

Abstract
Schizophrenia is a severe and highly heritable psychiatric disorder affecting approximately 1% of the population. Genome-wide association studies have identified 108 independent genetic loci with genome-wide significance but their functional importance has yet to be elucidated. Here, we develop a novel strategy based on network analysis of protein–protein interactions (PPI) to infer biological function associated with variants most strongly linked to illness risk. We show that the schizophrenia loci are strongly linked to synaptic transmission (P FWE < .001) and ion transmembrane transport (P FWE = .03), but not to ontological categories previously found to be shared across psychiatric illnesses. We demonstrate that brain expression of risk-linked genes within the identified processes is strongly modulated during birth and identify a set of synaptic genes consistently changed across multiple brain regions of adult schizophrenia patients. These results suggest synaptic function as a developmentally determined schizophrenia process supported by the illness’s most associated genetic variants and their PPI networks. The implicated genes may be valuable targets for mechanistic experiments and future drug development approaches.
Key words: genetics, pathway analysis, functional analysis, GWAS
Introduction
Schizophrenia is a severe and heritable psychiatric disorder with a lifetime risk of approximately 1%. Substantial genetic risk is conferred by a large number of common alleles with small effect sizes.1–5 The most recent large scale association study of about 150 000 individuals has identified 108 independent genetic loci linked to the disorder with genome-wide significance.5 However, the inference of affected biological pathways is a bottleneck for understanding their etiological relevance. Commonly used functional analysis approaches explore pathways for enrichment of illness-associated variants at a genome-wide level.6,7 Such analysis has recently been performed across psychiatric illnesses and has identified links to histone methylation, immune pathways and largely schizophrenia specific associations in synaptic pathways.6 However, the interpretation of etiological mechanisms and the relationship to illness risk is hampered by the large number and weak illness association of individual variants.
Therefore, functional analyses of variants with strong statistical support, such as genome-wide significance, would significantly aid interpretation but are challenging due to the small numbers of variants passing the significance threshold. For example, 2 commonly used enrichment methods did not find significantly enriched ontological categories among the 108 schizophrenia loci.5 In fact, functional analyses in the cancer field suggest that sets of genes derived statistically do not independently give insight into biological function.8 Rather, protein–protein interactions (PPI) were identified as a key element mapping genetic variation to biological processes. PPI networks have previously been associated with schizophrenia risk genes but functional analysis of such networks has thus far been limited to conventional enrichment analysis.9 For this application, inference of illness associated ontological categories depends on molecules indirectly linked to illness risk, ie, protein interaction partners of risk genes. Therefore, such analysis depends on the connectivity between the PPI network and the risk genes, which is not addressed by conventional enrichment methods. Risk genes with a high number of PPI partners within the same ontological category may lead to an overestimation of this category’s importance in relation to disease risk. Vice versa, such effects may reduce the power of identifying illness associations for other categories.
Building on substantial work by the Psychiatrics Genomics Consortium (PGC) and the consortium behind the BrainSpan atlas, we have developed a novel approach to infer risk associated ontological categories from a network of PPI and GeneOntology information. We aimed to (1) identify ontological categories linked to the 108 schizophrenia risk loci and to show that associations are not confounded by the connectivity between risk loci and the PPI network, (2) explore whether ontological categories previously linked to schizophrenia by genome-wide enrichment analyses mapped to the 108 loci, and (3) explore gene expression within identified risk categories across developmental stages and in adult schizophrenia patients. For the latter analyses, we investigated BrainSpan data comprising expression data across multiple brain areas from healthy donors from before birth until old age.10 Brain expression analyses in schizophrenia patients were based on post-mortem samples from BA9, BA10, BA46, the superior temporal cortex, the parietal cortex, the hippocampus and the associative striatum in 6 independent datasets.11–15 Such analysis may increase our understanding of the genetic underpinning of schizophrenia specific and shared biological risk mechanisms.
Methods
Network Construction
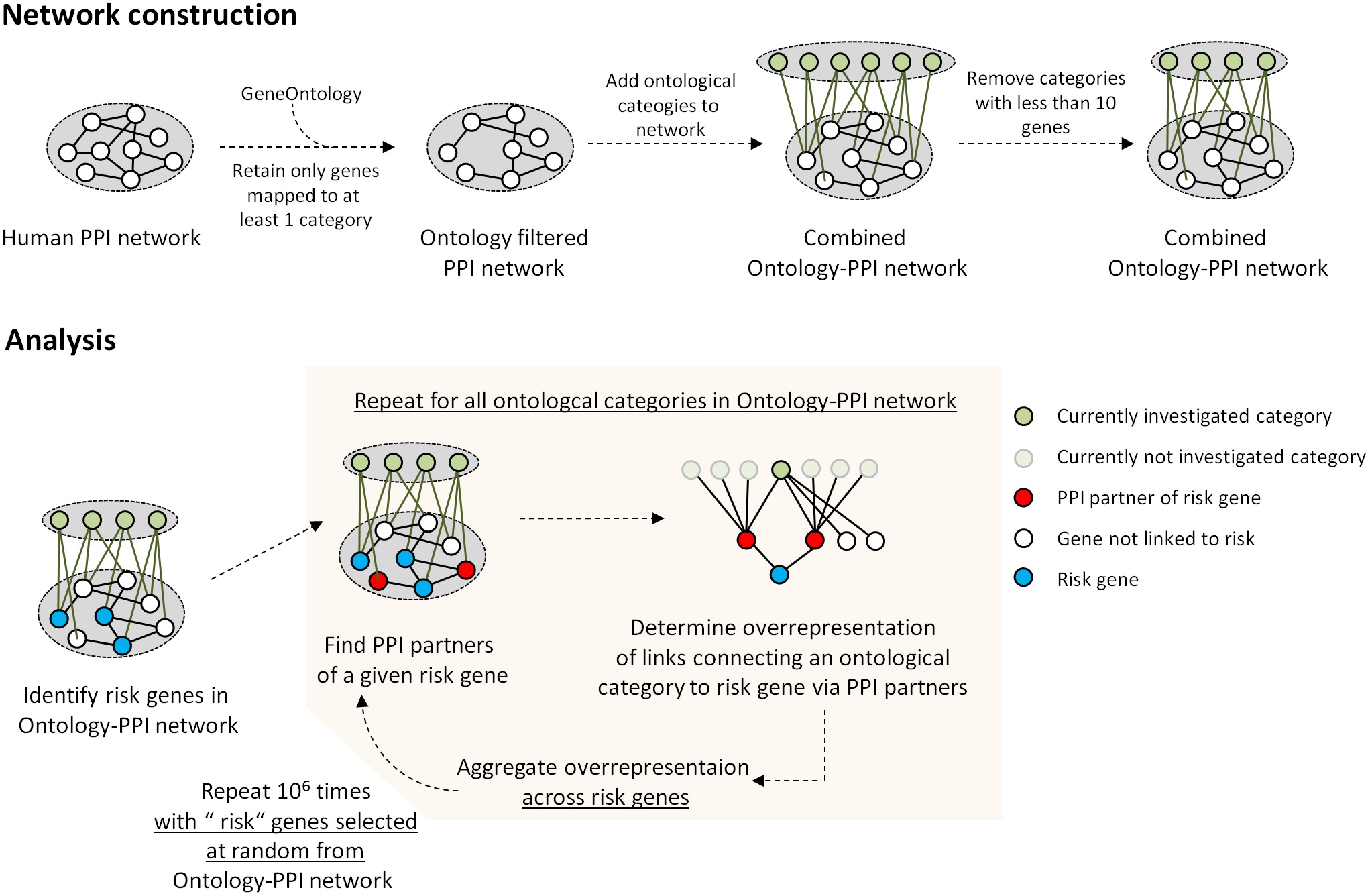
Analysis was based on a network incorporating PPI (BioGRID database vs 3.2.11616) and ontological category information from the Gene Ontology (mappings from 08.03.2014, derived from R library org.Hs.eg.db, vs 3.0.0, including the categories “biological process,” “molecular function,” and “cellular component; figure 1a). Our approach builds on work performed by PGC, as we are specifically focusing on the 108 genome-wide significant schizophrenia risk loci and the gene mapping identified by this consortium. As performed by the Network and Pathway Analysis Subgroup of the PGC,6 we focused analysis on ontological categories linked to at least 10 genes. Within the PPI network, only genes mapping to at least 1 ontological category were retained. The network was an unweighted, undirected graph with links representing either PPIs or ontological annotations. Further details on the construction of the network investigated here are described in the supplementary methods and a schematic overview of the procedure is shown in supplementary figure 1. This study received ethical approval from the local ethics committee (2013-842R-MA).
Fig. 1.

Overview of network based functional analysis. a) Schematic illustration of the network linking the PPI to ontological category information. b) Selection of the 44 loci mapping to single, unique genes with at least 1 link to the PPI network. c) Network illustration of the 2 significantly schizophrenia associated ontological categories “synaptic transmission” (GO:0007268) and “ion transmembrane transport” (GO:0034765). Nodes are colored depending on their predominant link to a given ontological category or its respectively associated proteins (red-colored proteins are shared between ontological categories and red-colored risk loci predominantly associated with such nodes).
Analysis of Risk-Locus Associated Biological Processes
To quantify the strength of association between ontological categories and risk loci, we determined the overrepresentation of links between proteins linked to a given ontological category and the schizophrenia risk loci. For a given category, we determined across all i risk loci the sum ∑i(b/d RISK) where b is the observed number of links between the category’s associated proteins and a given risk locus and d RISK is the risk locus’s degree. The locus’s degree is the number of links between the gene it maps to and all interaction partners in the PPI network. A given locus’s degree is proportional to the expected number of links between the locus and the category’s proteins, which is ∑d PPI * d RISK / (2*m), where ∑d PPI is the sum of the degrees of all proteins linked to a given ontological category and m is the total number of links in the PPI network. This expected number is equal to c * d RISK, where c is a constant since network configuration of the proteins linked to a given category, as well as the total number of links in the network do not change throughout the procedures applied here.
To estimate significance the overrepresentation estimate was compared against estimates obtained from (106) random selections of the same number of genes from the PPI network. The frequency of estimates at least as high as that from the real schizophrenia loci was used as empirical P value and adjusted for the Family-Wise-Error-Rate using Bonferroni’s method. Statistical analyses were performed considering as risk loci those 44 that mapped to single genes (out of a total of 108 loci with genome wide significance, figure 1b, supplementary tables S1 and S2) and whose gene product had at least 1 interaction partner in the PPI network. We explored whether schizophrenia associated results were confounded by properties of the investigated network, by confounders reported in a previous enrichment analysis of GWAS data17 or by the selection of loci mapping to single genes. Also, we tested whether risk associated ontological categories were specific for schizophrenia compared to rheumatoid arthritis (RA) and prostate cancer (PC) (supplementary methods). Analyses were performed using R (http://cran.r-project.org/).
Analysis of Gene Expression Across Developmental Stages
For analysis of brain expression across developmental stages, we build on work performed by the consortium behind the BrainSpan atlas. Data preprocessing was performed as previously described,10 data was quantile normalized using the R library limma, vs 3.22.1 and log2-transformed. After filtering, the dataset comprised expression information on 16 brain regions from 47 donors. Data was aggregated into age-groups and brain regions as described previously6 and expression was represented by the respective mean levels within groupings. We evaluated the change of expression throughout developmental stages using Spearman’s correlation analysis and due to the exploratory nature of this analysis, determined False Discovery Rate (FDR) adjusted P values according to the method of Benjamini and Hochberg.18
Preprocessing and Analysis of Gene Expression Data
For investigation of schizophrenia post-mortem brain expression, we utilized data from the GEO database and identified datasets by extensive keyword searches. Here, we focus on no-cerebellar datasets with at least 10 patient and control subjects per dataset. An overview of the datasets is shown in table 1. Expression was quantile normalized and log2 transformed. Univariate case-control differences were determined using Wilcoxon tests and P values adjusted according to the method of Benjamini and Hochberg.18
Table 1.
Overview Gene-Expression Datasets Used in This Study (SZ, Schizophrenia; HC, Healthy control)
| Dataset | Dataset Identifier | Reference | Brain Region | No. of Subjects (SZ / HC) |
|---|---|---|---|---|
| I | GSE17612 | Maycox et al11 | Anterior prefrontal cortex (Brodmann Area (BA) 10) | 28 / 23 |
| II | GSE12679 | Harris et al12 | Prefrontal cortex (BA 9) | 16 / 11 |
| III | GSE21935 | Barnes et al13 | Superior temporal cortex (BA22) | 23 / 18 |
| IV | GSE53987 | TA Lanz (presented at ACNP 2012) | Hippocampus | 15 / 18 |
| Prefrontal cortex (BA46) | 15 / 19 | |||
| Associative striatum | 18 / 18 | |||
| V | GSE35977 | Chen et al14 | Parietal cortex | 49 / 49 |
| VI | GSE21138 | Narayan et al15 | Prefrontal cortex (BA46) | 30 / 29 |
Results
The network investigated here comprised 13 934 proteins, 131 191 interactions between these, 2805 ontological categories and 161 900 links connecting ontological categories to proteins within the PPI network (figure 1a). Adjusting for the Family Wise Error Rate across all tested ontological categories (n = 2417) linked to the schizophrenia risk loci through the PPI network, we found that “synaptic transmission” (GO:0007268, z = 8.98, P raw < 10–6, P FWE < .001) and “ion transmembrane transport” (GO:0034765, z = 9.77, P raw = 1.1·10–5, P FWE = .03) were significantly linked to the schizophrenia risk loci. The networks relevant for these associations (figure 1c) were linked to a total of 15 out of 44 risk loci via 38 different interaction partners. Exploration of annotation quality of risk linked PPI partners showed that for GO:0007268, 87% of annotations were based on cited publications while 5% of annotations were based on computational methods. In contrast, for GO:0034765, 55% of genes were annotated computationally.
We aimed to exclude confounding due to the network’s configuration. For this, we compared schizophrenia loci to gene sets selected at random and found no significant associations (minimum P = .34) with several parameters detailed in the supplementary methods section. Similarly, illness associations were not affected by confounders known from functional analysis of GWAS data (minimum P = .22, see supplementary methods).17 Analysis focused on loci that mapped to single genes, which may introduce bias if such mapping is differentially associated with biological function. Therefore, we repeatedly selected 44 genes containing at least 1 single nucleotide polymorphism (SNP) from genetic regions non-overlapping with any other genes using gene boundaries extended by 20kb as applied for annotation of the 108 risk loci.5 This also identified “synaptic transmission” (GO:0007268, P raw < 10–6, P FWE < .001) and “ion transmembrane transport” (GO:0034765, P raw = 8.0·10–6, P FWE = .02), as well as “regulation of long-term synaptic plasticity” (GO:0048169; P raw = 1.2·10–5, P FWE = .03) and “positive regulation of synaptic transmission” (GO:0050806; P raw = 10–6, P FWE = .002) as significantly schizophrenia associated.
Associations of Ontological Categories Previously Linked to Schizophrenia
We explored potential associations between schizophrenia risk loci and ontological categories previously implicated by the PGC cross-disorder working group. Of the 5 categories most strongly associated with schizophrenia in a GWAS enrichment meta-analysis,6 4 had at least 10 links to the PPI network. The 2 ontological categories with the highest enrichment also showed significant associations to schizophrenia risk loci through the PPI network. These were “postsynaptic density” (GO:0014069; nominal P = .02) and “postsynaptic membrane” (GO:0045211; nominal P < .001). In contrast, the other 2 ontological categories (“dendritic spine,” GO:0043197, P = .66) and “histone H3-K4 methylation” (GO:0051568, P = .62), which was the most significantly enriched category across 5 psychiatric disorders, showed no significant associations in the present study. Similarly, calcium channel activity (GO:0005262), the most significant ontological category implicated by SNP-based meta-analysis GWAS across 5 psychiatric illnesses19 showed only a trend towards a significant association (P = .08).
Analysis of Disease Specificity and Positive Control
We aimed to assess the specificity of the identified schizophrenia associations, in particular to explore potential methodological bias that could lead to identification of the same risk associated ontological categories across different illnesses without strong genetic overlap. Therefore, we performed the same analysis on 25 and 41 risk SNPs linked to RA and PC, respectively. For both sets of risk genes, no significant associations were found. The ontological categories most strongly associated with PC and RA were “DNA-dependent ATPase activity” (GO:0008094, P raw = 4.1·10–5, P FWE = .08) and “T cell costimulation” (GO:0031295, P raw = 2.1·10–4, P FWE = .42), respectively.
As positive control, we applied our method on GWAS results investigating the polygenic basis of dyslipidemia in over 19 000 subjects.20 Thirty loci have been found to be associated with quantitative molecular traits of dyslipidemia and cumulative allelic dosages of risk alleles at these loci were associated in a stepwise fashion with lipoprotein concentrations (decrease for high-density lipoprotein [HDL] cholesterol and increase for low-density lipoprotein (LDL) cholesterol and triglycerides).20 Consistent with this, genome-wide enrichment analysis has found numerous associations with relevant lipid and lipoprotein pathways.17 To annotate a given locus, we use the gene in closest physical proximity to the index SNP or (if available) the gene within 500kb whose expression has been found to be most strongly modulated by the index SNP20 (supplementary table S3). After correction for the family-wise error rate across 1838 ontological categories, we found significant associations with “LDL particle receptor binding” (GO:0050750, P raw = 7.0·10–6, P FWE = .01) and “LDL particle” (GO:0034362, P raw = 1.5·10–5, P FWE = .03). Correcting for the FDR identified associations with 25 ontological categories with FDR < 0.05 that were predominantly associated with lipid and lipoprotein pathways (supplementary table S4). For added confirmation, we used only loci where a single gene was linked to the locus or the index SNP modulated expression of only 1 gene within 500kb. Using the resulting 12 genes and the 1333 linked ontological categories yielded very similar results (supplementary table S4).
Developmental Trajectory of Synaptic Gene Expression
To investigate the potential neurodevelopmental role of the identified ontological categories, we examined in different developmental time-points and brain regions the expression of 36 of the 38 interaction partners linked to schizophrenia risk loci through the PPI network using the BrainSpan dataset (no data was available for GRIP1 and GNG10).10 This dataset contains expression data from 16 brain regions in over 50 brains acquired across the human lifespan. We found that 22 of the investigated genes had a significant change in expression patterns across time (P FDR < .05, figure 2). Since expression appeared to be modulated most by birth, we split regional analyses considering separately samples taken before and after birth. This showed that after birth expression significantly increased with some regional specificity for neocortical regions.
Fig. 2.

Gene expression patterns throughout development stages and across brain regions. Genes were ordered using complete linkage hierarchical clustering on the Euclidean distance of their expression levels. Genes shown in bold show significant change throughout developmental stages (Spearman correlation, False Discovery Rate [FDR] < 0.05). Graphical layout adapted from ref.6
Expression Analysis of Synaptic Genes
To explore potential expression differences of genes part of the 2 ontological categories, we used 6 gene expression datasets from post mortem brains of schizophrenia patients and controls (table 1). Using all genes part of the 2 ontological categories, (total n = 333–450, depending on the dataset) we found significant differences between schizophrenia patients and controls (FDR < .05) for 55 and 4 genes in the hippocampus and prefrontal cortex of dataset IV, respectively, for 22 genes in the parietal cortex (dataset V) and 5 genes in the prefrontal cortex of dataset VI. Figure 3 shows the overlap between the identified gene sets and highlights the shared schizophrenia association of the potassium channel genes KCNK1 and KCNS3 across the hippocampus, the prefrontal and the parietal cortex. The relative overlap between gene sets with FDR < 0.05 was substantial. 20%, 100%, 55% and 60% of such genes in datasets IV (hippocampus), IV (BA46), V (parietal cortex), and VI (BA46), respectively, overlapped with the significant gene sets in the respective other 3 datasets. We used permutation testing to show that even the lowest overlap of 20% was very unlikely to occur by chance, considering all pairwise comparisons between the 4 datasets described above (P < .001). All genes with FDR < 0.05 are summarized in supplementary table S5.
Fig. 3.

Overlap of significant expression differences across datasets. Displayed genes showed expression changes with P FDR < .05. Arrows indicate whether expression was consistently increased (up arrow) or decreased (down arrow) across the datasets where a significant change in a given gene was observed.
Discussion
Here we showed that through consideration of the biological vicinity of risk genes, synaptic function emerges as a schizophrenia risk mechanism from genetic variants with the strongest risk association. This finding is consistent with genome wide enrichment analysis,6 exome sequencing results,21,22 the effect of schizophrenia associated copy number variants (CNVs) on postsynaptic signaling,23 as well as of psychiatric risk linked mutations on postsynaptic protein scaffolding.24 Consistent with this, postsynaptic function, a mostly schizophrenia specific pathway identified by enrichment analysis, was associated with schizophrenia risk loci. In contrast, histone methylation, the ontological category most strongly implicated across psychiatric illnesses, was not. This discrepancy may be due to the fact that our approach focuses on the most significant risk variants and differences compared to genome-wide analysis6 may suggest a divergence of biological risk mechanisms depending on the underlying strength of risk association. Quantitative assessment of such divergence may provide interesting insights into biological risk mechanisms shared across and specific for individual disorders. Despite this, our results do not allow conclusions about illness specificity as synaptic processes may also be implicated by risk variants of other illnesses and their respective PPI networks. The identification of synaptic function as a schizophrenia risk process is further consistent with numerous nongenetic studies.25–28 For example, histopathological investigations have found reduced spine densities in multiple brain regions of schizophrenia patients that are thought to contribute to altered neural circuitry,29–33 as well as to reduced cortical and hippocampal volumes found in schizophrenia patients.26 Also, impaired synaptic neurotransmitter release and neurotransmission associated protein expression have been found in schizophrenia.34,35 A synaptic pathology in schizophrenia is further consistent with findings in patients with encephalitis, where schizophrenia-like symptoms can be induced by IgG serum and CSF antibodies against N-methyl-D-aspartate glutamate receptors (NMDA-R).36 These antibodies selectively decrease NMDA-R clusters in postsynaptic dendrites37 and have been found in serum of up to 10% of schizophrenia patients.38–40 Interestingly, the induced symptom profile appears to depend on the intensity of the antibody effect on NMDA-R density, an effect similar to that observed for different doses of NMDA-R antagonists.38 While low doses cause psychosis, anxiety, agitation, memory disturbance, decreased responsiveness to pain, and speech reduction, higher doses have been reported to induce unresponsiveness with catatonic features, dyskinesias, autonomic instability, and seizures.38,41–43
In the present study, expression analysis indicated a substantial brain expression change of risk-protein linked interaction partners during birth and some regional specificity of this effect for neocortical brain regions. We also found significant brain expression differences within the identified synaptic categories across multiple datasets and brain regions in adult schizophrenia patients. The strongest differences were found in the hippocampus, the subcortical region showing the most pronounced structural change in MRI investigations of schizophrenia patients.44 Interestingly, the identified expression changes strongly overlapped across the hippocampus, parietal and prefrontal cortex, supporting synaptic alterations spanning across brain regions. Genes for the potassium channel proteins KCNK1 and KCNS3 showed consistent expression change across all of the aforementioned regions. Reduced expression of KCNS3 in prefrontal cortical parvalbumin neurons in schizophrenia and has been linked to impaired γ-oscillations that are thought to underlie the cognitive impairments in schizophrenia.45 Reduced KCNK1 expression in schizophrenia patients has also been detected in previous prefrontal cortex expression studies.46,47
The strong change of synaptic gene expression we observed during the perinatal period may suggest that this period is particularly susceptible for the manifestation of genetically determined alterations in synaptic processes that may then persist into adulthood. However, it should be taken into account that the present study provides only indirect evidence since it does not directly link genetic risk to protein expression during the perinatal period, and does not take into account other potentially relevant factors, such as post-translational modification, enzyme activity, molecular trafficking or schizophrenia specific differences in PPIs. Post mortem and neuroimaging findings have highlighted the etiological relevance of synaptic function and support the hypothesis of schizophrenia being a disorder induced by developmental synaptic disturbances that lead to reduced synaptic plasticity and connectivity.25,48 Future experiments will show how genetic variation in schizophrenia risk loci impacts on perinatal gene expression. Of particular interest are interactions with environmental factors previously linked to schizophrenia risk, such as perinatal and obstetric complications,49,50 season of birth51 or prenatal malnutrition.52 Obstetric complications have already been hypothesized to increase schizophrenia risk via premature synaptic pruning caused by fetal hypoxia.53 Similarly, even moderate malnutrition that does not impact on the offspring’s birth weight, has been shown to impair synaptic plasticity in rats.54
Limitations
Our study has several limitations. Since the network method is novel, our results should be seen as preliminary and need to be validated in independent samples and using alternative computational approaches. A limitation is our focus on risk loci that mapped to single genes, which omits a substantial amount of disorder-associated genetic variability. Such selection may be a biased representation of the schizophrenia risk architecture and although our results suggest that selection of loci mapping to single genes did not bias the identified ontological categories, we cannot exclude the possibility of residual bias. Future studies incorporating loci mapping to multiple genes may face the challenge that a higher gene number can impede inference of risk associated ontological categories, as such loci lead to stronger chance associations. Another limitation is that the data used in this study were not acquired for the objectives pursued here. Exome or whole genome sequencing data would have been more suitable in particular for mapping risk variants to genes. Another limitation is that inference was based on the content of a PPI network, whose underlying data mainly originated from nonpsychiatric research, which may have biased our results. A similar issue may affect ontological categories, since human annotations are known to be non-equally distributed across genes and this may reflect differences in research interest regarding the respective genes.55 Variants were selected based on a genome-wide significance threshold, which reduces the risk of false-positives but is not without limitations due its inability to account for factors such as study power or the number of likely true positives.56 Also, the current definition of “risk locus” relates to physical regions containing SNPs correlated with each of the 128 schizophrenia associated index SNPs with r 2 > .6 (or .5 in the case of the dyslipidemia GWAS analyses), which may miss etiologically relevant genes mapping to SNPs in lower linkage disequilibrium. We found no significant links between ontological categories and risk genes of PC or RA, which may suggest that due to the approximately 7-fold higher patient numbers used for determination of schizophrenia risk loci compared those of RA and the combination of PC risk loci across multiple independent GWAS studies, schizophrenia risk loci may currently be supported by higher statistical power and may therefore cover a higher portion of true positive associations. Also, analysis of RA and PC was based on published gene annotations, which may not sufficiently capture etiologically relevant genes. Annotation quality analysis highlighted that a large portion of risk linked PPI partners within the ontological category GO:0034765 was computationally generated, which has been associated with lower reliability compared to annotation based on experimental evidence.57 Finally, we did not observe significant differences in 2 prefrontal cortex datasets (BA 9 and 10), the superior temporal cortex (BA22) and the associative striatum. This may be due to regional specificity of the expression changes we identified across BA46, the hippocampus and the parietal cortex or due to the small sample numbers that make it difficult to detect more subtle changes.
Conclusions
Using a novel functional analysis method we have identified strong links between schizophrenia risk loci and synaptic function. In contrast to genome-wide analysis, this method allows identification of specific loci that mediate associations with a given risk mechanism and pinpoints sets of risk associated genes for further mechanistic investigations. Our results support the importance of PPI networks to map genetic risk to biological function. The assessment of such biological vicinity of risk genes may be a fruitful avenue for future investigations of schizophrenia associations in other data modalities, such as neuroimaging or polygenic analyses. Here, we found that brain expression of genes within the identified processes changes substantially during early development, consistent with the etiological hypothesis of developmentally determined synaptic alterations in schizophrenia. Our results link such synaptic processes with a small set of genes with strong risk association that may provide the basis for mechanistic experiments and which may be valuable targets for future drug development approaches.
Supplementary Material
Supplementary material is available at http://schizophreniabulletin.oxfordjournals.org.
Funding
This study was supported by the DFG Emmy-Noether-Program SCHW 1768/1-1.
Supplementary Material
Acknowledgments
A.M.-L. received the following incomes: Consultancy: Astra Zeneca, Elsevier, F. Hoffmann-La Roche, Gerson Lehrman Group, Lundbeck, Outcome Europe Sárl, Outcome Sciences, Roche Pharma, Servier International, Thieme Verlag; Lectures including travel fees: Abbott, Astra Zeneca, Aula Médica Congresos, BASF, Groupo Ferrer International, Janssen-Cilag, Lilly Deutschland, LVR Klinikum Düsseldorf, Servier Deutschland, Otsuka Pharmaceuticals, Boehringer Ingelheim; Grants: ECNP Neuropsychopharmacology Award, Prix ROGER DE SPOELBERCH. All other authors declare that there are no potential conflicts of interest.
References
- 1. Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003;60:1187–1192. [DOI] [PubMed] [Google Scholar]
- 2. Sullivan PF, Daly MJ, O’Donovan M. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet. 2012;13:537–551. doi:10.1038/nrg3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Purcell SM, Wray NR, Stone JL, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi:http://dx.doi.org/10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ripke S, O’Dushlaine C, Chambert K, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet. 2013;45:1150–1159. doi:10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ripke S, Neale BM, Corvin A, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci. 2015;18:199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perry JRB, McCarthy MI, Hattersley AT, Zeggini E, Weedon MN, Frayling TM. Interrogating type 2 diabetes genome-wide association data using a biological pathway-based approach. Diabetes. 2009;58:1463–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shi W, Bessarabova M, Dosymbekov D, et al. Functional analysis of multiple genomic signatures demonstrates that classification algorithms choose phenotype-related genes. Pharmacogenomics J. 2010;10:310–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Luo X, Huang L, Jia P, et al. Protein-protein interaction and pathway analyses of top schizophrenia genes reveal schizophrenia susceptibility. Schizophr Bull. 2014;40:39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kang HJ, Kawasawa YI, Cheng F, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maycox PR, Kelly F, Taylor A, et al. Analysis of gene expression in two large schizophrenia cohorts identifies multiple changes associated with nerve terminal function. Mol Psychiatry. 2009;14:1083–1094. [DOI] [PubMed] [Google Scholar]
- 12. Harris LW, Wayland M, Lan M, et al. The cerebral microvasculature in schizophrenia: a laser capture microdissection study. PLoS One. 2008;3:e3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barnes MR, Huxley-Jones J, Maycox PR, et al. Transcription and pathway analysis of the superior temporal cortex and anterior prefrontal cortex in schizophrenia. J Neurosci Res. 2011;89:1218–1227. [DOI] [PubMed] [Google Scholar]
- 14. Chen C, Cheng L, Grennan K, et al. Two gene co-expression modules differentiate psychotics and controls. Mol Psychiatry. 2013;18:1308–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Narayan S, Tang B, Head SR, et al. Molecular profiles of schizophrenia in the CNS at different stages of illness. Brain Res. 2008;1239:235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stark C, Breitkreutz B-J, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:D535–D539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ayellet VS, Groop L, Mootha VK, Daly MJ, Altshuler D. Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet. 2010;6:e1001058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57:289–300. doi:10.2307/2346101. [Google Scholar]
- 19. Group C, Consortium PG. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–1379. doi:10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kathiresan S, Willer CJ, Peloso GM, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009;41:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fromer M, Pocklington AJ, Kavanagh DH, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Purcell SM, Moran JL, Fromer M, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kirov E, Pocklington A, Holmans P, Ivanov D. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry. 2012;17:142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ting JT, Peça J, Feng G. Functional consequences of mutations in postsynaptic scaffolding proteins and relevance to psychiatric disorders. Annu Rev Neurosci. 2012;35:49–71. [DOI] [PubMed] [Google Scholar]
- 25. McGlashan TH, Hoffman RE. Schizophrenia as a disorder of developmentally reduced synaptic connectivity. Arch Gen Psychiatry. 2000;57:637–648. [DOI] [PubMed] [Google Scholar]
- 26. Glausier JR, Lewis DA. Dendritic spine pathology in schizophrenia. Neuroscience. 2013;251:90–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moyer CE, Shelton MA, Sweet RA. Dendritic spine alterations in schizophrenia. Neurosci Lett. 2015;601:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Beneyto M, Lewis DA. Insights into the neurodevelopmental origin of schizophrenia from postmortem studies of prefrontal cortical circuitry. Int J Dev Neurosci. 2011;29:295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Black JE, Kodish IM, Grossman AW, et al. Pathology of layer V pyramidal neurons in the prefrontal cortex of patients with schizophrenia. Am J Psychiatry. 2004;161:742–744. doi:10.1176/appi.ajp.161.4.742. [DOI] [PubMed] [Google Scholar]
- 30. Garey LJ, Ong WY, Patel TS, et al. Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J Neurol Neurosurg Psychiatry. 1998;65:446–453. doi:10.1136/jnnp.65.4.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57:65–73. doi:10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- 32. Kolluri N, Sun Z, Sampson AR, Lewis DA. Lamina-specific reductions in dendritic spine density in the prefrontal cortex of subjects with schizophrenia. Am J Psychiatry. 2005;162:1200–1202. [DOI] [PubMed] [Google Scholar]
- 33. Lewis DA, Pierri JN, Volk DW, Melchitzky DS, Woo TUW. Altered GABA neurotransmission and prefrontal cortical dysfunction in schizophrenia. Biol Psychiatry. 1999;46:616–626. [DOI] [PubMed] [Google Scholar]
- 34. Tsai G, Coyle JT. Glutamatergic mechanisms in schizophrenia. Annu Rev Pharmacol Toxicol. 2002;42:165–179. [DOI] [PubMed] [Google Scholar]
- 35. Hashimoto T, Arion D, Unger T, et al. Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2008;13:147–161. doi:http://dx.doi.org/10.1038/sj.mp.4002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hughes EG, Peng X, Gleichman AJ, et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci. 2010;30:5866–5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Steiner J, Walter M, Glanz W. Increased prevalence of diverse N-methyl-D-aspartate glutamate receptor antibodies in patients with an initial diagnosis of schizophrenia: specific relevance of IgG. JAMA. 2013;70:271–278. doi:10.1001/2013.jamapsychiatry.86. [DOI] [PubMed] [Google Scholar]
- 39. Steiner J, Teegen B, Schiltz K, Bernstein HG, Stoecker W, Bogerts B. Prevalence of N-methyl-D-aspartate receptor autoantibodies in the peripheral blood: healthy control samples revisited. JAMA Psychiatry. 2014;71:838–839. [DOI] [PubMed] [Google Scholar]
- 40. Zandi MS, Irani SR, Lang B, et al. Disease-relevant autoantibodies in first episode schizophrenia. J Neurol. 2011;258:686–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011;10:63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308. [DOI] [PubMed] [Google Scholar]
- 43. Weiner AL, Vieira L, McKay CA, Bayer MJ. Ketamine abusers presenting to the emergency department: a case series. J Emerg Med. 2000;18:447–451. [DOI] [PubMed] [Google Scholar]
- 44. van Erp TGM, Hibar DP, Rasmussen JM, et al. Subcortical brain volume abnormalities in 2028 individuals with schizophrenia and 2540 healthy controls via the ENIGMA consortium. Mol Psychiatry. 2016;21:547–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Georgiev D, Arion D, Enwright JF, et al. Lower gene expression for KCNS3 potassium channel subunit in parvalbumin-containing neurons in the prefrontal cortex in schizophrenia. Am J Psychiatry. 2014;171:62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mistry M, Gillis J, Pavlidis P. Genome-wide expression profiling of schizophrenia using a large combined cohort. Mol Psychiatry. 2013;18:215–225. doi:10.1038/mp.2011.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Arion D, Unger T, Lewis DA, Levitt P, Mirnics K. Molecular evidence for increased expression of genes related to immune and chaperone function in the prefrontal cortex in schizophrenia. Biol Psychiatry. 2007;62:711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stephan KE, Baldeweg T, Friston KJ. Synaptic plasticity and dysconnection in schizophrenia. Biol Psychiatry. 2006;59:929–939. [DOI] [PubMed] [Google Scholar]
- 49. Cannon M, Jones PB, Murray RM. Obstetric complications and schizophrenia: historical and meta-analytic review. Am J Psychiatry. 2002;159:1080–1092. [DOI] [PubMed] [Google Scholar]
- 50. Mittal VA, Ellman LM, Cannon TD. Gene-environment interaction and covariation in schizophrenia: the role of obstetric complications. Schizophr Bull. 2008;34:1083–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mortensen PB, Pedersen CB, Westergaard T, et al. Effects of family history and place and season of birth on the risk of schizophrenia. N Engl J Med. 1999;340:603–608. doi:10.1056/NEJM199902253400803. [DOI] [PubMed] [Google Scholar]
- 52. Susser E, St Clair D, He L. Latent effects of prenatal malnutrition on adult health: the example of schizophrenia. Ann N Y Acad Sci. 2008;1136:185–192. [DOI] [PubMed] [Google Scholar]
- 53. Rosso IM, Cannon TD, Huttunen T, Huttunen MO, Lönnqvist J, Gasperoni TL. Obstetric risk factors for early-onset schizophrenia in a Finnish birth cohort. Am J Psychiatry. 2000;157:801–807. [DOI] [PubMed] [Google Scholar]
- 54. Flores O, Pérez H, Valladares L, et al. Hidden prenatal malnutrition in the rat: role of β1-adrenoceptors on synaptic plasticity in the frontal cortex. J Neurochem. 2011;119:314–323. [DOI] [PubMed] [Google Scholar]
- 55. Gillis J, Pavlidis P. Assessing identity, redundancy and confounds in Gene Ontology annotations over time. Bioinformatics. 2013;29:476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McCarthy MI, Abecasis GR, Cardon LR, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–369. [DOI] [PubMed] [Google Scholar]
- 57. Skunca N, Altenhoff A, Dessimoz C. Quality of computationally inferred gene ontology annotations. PLoS Comput Biol. 2012;8:e1002533. doi:10.1371/journal.pcbi.1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}