

ABSTRACT
Chronic lymphocytic leukemia (CLL) is the most common form of leukemia that affects B lymphocytes in adults. Natural killer (NK) cells in CLL patients are intrinsically potent but display poor in situ effector functions. NKG2D is an activating receptor found on NK and CD8+ T cells and plays a role in immunosurveillance of CLL. In this study, we developed mono- and dual-targeting triplebodies utilizing a natural ligand for human NKG2D receptor (ULBP2) to retarget NK cells against tumor cells. Triplebodies in both formats showed better ability to induce NK-cell-dependent killing of target cells compared to bispecific counterparts. A mono-targeting triplebody ULBP2-aCD19-aCD19 successfully triggered NK cell effector functions against CLL cell line MEC1 and primary tumor cells in allogenic and autologous settings. Additionally, a dual-targeting triplebody ULBP2-aCD19-aCD33 specific for two distinct tumor-associated antigens was developed to target antigen loss variants, such as mixed lineage leukemia (MLL). Of note, this triplebody exhibited cytotoxic activity against CD19/CD33 double positive cells and retained its binding features even in the absence of one of the tumor antigens. Further, ULBP2-aCD19-aCD19 showed significant in vivo activity in immune-deficient (NSG) mouse model transplanted with CLL cell line as target cells and human immune cells as an effector population providing a proof-of-principle for this therapeutic concept.
KEYWORDS: CLL, CD19, immunoligand, NK cells, NKG2D, triplebody, ULBP2
Abbreviations
- CLL
chronic lymphocytic leukemia
- HSCT
hematopoietic stem cell transplantation
- IFNγ
interferon gamma
- IL-2
interleukin-2
- IL-15
interleukin-15
- IMAC
immobilized-metal-affinity chromatography
- KIR
killer cell immun-like receptor
- MLL
mixed lineage leukemia
- NCR
natural cytotoxicity receptor
- NK cells
natural killer cells
- NKG2D
natural killer group 2 member D
- NSG
NOD scid gamma
- PBMC
peripheral blood mononuclear cells
- scFv
single-chain variable fragment
- ULBP2
UL16-binding protein 2
Introduction
Chronic lymphocytic leukemia of B cells (B-CLL) represents the most common form of leukemia in the western world with very heterogeneous clinical prognosis.1,2 It is characterized by progressive outgrowth of monoclonal CD5+/CD19+ double positive B cells in peripheral blood, bone marrow as well as lymph nodes and spleen.3 Therapeutic monoclonal antibodies have positively contributed toward the management of CLL.2,4 A chimeric anti-CD20 antibody—rituximab—and a humanized anti-CD52 antibody—alemtuzumab—have been recently introduced for the treatment of progressive diseases.2,4 Initially, rituximab as a single agent did not improve overall response rate (ORR) in CLL; however, when combined with fludarabine, this chemo-immunotherapeutic regimen improved ORR and complete response rates (CR).2,4 Current chemo/immunotherapy and novel drugs including tyrosine kinase or Bcl-2 inhibitors result in durable remissions in a substantial proportion of patients. Nonetheless, severe side effects, drug resistance and relapse in CLL subgroups highlight a clear clinical need for novel treatment strategies.
The only curative therapy option is the hematopoietic stem cell transplantation (HSCT), for which most patients do not qualify due to old age or lack of fitness. Complete remissions in HSCT are achieved through the graft versus leukemia (GvL) effect5 mediated mainly by NK cells.6 NK cells utilize sets of “activating” and “inhibitory” receptors to sense various kinds of danger signals.7,8 The major activating receptors on NK cells include FcγRIIIa (CD16a), NKG2D and the natural cytotoxicity receptors (NCRs) such as NKp30, NKp44 and NKp46.7 The natural killer (NK) group 2 member D (NKG2D) receptor is a type-II transmembrane-anchored glycoprotein, which is found on the surface of NK cells, γ/δ T cells and cytotoxic CD8+ α/β T cells.9,10 Stimulation of NKG2D receptor directly activates NK cells and γ/δ T cells and provides costimulatory signals to CD8+ α/β T cells.9 Known ligands of the NKG2D receptor are the major histocompatibility complex class-I-related chains (MIC) A and B and the UL16-binding proteins (ULBP1-6).11
The role of NK cells in immunosurveillance of leukemia is well established, although the majority of studies show that NK cells display poor in situ effector functions in CLL patients. Outgrowth of malignant cells leading to low NK to CLL (effector:target) ratio is one of the main factors accountable for in situ resistance to NK cell effector functions.12 This is supported by ex vivo expansion of NK cells within the PBMC population from CLL patients, which enhances natural as well as antibody-dependent NK cell activity.3,12 Additionally, shedding of NK-cell-activating ligands from the surface of tumor cells is another important immune escape mechanism.1,13 Soluble NKG2D ligands including sMICA, sMICB and sULBP2 are of prognostic relevance in CLL.14
Despite these immune escape mechanisms, NK cells are the main effectors of rituximab-induced response in CLL.15 However, loss of CD20 antigen on CLL cells following rituximab treatment leads to expansion of antigen-loss variants resistant to rituximab.16-18 Functional polymorphisms of FcγRIIIa in humans are additional limitations that account for varying affinities of rituximab to the FcγRIIIA receptor and subsequent varying clinical responses in patients.19 To this end, novel recombinant proteins in various formats that exploit the basic concepts of antibodies to retarget NK cells, either via scFv (immune constructs) or via natural ligands (immunoligands) have been studied to overcome antibody-related limitations.19 We reported the first such immunoligand, ULBP2-BB4 (scFv against CD138), which successfully activated and retargeted NK cells through ULBP2 against CD138-positive multiple myeloma cells both in vitro and in vivo.20 Subsequently, two additional immunoligands in similar formats, ULBP2-aCEA (scFv against CEA)21 and ULBP2-aPSMA (scFv against PSMA)22 and immunoligands fused to other ligands,23-25 validated our approach. Several trispecific immunoconstructs targeting the FcγRIIIa receptor on NK cells have been developed and compared with their bispecific counterparts.26-28 An additional tumor-specific arm in such trispecific construct increased its avidity, leading to enhanced target cell killing.26-28 Moreover, increase in the construct size beyond the kidney exclusion limit of 65 kDa was an additional clinical advantage.29
Taking note of significant advantages of trispecific constructs over their bispecific counterparts, we designed mono-targeting and dual-targeting triplebodies (immunoligands with three binding moieties)—ULBP2-aCD19-aCD19 and ULBP2-aCD19-aCD33 with ULBP2 as a natural ligand to activate the NKG2D receptor on NK cells. ULBP2-aCD19-aCD19 contained two sets of CD19-specific scFv (aCD19) to target CD19-positive target cells with higher avidity and ULBP2-aCD19-aCD33 was developed to further validate the dual-targeting approach by a triplebody in this format. To our knowledge, these are the first triplebodies harboring a natural ligand for the NKG2D receptor to activate and retarget NK cells against antigen-positive tumor cells. Both triplebodies efficiently bound to all target moieties simultaneously and successfully retargeted NK cells to kill cancer cell lines in an antigen-specific manner. Further, ULBP2-aCD19-aCD19 could mediate NK-cell-dependent killing of primary CLL cells both in allogenic and autologous settings. Finally, ULBP2-aCD19-aCD19 proved significant in vivo ability to activate and retarget immune cells to kill transplanted MEC1 cells in a xenograft mouse model.
Results
Expression and purification of bi- and tri-specific immunoligands
Bispecific immunoligands and triplebodies carried ULBP2 at the N-terminus and contained the ULBP2-innate signal for secretion. Igκ leader sequence at the N-terminus of control constructs (aCD19scFv and aCD33scFv) enabled their secretion into the supernatant of transfected HEK293T cells (Fig. 1A). Gly/Ser linker of total 20 amino acids—(GGGGS)4x was used to link each moiety (Fig. 1A) as well as VH and VL domains of an scFv (not shown in the figure) to confer flexibility of the immunoligands. C-terminal end of ULBP2-aCD19 (U-19), ULBP2-aCD33 (U-33), ULBP2-aCD19-aCD19 (U-19-19) and ULBP2-aCD19-aCD33 (U-19-33) consisted of a c-Myc tag for detection and a 6xHis tag used for purification (Fig. 1A). All of these constructs were efficiently expressed and secreted by HEK293T cells. Between 400 µg and 700 µg of bispecific immunoligands and triplebodies and around 1 mg of control constructs were purified from around 1 L of cell culture supernatant. The immunoligand preparations were of high purity, expected size and monomeric nature as verified by anti-c-Myc Ab mediated immunostaining (Fig. 1B) and coomassie staining (Fig. 1C). U-19 and U-33 had the expected molecular weight of ≈64 kDa and ≈58 kDa, respectively, while U-19-19 and U-19-33 had the expected molecular weight of ≈91 kDa and ≈89 kDa, respectively.
Figure 1.
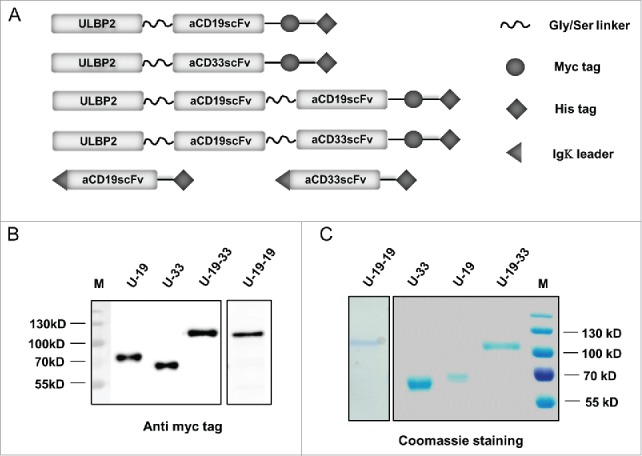
Schematic representation and expression of ULBP2-aCD19, ULBP2-aCD33, ULBP2-aCD19-aCD33 and ULBP2-aCD19-aCD19 immunoligands. (A) Cartoon illustrating bispecific immunoligands ULBP2-aCD19 (U-19) and ULBP2-aCD33 (U-33) and triplebodies ULBP2-aCD19-aCD33 (U-19-33) and ULBP2-aCD19-aCD19 (U-19-19) as well as control constructs: aCD19scFv and aCD33scFv. All four “test” immunoligands contain 15mer Gly-Ser linker (GGGGS)3x to provide flexibility and a c-Myc tag and 6xHis tag for purification/detection. An N-terminal part of ULBP2 facilitated secretion of all four immunoligands into the s/n of transfected cells. In contrast, Igκ leader sequence was placed 3′ of both control constructs (aCD19scFv and aCD33scFv) to facilitate their secretion and 6xHis tag was used for purification. (B and C) All constructs were expressed in eukaryotic cell line HEK293T and purified from the supernatant by affinity chromatography utilizing their 6xHis tag. All four (U-19, U-33, U-19-33 and U-19-19) immunoligands were separated on SDS-PAGE for protein gel blot staining (B) using anti c-Myc tag antibody or coomassie staining (C) to confirm the size and purity. Coomassie staining was also done to check the size and purity of both control constructs aCD19scFv and aCD33scFv (Data not shown).
Bispecific immunoligands and triplebodies bind specifically and simultaneously to all target moieties
MEC1 cells (CD19+) were used to assess the specificity of ULBP2-aCD19 and ULBP2-aCD19-aCD19. Their binding to MEC1 cells was detected by a c-Myc-tag-specific antibody (Fig. 2A; upper panel) as well as recombinant NKG2D receptor (Fig. 2A; lower panel), confirming the ability of immunoligands to bind to both target moieties CD19 and NKG2D simultaneously. Both immunoligands did not bind to CD19− HL60 control cells, thereby showing their antigen specificity (Fig. 2A; right “HL60”). Moreover, pre-incubating MEC1 cells with molar excess of the control construct aCD19scFv completely abolished the binding of both immunoligands (Fig. 2A; left “MEC1”) confirming the antigen-dependent specificities of both immunoligands.
Figure 2.
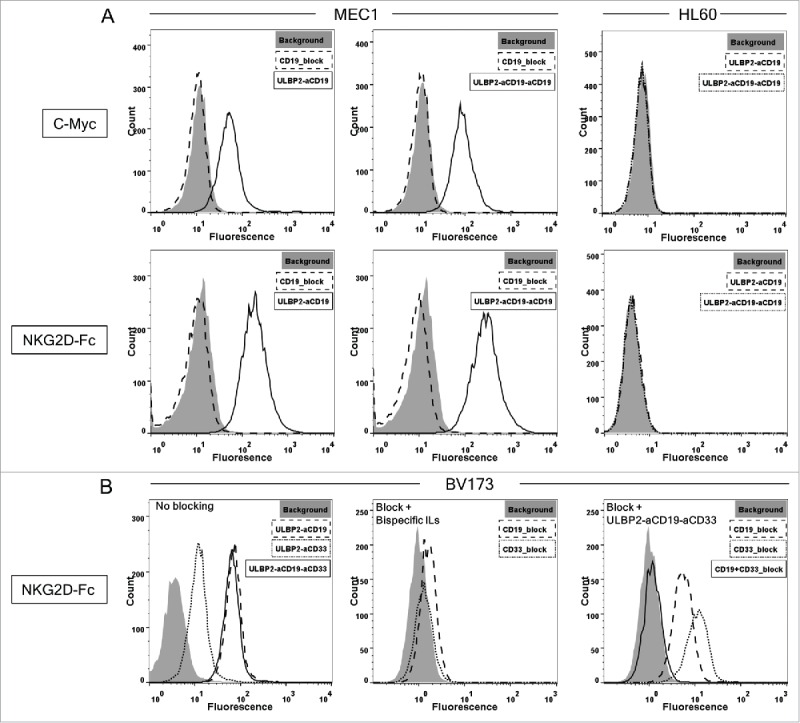
Specificity of ULBP2-aCD19, ULBP2-aCD33, ULBP2-aCD19-aCD33 and ULBP2-aCD19-aCD19 immunoligands to their respective target moieties. (A) CD19+ (MEC1) and CD19− (HL60) cell lines were incubated with 10 µg/mL of ULBP2-aCD19 or ULBP2-aCD19-aCD19 and binding was detected by either anti c-Myc tag antibody (upper panel—“c-Myc”) or recombinant human NKG2D receptor (lower panel—“NKG2D”) where the latter confirms that immunoligands can bind to CD19 and NKG2D simultaneously. Gray-filled area depicts the background staining. Pre-blocking of CD19 antigen by anti-CD19scFv (20 µg/mL) inhibited the binding of immunoligands (dashed line—“MEC1”). (B) CD19+CD33+ cell line BV173 was incubated with 10 µg/mL of ULBP2-aCD19, ULBP2-aCD33 and ULBP2-aCD19-aCD33 and binding was detected by recombinant human NKG2D receptor (“No blocking”). Pre-blocking of CD19 or CD33 antigen by respective blocking construct (20 µg/mL) prevented the binding of ULBP2-aCD19 and ULBP2-aCD33, respectively (“Block + Bispecific ILs”) but not of ULBP2-aCD19-aCD33 (“Block + ULBP2-aCD19-aCD33”). Only simultaneous blocking of both CD19 and CD33 antigens on BV173 cells could completely inhibit the binding of ULBP2-aCD19-aCD33 (“Block + ULBP2-aCD19-aCD33”).
Binding ability of ULBP2-aCD19-aCD33 was assessed using the double positive CD19+CD33+ BV173 cell line. Binding of ULBP2-aCD19-aCD33 and both bispecific counterparts was detected by recombinant NKG2D receptor (Fig. 2B; left). Lower binding of ULBP2-aCD33 to BV173 cells corresponds with low amount of CD33 antigens on this cell line compared to CD19 surface expression (Fig. S3 “BV173”). Pre-blocking of CD19 and CD33 antigens inhibited the binding of respective bispecific immunoligand, further approving their antigen specificities (Fig. 2B; middle). Dual specificity of ULBP2-aCD19-aCD33 to both tumor antigens (CD19 and CD33) was checked by pre-blocking of one or both antigens with their respective control construct (aCD19scFv and/or aCD33scFv). Partial blocking by either of the control constructs but complete blocking by both aCD19scFv and aCD33scFv verified that ULBP2-aCD19-aCD33 retained its individual specificity to all three target moieties including both antigens on BV173 cells (Fig. 2B; right). These individual specificities were independently assessed and proven by incubating ULBP2-aCD19-aCD33 to CD19+ MEC1 cells and detecting with recombinant CD33 and NKG2D receptors (Fig. 3, upper right). ULBP2-aCD33 failed to bind to CD19+ and CD33− MEC1 cells (Fig. 3, lower right), and binding of ULBP2-aCD19 was only detected by NKG2D receptor (Fig. 3, upper left). These experiments clearly showed that ULBP2-aCD19-aCD33 could simultaneously bind to all three binding partners.
Figure 3.
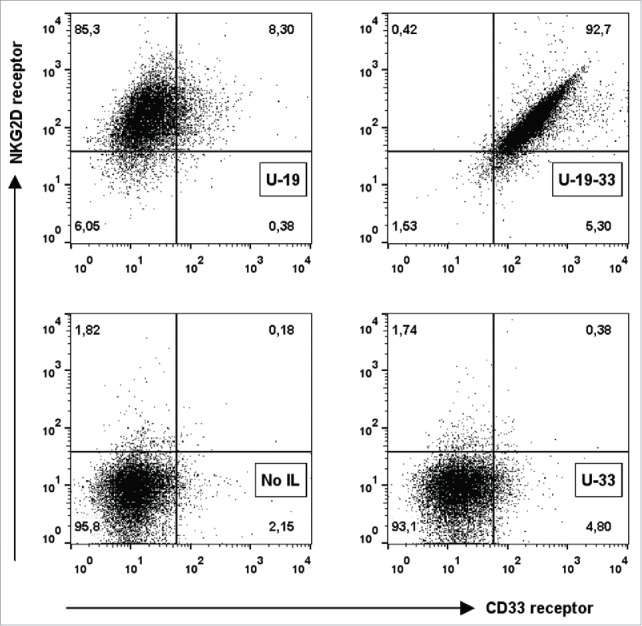
Simultaneous and specific antigen binding of a triplebody ULBP2-aCD19-aCD33. Simultaneous binding of ULBP2-aCD19-aCD33 to all three target moieties CD19, CD33 and NKG2D was detected. CD19+ MEC1 cell line was incubated with ULBP2-aCD19-aCD33 and its CD19 specific binding to MEC1 cells was detected using recombinant human NKG2D-Fc and CD33-FLAG receptors followed by AF647 labeled anti-Fc and PE labeled anti-FLAG antibodies, respectively (U-19-33). Binding of ULBP2-aCD19 to CD19 on MEC1 cells was detected only using NKG2D-Fc as it lacked anti-CD33scFv (U-19). ULBP2-aCD33 was used as a negative control that failed to bind CD19+ MEC1 cells (U-33) whereas NKG2D-Fc and CD33-FLAG background binding was minimal (No IL).
Triplebodies show better NK-cell-dependent killing of CLL and MLL target cells compared to bispecific counterparts
Next, immunoligand-mediated and NK-cell-dependent antigen-specific killing of target cells was investigated using primary NK cells from healthy donors and tumor cell lines. We specifically asked whether addition of a third arm, either targeting the same (as antiCD19 scFv) or a different (as antiCD33scFv) antigen, to the bispecific immunoligand is beneficial in terms of target cell killing. For this, NK cells were purified from PBMC of healthy donors and were used as effector population against target cells at various ratios. NK cell preparations from different donors were routinely stained for surface markers to characterize NKG2D expression, NK cell activation status (CD69 expression) and purity (by CD56 and CD16 co-expression as well as NKp46 expression) and preparations with ≥ 98% purity were used for experiments. IL-15 has previously shown to upregulate NKG2D expression on NK cells and hence purified NK cells were overnight incubated with IL-2 and IL-15.20 FACS-based toxicity assay was performed by incubating target cells (MEC1 or HL60) with purified NK cells with or without 10 nM of ULBP2-aCD19 and ULBP2-aCD19-aCD19 immunoligands for 3 h at indicated effector to target (E:T) ratios. Both immunoligands enhanced NK-cell-dependent killing of MEC1 cells in E:T-ratio-dependent manner (Fig. 4A, “MEC1”). This was antigen specific as these immunoligands failed to mediate this effect against the CD19-negative HL60 cell line (Fig. 4A, “HL60”). Further, ULBP2-aCD19-aCD19-mediated killing of MEC1 cells was higher compared to that mediated by ULBP2-aCD19 at all E:T ratios. This was also seen with a dual-targeting triplebody, which was raised against two different tumor antigens, CD19 and CD33. ULBP2-aCD19-aCD33 enhanced NK-cell-dependent killing of both BV173 and SEM cell lines at least by 2-fold at all E:T ratios. The killing induced by ULBP2-aCD19-aCD33 was antigen specific (Fig. S1B) as well as higher compared to that by both bispecific counterparts (Fig. 4A, “BV173” and “SEM”). Moreover, NK cell toxicity induced by immunoligands was also dependent on antigen density on the surface of target cells. ULBP2-aCD33-induced killing was less compared to ULBP2-aCD19 for both BV173 and SEM cell lines as the expression of CD33 is several folds less than CD19 on the surface of both cell lines (Fig. S3). Further, NK cell toxicity induced by both triplebodies was strictly NKG2D receptor dependent as blocking of the receptor completely abolished their effects (Fig. S7).
Figure 4.

Enhancement of primary NK cell effector functions by bispecific immunoligands and triplebodies. Cytotox assays. Primary NK cells were purified from peripheral blood mononuclear cells (PBMC) of healthy donors by negative selection and were cultured with IL-2 (200 U/mL) and IL-15 (10 ng/mL) overnight before the experiments on the following day. For all experiments, each N represents independent healthy donor. (A, Upper panel) Purified NK cells were co-incubated with DiR labeled CD19+ (MEC1) and CD19− (HL60) cell lines at indicated effector to target (E:T) ratios either alone (•) or in presence of 10 nM ULBP2-aCD19 (▪) or ULBP2-aCD19-aCD19 (▴) immunoligand for 3 h and dead target cells were measured by 7-AAD staining on FACS. (A, lower lane) Purified NK cells were co-incubated with DiR labeled CD19+CD33+ cell lines (BV173 and SEM) at indicated effector to target (E:T) ratios either alone (•) or in presence of 100 nM ULBP2-aCD19 (▪), ULBP2-aCD33 (▾) or ULBP2-aCD19-aCD33 (▴) immunoligand for 3 h and dead target cells were measured by 7-AAD staining on FACS. For simplicity, selected statistical significances are shown in comparison with “No construct” group (*p < 0.05; **p < 0.01). Error bars indicate SEM (MEC1 (N = 4), HL60 (N = 3), BV173 (N = 5) and SEM (N = 4)). (B) Degranulation assay. Purified NK cells were co-incubated with MEC1 and SEM cells at E:T ratio of 2.5:1 either alone or in presence of indicated immunoligand for 6 h and CD107a/LAMP-1 staining within NK cells (stained and gated with anti-CD56 and anti-NKp46 antibodies) were measured to determine degranulated NK cell population. Error bars indicate SEM and ** represents p < 0.01; *** represents p < 0.001 (MEC1 (N = 3) and SEM (N = 5)) (C) ELISA-based IFNγ assay. Left: Purified NK cells were co-incubated with MEC1 cells at E:T ratio of 1:1 either alone or in presence of 10 nM immunoligand for 24 h and supernatant was collected for IFNγ detection by ELISA. IFNγ secretion by MEC1 cells (with or without immunoligand) was carefully controlled and was found to be negative (data not shown). Experiments were conducted with two independent NK donors and one example is shown where error bars indicate SEM of duplicates. (C, right) Purified NK cells were cultured in plate pre-coated with indicated immunoligands for 48 h and supernatant was collected for IFNγ detection by ELISA. Experiments were conducted with three independent NK cell donors and one example is shown where error bars indicate SEM of duplicates.
These toxicity data were in line with the independent findings of FACS-based NK cell degranulation assay. NK cells, either alone or in presence of triplebodies, were incubated with target cells at an E:T ratio of 2.5:1, and surface expression of lysosomal-associated membrane protein-1 (LAMP-1 or CD107a) on NK cells was analyzed to confirm degranulation. Both triplebodies could increase the degranulating NK cell population in response to respective target cells MEC1 and SEM (Fig. 4B). In addition, the ability of immunoligands to stimulate IFNγ production was assessed in two different settings. IFNγ production was measured following 24 h coculture of NK and MEC1 cells, either alone or in presence of immunoligands at E:T ratio of 1:1. Assessment with two independent NK cell donors showed comparable enhancement of IFNγ production by NK cells by ULBP2-aCD19 and ULBP2-aCD19-aCD19 (one representative shown; Fig. 4C, left) while MEC1 cells alone or in presence of immunoligands did not produce IFNγ (data not shown). Similarly, 48 h NK cell culture in wells pre-coated with immunoligands augmented IFNγ secretion by NK cells from three independent donors (one representative shown; Fig. 4C, right). In both experimental settings control constructs (sole moieties against the target cell antigen) failed to induce IFNγ release, suggesting that increased IFNγ levels were due to the presence of ULBP2 (NKG2D ligand) and hence NKG2D dependent (Fig. 4C).
Subsequently, toxicity assays were also performed for ULBP2-aCD19-aCD19 using primary CLL cells as target cells and NK cells from healthy donors (allogeneic setting) (Fig. 5A) or the same CLL patients (autologous setting) (Fig. 5B and C) as effector population. In allogeneic experiments, presence of ULBP2-aCD19-aCD19 increased NK-cell-dependent killing of primary CLL cells from early (Binet A—Pt #1) and late stage (Binet C—Pt #2) CLL patients in the range of 1.5-fold–2-fold at all E:T ratios (Fig. 5A). Cumulative analysis of five CLL patients also showed that the effect mediated by ULBP2-aCD19-aCD19 was significantly higher than background killing (NK) of all but one E:T ratio (1.25:1) (Fig. S6). Moreover, in accordance with previous experiments with cell lines, ULBP2-aCD19-aCD19 maintained its higher NK-cell-dependent killing capacity than ULBP2-aCD19. Since, these experiments were performed in allogeneic setting, killer cell immunoglobulin-like receptors (KIR) mismatch between effector and target cells would have contributed in target cell killing. Therefore, ability of ULBP2-aCD19-aCD19 to retarget patient's own effector cells against tumor cells was tested. In this autologous setting, presence of ULBP2-aCD19-aCD19 enhanced NK-cell-dependent killing of CLL cells from both patients (Pt #3 and #4) despite self-tolerance (Fig. 5B) and this cytotoxic effect became more prominent with increase in NK to CLL ratio (e.g. E:T = 8:1). To attain such high NK to CLL ratio within PBMC of CLL patients is unlikely due to high CLL burden. Therefore, it was of interest to test the effect of immunoligand without altering patient's innate E:T ratio. To this end, PBMCs from Pt #3 to Pt #4 were incubated alone or in presence of ULBP2-aCD19-aCD19 for 48 h and CLL cell count was measured. Both patients had Binet A stage CLL at the time of sample collection but pt #3 had higher NK:CLL ratio (0.05:1) compared to pt #4 (0.009:1). Presence of ULBP2-aCD19-aCD19 reduced CLL count within PBMC from pt #3 following 48 h, which also correlated with higher 7AAD-positive CLL cells while CLL cell count within PBMC of pt #4 remained unchanged despite the presence of immunoligand (Fig. S2B).
Figure 5.

ULBP2-aCD19-aCD19 mediates NK-cell-dependent killing of primary CLL cells in allogenic and autologous settings. (A) Allogeneic setting. Purified NK cells from healthy donors were co-incubated with DiR labeled primary CLL cells either alone (•) or in presence of 10 nM ULBP2-aCD19 (▪) or ULBP2-aCD19-aCD19 (▴) immunoligands for 3 h and dead target cells were measured by 7-AAD staining on FACS. Two representative results (Pt #1 and Pt #2) from four independent experiments with different CLL patients are shown. NK cells for each experiment were obtained from different healthy donors. (B) Autologous setting. NK cells were purified from PBMC of CLL patients (Pt #3 and Pt #4) by negative selection and were cultured with IL-2 (200 U/mL) and IL-15 (10 ng/mL) overnight and on the next day were co-incubated with DiR labeled CLL cells from the same patient either alone (•) or in presence of 10 nM ULBP2-aCD19-aCD19 (▴) for 3 h before measuring 7-AAD staining on FACS. (C) Innate NK:CLL ratio. Isolated PBMC from CLL patients (Pt #3 and Pt #4) were cultured alone or in presence of 10 nM ULBP2-aCD19-aCD19 for 48 h and CLL cell count was determined by gating for CD20+/CD5+ CLL cells.
ULBP2-aCD19-aCD19 prevents tumor growth in MEC1 xenograft mouse model
Finally, antitumor activity of ULBP2-aCD19-aCD19 (U-19-19) was assessed in MEC1 xenografted NSG mice (Fig. 6). MEC1 cells were transplanted by a single subcutaneously (s.c.) injection to all mice (N = 21) and 2 d later isolated peripheral blood mononuclear cells (PBMC) from a healthy donor were injected either alone (N = 4) or along with a nonspecific construct (N = 3) or ULBP2-aCD19-aCD19 (N = 7) via tail vein while control mice were left untreated (N = 7). Tumor growth was monitored by measuring the tumor volume periodically and the status until day 28 is shown in Fig. 6A. Treatment with PBMC either alone or with an irrelevant construct (PBMC ± U-PSMA) delayed the tumor occurrence and growth compared to the control group (no treatment) mice; however, these differences were not significant at any given time point. On the other hand, treatment with PBMC and ULBP2-aCD19-aCD19 (PBMC + U-19-19) completely restricted the tumor growth in these mice and this effect was significant compared to the “no treatment” group (Fig. 6A). Tumor growth was monitored for the total duration of 51 d following transplantation and mice were either sacrificed upon reaching 1,000 m3 tumor volume or censored at the end of the study in case of no tumor sign (Fig. 6B). Despite late onset of tumor in “PBMC ± U-PSMA” mice, progression in these mice was quicker than “no treatment” group. All the mice in both of these groups (total N =14) were sacrificed between day 28 and day 44 following tumor transplantation with no significant difference (p = 0.56). In contrast, 7/7 mice in “PBMC + U-19-19” group survived without any evident tumor sign throughout the study and were censored at the end. Interestingly, tumor occurrence and initial progression in three mice treated with PBMC + ULBP2-PSMA (non-specific construct) was delayed compared to that in four mice treated with PBMC alone (Fig. 6C). However, this initial discrepancy was not maintained as evident by the quicker subsequent progression of tumor in “PBMC + U-PSMA” group and comparable outcome within this control group (Fig. 6D).
Figure 6.

Antitumor activity of ULBP2-aCD19-aCD19 in an immunodeficient (NSG) mouse model. 5 × 106 MEC1 cells were subcutaneously (s.c.) transplanted into 21 NSG mice. Two days later, 4 × 105 PBMC from a healthy donor together with 15 µg of ULBP2-aCD19-aCD19 were injected (i.v.) into seven mice while another seven mice were left untreated (control group). Remaining seven mice were either treated with 4 × 105 PBMC alone (four mice) or in combination with 15 µg of a non-specific immunoligand ULBP2-aPSMA (three mice). (A) Tumor growth for each mouse was measured at regular interval using the formula for tumor volume (L × W × H/2). Results show the measurements until day 28 (when all the mice were alive) and are depicted as mean (SEM) of seven mice in “no treatment” (•), “PBMC ± U-PSMA” (▪) and “ULBP2 + U-19-19” (▴) group, respectively. For simplicity, statistical significance between “No treatment” and “PBMC + U-19-19” groups are shown. (*p < 0.05; **p < 0.01). (B) Mice were sacrificed when the tumor volume reached 1,000 mm3. The study was continued for 51 d since the tumor challenge with no signs of tumor development in “U-19-19” treatment group and mice in this group were censored at the end of the study. Survival curve of “PBMC + U-19-19” group was significantly different from “PBMC ± U-PSMA” (p = 0.0003) and “No treatment” group (p = 0.0002) while there was no significant difference between “PBMC ± U-PSMA” and “No treatment” groups (p = 0.568). (Two tailed p value < 0.0167 was considered as significant; 0.0167 was Bonferroni-corrected threshold). (C) Tumor size in individual mouse at day 32 showing late onset of tumor growth in mice treated with PBMC + U-PSMA (non-specific immunoligand; ▴) compared to PBMC (▪) treated mice (*p < 0.05; **p < 0.01). (D) Days required for tumor volume to reach 1,000 mm3 in individual mouse in all control groups. Despite late onset of tumor growth in three mice within “PBMC + U-PSMA” (▴) group, tumor in these mice progressed much quicker and required similar time-point as “no treatment” (•) and “PBMC” (▪) groups to reach 1,000 mm3.
Discussion
In this study, we reported triplebodies ULBP2-aCD19-aCD19 and ULBP2-aCD19-aCD33 with targeting of either a single antigen (CD19) or two different antigens (CD19 and CD33), respectively. Both immunoligands utilized a natural ligand ULBP2 to retarget NK cells via NKG2D receptor against respective tumor cell lines in an antigen-specific manner. Main findings of the study confirmed the superior cytotoxic potential of triplebody format over its bispecific counterpart. ULBP2-aCD19-aCD19 further showed promising induction of NK-cell-dependent killing of primary CLL cells and transplanted CLL cell line in NSG mice, thereby approving its further preclinical development.
Accumulating evidences have been reviewed showing the importance of NKG2D receptor in tumor immune surveillance and in designing novel immunotherapeutic approaches.19,30 So far, these novel approaches mainly involved bispecific recombinant proteins by our group and others to exploit NKG2D-dependent NK cell activation against tumor.19-21 In this study, our strategy was to design NKG2D-triggering triplebodies that involved targeting of CD19 antigen or CD19/CD33 in combination. ULBP2-aCD19-aCD19 targets CD19 on CLL cells by two subsequent antiCD19 scFvs separated by 20 amino acid long Gly/Ser linker. CD19, a 95 kDa transmembrane glycoprotein, is an attractive and a clinically validated target for antibody-derived therapeutic agents against malignant B cells such as CLL.31-34 It is exclusively expressed on all developmental stages of B cells except on terminally differentiated plasma cells and unlike CD20 is not shed from the cell surface upon treatment.31,32
A CD19-specific triplebody in a single polypeptide format targeting CD16 receptor on effector cells was previously developed.35 This triplebody displayed greater avidity for CD19+ target cells and subsequent enhanced cytotoxic potential compared to the bispecific counterpart.35 In this study, we also showed for both triplebodies higher NK-cell-dependent killing of all respective target cell lines (MEC1, BV173 or SEM) at all E:T ratios when compared to the bispecific counterparts. Interestingly, using primary CLL cells as targets, we observed that ULBP2-aCD19-aCD33 was more effective than ULBP2-aCD19, although CLL cells (CD19+) do not express CD33 (Fig. S4). One explanation for this enhanced effect is the stronger and qualitatively more effective synapse formation between NK and target cells owing to an additional antiCD19 or antiCD33 scFv in the triplebody. In addition to stronger NKG2D stimulation by triplebodies, this may have facilitated interaction between other NK cell activating receptors on NK cells and their cognate ligands on target cells. A further advantage of a dual-targeting format is its ability to bind target cells even if one of the antigens is lost from the surface. Antigen loss following a targeted therapy is observed in both CLL and MLL patients.17,18,36 As a proof-of-principle, ULBP2-aCD19-aCD33 retained its specificity for target cells even if CD19 or CD33 antigen was pre-blocked on the surface of BV173 to mimic antigen loss variants. This feature of a dual-targeting format can be utilized to generate dual-targeting triplebodies for CLL or any other malignancies.
We further demonstrated that immunoligands induced IFNγ secretion by NK cells only in the presence of target cells or when immobilized to the wells, making non-specific NK cell activation rather unlikely. Such NK cell activation was strictly NKG2D dependent, as control constructs lacking ULBP2 could not induce IFNγ secretion from NK cells. This was in line with our previous work with the bispecific immunoligands ULBP2-BB4 and ULBP2-PSMA, both having similar format to the immunoligands used in this study, while, shortening of Gly/Ser linker to a 2mer (Gly-Ser) in ULBP2-PSMA could induce IFNγ even in soluble form.20,22
Purified NK cells used for in vitro experiments in this study were primed with IL-15, as resting NK cells were found to be less responsive to NKG2D stimulation by immunoligands (data not shown). It is well established that IL-15, a growth factor responsible for NK cell survival and differentiation, also induces NKG2D surface expression on NK cells and primes key components of NKG2D signaling pathway such as DAP10.37,38 In contrast to the short in vitro experiments with purified NK cells, the animal study with isolated PBMC was not dependent on prior or external IL-15 priming. ULBP2-aCD19-aCD19 when injected along with freshly isolated resting human PBMC completely abolished MEC1 tumor growth in all seven mice. Among PBMC, T cells and monocytes are the main source of IL-2 and IL-15, respectively, which may have augmented the activity of the immunoligand. Moreover, involvement of other NKG2D-positive effector cells cannot be ruled out. We previously reported the activation of NKT and T cells from spleen and peripheral blood of a syngeneic mouse model by ULBP2-aCEA immunoligand.21 Although, γ/δ T cells represent a small subset of T cells, NKG2D co-stimulation on Vγ9Vδ2 T cells promotes secretion of IFNγ, TNFα and cytotoxicity.39 Recently, recombinant antibodies could successfully retarget γ/δ T cell effector functions against HER2-positive tumor cells.40,41 Similarly, co-stimulation of NKG2D on CD8+ T cells decreased expression of anti-inflammatory cytokines IL-10, IL-9, IL-13 while induced cytotoxicity as well as expression of pro-inflammatory cytokines including IFNγ and TNFα.42,43 Taking together, we believe that ULBP2-aCD19-aCD19 not only stimulated NK cells but might have also retargeted NKT, γ/δ T and CD8+ T cells in mice to promote pro-inflammatory environment and deliver direct toxicity to target cells. This effect was strictly dependent on ULBP2-aCD19-aCD19 as PBMC alone or with non-specific immunoligand (ULBP2-PSMA) delayed the tumor occurrence but failed to restrict it. Interestingly, tumor occurrence and initial progression in three mice treated with PBMC + ULBP2-PSMA was delayed compared to four mice treated with PBMC alone. Non-specific construct used in this experiment was ULBP2-PSMA with a short 2mer (Gly-Ser) linker that requires antigen-positive cells to activate and degranulate NK cells but can induce IFNγ secretion from NK cells even in soluble form.22 IFNγ is a pro-inflammatory cytokine with direct antitumor and diverse immune stimulatory properties.44 In addition, the effect of this immunoligand on other NKG2D-positive immune cells (NKT, γ/δ T cells and CD8+ α/β T cells) is not studied. It is possible that ULBP2-PSMA induced IFNγ secretion by NK cells and/or activation of other NKG2D-positive immune cells may have contributed to delayed tumor growth which eventually progressed in the absence of NK cell degranulation.
ULBP2-aCD19-aCD19 showed promising antitumor activity against primary CLL cells, both in allogeneic and autologous settings. An allogeneic setting takes advantage of incompatibility between KIRs on (healthy) donor NK cells and MHC class I on target cells.45 ULBP2-aCD19-aCD19 could successfully exploit this NK-CLL incompatibility by enhancing NK-cell-dependent killing of primary CLL cells from the patients at different stages (Binet A and C shown). For autologous setting, two patients presented with Binet A stage CLL but with varying proportions of NK and CLL cell compartments were tested. ULBP2-aCD19-aCD19 could reduce the CLL cell number within the PBMC sample from patient 3 (Pt #3) who had lower CLL burden and hence higher NK:CLL ratio (0.05:1) but not from patient 4 (Pt #4) with higher CLL burden and lower NK:CLL ratio (0.009:1). In CLL patients, high burden of malignant B cells in peripheral blood naturally reduces NK cell proportion and negatively affects the natural and antibody-dependent cytotoxicity of the latter.3,12 Laprevotte et al. reported the median NK:B cells ratio as 0.02:1 in CLL patients and further showed that IL-15 could expand patients' NK cells, which in turn elevated CLL cell depletion by anti-CD20 antibodies (Rituximab and GA101).12 To check whether increasing NK:B cells ratio also enhances the killing activity of the immunoligand, we purified NK cells from both patients 3 and 4 and incubated with purified CLL cells in autologous setting. Within 3 h of co-incubation, ULBP2-aCD19-aCD19 increased NK-cell-dependent killing of CLL cells from both patients in E:T-ratio-dependent manner while bypassing self-inhibition. Together, these data suggest that ULBP2-aCD19-aCD19 is a potential therapeutic agent for CLL patients with high NK to CLL cell ratio. Thus, an application in patients with early stage CLL or those who have been treated with chemoimmunotherapy or HSCT to deplete the CLL bulk might be a promising treatment approach. Adoptive transfer of autologous or allogeneic NK cells is a well-sought therapeutic option for hematological malignancies.46,47 Ex vivo expansion of NK and NKT cells from healthy donors and CLL patients have shown exciting results, where Guven et al. reported even better expansion rates for functionally active NK cells from CLL patients despite initial low numbers.3,48 However, possible side effects of immunoligand-associated NK cell activation in patients such as autoimmune reactions toward healthy B cells should be considered. Limitations of autologous and allogeneic NK cell infusions have been discussed elsewhere49 and must be pointed out in the context of adoptive transfer of NK cells with immunoligand. Inherent NKG2D receptor expression on NK cells from CLL patients is another crucial prerequisite for this clinical option. We confirmed that NK cells from CLL patients express NKG2D receptor on the surface and the expression can be upregulated by cytokines such as IL2 alone or IL2+IL15 (Fig. S5).
Finally, soluble ligands of NKG2D present in the blood of CLL patients may interfere with NKG2D-dependent target cell killing.13 To exclude this, we analyzed target cell killing in the absence or presence of patient serum. We used two patients samples from advanced-stage CLL (Binet B and Binet C) having total 537 pg/mL and 670 pg/mL of soluble NKG2D ligands (ULBP2, MIC-A and B), respectively.13 Toxicity assays in the presence of CLL serum revealed that ULBP2-aCD19-aCD19 could enhance NK-cell-dependent MEC1 toxicity despite the NK cell inhibition by CLL serum (Fig. S8).
We believe that coating of ex vivo expanded NK cells or NK92 cell line with ULBP2-aCD19-aCD19 would further enhance their effectiveness and it would be interesting to assess the therapeutic ability of this combinatory strategy against CLL.
Materials and methods
Cloning, expression and purification of bi- and tri-specific immunoligands
Cloning, expression and purification of a bispecific immunoligand ULBP2-BB4 have been described in detail previously.20 CD19-specific single chain (scFv) was derived from 4G7 hybridoma (CD19, IgG1) and CD33-specific scFv were kind gifts by Dr Georg Fey (University of Erlangen-Nuernberg, Erlangen, Germany). The sequence of CD19 and CD33-specific scFv was optimized for better expression in eukaryotic system (OptimumGene™ algorithm, Genscript Ltd.). Optimized aCD19 and aCD33 scFv were restricted with SfiI and NotI and cloned into the eukaryotic expression vector pL (ULBP2-BB4) by replacing single chain BB4 to generate pL (ULBP2-aCD19) and pL (ULBP2-aCD33), respectively. Expression vector pL (previously designated as pMS) has been described previously.20,50 In brief, pL is a derivative of the pSecTag2 plasmid (Invitrogen) that contains the IVS/IRES-EGFP sequence of the pIRES-EGFP plasmid (Clontech). Further, both aCD19 and aCD33 scFvs were PCR amplified to replace SfiI by NotI restriction site at the 5′ end to generate respective scFv flanked by NotI sites. These PCR products were cloned into a linearized pL (ULBP2-aCD19) to generate pL (ULBP2-aCD19-aCD19) and pL (ULBP2-aCD19-aCD33). Cloning of control constructs pL (aCD19) and pL (aCD33) was accomplished by PCR amplifying each scFv, thereby adding NheI site followed by human Ig kappa (Igκ) light-chain signal peptide in front of aCD19 and aCD33 scFv sequence, respectively, and cloning of each resulting PCR product into an empty pL vector.
For expression of cloned constructs, HEK293T cells were transfected using Lipofectamine 2000 (Invitrogen) according to manufacturer's instructions. In brief, 4 µg of plasmid DNA and 10 µL of Lipofectamine were used to transfect 1 × 106 HEK293T cells in a six-well tissue-culture plate. Two days after transfection, cells were transferred to a 75 cm2 medium flask and were grown in RPMI-1640 supplemented with 200 µg/mL Zeocin (Invitrogen) for selection of stable clones. Following 2–3 weeks of selection and cultivation, stably transfected polyclonal clones were visible under fluorescence microscope, as all constructs were expressed simultaneously with enhanced green fluorescent protein (EGFP) encoded by a bicistronic mRNA. For more efficient productivity, transfected HEK293T cells were further cultivated in serum-free CD293 medium (Invitrogen) and supernatant was collected every 2–3 d for up to one month. The purification of his-tagged recombinant constructs was achieved by immobilized-metal-affinity chromatography (IMAC) using Ni-NTA-Sepharose (Qiagen) according to manufacturer's protocol. The size, purity and immunoreactivity (against the c-Myc-tag-specific monoclonal antibody) of all immunoligands were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and protein gel blotting.
Cell lines
Human embryonic kidney-derived cell line 293T (HEK293T), chronic B cell leukemia cell line MEC1, pro-B ALL-derived cell line SEM, pre-B phenotypic cell line BV173 and acute myeloid leukemia cell line HL60 were purchased from DSMZ. HEK293T was cultivated in Dulbecco´s MEM (Invitrogen), SEM was cultivated in Iscove's MDM (Invitrogen) while MEC1, HL60 and BV173 were cultivated in RPMI-1640 medium (Invitrogen) at 37°C in 5% CO2 atmosphere. All mediums were supplemented with 10% (vol/vol) heat-inactivated fetal calf serum (Invitrogen) and 50 µg/mL penicillin, 50 µg/mL streptomycin and 2 mM L-glutamine.
Primary natural killer (NK) cells and chronic lymphocytic leukemia (CLL) cells
PBMCs of healthy donors were isolated from buffy coats using Ficoll-Paques density gradient centrifugation. These PBMC preparations were used to purify NK cells by negative selection using NK cell isolation kit (Miltenyi) and autoMACS Pro Separator (Miltenyi, Bergisch-Gladbach, Germany) according to manufacturer's instructions. Purified polyclonal NK cells were cultivated overnight at 37°C in 5% CO2 atmosphere in Iscove's Modified Dulbecco's Medium (IMDM) supplemented with 10% (vol/vol) heat-inactivated fetal calf serum (Invitrogen) and 50 µg/mL penicillin, 50 µg/mL streptomycin, 2 mM L-glutamine and recombinant human IL-2 (200 U/mL) + recombinant human IL-15 (10 ng/mL). Purified NK cell preparations were stained with fluorescein isothiocyanate labeled antibodies against NKG2D (clone 1D11, Abcam), CD69 (clone FN30, BioLegend) and CD16 (clone 3G8, BioLegend) as well as PE-labeled antibodies against CD56 (clone MY31, BD), and NKp46 (clone 9E2, BioLegend). APC-conjugated antibody against CD3 (clone UCHT1, BD) was used to detect any contaminating T or NKT cells and preparations containing ≥ 98% NK cells were used for experiments. Freshly purified CLL cells were cultured overnight in Panserin 411S (Pan-Biotech, Germany). Sampling of blood samples (CLL patients and healthy donors) was approved by the local ethics committee of the University of Cologne under reference numbers 11-140 and 08-275, and donors provided written consent in accordance with the Declaration of Helsinki.
Flow cytometry
Binding of ULBP2-aCD19 and ULBP2-aCD19-aCD19 (10 µg/mL) was detected using c-Myc-tag-specific mouse monoclonal antibody, which was subsequently visualized by PE-conjugated secondary goat-anti-mouse Ab. Alternatively, bound ULBP2-aCD19 and ULBP2-aCD19-aCD19 (10 µg/mL) as well as ULBP2-aCD33 and ULBP2-aCD19-aCD33 (10 µg/mL) were also detected using recombinant human NKG2D-Fc receptor (R&D systems, Cat. 1299-NK), which was visualized using Cy3-conjugated anti-Fc antibody. For blocking experiments, cells were pre-incubated with 20 µg/mL of anti-CD19 and/or anti-CD33 scFv before incubating with bi- or tri-specific construct. For detection of simultaneous binding of a dual-targeting triplebody ULBP2-aCD19-aCD33, immunoligands were incubated with CD19+ MEC1 cells and bound immunoligands were detected using recombinant human NKG2D-Fc receptor and recombinant human CD33-FLAG (Origene, Cat. TP307023). Bound CD33-FLAG receptor was in turn detected using FLAG/DDK tag-specific PE-labeled antibody (clone poly9236, BioLegend). All incubations were carried out on ice for 30 min (and in dark for fluorochrome-conjugated antibodies) and cells were washed twice with phosphate-buffered saline (PBS) containing 0.2% bovine serum albumin (BSA) and 0.2% NaN3.
Cytotoxicity and degranulation assay
Target cell lines MEC1, HL60, BV173, SEM and primary CLL cells were labeled with membrane dye DiR (Invitrogen) according to manufacturer's instructions. Purified NK cells were co-incubated with labeled target cells in “U” bottom 96-well plates at indicated E:T ratios with or without indicated immunoligands for 3 h. Final concentration of 10 nM and 100 nM of immunoligands were used in cytotox assays with MEC1, HL60 and BV173, SEM cells as target cells, respectively. At the end of co-culture, cells were stained with 7-AAD (BioLegend) and measured by flow cytometry (Gallios, Beckman Coulter). Gating on 7-AAD-positive population within DiR-positive population indicated dead target cells in percentage (%) (Fig. S1A). For autologous cytotox assay using patient's innate NK:CLL ratio, PBMC were isolated from the peripheral blood of CLL patients and were cultured alone or in presence of 10 nM ULBP2-aCD19-aCD19 for 48 h. Fixed amount of counting beads (Thermo Fisher, Cat. C36950) were added in each sample and relative CLL cell count and 7-AAD-positive cells were determined by gating for CD20+/CD5+ CLL cells (Fig. S2A).
NK cell degranulation was assessed by flow cytometric analysis of the cell surface expression of the lysosomal protein CD107a. In brief, indicated target cell line and purified NK cells were co-incubated in “U” bottom 96-well plates at 2.5:1—E:T ratio with or without ULBP2-aCD19-aCD19 (10 nM) or ULBP2-aCD19-aCD33 (100 nM) for 6 h. After 2 h of incubation, monensin (BioLegend) was added at a final concentration of 2 µM in every well and co-culture was continued for 4 h. PE-labeled anti-human CD107a mAb (clone H4A3, BioLegend) was present in the medium throughout the 6 h incubation period, because CD107a that has been externalized by NK cells upon degranulation is rapidly re-internalized. At the end of the incubation, cells were stained with Alexa fluor 647 (AF647)-labeled anti-NKp46 (clone 9E2, BioLegend) and BV421-labeled anti-CD56 (clone HCD56, BioLegend) antibodies for 30 min on ice. Cells were then washed thrice with PBS containing 0.2 % BSA and 0.2 % NaN3 and were measured by flow cytometry. Gating on CD107a-positive cells within NKp46/CD56 double positive NK population revealed degranulated NK cells.
IFNγ detection
Primary NK cells from healthy donors were incubated overnight with IL-2 (200 U/mL) and IL-15 (10 ng/mL) in IMDM, as stated before. Next day, purified NK cells were either cultured in 96-well maxisorp plates (pre-coated with 10 µg/mL of ULBP2-aCD19, ULBP2-aCD33, ULBP2-aCD19-aCD33 or control constructs) for 48 h or co-cultured with MEC1 cells in “U” bottom 96-well plates at 1:1 E:T ratio with or without 10 nM of ULBP2-aCD19 or ULBP2-aCD19-aCD19 for 24 h. Following respective time-points, supernatant was collected and used to detect human IFNγ by ELISA according to manufacturer's protocol (BioLegend).
Xenograft tumor model
21 five-week old NSG mice were purchased from Jackson laboratories and maintained under sterile conditions. 5 × 106 MEC1 cells suspended in 100 µL HBSS buffer were injected s.c. into all 21 mice, which were randomly allocated to three groups, each consisting of seven mice. PBMCs of a single healthy donor were isolated from buffy coats using Ficoll-Paques density gradient centrifugation. Two days later, seven mice received 15 µg of ULBP2-aCD19-aCD19 along with 4 × 105 PBMC in 100 µL HBSS buffer through an intravenous (i.v.) injection while the other seven mice were further divided into two groups of four and three mice. A group of four mice received an i.v. injection of 4 × 105 PBMC alone in 100 µL HBSS buffer and other three mice received an i.v. injection of 15 µg ULBP2-PSMA (nonspecific immunoligand) along with 4 × 105 PBMC in 100 µL HBSS buffer. The remaining seven mice were left untreated and served as controls. Tumor development was measured periodically and the tumor volume was determined by the formula: (length × width × height)/2. The animals were sacrificed when the tumor volume reached 1,000 mm3 or at the end of the study period in case of no tumor sign. Animal experiments were approved from local authorities (State Northrhine-Westfalia, LANUV, approval no. 84-02.04.2012.A216).
Statistical analysis
Data were analyzed with GraphPad Prism 6 (San Diego, CA). Paired t-test was performed to compare two groups in NK cell degranulation assay. For comparisons involving three or more groups, one-way ANOVA test was performed and p values were corrected by Tukey's multiple comparisons test. Kaplan–Meier survival at fixed time points was compared using log-rank (Mantel–Cox) test. A two tailed p < 0.05 indicated statistical significance. Error bars are presented as mean plus SEM.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Anne Krüßmann and Silke Modersohn for their expert technical assistance. We thank Professor Georg Fey for many helpful discussions.
Funding
This study was supported by the Deutsche José Carreras Leukämie-Stiftung e.V. grant DJCLS R14/08 to E.P.v.S., by the Deutsche Forschungsgemeinschaft (KFO286, TP4 to E.P.v.S.) and by the Deutsche Krebshilfe (grant 109751 to E.P.v.S. and UK).
References
- 1.Shatnyeva OM, Hansen HP, Reiners KS, Sauer M, Vyas M, von Strandmann EP. DNA damage response and evasion from immunosurveillance in CLL: new options for NK cell-based immunotherapies. Front Genet 2015; 6:11; PMID:25699074; http://dx.doi.org/ 10.3389/fgene.2015.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Le Garff-Tavernier M, Decocq J, de Romeuf C, Parizot C, Dutertre CA, Chapiro E, Davi F, Debré P, Prost JF, Teillaud JL et al.. Analysis of CD16+CD56dim NK cells from CLL patients: evidence supporting a therapeutic strategy with optimized anti-CD20 monoclonal antibodies. Leukemia 2011; 25:101-9; PMID:20975664; http://dx.doi.org/ 10.1038/leu.2010.240 [DOI] [PubMed] [Google Scholar]
- 3.Guven H, Gilljam M, Chambers BJ, Ljunggren HG, Christensson B, Kimby E, Dilber MS. Expansion of natural killer (NK) and natural killer-like T (NKT)-cell populations derived from patients with B-chronic lymphocytic leukemia (B-CLL): a potential source for cellular immunotherapy. Leukemia 2003; 17:1973-80; PMID:14513047; http://dx.doi.org/ 10.1038/sj.leu.2403083 [DOI] [PubMed] [Google Scholar]
- 4.Gentile M, Vigna E, Mazzone C, Lucia E, Recchia A, Morabito L, Bisconte M, Gentile C, Morabito F. Rituximab for the treatment of patients with chronic lymphocytic leukemia. Cancer Manag Res 2010; 2:71-81; PMID:21188098; http://dx.doi.org/ 10.2147/CMAR.S5621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Veuillen C, Aurran-Schleinitz T, Castellano R, Rey J, Mallet F, Orlanducci F, Pouyet L, Just-Landi S, Coso D, Ivanov V et al.. Primary B-CLL resistance to NK cell cytotoxicity can be overcome in vitro and in vivo by priming NK cells and monoclonal antibody therapy. J Clin Immunol 2012; 32:632-46; PMID:22318393; http://dx.doi.org/ 10.1007/s10875-011-9624-5 [DOI] [PubMed] [Google Scholar]
- 6.Maki G, Hayes GM, Naji A, Tyler T, Carosella ED, Rouas-Freiss N, Gregory SA. NK resistance of tumor cells from multiple myeloma and chronic lymphocytic leukemia patients: implication of HLA-G. Leukemia 2008; 22:998-1006; PMID:18288133; http://dx.doi.org/ 10.1038/leu.2008.15 [DOI] [PubMed] [Google Scholar]
- 7.Gismondi A, Stabile H, Nisti P, Santoni A. Effector functions of natural killer cell subsets in the control of hematological malignancies. Front Immunol 2015; 6:567; PMID:26594216; http://dx.doi.org/ 10.3389/fimmu.2015.00567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol 2008; 9:495-502; PMID:18425106; http://dx.doi.org/ 10.1038/ni1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999; 285:727-9; PMID:10426993; http://dx.doi.org/ 10.1126/science.285.5428.727 [DOI] [PubMed] [Google Scholar]
- 10.Das H, Groh V, Kuijl C, Sugita M, Morita CT, Spies T, Bukowski JF. MICA engagement by human Vgamma2Vdelta2 T cells enhances their antigen-dependent effector function. Immunity 2001; 15:83-93; PMID:11485740; http://dx.doi.org/ 10.1016/S1074-7613(01)00168-6 [DOI] [PubMed] [Google Scholar]
- 11.Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol 2003; 3:781-90; PMID:14523385; http://dx.doi.org/ 10.1038/nri1199 [DOI] [PubMed] [Google Scholar]
- 12.Laprevotte E, Voisin G, Ysebaert L, Klein C, Daugrois C, Laurent G, Fournie JJ, Quillet-Mary A. Recombinant human IL-15 trans-presentation by B leukemic cells from chronic lymphocytic leukemia induces autologous NK cell proliferation leading to improved anti-CD20 immunotherapy. J Immunol 2013; 191:3634-40; PMID:23997218; http://dx.doi.org/ 10.4049/jimmunol.1300187 [DOI] [PubMed] [Google Scholar]
- 13.Reiners KS, Topolar D, Henke A, Simhadri VR, Kessler J, Sauer M, Bessler M, Hansen HP, Tawadros S, Herling M et al.. Soluble ligands for NK cell receptors promote evasion of chronic lymphocytic leukemia cells from NK cell anti-tumor activity. Blood 2013; 121:3658-65; PMID:23509156; http://dx.doi.org/ 10.1182/blood-2013-01-476606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nuckel H, Switala M, Sellmann L, Horn PA, Durig J, Duhrsen U, Küppers R, Grosse-Wilde H, Rebmann V. The prognostic significance of soluble NKG2D ligands in B-cell chronic lymphocytic leukemia. Leukemia 2010; 24:1152-9; PMID:20428196; http://dx.doi.org/ 10.1038/leu.2010.74 [DOI] [PubMed] [Google Scholar]
- 15.Ysebaert L, Gross E, Kuhlein E, Blanc A, Corre J, Fournie JJ, Laurent G, Quillet-Mary A. Immune recovery after fludarabine-cyclophosphamide-rituximab treatment in B-chronic lymphocytic leukemia: implication for maintenance immunotherapy. Leukemia 2010; 24:1310-6; PMID:20463751; http://dx.doi.org/ 10.1038/leu.2010.89 [DOI] [PubMed] [Google Scholar]
- 16.O'Brien SM, Kantarjian H, Thomas DA, Giles FJ, Freireich EJ, Cortes J, Lerner S, Keating MJ. Rituximab dose-escalation trial in chronic lymphocytic leukemia. J Clin Oncol 2001; 19:2165-70; PMID:11304768 [DOI] [PubMed] [Google Scholar]
- 17.Beers SA, French RR, Chan HT, Lim SH, Jarrett TC, Vidal RM, Wijayaweera SS, Dixon SV, Kim H, Cox KL et al.. Antigenic modulation limits the efficacy of anti-CD20 antibodies: implications for antibody selection. Blood 2010; 115:5191-201; PMID:20223920; http://dx.doi.org/ 10.1182/blood-2010-01-263533 [DOI] [PubMed] [Google Scholar]
- 18.Beum PV, Kennedy AD, Williams ME, Lindorfer MA, Taylor RP. The shaving reaction: rituximab/CD20 complexes are removed from mantle cell lymphoma and chronic lymphocytic leukemia cells by THP-1 monocytes. J Immunol 2006; 176:2600-9; PMID:16456022; http://dx.doi.org/ 10.4049/jimmunol.176.4.2600 [DOI] [PubMed] [Google Scholar]
- 19.Vyas M, Koehl U, Hallek M, von Strandmann EP. Natural ligands and antibody-based fusion proteins: harnessing the immune system against cancer. Trends Mol Med 2014; 20:72-82; PMID:24268686; http://dx.doi.org/ 10.1016/j.molmed.2013.10.006 [DOI] [PubMed] [Google Scholar]
- 20.von Strandmann EP, Hansen HP, Reiners KS, Schnell R, Borchmann P, Merkert S, Simhadri VR, Draube A, Reiser M, Purr I et al.. A novel bispecific protein (ULBP2-BB4) targeting the NKG2D receptor on natural killer (NK) cells and CD138 activates NK cells and has potent antitumor activity against human multiple myeloma in vitro and in vivo. Blood 2006; 107:1955-62; PMID:16210338; http://dx.doi.org/ 10.1182/blood-2005-05-2177 [DOI] [PubMed] [Google Scholar]
- 21.Rothe A, Jachimowicz RD, Borchmann S, Madlener M, Kessler J, Reiners KS, Sauer M, Hansen HP, Ullrich RT, Chatterjee S et al.. The bispecific immunoligand ULBP2-aCEA redirects natural killer cells to tumor cells and reveals potent anti-tumor activity against colon carcinoma. Int J Cancer 2014; 134:2829-40; PMID:24242212; http://dx.doi.org/ 10.1002/ijc.28609 [DOI] [PubMed] [Google Scholar]
- 22.Jachimowicz RD, Fracasso G, Yazaki PJ, Power BE, Borchmann P, Engert A, Hansen HP, Reiners KS, Marie M, von Strandmann EP et al.. Induction of in vitro and in vivo NK cell cytotoxicity using high-avidity immunoligands targeting prostate-specific membrane antigen in prostate carcinoma. Mol Cancer Ther 2011; 10:1036-45; PMID:21525185; http://dx.doi.org/ 10.1158/1535-7163.MCT-10-1093 [DOI] [PubMed] [Google Scholar]
- 23.Kellner C, Gunther A, Humpe A, Repp R, Klausz K, Derer S, Valerius T, Ritgen M, Brüggemann M, van de Winkel JG et al.. Enhancing natural killer cell-mediated lysis of lymphoma cells by combining therapeutic antibodies with CD20-specific immunoligands engaging NKG2D or NKp30. Oncoimmunology 2016; 5:e1058459; PMID:26942070; http://dx.doi.org/ 10.1080/2162402X.2015.1058459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kellner C, Maurer T, Hallack D, Repp R, van de Winkel JG, Parren PW, Valerius T, Humpe A, Gramatzki M, Peipp M. Mimicking an induced self phenotype by coating lymphomas with the NKp30 ligand B7-H6 promotes NK cell cytotoxicity. J Immunol 2012; 189:5037-46; PMID:23066150; http://dx.doi.org/ 10.4049/jimmunol.1201321 [DOI] [PubMed] [Google Scholar]
- 25.Peipp M, Derer S, Lohse S, Staudinger M, Klausz K, Valerius T, Gramatzki M, Kellner C. HER2-specific immunoligands engaging NKp30 or NKp80 trigger NK-cell-mediated lysis of tumor cells and enhance antibody-dependent cell-mediated cytotoxicity. Oncotarget 2015; 6:32075-88; PMID:26392331; http://dx.doi.org/ 10.18632/oncotarget.5135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schubert I, Saul D, Nowecki S, Mackensen A, Fey GH, Oduncu FS. A dual-targeting triplebody mediates preferential redirected lysis of antigen double-positive over single-positive leukemic cells. MAbs 2014; 6:286-96; PMID:24135631; http://dx.doi.org/ 10.4161/mabs.26768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schubert I, Kellner C, Stein C, Kugler M, Schwenkert M, Saul D, Mentz K, Singer H, Stockmeyer B, Hillen W et al.. A single-chain triplebody with specificity for CD19 and CD33 mediates effective lysis of mixed lineage leukemia cells by dual targeting. MAbs 2011; 3:21-30; PMID:21081841; http://dx.doi.org/ 10.4161/mabs.3.1.14057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kugler M, Stein C, Kellner C, Mentz K, Saul D, Schwenkert M, Schubert I, Singer H, Oduncu F, Stockmeyer B et al.. A recombinant trispecific single-chain Fv derivative directed against CD123 and CD33 mediates effective elimination of acute myeloid leukaemia cells by dual targeting. Br J Haematol 2010; 150:574-86; PMID:20636437; http://dx.doi.org/ 10.1111/j.1365-2141.2010.08300.x [DOI] [PubMed] [Google Scholar]
- 29.Weidle UH, Tiefenthaler G, Weiss EH, Georges G, Brinkmann U. The intriguing options of multispecific antibody formats for treatment of cancer. Cancer Genom Proteom 2013; 10:1-18; PMID:23382582 [PubMed] [Google Scholar]
- 30.Coudert JD, Held W. The role of the NKG2D receptor for tumor immunity. Semin Cancer Biol 2006; 16:333-43; PMID:16914326; http://dx.doi.org/ 10.1016/j.semcancer.2006.07.008 [DOI] [PubMed] [Google Scholar]
- 31.Robak T. Current and emerging monoclonal antibody treatments for chronic lymphocytic leukemia: state of the art. Expert Rev Hematol 2014; 7:841-57; PMID:25249370; http://dx.doi.org/ 10.1586/17474086.2014.963048 [DOI] [PubMed] [Google Scholar]
- 32.Kellner C, Bruenke J, Horner H, Schubert J, Schwenkert M, Mentz K, Barbin K, Stein C, Peipp M, Stockmeyer B et al.. Heterodimeric bispecific antibody-derivatives against CD19 and CD16 induce effective antibody-dependent cellular cytotoxicity against B-lymphoid tumor cells. Cancer Lett 2011; 303:128-39; PMID:21339041; http://dx.doi.org/ 10.1016/j.canlet.2011.01.020 [DOI] [PubMed] [Google Scholar]
- 33.Lorentzen CL, Straten PT. CD19-chimeric antigen receptor T cells for treatment of chronic lymphocytic leukaemia and acute lymphoblastic leukaemia. Scand J Immunol 2015; 82:307-19; PMID:26099639; http://dx.doi.org/ 10.1111/sji.12331 [DOI] [PubMed] [Google Scholar]
- 34.Newman MJ, Benani DJ. A review of blinatumomab, a novel immunotherapy. J Oncol Pharm Pract 2016; 22:639-45; PMID:26607163; http://dx.doi.org/ 10.1177/1078155215618770 [DOI] [PubMed] [Google Scholar]
- 35.Kellner C, Bruenke J, Stieglmaier J, Schwemmlein M, Schwenkert M, Singer H, Mentz K, Peipp M, Lang P, Oduncu F et al.. A novel CD19-directed recombinant bispecific antibody derivative with enhanced immune effector functions for human leukemic cells. J Immunother 2008; 31:871-84; PMID:18833000; http://dx.doi.org/ 10.1097/CJI.0b013e318186c8b4 [DOI] [PubMed] [Google Scholar]
- 36.Rayes A, McMasters RL, O'Brien MM. Lineage switch in MLL-rearranged infant leukemia following CD19-directed therapy. Pediatr Blood Cancer 2016; 63:1113-5; PMID:26914337; http://dx.doi.org/ 10.1002/pbc.25953 [DOI] [PubMed] [Google Scholar]
- 37.Horng T, Bezbradica JS, Medzhitov R. NKG2D signaling is coupled to the interleukin 15 receptor signaling pathway. Nat Immunol 2007; 8:1345-52; PMID:17952078; http://dx.doi.org/ 10.1038/ni1524 [DOI] [PubMed] [Google Scholar]
- 38.Zhang C, Zhang J, Niu J, Zhang J, Tian Z. Interleukin-15 improves cytotoxicity of natural killer cells via up-regulating NKG2D and cytotoxic effector molecule expression as well as STAT1 and ERK1/2 phosphorylation. Cytokine 2008; 42:128-36; PMID:18280748; http://dx.doi.org/ 10.1016/j.cyto.2008.01.003 [DOI] [PubMed] [Google Scholar]
- 39.Gogoi D, Chiplunkar SV. Targeting gamma delta T cells for cancer immunotherapy: bench to bedside. Indian J Med Res 2013; 138:755-61; PMID:24434328 [PMC free article] [PubMed] [Google Scholar]
- 40.Oberg HH, Kellner C, Gonnermann D, Peipp M, Peters C, Sebens S, Kabelitz D, Wesch D. Gammadelta T cell activation by bispecific antibodies. Cell Immunol 2015; 296:41-9; PMID:25979810; http://dx.doi.org/ 10.1016/j.cellimm.2015.04.009 [DOI] [PubMed] [Google Scholar]
- 41.Oberg HH, Peipp M, Kellner C, Sebens S, Krause S, Petrick D, Adam-Klages S, Röcken C, Becker T, Vogel I et al.. Novel bispecific antibodies increase gammadelta T-cell cytotoxicity against pancreatic cancer cells. Cancer Res 2014; 74:1349-60; PMID:24448235; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-0675 [DOI] [PubMed] [Google Scholar]
- 42.Barber A, Sentman CL. NKG2D receptor regulates human effector T-cell cytokine production. Blood 2011; 117:6571-81; PMID:21518928; http://dx.doi.org/ 10.1182/blood-2011-01-329417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Upshaw JL, Leibson PJ. NKG2D-mediated activation of cytotoxic lymphocytes: unique signaling pathways and distinct functional outcomes. Semin Immunol 2006; 18:167-75; PMID:16723257; http://dx.doi.org/ 10.1016/j.smim.2006.03.001 [DOI] [PubMed] [Google Scholar]
- 44.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 2004; 75:163-89; PMID:14525967; http://dx.doi.org/ 10.1189/jlb.0603252 [DOI] [PubMed] [Google Scholar]
- 45.Lim O, Jung MY, Hwang YK, Shin EC. Present and future of allogeneic natural killer cell therapy. Front Immunol 2015; 6:286; PMID:26089823; http://dx.doi.org/ 10.3389/fimmu.2015.00286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kater AP, van Oers MH, Kipps TJ. Cellular immune therapy for chronic lymphocytic leukemia. Blood 2007; 110:2811-8; PMID:17638850; http://dx.doi.org/ 10.1182/blood-2007-01-068932 [DOI] [PubMed] [Google Scholar]
- 47.Dahlberg CI, Sarhan D, Chrobok M, Duru AD, Alici E. Natural killer cell-based therapies targeting cancer: possible strategies to gain and sustain anti-tumor activity. Front Immunol 2015; 6:605; PMID:26648934; http://dx.doi.org/ 10.3389/fimmu.2015.00605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carlens S, Gilljam M, Chambers BJ, Aschan J, Guven H, Ljunggren HG, Christensson B, Dilber MS. A new method for in vitro expansion of cytotoxic human CD3-CD56+ natural killer cells. Hum Immunol 2001; 62:1092-8; PMID:11600215; http://dx.doi.org/ 10.1016/S0198-8859(01)00313-5 [DOI] [PubMed] [Google Scholar]
- 49.Cheng M, Chen Y, Xiao W, Sun R, Tian Z. NK cell-based immunotherapy for malignant diseases. Cell Mol Immunol 2013; 10:230-52; PMID:23604045; http://dx.doi.org/ 10.1038/cmi.2013.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stocker M, Tur MK, Sasse S, Krussmann A, Barth S, Engert A. Secretion of functional anti-CD30-angiogenin immunotoxins into the supernatant of transfected 293T-cells. Protein Expr Purif 2003; 28:211-9; PMID:12699683; http://dx.doi.org/ 10.1016/S1046-5928(02)00709-X [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.