski M. Effects of sensitization of trigeminovascular neurons to triptan therapy during migraine. Rev Neurol (Paris) 2005;161(6-7):658–660. doi: 10.1016/s0035-3787(05)85109-4. [DOI] [PubMed] [Google Scholar]
- 19.Burstein R, Yarnitsky D, Goor-Aryeh I, Ransil BJ, Bajwa ZH. An association between migraine and cutaneous allodynia. Ann Neurol. 2000;47(5):614–624. [PubMed] [Google Scholar]
- 20.Carson L, Lewis D, Tsou M, McGuire E, Surran B, Miller C, Vu TA. Abdominal migraine: an under-diagnosed cause of recurrent abdominal pain in children. Headache. 2011;51(5):707–712. doi: 10.1111/j.1526-4610.2011.01855.x. [DOI] [PubMed] [Google Scholar]
- 21.Chen L, Malarick C, Seefeld L, Wang S, Houghton M, Mao J. Altered quantitative sensory testing outcome in subjects with opioid therapy. Pain. 2009;143(1-2):65–70. doi: 10.1016/j.pain.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi JI, Koehrn FJ, Sorkin LS. Carrageenan induced phosphorylation of Akt is dependent on neurokinin-1 expressing neurons in the superficial dorsal horn. Mol Pain. 2012;8:4. doi: 10.1186/1744-8069-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chu LF, Angst MS, Clark D. Opioid-induced hyperalgesia in humans: molecular mechanisms and clinical considerations. Clin J Pain. 2008;24(6):479–496. doi: 10.1097/AJP.0b013e31816b2f43. [DOI] [PubMed] [Google Scholar]
- 24.Classey JD, Bartsch T, Goadsby PJ. Distribution of 5-HT(1B), 5-HT(1D) and 5-HT(1F) receptor expression in rat trigeminal and dorsal root ganglia neurons: relevance to the selective anti-migraine effect of triptans. Brain Res. 2010;1361:76–85. doi: 10.1016/j.brainres.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 25.Dahlof CG, Dodick D, Dowson AJ, Pascual J. How does almotriptan compare with other triptans? A review of data from placebo-controlled clinical trials. Headache. 2002;42(2):99–113. doi: 10.1046/j.1526-4610.2002.02025.x. [DOI] [PubMed] [Google Scholar]
- 26.De Felice M, Ossipov MH, Wang R, Dussor G, Lai J, Meng ID, Chichorro J, Andrews JS, Rakhit S, Maddaford S, Dodick D, Porreca F. Triptan-induced enhancement of neuronal nitric oxide synthase in trigeminal ganglion dural afferents underlies increased responsiveness to potential migraine triggers. Brain. 2010;133(Pt 8):2475–2488. doi: 10.1093/brain/awq159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Felice M, Ossipov MH, Wang R, Lai J, Chichorro J, Meng I, Dodick DW, Vanderah TW, Dussor G, Porreca F. Triptan-induced latent sensitization: a possible basis for medication overuse headache. Ann Neurol. 2010;67(3):325–337. doi: 10.1002/ana.21897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diener HC, Limmroth V. Medication-overuse headache: a worldwide problem. Lancet Neurol. 2004;3(8):475–483. doi: 10.1016/S1474-4422(04)00824-5. [DOI] [PubMed] [Google Scholar]
- 29.Dina OA, Khasar SG, Gear RW, Levine JD. Activation of Gi induces mechanical hyperalgesia poststress or inflammation. Neuroscience. 2009;160(2):501–507. doi: 10.1016/j.neuroscience.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferrari LF, Bogen O, Chu C, Levine JD. Peripheral administration of translation inhibitors reverses increased hyperalgesia in a model of chronic pain in the rat. J Pain. 2013;14(7):731–738. doi: 10.1016/j.jpain.2013.01.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferrari LF, Bogen O, Levine JD. Second messengers mediating the expression of neuroplasticity in a model of chronic pain in the rat. J Pain. 2014;15(3):312–320. doi: 10.1016/j.jpain.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferrari LF, Bogen O, Reichling DB, Levine JD. Accounting for the delay in the transition from acute to chronic pain: axonal and nuclear mechanisms. J Neurosci. 2015;35(2):495–507. doi: 10.1523/JNEUROSCI.5147-13.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferrari LF, Levine E, Levine JD. Role of a novel nociceptor autocrine mechanism in chronic pain. Eur J Neurosci. 2013;37(10):1705–1713. doi: 10.1111/ejn.12145. [DOI] [PubMed] [Google Scholar]
- 34.Ferrari LF, Levine JD. Plasma membrane mechanisms in a preclinical rat model of chronic pain. J Pain. 2015;16(1):60–66. doi: 10.1016/j.jpain.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferrari MD, Goadsby PJ, Roon KI, Lipton RB. Triptans (serotonin, 5-HT1B/1D agonists) in migraine: detailed results and methods of a meta-analysis of 53 trials. Cephalalgia. 2002;22(8):633–658. doi: 10.1046/j.1468-2982.2002.00404.x. [DOI] [PubMed] [Google Scholar]
- 36.Ferrari MD, Roon KI, Lipton RB, Goadsby PJ. Oral triptans (serotonin 5-HT(1B/1D) agonists) in acute migraine treatment: a meta-analysis of 53 trials. Lancet. 2001;358(9294):1668–1675. doi: 10.1016/S0140-6736(01)06711-3. [DOI] [PubMed] [Google Scholar]
- 37.Gerhardt CC, van Heerikhuizen H. Functional characteristics of heterologously expressed 5-HT receptors. Eur J Pharmacol. 1997;334(1):1–23. doi: 10.1016/s0014-2999(97)01115-1. [DOI] [PubMed] [Google Scholar]
- 38.Ghiotto N, Sances G, Galli F, Tassorelli C, Guaschino E, Sandrini G, Nappi G. Medication overuse headache and applicability of the ICHD-II diagnostic criteria: 1-year follow-up study (CARE I protocol) Cephalalgia. 2009;29(2):233–243. doi: 10.1111/j.1468-2982.2008.01712.x. [DOI] [PubMed] [Google Scholar]
- 39.Goadsby PJ. The pharmacology of headache. Prog Neurobiol. 2000;62(5):509–525. doi: 10.1016/s0301-0082(00)00010-1. [DOI] [PubMed] [Google Scholar]
- 40.Goadsby PJ, Lipton RB, Ferrari MD. Migraine--current understanding and treatment. N Engl J Med. 2002;346(4):257–270. doi: 10.1056/NEJMra010917. [DOI] [PubMed] [Google Scholar]
- 41.Green AL, Gu P, De Felice M, Dodick D, Ossipov MH, Porreca F. Increased susceptibility to cortical spreading depression in an animal model of medication-overuse headache. Cephalalgia. 2013;34(8):594–604. doi: 10.1177/0333102413515344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hamblin MW, Metcalf MA. Primary structure and functional characterization of a human 5-HT1D-type serotonin receptor. Mol Pharmacol. 1991;40(2):143–148. [PubMed] [Google Scholar]
- 43.Hargreaves RJ, Shepheard SL. Pathophysiology of migraine--new insights. Can J Neurol Sci. 1999;26(3):S12–19. doi: 10.1017/s0317167100000147. [DOI] [PubMed] [Google Scholar]
- 44.Hay JL, White JM, Bochner F, Somogyi AA, Semple TJ, Rounsefell B. Hyperalgesia in opioid-managed chronic pain and opioid-dependent patients. J Pain. 2009;10(3):316–322. doi: 10.1016/j.jpain.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 45.Headache Classification C, Olesen J, Bousser MG, Diener HC, Dodick D, First M, Goadsby PJ, Gobel H, Lainez MJ, Lance JW, Lipton RB, Nappi G, Sakai F, Schoenen J, Silberstein SD, Steiner TJ. New appendix criteria open for a broader concept of chronic migraine. Cephalalgia. 2006;26(6):742–746. doi: 10.1111/j.1468-2982.2006.01172.x. [DOI] [PubMed] [Google Scholar]
- 46.Hou M, Kanje M, Longmore J, Tajti J, Uddman R, Edvinsson L. 5-HT(1B) and 5-HT(1D) receptors in the human trigeminal ganglion: co-localization with calcitonin gene-related peptide, substance P and nitric oxide synthase. Brain Res. 2001;909(1-2):112–120. doi: 10.1016/s0006-8993(01)02645-2. [DOI] [PubMed] [Google Scholar]
- 47.Humphrey PP. The discovery of a new drug class for the acute treatment of migraine. Headache. 2007;47(1):S10–19. doi: 10.1111/j.1526-4610.2007.00672.x. [DOI] [PubMed] [Google Scholar]
- 48.Johnson JA, Gray MO, Chen CH, Mochly-Rosen D. A protein kinase C translocation inhibitor as an isozyme-selective antagonist of cardiac function. J Biol Chem. 1996;271(40):24962–24966. doi: 10.1074/jbc.271.40.24962. [DOI] [PubMed] [Google Scholar]
- 49.Johnston MM, Rapoport AM. Triptans for the management of migraine. Drugs. 2010;70(12):1505–1518. doi: 10.2165/11537990-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 50.Joseph EK, Bogen O, Alessandri-Haber N, Levine JD. PLC-beta 3 signals upstream of PKC epsilon in acute and chronic inflammatory hyperalgesia. Pain. 2007;132(1-2):67–73. doi: 10.1016/j.pain.2007.01.027. [DOI] [PubMed] [Google Scholar]
- 51.Joseph EK, Chen X, Bogen O, Levine JD. Oxaliplatin acts on IB4-positive nociceptors to induce an oxidative stress-dependent acute painful peripheral neuropathy. J Pain. 2008;9(5):463–472. doi: 10.1016/j.jpain.2008.01.335. [DOI] [PubMed] [Google Scholar]
- 52.Joseph EK, Levine JD. Hyperalgesic priming is restricted to isolectin B4-positive nociceptors. Neuroscience. 2010;169(1):431–435. doi: 10.1016/j.neuroscience.2010.04.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Joseph EK, Parada CA, Levine JD. Hyperalgesic priming in the rat demonstrates marked sexual dimorphism. Pain. 2003;105(1-2):143–150. doi: 10.1016/s0304-3959(03)00175-1. [DOI] [PubMed] [Google Scholar]
- 54.Kakisaka Y, Wakusawa K, Haginoya K, Uematsu M, Tsuchiya S. Abdominal migraine associated with ecchymosis of the legs and buttocks: does the symptom imply an unknown mechanism of migraine? Tohoku J Exp Med. 2010;221(1):49–51. doi: 10.1620/tjem.221.49. [DOI] [PubMed] [Google Scholar]
- 55.Kayser V, Aubel B, Hamon M, Bourgoin S. The antimigraine 5-HT 1B/1D receptor agonists, sumatriptan, zolmitriptan and dihydroergotamine, attenuate pain-related behaviour in a rat model of trigeminal neuropathic pain. Br J Pharmacol. 2002;137(8):1287–1297. doi: 10.1038/sj.bjp.0704979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Khasabov SG, Rogers SD, Ghilardi JR, Peters CM, Mantyh PW, Simone DA. Spinal neurons that possess the substance P receptor are required for the development of central sensitization. J Neurosci. 2002;22(20):9086–9098. doi: 10.1523/JNEUROSCI.22-20-09086.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Khasar SG, Burkham J, Dina OA, Brown AS, Bogen O, Alessandri-Haber N, Green PG, Reichling DB, Levine JD. Stress induces a switch of intracellular signaling in sensory neurons in a model of generalized pain. J Neurosci. 2008;28(22):5721–5730. doi: 10.1523/JNEUROSCI.0256-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO. A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron. 1999;24(1):253–260. doi: 10.1016/s0896-6273(00)80837-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kras JV, Weisshaar CL, Pall PS, Winkelstein BA. Pain from intra-articular NGF or joint injury in the rat requires contributions from peptidergic joint afferents. Neurosci Lett. 2015;604:193–198. doi: 10.1016/j.neulet.2015.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee M, Silverman SM, Hansen H, Patel VB, Manchikanti L. A comprehensive review of opioid-induced hyperalgesia. Pain Physician. 2011;14(2):145–161. [PubMed] [Google Scholar]
- 61.Ma QP, Hill R, Sirinathsinghji D. Colocalization of CGRP with 5-HT1B/1D receptors and substance P in trigeminal ganglion neurons in rats. Eur J Neurosci. 2001;13(11):2099–2104. doi: 10.1046/j.0953-816x.2001.01586.x. [DOI] [PubMed] [Google Scholar]
- 62.Mantyh PW, Rogers SD, Honore P, Allen BJ, Ghilardi JR, Li J, Daughters RS, Lappi DA, Wiley RG, Simone DA. Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance P receptor. Science. 1997;278(5336):275–279. doi: 10.1126/science.278.5336.275. [DOI] [PubMed] [Google Scholar]
- 63.Mao J. Opioid-induced abnormal pain sensitivity: implications in clinical opioid therapy. Pain. 2002;100(3):213–217. doi: 10.1016/S0304-3959(02)00422-0. [DOI] [PubMed] [Google Scholar]
- 64.Maroteaux L, Saudou F, Amlaiky N, Boschert U, Plassat JL, Hen R. Mouse 5HT1B serotonin receptor: cloning, functional expression, and localization in motor control centers. Proc Natl Acad Sci U S A. 1992;89(7):3020–3024. doi: 10.1073/pnas.89.7.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mercadante S, Arcuri E. Hyperalgesia and opioid switching. Am J Hosp Palliat Care. 2005;22(4):291–294. doi: 10.1177/104990910502200411. [DOI] [PubMed] [Google Scholar]
- 66.Nishiguchi J, Sasaki K, Seki S, Chancellor MB, Erickson KA, de Groat WC, Kumon H, Yoshimura N. Effects of isolectin B4-conjugated saporin, a targeting cytotoxin, on bladder overactivity induced by bladder irritation. Eur J Neurosci. 2004;20(2):474–482. doi: 10.1111/j.1460-9568.2004.03508.x. [DOI] [PubMed] [Google Scholar]
- 67.Oladosu FA, Maixner W, Nackley AG. Alternative Splicing of G Protein-Coupled Receptors: Relevance to Pain Management. Mayo Clin Proc. 2015;90(8):1135–1151. doi: 10.1016/j.mayocp.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Olesen J, Burstein R, Ashina M, Tfelt-Hansen P. Origin of pain in migraine: evidence for peripheral sensitisation. Lancet Neurol. 2009;8(7):679–690. doi: 10.1016/S1474-4422(09)70090-0. [DOI] [PubMed] [Google Scholar]
- 69.Parada CA, Reichling DB, Levine JD. Chronic hyperalgesic priming in the rat involves a novel interaction between cAMP and PKCepsilon second messenger pathways. Pain. 2005;113(1-2):185–190. doi: 10.1016/j.pain.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 70.Parada CA, Yeh JJ, Joseph EK, Levine JD. Tumor necrosis factor receptor type-1 in sensory neurons contributes to induction of chronic enhancement of inflammatory hyperalgesia in rat. Eur J Neurosci. 2003;17(9):1847–1852. doi: 10.1046/j.1460-9568.2003.02626.x. [DOI] [PubMed] [Google Scholar]
- 71.Pierce PA, Xie GX, Levine JD, Peroutka SJ. 5-Hydroxytryptamine receptor subtype messenger RNAs in rat peripheral sensory and sympathetic ganglia: a polymerase chain reaction study. Neuroscience. 1996;70(2):553–559. doi: 10.1016/0306-4522(95)00329-0. [DOI] [PubMed] [Google Scholar]
- 72.Potrebic S, Ahn AH, Skinner K, Fields HL, Basbaum AI. Peptidergic nociceptors of both trigeminal and dorsal root ganglia express serotonin 1D receptors: implications for the selective antimigraine action of triptans. J Neurosci. 2003;23(34):10988–10997. doi: 10.1523/JNEUROSCI.23-34-10988.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reichling DB, Levine JD. Critical role of nociceptor plasticity in chronic pain. Trends Neurosci. 2009;32(12):611–618. doi: 10.1016/j.tins.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rios C, Gomes I, Devi LA. mu opioid and CB1 cannabinoid receptor interactions: reciprocal inhibition of receptor signaling and neuritogenesis. Br J Pharmacol. 2006;148(4):387–395. doi: 10.1038/sj.bjp.0706757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ruat M, Traiffort E, Leurs R, Tardivel-Lacombe J, Diaz J, Arrang JM, Schwartz JC. Molecular cloning, characterization, and localization of a high-affinity serotonin receptor (5-HT7) activating cAMP formation. Proc Natl Acad Sci U S A. 1993;90(18):8547–8551. doi: 10.1073/pnas.90.18.8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Silberstein SD. Practice parameter: evidence-based guidelines for migraine headache (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000;55(6):754–762. doi: 10.1212/wnl.55.6.754. [DOI] [PubMed] [Google Scholar]
- 77.Silberstein SD, Olesen J, Bousser MG, Diener HC, Dodick D, First M, Goadsby PJ, Gobel H, Lainez MJ, Lance JW, Lipton RB, Nappi G, Sakai F, Schoenen J, Steiner TJ International Headache S. The International Classification of Headache Disorders, 2nd Edition (ICHD-II)--revision of criteria for 8.2 Medication-overuse headache. Cephalalgia. 2005;25(6):460–465. doi: 10.1111/j.1468-2982.2005.00878.x. [DOI] [PubMed] [Google Scholar]
- 78.Taiwo YO, Bjerknes LK, Goetzl EJ, Levine JD. Mediation of primary afferent peripheral hyperalgesia by the cAMP second messenger system. Neuroscience. 1989;32(3):577–580. doi: 10.1016/0306-4522(89)90280-7. [DOI] [PubMed] [Google Scholar]
- 79.Taiwo YO, Heller PH, Levine JD. Mediation of serotonin hyperalgesia by the cAMP second messenger system. Neuroscience. 1992;48(2):479–483. doi: 10.1016/0306-4522(92)90507-x. [DOI] [PubMed] [Google Scholar]
- 80.Taiwo YO, Levine JD. Prostaglandin effects after elimination of indirect hyperalgesic mechanisms in the skin of the rat. Brain Res. 1989;492(1-2):397–399. doi: 10.1016/0006-8993(89)90928-1. [DOI] [PubMed] [Google Scholar]
- 81.Taiwo YO, Levine JD. Serotonin is a directly-acting hyperalgesic agent in the rat. Neuroscience. 1992;48(2):485–490. doi: 10.1016/0306-4522(92)90508-y. [DOI] [PubMed] [Google Scholar]
- 82.Tepper SJ, Rapoport AM, Sheftell FD. Mechanisms of action of the 5-HT1B/1D receptor agonists. Arch Neurol. 2002;59(7):1084–1088. doi: 10.1001/archneur.59.7.1084. [DOI] [PubMed] [Google Scholar]
- 83.Tfelt-Hansen P. Efficacy and adverse events of subcutaneous, oral, and intranasal sumatriptan used for migraine treatment: a systematic review based on number needed to treat. Cephalalgia. 1998;18(8):532–538. doi: 10.1046/j.1468-2982.1998.1808532.x. [DOI] [PubMed] [Google Scholar]
- 84.Tfelt-Hansen P, De Vries P, Saxena PR. Triptans in migraine: a comparative review of pharmacology, pharmacokinetics and efficacy. Drugs. 2000;60(6):1259–1287. doi: 10.2165/00003495-200060060-00003. [DOI] [PubMed] [Google Scholar]
- 85.Tipton AF, Tarash I, McGuire B, Charles A, Pradhan AA. The effects of acute and preventive migraine therapies in a mouse model of chronic migraine. Cephalalgia. 2015 doi: 10.1177/0333102415623070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Van Sande J, Allgeier A, Massart C, Czernilofsky A, Vassart G, Dumont JE, Maenhaut C. The human and dog 5-HT1D receptors can both activate and inhibit adenylate cyclase in transfected cells. Eur J Pharmacol. 1993;247(2):177–184. doi: 10.1016/0922-4106(93)90075-k. [DOI] [PubMed] [Google Scholar]
- 87.Vierck CJ, Jr, Kline RH, Wiley RG. Intrathecal substance p-saporin attenuates operant escape from nociceptive thermal stimuli. Neuroscience. 2003;119(1):223–232. doi: 10.1016/s0306-4522(03)00125-8. [DOI] [PubMed] [Google Scholar]
- 88.Villalon CM, Heiligers JP, Centurion D, De Vries P, Saxena PR. Characterization of putative 5-HT7 receptors mediating tachycardia in the cat. Br J Pharmacol. 1997;121(6):1187–1195. doi: 10.1038/sj.bjp.0701260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vulchanova L, Olson TH, Stone LS, Riedl MS, Elde R, Honda CN. Cytotoxic targeting of isolectin IB4-binding sensory neurons. Neuroscience. 2001;108(1):143–155. doi: 10.1016/s0306-4522(01)00377-3. [DOI] [PubMed] [Google Scholar]
- 90.Wang X, Fang Y, Liang J, Yan M, Hu R, Pan X. 5-HT7 receptors are involved in neurogenic dural vasodilatation in an experimental model of migraine. J Mol Neurosci. 2014;54(2):164–170. doi: 10.1007/s12031-014-0268-9. [DOI] [PubMed] [Google Scholar]
- 91.Wang X, Fang Y, Liang J, Yin Z, Miao J, Luo N. Selective inhibition of 5-HT7 receptor reduces CGRP release in an experimental model for migraine. Headache. 2010;50(4):579–587. doi: 10.1111/j.1526-4610.2010.01632.x. [DOI] [PubMed] [Google Scholar]
- 92.Weisshaar CL, Winkelstein BA. Ablating spinal NK1-bearing neurons eliminates the development of pain and reduces spinal neuronal hyperexcitability and inflammation from mechanical joint injury in the rat. J Pain. 2014;15(4):378–386. doi: 10.1016/j.jpain.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wiley RG, Kline RHt, Vierck CJ., Jr Anti-nociceptive effects of selectively destroying substance P receptor-expressing dorsal horn neurons using [Sar9,Met(O2)11]-substance P-saporin: behavioral and anatomical analyses. Neuroscience. 2007;146(3):1333–1345. doi: 10.1016/j.neuroscience.2007.01.066. [DOI] [PubMed] [Google Scholar]
- 94.Wotherspoon G, Priestley JV. Expression of the 5-HT1B receptor by subtypes of rat trigeminal ganglion cells. Neuroscience. 2000;95(2):465–471. doi: 10.1016/s0306-4522(99)00465-0. [DOI] [PubMed] [Google Scholar]
- 95.Zgombick JM, Borden LA, Cochran TL, Kucharewicz SA, Weinshank RL, Branchek TA. Dual coupling of cloned human 5-hydroxytryptamine1D alpha and 5-hydroxytryptamine1D beta receptors stably expressed in murine fibroblasts: inhibition of adenylate cyclase and elevation of intracellular calcium concentrations via pertussis toxin-sensitive G protein(s) Mol Pharmacol. 1993;44(3):575–582. [PubMed] [Google Scholar]
- 96.Zgombick JM, Weinshank RL, Macchi M, Schechter LE, Branchek TA, Hartig PR. Expression and pharmacological characterization of a canine 5-hydroxytryptamine1D receptor subtype. Mol Pharmacol. 1991;40(6):1036–1042. [PubMed] [Google Scholar]