

Abstract
Recently, adipocytes have been recognized as actively participating in local and systemic immune responses via the secretion of peptides detectable in relevant levels in the systemic circulation, the so-called “adipo(cyto)kines”. Multiple studies appearing within the last 10-15 years have focused on the possible impact of adipose tissue depots on inflammatory bowel disease (IBD). Consequently, various hypotheses regarding the role of different adipokines in inflammatory diseases in general and in intestinal inflammatory processes in particular have been developed and have been further refined in recent years. After a focused summary of the data reported concerning the impact of visceral adipose tissue on IBD, such as Crohn’s disease and ulcerative colitis, our review focuses on recent developments indicating that adipocytes as part of the innate immune system actively participate in antimicrobial host defenses in the context of intestinal bacterial translocation, which are of utmost importance for the homeostasis of the whole organism. Modulators of adipose tissue function and regulators of adipokine secretion, as well as modifiers of adipocytic pattern recognition molecules, might represent future potential drug targets in IBD.
Keywords: Inflammatory bowel disease, Crohn’s disease, ulcerative colitis, adipose tissue, adipokines, leptin, adiponectin, innate immunity
Introduction
Recently, adipocytes have been recognized to actively participate in systemic immune responses via the secretion of peptides detectable in relevant levels in the systemic circulation, the so-called “adipo(cyto)kines” [1-3]. Multiple original studies and review articles appearing within the last 10-15 years have focused on the possible impact of adipose tissue depots on inflammatory bowel disease (IBD). A recent search of the PubMed database (in April 2016) using the search terms “inflammatory bowel disease AND fat (adipose) tissue” resulted in 316 items. Publications in this field are constantly growing in number and increasingly concentrate on adipokines. The search terms “inflammatory bowel disease AND adipokines” returned 132 publications. Leptin is the most prominent adipokine in the field of IBD research, as 83 of these 132 publications were related to leptin, followed by adiponectin, resistin, visfatin and others. Because of the growing body of data, the number of review articles in this field is also increasing, with a total number of 45 review articles returned when the search terms “review AND inflammatory bowel disease AND adipose tissue” were used. Among these 45 articles, 15 focus on adipokines/secreted factors, 9 on mesenchymal stem cells/fibrosis, 4 on obesity/muscle/exercise, 3 on peroxisome proliferator-activated receptor γ (PPARγ), 3 on the innate immunity of adipocytes, 2 on metabolism/fatty acids, 2 on neurotransmitters/neuropeptides, and 7 on other or mixed topics.
Various hypotheses regarding the role of different adipokines in inflammatory diseases in general and in intestinal inflammatory processes in particular have been developed and have been further refined in recent years. It is not the intent of this review to reiterate these developments in detail. Rather, after a focused summary of the data reported on the impact of visceral adipose tissue (VAT) on IBD, such as Crohn’s disease and ulcerative colitis, our manuscript will focus on recent developments indicating that, in the context of intestinal bacterial translocation, adipocytes actively participate as part of the innate immune system in antimicrobial host defenses, which are of utmost importance for the homeostasis of the whole organism.
Interestingly, inflammatory visceral fat hypertrophy (also named “creeping fat”) is indicative for Crohn’s disease to an extent that it has been proposed as a useful diagnostic marker in the differential diagnosis of IBD from other intestinal inflammations such as intestinal tuberculosis [4]. In computed tomography scans, the increase in submucosal fat in patients with Crohn’s disease of longer duration leads to a characteristic “fat halo sign” [5].
Early studies described an inflammatory reaction of hypertrophic mesenteric adipose tissue (MAT) in patients with Crohn’s disease, characterized by increased concentrations of PPARγ and tumor necrosis factor α (TNFα) within the mesenteric fat depot [6]. Similar results were found in experimental 2,4,6-trinitrobenzenesulfonic acid (TNBS)-induced colitis in mice [7], while infliximab treatment restored MAT PPARγ expression to baseline in these mice [8]. Interestingly, the angiotensin II type 1 receptor blocker and PPARγ agonist telmisartan ameliorated spontaneous colitis in interleukin-10-deficient (IL-10-/-) mice, restoring VAT morphology and adipokine secretion to a non-colitic phenotype [9]. Notably, mice deficient in Toll-like receptor 9 (TLR9) signaling (TLR9-/-), resistant to chronic dextran sulfate sodium (DSS)-induced colitis, exhibit an altered adipokine expression profile in VAT compared to wild-type mice [10].
Remarkably, PPARγ is the key transcriptional regulator in the terminal differentiation of adipocytes from mesenchymal stem cells [11,12]. These data imply an intricate relationship between intestinal inflammation and adjacent VAT, more than merely being a passive “bystander”.
Obesity and IBD: bystanders, mutual friends, or enemies?
Generally, obesity has been postulated to contribute to the onset and progression of various autoimmune diseases in humans [13]. While earlier reports indicated that the prevalence of obesity was lower in patients suffering from IBD than in the general population [14,15], the overall incidence of obesity in these patients has recently been increasing and is currently estimated to be around 25-30%, similar to the rate in the general population [16,17]. Interestingly, bariatric surgery has been observed to improve intestinal inflammation in patients with IBD [18], although other reports caution against a potentially deleterious effect [19,20]. While earlier reports hinted at a potentially worse disease course in obese as compared to normal-weight IBD patients [15,21], other authors found less severe disease in obese patients [22]. Diet-induced obesity worsens TNBS-induced experimental colitis in mice [23] as well as spontaneous intestinal inflammation in multidrug resistance protein 1a deficient mice [24]. In IBD patients, however, the large “IBD in EPIC Study”, which included more than 300,000 participants, found no association between IBD and obesity, measured by body mass index (BMI) [25]. It should be noted that BMI is only a crude index of obesity, and this may be partly responsible for the lack of association in this large study. In summary, the impact of obesity on IBD remains to be clarified.
Do drugs that induce weight loss in obese patients [26,27] impact on the incidence, prevalence and/or clinical course of IBD? No data in this regard are available on sympathomimetic drugs approved for short-term use (amfepramone, benzphetamine, diethylpropion, phendimetrazine, phentermine, ephedrine, caffeine). Interestingly, bupropion, an antidepressant used in the pharmacotherapy of obesity, has shown beneficial effects on intestinal ischemia/reperfusion injury in rats [28], while the opioid antagonist naltrexone effectively reduced intestinal inflammation in human Crohn’s disease, as well as in rodent models of colitis [29-31]. However, these observations were mostly short-term effects, while the weight-lowering effects of these compounds are expected with longer-term use. Thus, reduced intestinal inflammation in these models is not likely to be causally linked to weight loss or reduced adipocyte numbers.
For long-term treatment of obesity aimed at inducing weight loss, orlistat, lorcaserin and the combination phentermine/topiramate have been approved [26,27]; however, no data are available regarding the impact of these compounds on IBD. Interestingly, glucagon-like peptide-2 (GLP-2) had beneficial effects in rodent enteritis models [32,33], and inhibition of the GLP-diminishing enzyme dipeptidyl peptidase 4 had beneficial effects in acute DSS-induced colitis in mice [34]. No data are available on GLP-1-analogs in autoimmune intestinal inflammation.
Importantly, metformin has been demonstrated to inhibit TNFα-induced proinflammatory cytokine induction in colonic epithelial cells via nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway inhibition in vitro [35]. Metformin treatment, via signal transducer and activator of transcription 3/IL-17 inhibition, ameliorated murine acute DSS-induced colitis as well as chronic colitis in IL-10-/- mice [35,36]. Additionally, metformin treatment reduced colitis-associated tumorigenesis in rats and mice [35,37]. However, no data are available on metformin treatment in patients suffering from IBD.
There is a wealth of data proving the beneficial effect of PPARγ agonists (thiazolidinediones, “glitazones”) in IBD. PPARγ receptors were originally described in adipose tissue, and agonistic compounds have long been used as insulin-sensitizing, antidiabetic agents in patients [38,39]. Observational studies did not find a significant reduction in ulcerative colitis flares in patients receiving thiazolidinediones as compared to other oral antidiabetic drugs [40]; however, the systemic and local administration of rosiglitazone had beneficial effects in ulcerative colitis patients [41-43].
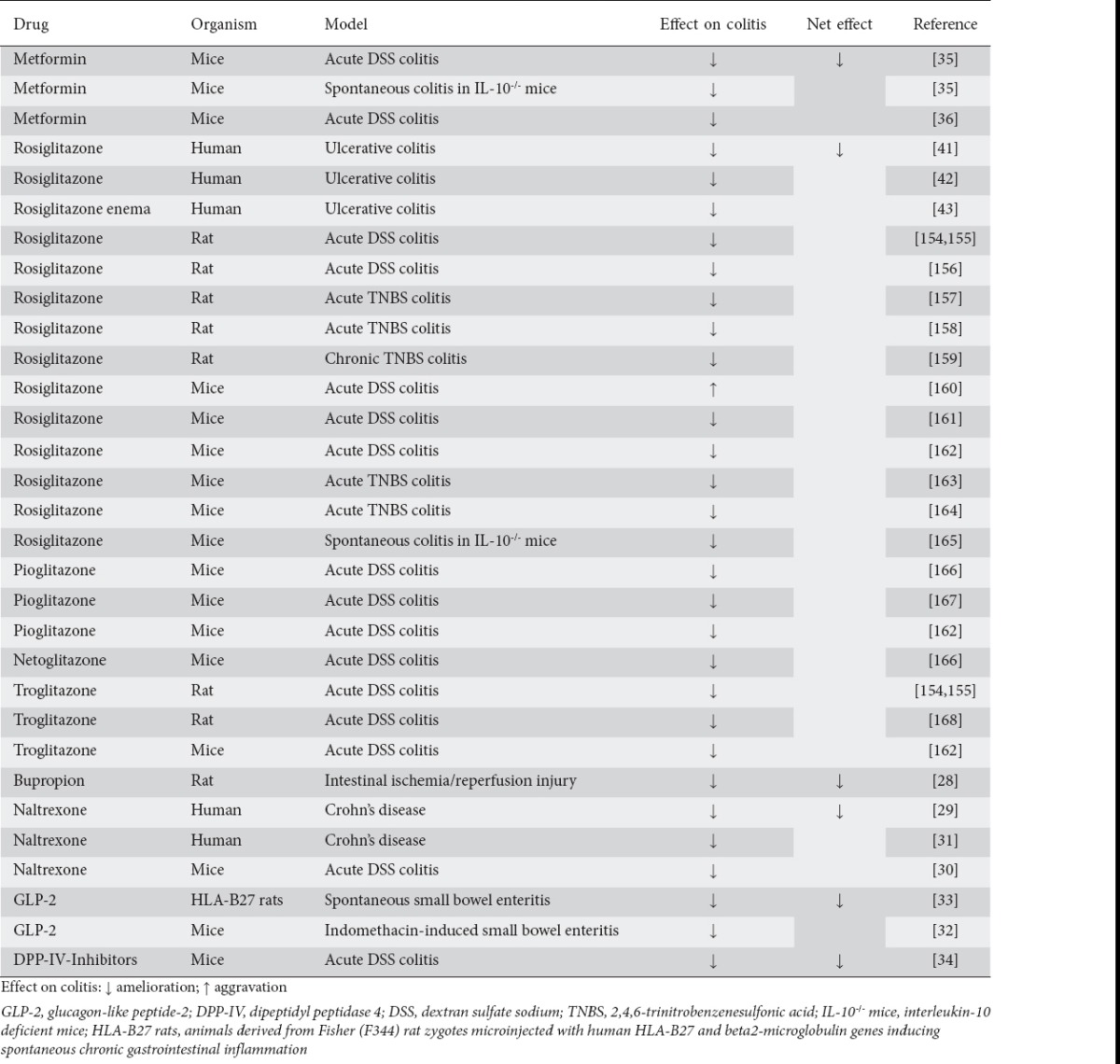
In summary, several insulin-sensitizing and weight-lowering drugs have pleiotropic, beneficial effects on autoimmune intestinal inflammation (Table 1). However, data in the literature on the impact of obesity in general on IBD are contradictory. A closer look at possible underlying mechanisms might shed some light on these discrepancies. How adipocytes, as key players in obesity, might impact on the intestine remains to be elucidated.
Table 1.
Summary of studies investigating the efficacy of antidiabetic and weight-lowering drugs in intestinal inflammation
Adipocytes in IBD: is it all about adipokines?
Historically seen as passive bystanders, adipocytes have increasingly been recognized as active participants in a variety of physiological reactions, including the immune system, via the expression and secretion of multiple hormone-like factors with auto- and paracrine effects, the so-called adipokines. Various different adipokines with pleiotropic roles have been described in this regard, many of which are found in significant concentrations in the systemic circulation [1-3,44-46]. These adipokines show correlations with the activity of a variety of autoimmune as well as infectious diseases [13,47]. Given the ill-defined role of obesity in IBD, it was an obvious necessity to investigate the role of defined adipokines and the potential diagnostic and pathophysiological value of systemic serum levels of adipokines in patients suffering from IBD, and to correlate these findings with observations made in different mouse models of (autoimmune) intestinal inflammation.
One of the first adipokines to be investigated in relation to intestinal inflammation was leptin, in 2002. Based on the observation that leptin-deficient animals, as well as humans, exhibit defective T-cell-function, Siegmund et al sought to determine whether experimentally induced colitis (DSS- and TNBS-induced colitis models) was affected in leptin-deficient (ob/ob) mice. They found that intestinal inflammation in both experimentally induced colitis models was significantly attenuated in ob/ob mice [48]. These results were mirrored by a markedly reduced inflammatory activity in oxazolone-induced colitis in mice and in a mouse model of infectious diarrhea (Clostridium difficile toxin A-induced enteritis) [49,50]. Similar observations were made regarding the DSS-induced colitis model, the Clostridium difficile toxin A-induced enteritis model, and the infectious Clostridium difficile-induced colitis model in both leptin-receptor-deficient (db/db) mice [50-52] and leptin receptor-mutant mice [52]. On the other hand, intrarectal leptin administration induced mucosal inflammation in mice [53]. Pathophysiologically, leptin proved to be an important inducer of various proinflammatory cytokines and played a regulatory role in T cell polarization [49].
Remarkably, no differences were demonstrated in the CD4+CD45RBhigh transfer model of colitis [54,55] when cells from ob/ob mice were transferred, indicating that leptin’s impact on colitis is not mediated by T cell secretion. Rather, when CD4+CD45RBhigh cells of db/db mice were used in this transfer model of colitis, the receptor animals showed a delayed colitis onset, indicating that leptin does act on T cells under these circumstances. It is noteworthy that no differences were noted in the course of spontaneous colitis in ob/ob x IL-10-/- mice compared to wild-type (wt)/wt x IL-10-/- mice, indicating that leptin does not impact on intestinal inflammation in this model. Table 2 summarizes the results of experimental animal studies that investigated leptin’s role in different models of intestinal inflammation.
Table 2.
Summary of experimental animal studies investigating the role of leptin and leptin receptor signaling in intestinal inflammation
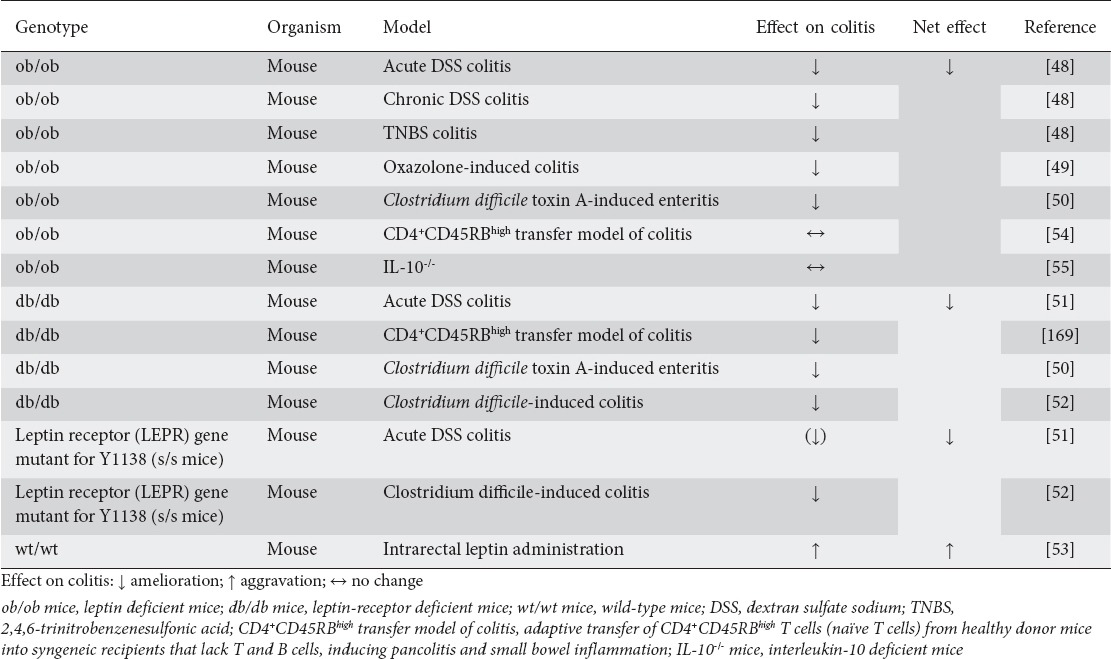
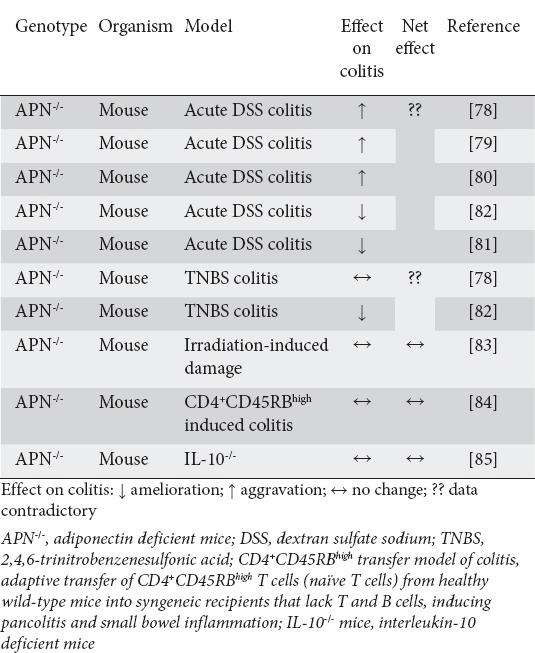
Table 3.
Summary of experimental animal studies investigating the role of adiponectin in intestinal inflammation
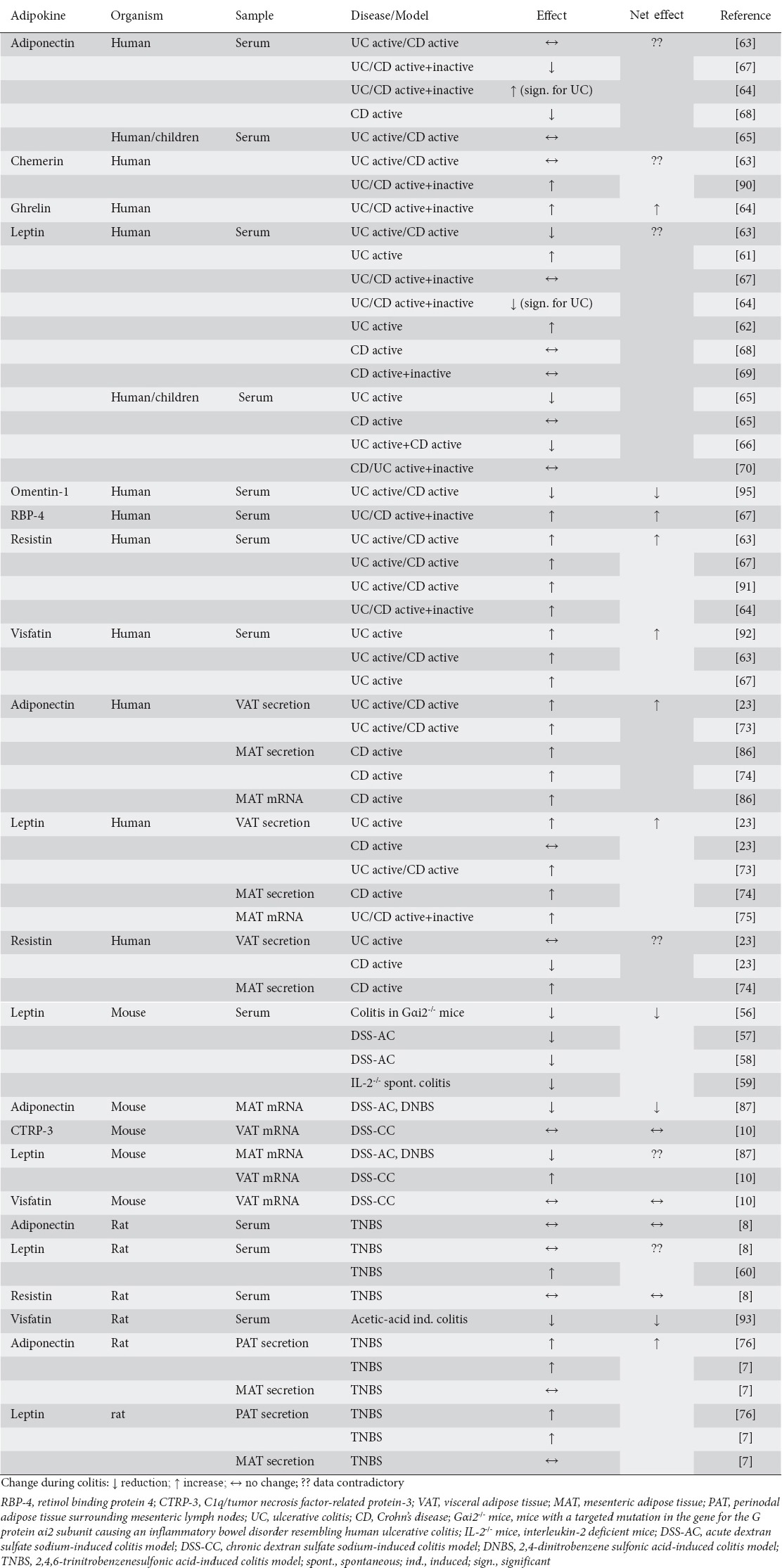
Interestingly, systemic leptin serum levels in mice seem to be reduced in different models of colitis [56-59], while other groups found increased versus unchanged serum levels in other colitis models in rats [8,60]. Studies investigating systemic leptin serum levels in patients suffering from IBD reported contradictory results, summarized in Table 4; while some groups found increased leptin concentrations in the systemic circulation in active IBD [61,62], others reported reduced [63-66] or unchanged systemic leptin levels [65,67-70], in part depending on the disease subgroup (Crohn’s disease versus ulcerative colitis). However, separate disease subgroups and different patient cohorts were not sufficient to explain the varying results. Another potential source of heterogeneity between studies is the different treatment status of the IBD patients who were included; however, data from truly treatment naïve patients are sparse.
Table 4.
Summary of observational studies investigating visceral adipose tissue adipokine expression, secretion and/or systemic levels in autoimmune intestinal inflammation
It has been demonstrated that adipocytes are the main contributors to systemic leptin serum levels [71,72]. However, keeping in mind that data in the literature on the impact of obesity in general on IBD are contradictory, generalized fat hypertrophy might not completely reflect the changes relevant in IBD in humans. Rather, the observed localized hypertrophy of fat tissue, particularly MAT, which is accompanied by proinflammatory changes within these fat depots [6], hints at possible paracrine effects of adipokines produced and secreted by mesenteric adipocytes. Remarkably, all studies investigating the leptin secretion/mRNA induction in MAT/VAT in IBD patients found unequivocally increased levels [23,73-75]. These results are mirrored by studies in MAT/VAT in rodent models of colitis [7,10,76]. In summary, leptin produced by MAT/VAT is upregulated and seems to act in a paracrine manner on the intestine as a proinflammatory mediator in patients suffering from IBD as well as in rodent models of colitis.
One of the first proteins demonstrated to be synthesized and secreted in large quantities almost exclusively by adipocytes, and as such one of the first “adipokines”, was adipocyte complement-related protein of 30 kDa (Acrp30), later termed “adiponectin” in 1995 [77]. Structurally, adiponectin has a globular head domain similar to complement factor C1q, as well as a collagen-like domain, and forms different higher-molecular secondary and tertiary structures. It was demonstrated to be detectable in significant quantities in the systemic circulation [77].
Studies by Nishihara and colleagues in 2006 demonstrated that acute DSS-induced colitis was significantly aggravated in adiponectin-deficient (APN-/-) mice, indicating that adiponectin had a protective effects on colitis in this model [78]. Other studies were able to reproduce these results [79,80]. Surprisingly, Fayad et al reported in 2007 that both acute DSS-induced colitis and acute TNBS-induced colitis were ameliorated in APN-/- mice [81,82], whereas Nishihara had found that TNBS-induced colitis remained unchanged in APN-/- mice [78]. Although differences in the concentration of DSS/TNBS chosen in these models have been held accountable for some of the observed inconsistencies, the impact of adiponectin on acute intestinal inflammation in mouse models remains elusive. No impact of adiponectin deficiency could be demonstrated on acute irradiation-induced small-intestinal damage [83], nor on mouse models of chronic autoimmune intestinal inflammation (CD4+CD45RBhigh induced colitis model, APN-/- x IL-10-/-) [84,85]. Table 3 summarizes the results of experimental animal studies that investigated adiponectin’s role in different models of intestinal inflammation.
Furthermore, the data regarding systemic serum levels of adiponectin in human IBD are also quite heterogeneous (Table 4). While some authors reported systemic serum levels to be unchanged in active Crohn’s disease and ulcerative colitis [63,65], others found reduced [67,68] or increased [64] adiponectin concentrations in the systemic circulation. Remarkably, as with the observations concerning leptin discussed above, all studies investigating the adiponectin secretion/mRNA induction in MAT/VAT in IBD patients found unequivocally increased levels [23,73,74,86]. However, in contrast to the data for leptin in animal models, these results were not completely mirrored by rodent models of colitis, where the data relating to MAT/VAT are contradictory (adiponectin levels decreased [87] versus increased [7,76]). These observations do implicate adiponectin, like leptin, as an important local mediator produced by mesenteric/visceral adipocytes and potentially impacting on the intestine in a paracrine manner in patients suffering from IBD, as well as in rodent models of colitis. However, as opposed to the clearly proinflammatory role of leptin, the exact impact of adiponectin on inflammation (pro-/anti-inflammatory) remains elusive.
Multiple other adipokines have been investigated in human autoimmune intestinal inflammation and in various rodent models of colitis. For example, systemic chemerin levels are elevated in experimental DSS-induced colitis in mice and intraperitoneal chemerin treatment exacerbates the disease, while treatment with anti-chemerin antibodies ameliorates colitis in this model [88,89]. However, data from serum samples of patients with Crohn’s disease or ulcerative colitis are contradictory [63,90]. Systemic resistin levels seem to be increased in patients with active IBD [63,64,67,91]; however, studies specifically targeting resistin expression/secretion in VAT in these patients found conflicting results [23,74]. Visfatin serum levels were increased in IBD patients [63,67,92], but this result could not be reproduced in rodent models of colitis [10,93]. The new adipokine C1q/TNF-related protein-3 (CTRP-3) is synthesized and secreted by MAT and ameliorates lipopolysaccharide (LPS)-induced IL-8 secretion, while reducing the basal expression of transforming growth factor β, connective tissue growth factor and collagen I in Crohn’s disease colonic lamina propria fibroblasts [94]. However, no difference was observed in VAT CTRP-3 mRNA expression between chronic DSS-induced colitis in mice compared to control mice [10]. Finally, there are reports that systemic serum levels of retinol-binding protein 4 [67] and ghrelin [64] are increased, while omentin-1 [95] is reduced in IBD patients. Table 4 summarizes the observational studies reported to date that investigated VAT expression, secretion and systemic adipokine levels in human IBD as well as in rodent models of intestinal inflammation.
Abdominal fat depots in IBD
Interestingly, diet-induced increased VAT mass in mice worsens the course of experimental TNBS-induced colitis, possibly via reduced intestinal epithelial cell adiponectin-receptor 1 expression [23]. A higher visceral-to-subcutaneous fat ratio is associated with and predictive for postoperative surgical morbidity in Crohn’s disease patients, whereas BMI is not [96,97]. In patients with Crohn’s disease, a higher visceral fat area is predictive of postoperative recurrence [98] and is associated with strictures and fistulas [99]. Pediatric IBD patients had 33% more VAT volume than age- and BMI-matched controls, and in these patients VAT was associated with complications of the disease course, for example fistulas, fibrosis and need for hospitalization [100]. In contrast, however, other studies found that an increased subcutaneous-to-visceral fat volume was predictive for postoperative complications after bowel resection in Crohn’s disease [101].
It should be noted that earlier reports found increased proinflammatory cytokine expression in MAT during TNBS-induced colonic inflammation in mice, which seemed to be mediated via increased substance P-induced neurokinin 1 receptor expression [102]. Studies in human IBD revealed that VAT in Crohn’s disease patients had a more proinflammatory gene expression profile as compared to ulcerative colitis [103]. Consequently, mesenteric fat has been proposed as a key player in Crohn’s disease [104]. Hypertrophic VAT adjacent to inflammatory lesions in Crohn’s disease patients exhibits proinflammatory gene expression; however, even VAT at sites distant from inflammatory lesions shows upregulated expression of genes involved in inflammation and immunity, similar to the changes observed in the VAT of obese patients [105]. In mice, both a high-fat diet and DSS-induced colitis induce similar inflammatory changes in VAT [106]. Interestingly, infliximab therapy in Crohn’s disease patients leads to an 18% increase in total abdominal fat, although the metabolic profile of these patients (glycemia, lipid profile) does not worsen [107]. Since VAT has been shown to be a key contributor to metabolic disturbances in obese patients [108-110], these results hint at potentially different reactions of VAT in autoimmune intestinal inflammation as opposed to the proinflammatory changes within the adipose tissue seen in obese patients.
Importantly, studies in human IBD revealed that VAT in Crohn’s disease with a proinflammatory gene expression profile is significantly more colonized by intestinal commensal bacteria of type Enterococcus faecalis [103]. In Crohn’s disease, as opposed to ulcerative colitis, translocation of intestinal bacteria to mesenteric fat depots has been demonstrated, leading to increased C-reactive protein secretion in systemically relevant levels in these adipocytes [111]. Notably, ovalbumin peptides translocate to MAT in healthy mice, which is not increased in experimental DSS-induced colitis. However, viable translocation of bacteria to MAT does not occur in healthy animals, but is increased in chronic DSS-induced colitis [112].
The importance of an intact intestinal barrier function for (intestinal) homeostasis has long been recognized, as has the key importance of intestinal (commensal) microbiota in the development of intestinal bowel diseases [113-116]. Together with accumulating data indicating innate immune responses in adipocytes [117-119], VAT is increasingly recognized as a key player in the maintenance of homeostasis. MAT from Crohn’s disease patients, as well as from mice suffering from TNBS-induced colitis, shows increased expression levels of PPARγ, farnesoid X receptor, leptin and adiponectin, which can be abrogated by probiotic treatment that reduces inflammatory activity [120]. Importantly, even without an inflammatory stimulus, VAT proinflammatory reactions are of physiological relevance in basal (intestinal) homeostasis, since an adipocyte-specific reduction of proinflammatory signaling in mice (aP2-dnTNF, a mouse model expressing a dominant-negative version of TNFα under the control of the ap2-promoter; aP2-RID, a mouse model expressing the adenoviral RIDα/β protein complex, which inhibits a number of proinflammatory signaling pathways under the control of the ap2-promoter; and TRE-IκBα, a mouse model expressing a mutated functional human IκBα, an inhibitor of the NF-κB pathway, under the control of a tet-responsive element) leads to increased lipid accumulation, glucose intolerance and systemic inflammation associated with impaired intestinal barrier function via impaired “healthy” adipose tissue expansion and remodeling [121]. VAT and leptin increase intestinal barrier permeability in rats in vivo without obvious inflammatory reactions in the intestine, and in colonic epithelial cells in co-culture experiments in vitro via a RhoA-ROCK-dependent pathway (RhoA, small GTPase protein of the Rho family, associated with cytoskeleton regulation; and ROCK, rho-associated, coiled-coil containing protein kinase) [122]. Other studies demonstrated that increased VAT is also associated with increased intestinal barrier permeability in healthy women [123].
In summary, accumulating data indicate that, rather than a general increase in fat, local fat depots, particularly the visceral fat depot, actively participate in intestinal inflammatory reactions [124-127]. These results argue against a systemic, but in favor of a localized auto- and paracrine effect of adipose tissue on the intestine. While an unequivocal causal link between adipocytes, insulin sensitivity and intestinal inflammation is missing thus far, these observations provide a rationale for a mutual interplay between VAT and the intestine with its different compartments. Especially in the light of recently emerging exciting data linking visceral obesity and intestinal barrier function [121-123], a much deeper analysis of innate immune signaling in visceral adipocytes in conditions of intestinal health and disease is needed to further characterize the important role of VAT in intestinal homeostasis, and more generally their key role in the homeostasis of the whole organism.
VAT as part of the innate immune system in IBD
Besides its established role as an endocrine organ [128,129], the adipose tissue can be regarded as part of the innate immune system [2,3,44,130], being activated by inflammatory or infectious processes. Moreover, the adipose tissue expresses the whole machinery of inflammation and innate immune activation, including classical cytokines (IL-1, IL-6, TNFα), chemokines [monocyte chemoattractant protein-1 (MCP-1); C-C motif chemokine ligand 2], complement components (C1q, C3a), TLRs, nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), and C1q/TNF-related proteins [130]. Thus, VAT could link innate immune reactions during gut inflammation to adjacent adipose tissue alterations such as creeping fat [131-134].
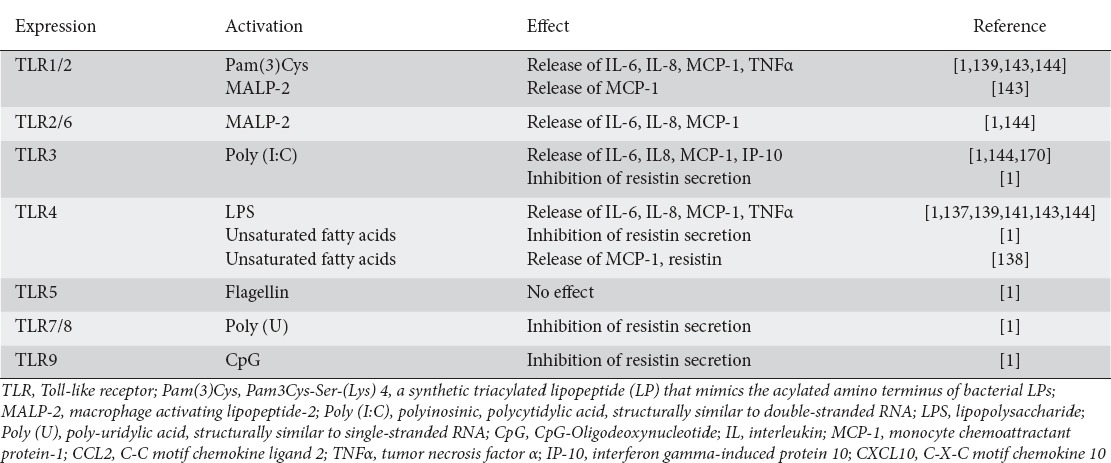
TLRs are among the most prominent sensing molecules (pattern recognition receptors) recognizing molecular patterns (pathogen-associated molecular patterns, PAMPs) derived from bacteria and viruses [135,136]. The groups led by Shapiro and Scherer [137] were the first to describe a prominent role of the TLR4 and TLR2 system in adipocytes. Following these reports, adipocytes have been shown [1,137-142] to express all known TLRs from TLR1 to TLR9, apart from TLR5 (flagellin receptor), and functionality has been demonstrated for all of them [1,143]. Table 5 summarizes the currently available data on the expression of TLRs and their functional activation by specific ligands in adipocytes. These data are complicated by the fact that adapter molecule utilization may differ in adipocytes when compared to classical immune cells. For example, Poly(I:C) signaling via TLR3 requires a Toll/IL-1 receptor-domain-containing adapter-inducing interferon(TRIF)-independent but myeloid differentiation primary response gene 88 (MyD88)-dependent route [144]. In contrast, LPS signaling via TLR4 requires MyD88, myelin and lymphocyte protein (Mal) and TRIF, whereas Pam3Cys signaling via TLR2 requires MyD88 and Mal, but not TRIF [144]. It has been proposed that the TLR system in adipocytes orchestrates the complex process of energy utilization in the context of immune responses and immune activation [144,145]. However, since increased intestinal permeability leads to a direct exposure of adjacent VAT to microbial products, these data at the same time provide the molecular basis for IBD-associated inflammation of VAT.
Table 5.
Summary of data on Toll-like receptor (TLR) expression and activation by specific ligands in adipocytes
In contrast to TLRs, the NOD-1 and NOD-2 proteins represent cytoplasmic receptors (NLRs) of the innate immune response. Since NOD-1 and NOD-2 are expressed in adipose tissue [146,147], these molecules not only might be involved in the pathogenesis of type 2 diabetes mellitus and obesity-related inflammation [136,147], but also might be activated in response to intestinal inflammation and microbial translocation. Activation of NOD proteins by NOD-1-specific ligands in adipocytes causes NF-κB p65 nuclear translocation and subsequent MCP-1, IL-6 and IL-8 production [147]. These results provide the molecular basis for the hypothesis that direct exposure of intestinal adipocytes to bacterial peptidoglycans could start the process of adipose tissue inflammation during gut inflammation. NLR protein-3 (NLRP3) represents an innate immune sensor, and its activation by microbial or endogenous danger signals causes caspase-1 activation and production of proinflammatory cytokines such as IL-1 and IL-18 [148]. NLRP3 activation by endoplasmic reticulum stress in adipocytes increases IL-1 expression and secretion [149], whereas repressors of NLRP3 reduce adipose tissue inflammation [150]. In mice deficient in NLRP3 expression, the defective inflammasome compartment was accompanied by reduced MCP-1 expression in adipocytes [148]. However, in spite of the well-known proinflammatory effects of a high-fat diet, such as adipose tissue macrophage infiltration, NLRP3 expression was not modified by a high-fat diet [142].
In summary, intestinal adipocytes residing within the VAT adjacent to inflamed gut express major functional components of the innate immune recognition system, such as TLRs and NODs/NLRPs. Consequently, visceral adipocytes are able to sense a wide variety of microbial components that cross the disturbed intestinal barrier seen in IBD [131]. Thus, the observed inflammatory transformation of VAT seems to be a consequence, rather than the cause of IBD. The physiological meaning behind this mechanism is most likely to provide an additional antimicrobial barrier surrounding the affected gut. This adipose tissue barrier might reduce the risk of intestinal perforation, bacterial translocation to the peritoneum and finally, systemic inflammation and sepsis [112]. Remarkably, a recent study published in Science in 2015 demonstrated that adipocytes protect against invasive bacterial infection by secreting antimicrobial peptides such as cathelicidin [117,151]. Thus, whereas VAT mass is clearly associated with obesity-related inflammation, insulin resistance and type 2 diabetes mellitus [152], intra-abdominal fat could be of benefit in IBD [103,112]. Whether viable bacteria are able to reside within adipocytes, and if so for how long, remains an additional important and unsolved question [103,153].
Fig. 1 depicts this hypothesis regarding the role of inflamed VAT in IBD. Because of the increased permeability of the mucus and epithelium, microbes and toxins are able to cross the mucus and epithelial barriers, which represent the first and second barrier. Subsequently, PAMPs are recognized by TLRs and NLRs in lamina propria mononuclear cells and in adipose tissue. Activation of TLRs and NLRs causes adipose tissue inflammation, hypertrophy and finally the formation of creeping fat. This altered adipose tissue releases cytokines, adipokines, chemokines, complement factors, antimicrobial peptides, and CTRPs, thus providing an additional barrier of local defense.
Figure 1.
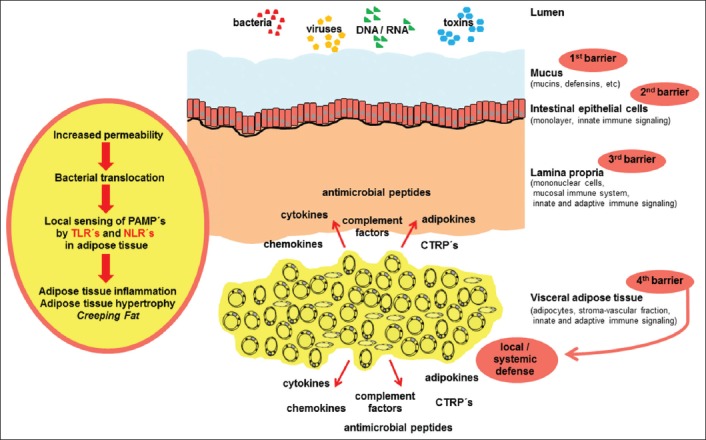
Current hypothetical view of the role of visceral adipose tissue in inflammatory bowel disease
The intestinal mucus provides a first barrier against luminal toxins and invading pathogens. In the context of intestinal inflammation and increased permeability of the mucus and epithelium, microbes and toxins are able to cross the epithelial and mucus barriers. Subsequently, lamina propria mononuclear cells interact with these compounds. When invasion cannot be contained by the mucosal compartment, pathogen associated molecular patterns (PAMPs) are recognized by Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain-like receptors (NLRs) in adipose tissue. Activation of TLRs and NLRs causes adipose tissue inflammation, hypertrophy and the formation of creeping fat. This altered adipose tissue releases cytokines, adipokines, chemokines, complement factors, antimicrobial peptides and C1q/tumor necrosis factor-related proteins (CTRPs), thus providing a “fourth barrier” of local defense. This mechanism might protect against local gut perforation, local peritonitis, systemic inflammation, and sepsis
Concluding remarks
Current data suggest an intricate relationship between intestinal inflammation and adjacent VAT depots. During intestinal inflammatory conditions, VAT is not merely a passive “bystander”, but actively participates in immune responses via the secretion of fat-derived hormones, the so-called adipokines. Furthermore, adipocytes, via the expression of pattern recognition receptors, actively participate in anti-microbial host defenses in the context of intestinal bacterial translocation, as part of the innate immune system. Thus, VAT constitutes a barrier against invading pathogens and contributes to the homeostasis of the whole organism (Fig. 1). This mechanism could protect the organism from local gut perforation, local peritonitis, systemic inflammation and sepsis. Today, many questions in this innovative field remain unanswered. However, with the rapidly growing body of evidence, modulators of adipose tissue function and regulators of adipokine secretion, as well as modifiers of adipocytic pattern recognition molecules, might turn out to be future drug targets in the treatment of IBD.
Acknowledgment
The support of Dr. Andreas Schmid and Dr. Jutta Schlegel, from the Department of Internal Medicine III, University of Giessen, Germany, who provided a focused literature search for parts of the article, is greatly appreciated.
Biography
Giessen University Hospital, Germany
Footnotes
Conflict of Interest: None
References
- 1.Kopp A, Buechler C, Neumeier M, et al. Innate immunity and adipocyte function: ligand-specific activation of multiple Toll-like receptors modulates cytokine, adipokine, and chemokine secretion in adipocytes. Obesity. 2009;17:648–656. doi: 10.1038/oby.2008.607. [DOI] [PubMed] [Google Scholar]
- 2.Schäffler A, Schölmerich J, Salzberger B. Adipose tissue as an immunological organ: Toll-like receptors, C1q/TNFs and CTRPs. Trends Immunol. 2007;28:393–399. doi: 10.1016/j.it.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 3.Schäffler A, Buechler C. CTRP family: linking immunity to metabolism. Trends Endocrinol Metab. 2012;23:194–204. doi: 10.1016/j.tem.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Ko JK, Lee HL, Kim JO, et al. Visceral fat as a useful parameter in the differential diagnosis of Crohn’s disease and intestinal tuberculosis. Intest Res. 2014;12:42–47. doi: 10.5217/ir.2014.12.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amitai MM, Arazi-Kleinman T, Avidan B, et al. Fat halo sign in the bowel wall of patients with Crohn’s disease. Clin Radiol. 2007;62:994–997. doi: 10.1016/j.crad.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 6.Desreumaux P, Ernst O, Geboes K, et al. Inflammatory alterations in mesenteric adipose tissue in Crohn’s disease. Gastroenterology. 1999;117:73–81. doi: 10.1016/s0016-5085(99)70552-4. [DOI] [PubMed] [Google Scholar]
- 7.Gambero A, Maróstica M, Abdalla Saad MJ, Pedrazzoli J Jr. Mesenteric adipose tissue alterations resulting from experimental reactivated colitis. Inflamm Bowel Dis. 2007;13:1357–1364. doi: 10.1002/ibd.20222. [DOI] [PubMed] [Google Scholar]
- 8.Clemente TR, Dos Santos AN, Sturaro JN, et al. Infliximab modifies mesenteric adipose tissue alterations and intestinal inflammation in rats with TNBS-induced colitis. Scand J Gastroenterol. 2012;47:943–950. doi: 10.3109/00365521.2012.688213. [DOI] [PubMed] [Google Scholar]
- 9.Li Y, Zuo L, Zhu W, et al. Telmisartan attenuates the inflamed mesenteric adipose tissue in spontaneous colitis by mechanisms involving regulation of neurotensin/microRNA-155 pathway. Biochem Pharmacol. 2015;93:461–469. doi: 10.1016/j.bcp.2014.12.020. [DOI] [PubMed] [Google Scholar]
- 10.Karrasch T, Schmid A, Kopp A, Obermeier F, Hofmann C, Schäffler A. Impact of toll-like-receptor-9 (TLR9) deficiency on visceral adipose tissue adipokine expression during chronic DSS-induced colitis in mice. Exp Clin Endocrinol Diabetes. 2015;123:80–87. doi: 10.1055/s-0034-1398502. [DOI] [PubMed] [Google Scholar]
- 11.Cristancho AG, Lazar MA. Forming functional fat: a growing understanding of adipocyte differentiation. Nat Rev Mol Cell Biol. 2011;12:722–734. doi: 10.1038/nrm3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.García-Alonso V, Clària J. Prostaglandin E2 signals white-to-brown adipogenic differentiation. Adipocyte. 2014;3:290–296. doi: 10.4161/adip.29993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Versini M, Jeandel PY, Rosenthal E, Shoenfeld Y. Obesity in autoimmune diseases: not a passive bystander. Autoimmun Rev. 2014;13:981–1000. doi: 10.1016/j.autrev.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 14.Steed H, Walsh S, Reynolds N. A brief report of the epidemiology of obesity in the inflammatory bowel disease population of Tayside, Scotland. Obes Facts. 2009;2:370–372. doi: 10.1159/000262276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blain A, Cattan S, Beaugerie L, Carbonnel F, Gendre JP, Cosnes J. Crohn’s disease clinical course and severity in obese patients. Clin Nutr. 2002;21:51–57. doi: 10.1054/clnu.2001.0503. [DOI] [PubMed] [Google Scholar]
- 16.Long MD, Crandall WV, Leibowitz IH, et al. ImproveCareNow Collaborative for Pediatric IBD Prevalence and epidemiology of overweight and obesity in children with inflammatory bowel disease. Inflamm Bowel Dis. 2011;17:2162–2168. doi: 10.1002/ibd.21585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seminerio JL, Koutroubakis IE, Ramos-Rivers C, et al. Impact of obesity on the management and clinical course of patients with inflammatory bowel disease. Inflamm Bowel Dis. 2015;21:2857–2863. doi: 10.1097/MIB.0000000000000560. [DOI] [PubMed] [Google Scholar]
- 18.Aminian A, Andalib A, Ver MR, Corcelles R, Schauer PR, Brethauer SA. Outcomes of bariatric surgery in patients with inflammatory bowel disease. Obes Surg. 2016;26:1186–1190. doi: 10.1007/s11695-015-1909-y. [DOI] [PubMed] [Google Scholar]
- 19.Ahn LB, Huang CS, Forse RA, Hess DT, Andrews C, Farraye FA. Crohn’s disease after gastric bypass surgery for morbid obesity: is there an association? Inflamm Bowel Dis. 2005;11:622–624. doi: 10.1097/01.mib.0000165113.33557.3a. [DOI] [PubMed] [Google Scholar]
- 20.Manguso F, Picascia S, Balzano A. Ulcerative colitis exacerbating after placement of intragastric balloon for the treatment of obesity. Inflamm Bowel Dis. 2008;14:872–873. doi: 10.1002/ibd.20373. [DOI] [PubMed] [Google Scholar]
- 21.Hass DJ, Brensinger CM, Lewis JD, Lichtenstein GR. The impact of increased body mass index on the clinical course of Crohn’s disease. Clin Gastroenterol Hepatol. 2006;4:482–488. doi: 10.1016/j.cgh.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 22.Flores A, Burstein E, Cipher DJ, Feagins LA. Obesity in inflammatory bowel disease: a marker of less severe disease. Dig Dis Sci. 2015;60:2436–2445. doi: 10.1007/s10620-015-3629-5. [DOI] [PubMed] [Google Scholar]
- 23.Sideri A, Stavrakis D, Bowe C, et al. Effects of obesity on severity of colitis and cytokine expression in mouse mesenteric fat. Potential role of adiponectin receptor 1. Am J Physiol Gastrointest Liver Physiol. 2015;308:G591–G604. doi: 10.1152/ajpgi.00269.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paik J, Fierce Y, Treuting PM, Brabb T, Maggio-Price L. High-fat diet-induced obesity exacerbates inflammatory bowel disease in genetically susceptible Mdr1a-/- male mice. J Nutr. 2013;143:1240–1247. doi: 10.3945/jn.113.174615. [DOI] [PubMed] [Google Scholar]
- 25.Chan SS, Luben R, Olsen A, et al. Body mass index and the risk for Crohn’s disease and ulcerative colitis: data from a European Prospective Cohort Study (The IBD in EPIC Study) Am J Gastroenterol. 2013;108:575–582. doi: 10.1038/ajg.2012.453. [DOI] [PubMed] [Google Scholar]
- 26.Cheung BM, Cheung TT, Samaranayake NR. Safety of antiobesity drugs. Ther Adv Drug Saf. 2013;4:171–181. doi: 10.1177/2042098613489721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodríguez JE, Campbell KM. Past, present, and future of pharmacologic therapy in obesity. Prim Care. 2016;43:61–67. doi: 10.1016/j.pop.2015.08.011. viii. [DOI] [PubMed] [Google Scholar]
- 28.Cámara-Lemarroy CR, Guzmán-de la Garza FJ, Cordero-Pérez P, et al. Bupropion reduces the inflammatory response and intestinal injury due to ischemia-reperfusion. Transplant Proc. 2013;45:2502–2505. doi: 10.1016/j.transproceed.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 29.Smith JP, Field D, Bingaman SI, Evans R, Mauger DT. Safety and tolerability of low-dose naltrexone therapy in children with moderate to severe Crohn’s disease: a pilot study. J Clin Gastroenterol. 2013;47:339–345. doi: 10.1097/MCG.0b013e3182702f2b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matters GL, Harms JF, McGovern C, et al. The opioid antagonist naltrexone improves murine inflammatory bowel disease. J Immunotoxicol. 2008;5:179–187. doi: 10.1080/15476910802131469. [DOI] [PubMed] [Google Scholar]
- 31.Smith JP, Bingaman SI, Ruggiero F, et al. Therapy with the opioid antagonist naltrexone promotes mucosal healing in active Crohn’s disease: a randomized placebo-controlled trial. Dig Dis Sci. 2011;56:2088–2097. doi: 10.1007/s10620-011-1653-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boushey RP, Yusta B, Drucker DJ. Glucagon-like peptide 2 decreases mortality and reduces the severity of indomethacin-induced murine enteritis. Am J Physiol. 1999;277:E937–E947. doi: 10.1152/ajpendo.1999.277.5.E937. [DOI] [PubMed] [Google Scholar]
- 33.Alavi K, Schwartz MZ, Palazzo JP, Prasad R. Treatment of inflammatory bowel disease in a rodent model with the intestinal growth factor glucagon-like peptide-2. J Pediatr Surg. 2000;35:847–851. doi: 10.1053/jpsu.2000.6861. [DOI] [PubMed] [Google Scholar]
- 34.Ban H, Bamba S, Imaeda H, et al. The DPP-IV inhibitor ER-319711 has a proliferative effect on the colonic epithelium and a minimal effect in the amelioration of colitis. Oncol Rep. 2011;25:1699–1703. doi: 10.3892/or.2011.1223. [DOI] [PubMed] [Google Scholar]
- 35.Koh SJ, Kim JM, Kim IK, Ko SH, Kim JS. Anti-inflammatory mechanism of metformin and its effects in intestinal inflammation and colitis-associated colon cancer. J Gastroenterol Hepatol. 2014;29:502–510. doi: 10.1111/jgh.12435. [DOI] [PubMed] [Google Scholar]
- 36.Lee SY, Lee SH, Yang EJ, et al. Metformin ameliorates inflammatory bowel disease by suppression of the STAT3 signaling pathway and regulation of the between Th17/Treg balance. PLoS One. 2015;10:e0135858. doi: 10.1371/journal.pone.0135858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li W, Wang QL, Liu X, et al. Combined use of vitamin D3 and metformin exhibits synergistic chemopreventive effects on colorectal neoplasia in rats and mice. Cancer Prev Res (Phila) 2015;8:139–148. doi: 10.1158/1940-6207.CAPR-14-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Speca S, Dubuquoy L, Desreumaux P. Peroxisome proliferator-activated receptor gamma in the colon: inflammation and innate antimicrobial immunity. J Clin Gastroenterol. 2014;48(Suppl 1):S23–S27. doi: 10.1097/MCG.0000000000000253. [DOI] [PubMed] [Google Scholar]
- 39.Croasdell A, Duffney PF, Kim N, Lacy SH, Sime PJ, Phipps RP. PPARγand the innate immune system mediate the resolution of inflammation. PPAR Res. 2015;2015:549691. doi: 10.1155/2015/549691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lund JL, Stürmer T, Porter CQ, Sandler RS, Kappelman MD. Thiazolidinedione use and ulcerative colitis-related flares: an exploratory analysis of administrative data. Inflamm Bowel Dis. 2011;17:787–794. doi: 10.1002/ibd.21348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lewis JD, Lichtenstein GR, Deren JJ, et al. Rosiglitazone for Ulcerative Colitis Study Group Rosiglitazone for active ulcerative colitis: a randomized placebo-controlled trial. Gastroenterology. 2008;134:688–695. doi: 10.1053/j.gastro.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liang HL, Ouyang Q. A clinical trial of combined use of rosiglitazone and 5-aminosalicylate for ulcerative colitis. World J Gastroenterol. 2008;14:114–119. doi: 10.3748/wjg.14.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pedersen G, Brynskov J. Topical rosiglitazone treatment improves ulcerative colitis by restoring peroxisome proliferator-activated receptor-gamma activity. Am J Gastroenterol. 2010;105:1595–1603. doi: 10.1038/ajg.2009.749. [DOI] [PubMed] [Google Scholar]
- 44.Schäffler A1, Müller-Ladner U, Schölmerich J, Büchler C. Role of adipose tissue as an inflammatory organ in human diseases. Endocr Rev. 2006;27:449–467. doi: 10.1210/er.2005-0022. [DOI] [PubMed] [Google Scholar]
- 45.Schäffler A, Schölmerich J, Büchler C. Mechanisms of disease: adipocytokines and visceral adipose tissue-emerging role in intestinal and mesenteric diseases. Nat Clin Pract Gastroenterol Hepatol. 2005;2:103–111. doi: 10.1038/ncpgasthep0090. [DOI] [PubMed] [Google Scholar]
- 46.Schäffler A, Schölmerich J, Büchler C. Mechanisms of disease: adipocytokines and visceral adipose tissue-emerging role in nonalcoholic fatty liver disease. Nat Clin Pract Gastroenterol Hepatol. 2005;2:273–280. doi: 10.1038/ncpgasthep0186. [DOI] [PubMed] [Google Scholar]
- 47.Karlsson EA, Beck MA. The burden of obesity on infectious disease. Exp Biol Med. 2010;235:1412–1424. doi: 10.1258/ebm.2010.010227. [DOI] [PubMed] [Google Scholar]
- 48.Siegmund B, Lehr HA, Fantuzzi G. Leptin: a pivotal mediator of intestinal inflammation in mice. Gastroenterology. 2002;122:2011–2025. doi: 10.1053/gast.2002.33631. [DOI] [PubMed] [Google Scholar]
- 49.Batra A, Okur B, Glauben R, et al. Leptin: a critical regulator of CD4+T-cell polarization in vitro and in vivo. Endocrinol. 2010;151:56–62. doi: 10.1210/en.2009-0565. [DOI] [PubMed] [Google Scholar]
- 50.Mykoniatis A, Anton PM, Wlk M, et al. Leptin mediates Clostridium difficile toxin A-induced enteritis in mice. Gastroenterology. 2003;124:683–691. doi: 10.1053/gast.2003.50101. [DOI] [PubMed] [Google Scholar]
- 51.Gove ME, Rhodes DH, Pini M, et al. Role of leptin receptor-induced STAT3 signaling in modulation of intestinal and hepatic inflammation in mice. J Leukoc Biol. 2009;85:491–496. doi: 10.1189/jlb.0808508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Madan R, Guo X, Naylor C, et al. Role of leptin-mediated colonic inflammation in defense against Clostridium difficile colitis. Infect Immun. 2014;82:341–349. doi: 10.1128/IAI.00972-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sitaraman S, Liu X, Charrier L, et al. Colonic leptin: source of a novel proinflammatory cytokine involved in IBD. FASEB J. 2004;18:696–698. doi: 10.1096/fj.03-0422fje. [DOI] [PubMed] [Google Scholar]
- 54.Fantuzzi G, Sennello JA, Batra A, et al. Defining the role of T cell-derived leptin in the modulation of hepatic or intestinal inflammation in mice. Clin Exp Immunol. 2005;142:31–38. doi: 10.1111/j.1365-2249.2005.02898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Siegmund B, Sennello JA, Lehr HA, et al. Development of intestinal inflammation in double IL-10- and leptin-deficient mice. J Leukoc Biol. 2004;76:782–786. doi: 10.1189/jlb.0404239. [DOI] [PubMed] [Google Scholar]
- 56.Hultgren OH, Ohman L, Hörnquist EH. Decreased leptin production in mice after onset of ulcerative colitis-like disease. Scand J Gastroenterol. 2004;39:1166–1167. doi: 10.1080/00365520410007962. [DOI] [PubMed] [Google Scholar]
- 57.Deboer MD, Li Y. Puberty is delayed in male mice with dextran sodium sulfate colitis out of proportion to changes in food intake, body weight, and serum levels of leptin. Pediatr Res. 2011;69:34–39. doi: 10.1203/PDR.0b013e3181ffee6c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.DeBoer MD, Li Y, Cohn S. Colitis causes delay in puberty in female mice out of proportion to changes in leptin and corticosterone. J Gastroenterol. 2010;45:277–284. doi: 10.1007/s00535-009-0192-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gaetke LM, Oz HS, de Villiers WJ, Varilek GW, Frederich RC. The leptin defense against wasting is abolished in the IL-2-deficient mouse model of inflammatory bowel disease. J Nutr. 2002;132:893–896. doi: 10.1093/jn/132.5.893. [DOI] [PubMed] [Google Scholar]
- 60.Barbier M, Cherbut C, Aubé AC, Blottière HM, Galmiche JP. Elevated plasma leptin concentrations in early stages of experimental intestinal inflammation in rats. Gut. 1998;43:783–790. doi: 10.1136/gut.43.6.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Biesiada G, Czepiel J, Ptak-Belowska A, et al. Expression and release of leptin and proinflammatory cytokines in patients with ulcerative colitis and infectious diarrhea. J Physiol Pharmacol. 2012;63:471–481. [PubMed] [Google Scholar]
- 62.Tuzun A, Uygun A, Yesilova Z, et al. Leptin levels in the acute stage of ulcerative colitis. J Gastroenterol Hepatol. 2004;19:429–432. doi: 10.1111/j.1440-1746.2003.03300.x. [DOI] [PubMed] [Google Scholar]
- 63.Waluga M, Hartleb M, Boryczka G, Kukla M, Zwirska-Korczala K. Serum adipokines in inflammatory bowel disease. World J Gastroenterol. 2014;20:6912–6917. doi: 10.3748/wjg.v20.i22.6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Karmiris K, Koutroubakis IE, Xidakis C, Polychronaki M, Voudouri T, Kouroumalis EA. Circulating levels of leptin, adiponectin, resistin, and ghrelin in inflammatory bowel disease. Inflamm Bowel Dis. 2006;12:100–105. doi: 10.1097/01.MIB.0000200345.38837.46. [DOI] [PubMed] [Google Scholar]
- 65.Chouliaras G, Panayotou I, Margoni D, et al. Circulating leptin and adiponectin and their relation to glucose metabolism in children with Crohn’s disease and ulcerative colitis. Pediatr Res. 2013;74:420–426. doi: 10.1038/pr.2013.114. [DOI] [PubMed] [Google Scholar]
- 66.Aurangzeb B, Leach ST, Lemberg DA, Day AS. Assessment of nutritional status and serum leptin in children with inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2011;52:536–541. doi: 10.1097/MPG.0b013e3181f87a95. [DOI] [PubMed] [Google Scholar]
- 67.Valentini L, Wirth EK, Schweizer U, et al. Circulating adipokines and the protective effects of hyperinsulinemia in inflammatory bowel disease. Nutrition. 2009;25:172–181. doi: 10.1016/j.nut.2008.07.020. [DOI] [PubMed] [Google Scholar]
- 68.Rodrigues VS, Milanski M, Fagundes JJ, et al. Serum levels and mesenteric fat tissue expression of adiponectin and leptin in patients with Crohn’s disease. Clin Exp Immunol. 2012;170:358–364. doi: 10.1111/j.1365-2249.2012.04660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nishi Y, Isomoto H, Ueno H, et al. Plasma leptin and ghrelin concentrations in patients with Crohn’s disease. World J Gastroenterol. 2005;11:7314–7317. doi: 10.3748/wjg.v11.i46.7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hoppin AG, Kaplan LM, Zurakowski D, Leichtner AM, Bousvaros A. Serum leptin in children and young adults with inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 1998;26:500–505. doi: 10.1097/00005176-199805000-00003. [DOI] [PubMed] [Google Scholar]
- 71.Frederich RC, Löllmann B, Hamann A, et al. Expression of ob mRNA and its encoded protein in rodents. Impact of nutrition and obesity. J Clin Invest. 1995;96:1658–1663. doi: 10.1172/JCI118206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Considine RV, Sinha MK, Heiman ML, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 73.Ponemone V, Keshavarzian A, Brand MI, et al. Apoptosis and inflammation: role of adipokines in inflammatory bowel disease. Clin Transl Gastroenterol. 2010;1:e1. doi: 10.1038/ctg.2010.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Paul G, Schäffler A, Neumeier M, et al. Profiling adipocytokine secretion from creeping fat in Crohn’s disease. Inflamm Bowel Dis. 2006;12:471–477. doi: 10.1097/00054725-200606000-00005. [DOI] [PubMed] [Google Scholar]
- 75.Barbier M, Vidal H, Desreumaux P, et al. Overexpression of leptin mRNA in mesenteric adipose tissue in inflammatory bowel diseases. Gastroenterol Clin Biol. 2003;27:987–991. [PubMed] [Google Scholar]
- 76.Acedo SC, Gotardo EM, Lacerda JM, de Oliveira CC, de Oliveira Carvalho P, Gambero A. Perinodal adipose tissue and mesenteric lymph node activation during reactivated TNBS-colitis in rats. Dig Dis Sci. 2011;56:2545–2552. doi: 10.1007/s10620-011-1644-8. [DOI] [PubMed] [Google Scholar]
- 77.Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem. 1995;270:26746–26749. doi: 10.1074/jbc.270.45.26746. [DOI] [PubMed] [Google Scholar]
- 78.Nishihara T, Matsuda M, Araki H, et al. Effect of adiponectin on murine colitis induced by dextran sulfate sodium. Gastroenterology. 2006;131:853–861. doi: 10.1053/j.gastro.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 79.Saxena A, Chumanevich A, Fletcher E, et al. Adiponectin deficiency: role in chronic inflammation induced colon cancer. Biochim Biophys Acta. 2012;1822:527–536. doi: 10.1016/j.bbadis.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Saxena A, Fletcher E, Larsen B, Baliga MS, Durstine JL, Fayad R. Effect of exercise on chemically-induced colitis in adiponectin deficient mice. J Inflamm (Lond) 2012;9:30. doi: 10.1186/1476-9255-9-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kaur K, Saxena A, Larsen B, et al. Mucus mediated protection against acute colitis in adiponectin deficient mice. J Inflamm (Lond) 2015;12:35. doi: 10.1186/s12950-015-0079-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fayad R, Pini M, Sennello JA, et al. Adiponectin deficiency protects mice from chemically induced colonic inflammation. Gastroenterology. 2007;132:601–614. doi: 10.1053/j.gastro.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 83.Ponemone V, Fayad R, Gove ME, Pini M, Fantuzzi G. Effect of adiponectin deficiency on intestinal damage and hematopoietic responses of mice exposed to gamma radiation. Mutat Res. 2010;690:102–107. doi: 10.1016/j.mrfmmm.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gove ME, Pini M, Fayad R, Cabay RJ, Fantuzzi G. Adiponectin deficiency modulates adhesion molecules expression and cytokine production but does not affect disease severity in the transfer model of colitis. Cytokine. 2009;47:119–125. doi: 10.1016/j.cyto.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pini M, Gove ME, Fayad R, Cabay RJ, Fantuzzi G. Adiponectin deficiency does not affect development and progression of spontaneous colitis in IL-10 knockout mice. Am J Physiol Gastrointest Liver Physiol. 2009;296 doi: 10.1152/ajpgi.90593.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yamamoto K, Kiyohara T, Murayama Y, et al. Production of adiponectin, an anti-inflammatory protein, in mesenteric adipose tissue in Crohn’s disease. Gut. 2005;54:789–796. doi: 10.1136/gut.2004.046516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Olivier I, Theodorou V, Valet P, Castan-Laurell I, Ferrier L, Eutamène H. Modifications of mesenteric adipose tissue during moderate experimental colitis in mice. Life Sci. 2014;94:1–7. doi: 10.1016/j.lfs.2013.09.028. [DOI] [PubMed] [Google Scholar]
- 88.Buechler C. Chemerin, a novel player in inflammatory bowel disease. Cell Mol Immunol. 2014;11:315–316. doi: 10.1038/cmi.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lin Y, Yang X, Yue W, et al. Chemerin aggravates DSS-induced colitis by suppressing M2 macrophage polarization. Cell Mol Immunol. 2014;11:355–366. doi: 10.1038/cmi.2014.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Weigert J, Obermeier F, Neumeier M, et al. Circulating levels of chemerin and adiponectin are higher in ulcerative colitis and chemerin is elevated in Crohn’s disease. Inflamm Bowel Dis. 2010;16:630–637. doi: 10.1002/ibd.21091. [DOI] [PubMed] [Google Scholar]
- 91.Konrad A, Lehrke M, Schachinger V, et al. Resistin is an inflammatory marker of inflammatory bowel disease in humans. Eur J Gastroenterol Hepatol. 2007;19:1070–1074. doi: 10.1097/MEG.0b013e3282f16251. [DOI] [PubMed] [Google Scholar]
- 92.Dogan S, Guven K, Celikbilek M, Deniz K, Saraymen B, Gursoy S. Serum visfatin levels in ulcerative colitis. J Clin Lab Anal. 2015 Dec 14; doi: 10.1002/jcla.21901. doi:10.1002/jcla.21901. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kolgazi M, Uslu U, Yuksel M, Velioglu-Ogunc A, Ercan F, Alican I. The role of cholinergic anti-inflammatory pathway in acetic acid-induced colonic inflammation in the rat. Chem Biol Interact. 2013;205:72–80. doi: 10.1016/j.cbi.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 94.Hofmann C, Chen N, Obermeier F, et al. C1q/TNF-related protein-3 (CTRP-3) is secreted by visceral adipose tissue and exerts antiinflammatory and antifibrotic effects in primary human colonic fibroblasts. Inflamm Bowel Dis. 2011;17:2462–2471. doi: 10.1002/ibd.21647. [DOI] [PubMed] [Google Scholar]
- 95.Yin J, Hou P, Wu Z, Nie Y. Decreased levels of serum omentin-1 in patients with inflammatory bowel disease. Med Sci Monit. 2015;21:118–122. doi: 10.12659/MSM.892081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Connelly TM, Juza RM, Sangster W, Sehgal R, Tappouni RF, Messaris E. Volumetric fat ratio and not body mass index is predictive of ileocolectomy outcomes in Crohn’s disease patients. Dig Surg. 2014;31:219–224. doi: 10.1159/000365359. [DOI] [PubMed] [Google Scholar]
- 97.Ding Z, Wu XR, Remer EM, et al. Association between high visceral fat area and postoperative complications in patients with Crohn’s disease following primary surgery. Colorectal Dis. 2016;18:163–172. doi: 10.1111/codi.13128. [DOI] [PubMed] [Google Scholar]
- 98.Mandanas S, Margaritidou E, Christoforidou V, et al. Breast metastasis from medullary thyroid carcinoma in a male patient: case report and review of the literature. Rare Tumors. 2015;7:5765. doi: 10.4081/rt.2015.5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Erhayiem B, Dhingsa R, Hawkey CJ, Subramanian V. Ratio of visceral to subcutaneous fat area is a biomarker of complicated Crohn’s disease. Clin Gastroenterol Hepatol. 2011;9:684–687.e1. doi: 10.1016/j.cgh.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 100.Uko V, Vortia E, Achkar JP, et al. Impact of abdominal visceral adipose tissue on disease outcome in pediatric Crohn’s disease. Inflamm Bowel Dis. 2014;20:2286–2291. doi: 10.1097/MIB.0000000000000200. [DOI] [PubMed] [Google Scholar]
- 101.Stidham RW, Waljee AK, Day NM, et al. Body fat composition assessment using analytic morphomics predicts infectious complications after bowel resection in Crohn’s disease. Inflamm Bowel Dis. 2015;21:1306–1313. doi: 10.1097/MIB.0000000000000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Karagiannides I, Kokkotou E, Tansky M, et al. Induction of colitis causes inflammatory responses in fat depots: evidence for substance P pathways in human mesenteric preadipocytes. Proc Natl Acad Sci USA. 2006;103:5207–5212. doi: 10.1073/pnas.0600821103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zulian A, Cancello R, Ruocco C, et al. Differences in visceral fat and fat bacterial colonization between ulcerative colitis and Crohn’s disease. An in vivo and in vitro study. PLoS One. 2013;8:e78495. doi: 10.1371/journal.pone.0078495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Siegmund B. Mesenteric fat in Crohn’s disease: the hot spot of inflammation? Gut. 2012;61:3–5. doi: 10.1136/gutjnl-2011-301354. [DOI] [PubMed] [Google Scholar]
- 105.Zulian A, Cancello R, Micheletto G, et al. Visceral adipocytes: old actors in obesity and new protagonists in Crohn’s disease? Gut. 2012;61:86–94. doi: 10.1136/gutjnl-2011-300391. [DOI] [PubMed] [Google Scholar]
- 106.Li H, Lelliott C, Håkansson P, et al. Intestinal, adipose, and liver inflammation in diet-induced obese mice. Metab Clin Exp. 2008;57:1704–1710. doi: 10.1016/j.metabol.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 107.Parmentier-Decrucq E, Duhamel A, Ernst O, et al. Effects of infliximab therapy on abdominal fat and metabolic profile in patients with Crohn’s disease. Inflamm Bowel Dis. 2009;15:1476–1484. doi: 10.1002/ibd.20931. [DOI] [PubMed] [Google Scholar]
- 108.Montague CT, O’Rahilly S. The perils of portliness: causes and consequences of visceral adiposity. Diabetes. 2000;49:883–888. doi: 10.2337/diabetes.49.6.883. [DOI] [PubMed] [Google Scholar]
- 109.Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr Rev. 2000;21:697–738. doi: 10.1210/edrv.21.6.0415. [DOI] [PubMed] [Google Scholar]
- 110.Shah RV, Murthy VL, Abbasi SA, et al. Visceral adiposity and the risk of metabolic syndrome across body mass index: the MESA Study. JACC Cardiovasc Imaging. 2014;7:1221–1235. doi: 10.1016/j.jcmg.2014.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Peyrin-Biroulet L, Gonzalez F, Dubuquoy L, et al. Mesenteric fat as a source of C reactive protein and as a target for bacterial translocation in Crohn’s disease. Gut. 2012;61:78–85. doi: 10.1136/gutjnl-2011-300370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Batra A, Heimesaat MM, Bereswill S, et al. Mesenteric fat - control site for bacterial translocation in colitis? Mucosal Immunol. 2012;5:580–591. doi: 10.1038/mi.2012.33. [DOI] [PubMed] [Google Scholar]
- 113.Packey CD, Sartor RB. Commensal bacteria, traditional and opportunistic pathogens, dysbiosis and bacterial killing in inflammatory bowel diseases. Curr Opin Infect Dis. 2009;22:292–301. doi: 10.1097/QCO.0b013e32832a8a5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sartor RB. Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol. 2006;3:390–407. doi: 10.1038/ncpgasthep0528. [DOI] [PubMed] [Google Scholar]
- 115.Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014;14:141–153. doi: 10.1038/nri3608. [DOI] [PubMed] [Google Scholar]
- 116.Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang LJ, Guerrero-Juarez CF, Hata T, et al. Innate immunity. Dermal adipocytes protect against invasive Staphylococcus aureus skin infection. Science. 2015;347:67–71. doi: 10.1126/science.1260972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yu L, Yan K, Liu P, et al. Pattern recognition receptor-initiated innate antiviral response in mouse adipose cells. Immunol Cell Biol. 2014;92:105–115. doi: 10.1038/icb.2013.66. [DOI] [PubMed] [Google Scholar]
- 119.Nakatsuji T, Chiang HI, Jiang SB, Nagarajan H, Zengler K, Gallo RL. The microbiome extends to subepidermal compartments of normal skin. Nat Commun. 2013;4:1431. doi: 10.1038/ncomms2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mencarelli A, Distrutti E, Renga B, et al. Probiotics modulate intestinal expression of nuclear receptor and provide counter-regulatory signals to inflammation-driven adipose tissue activation. PLoS One. 2011;6:e22978. doi: 10.1371/journal.pone.0022978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wernstedt Asterholm I, Tao C, Morley TS, et al. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 2014;20:103–118. doi: 10.1016/j.cmet.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Le Dréan G, Haure-Mirande V, Ferrier L, et al. Visceral adipose tissue and leptin increase colonic epithelial tight junction permeability via a RhoA-ROCK-dependent pathway. FASEB J. 2014;28:1059–1070. doi: 10.1096/fj.13-234203. [DOI] [PubMed] [Google Scholar]
- 123.Gummesson A, Carlsson LM, Storlien LH, et al. Intestinal permeability is associated with visceral adiposity in healthy women. Obesity (Silver Spring) 2011;19:2280–2282. doi: 10.1038/oby.2011.251. [DOI] [PubMed] [Google Scholar]
- 124.Karmiris K, Koutroubakis IE, Kouroumalis EA. The emerging role of adipocytokines as inflammatory mediators in inflammatory bowel disease. Inflamm Bowel Dis. 2005;11:847–855. doi: 10.1097/01.mib.0000178915.54264.8f. [DOI] [PubMed] [Google Scholar]
- 125.Fink C, Karagiannides I, Bakirtzi K, Pothoulakis C. Adipose tissue and inflammatory bowel disease pathogenesis. Inflamm Bowel Dis. 2012;18:1550–1557. doi: 10.1002/ibd.22893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gonçalves P, Magro F, Martel F. Metabolic inflammation in inflammatory bowel disease: crosstalk between adipose tissue and bowel. Inflamm Bowel Dis. 2015;21:453–467. doi: 10.1097/MIB.0000000000000209. [DOI] [PubMed] [Google Scholar]
- 127.Kredel L, Batra A, Siegmund B. Role of fat and adipokines in intestinal inflammation. Curr Opin Gastroenterol. 2014;30:559–565. doi: 10.1097/MOG.0000000000000116. [DOI] [PubMed] [Google Scholar]
- 128.McGown C, Birerdinc A, Younossi ZM. Adipose tissue as an endocrine organ. Clin Liver Dis. 2014;18:41–58. doi: 10.1016/j.cld.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 129.Booth A, Magnuson A, Fouts J, Foster MT. Adipose tissue: an endocrine organ playing a role in metabolic regulation. Horm Mol Biol Clin Investig. 2016;26:25–42. doi: 10.1515/hmbci-2015-0073. [DOI] [PubMed] [Google Scholar]
- 130.Schäffler A, Schölmerich J. Innate immunity and adipose tissue biology. Trends Immunol. 2010;31:228–235. doi: 10.1016/j.it.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 131.Bertin B, Desreumaux P, Dubuquoy L. Obesity, visceral fat and Crohn’s disease. Curr Opin Clin Nutr Metab Care. 2010;13:574–580. doi: 10.1097/MCO.0b013e32833cf0f4. [DOI] [PubMed] [Google Scholar]
- 132.Schaffler A, Herfarth H. Creeping fat in Crohn’s disease: travelling in a creeper lane of research? Gut. 2005;54:742–744. doi: 10.1136/gut.2004.061531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Schäffler A1, Schölmerich J, Büchler C. Mechanisms of disease: adipocytokines and visceral adipose tissue-emerging role in intestinal and mesenteric diseases. Nat Clin Pract Gastroenterol Hepatol. 2005;2:103–111. doi: 10.1038/ncpgasthep0090. [DOI] [PubMed] [Google Scholar]
- 134.Watanabe Y, Nakamura T, Ishikawa S, et al. The radioprotective 105/MD-1 complex contributes to diet-induced obesity and adipose tissue inflammation. Diabetes. 2012;61:1199–1209. doi: 10.2337/db11-1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Majewska M, Szczepanik M. The role of Toll-like receptors (TLR) in innate and adaptive immune responses and their function in immune response regulation. Postepy Hig Med Dosw (Online) 2006;60:52–63. [PubMed] [Google Scholar]
- 136.Prajapati B, Jena PK, Rajput P, Purandhar K, Seshadri S. Understanding and modulating the Toll like Receptors (TLRs) and NOD like Receptors (NLRs) cross talk in type 2 diabetes. Curr Diabetes Rev. 2014;10:190–200. doi: 10.2174/1573399810666140515112609. [DOI] [PubMed] [Google Scholar]
- 137.Lin Y, Lee H, Berg AH, Lisanti MP, Shapiro L, Scherer PE. The lipopolysaccharide-activated toll-like receptor (TLR)-4 induces synthesis of the closely related receptor TLR-2 in adipocytes. J Biol Chem. 2000;275:24255–24263. doi: 10.1074/jbc.M002137200. [DOI] [PubMed] [Google Scholar]
- 138.Schaeffler A, Gross P, Buettner R, et al. Fatty acid-induced induction of Toll-like receptor-4/nuclear factor-kappaB pathway in adipocytes links nutritional signalling with innate immunity. Immunology. 2009;126:233–245. doi: 10.1111/j.1365-2567.2008.02892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Vitseva OI, Tanriverdi K, Tchkonia TT, et al. Inducible Toll-like receptor and NF-kappaB regulatory pathway expression in human adipose tissue. Obesity (Silver Spring) 2008;16:932–937. doi: 10.1038/oby.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Khazen W, M’Bika JP, Collinet M, et al. Differentiation-dependent expression of interferon gamma and toll-like receptor 9 in 3T3-F442A adipocytes. Biochimie. 2007;89:669–675. doi: 10.1016/j.biochi.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 141.Creely SJ, McTernan PG, Kusminski CM, et al. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am J Physiol Endocrinol Metab. 2007;292:E740–E747. doi: 10.1152/ajpendo.00302.2006. [DOI] [PubMed] [Google Scholar]
- 142.Betanzos-Cabrera G, Estrada-Luna D, Belefant-Miller H, Cancino-Díaz JC. Mice fed with a high fat diet show a decrease in the expression of “toll like receptor (TLR)2 and TLR6 mRNAs in adipose and hepatic tissues. Nutr Hosp. 2012;27:1196–1203. doi: 10.3305/nh.2012.27.4.5842. [DOI] [PubMed] [Google Scholar]
- 143.Kopp A, Buechler C, Bala M, Neumeier M, Schölmerich J, Schäffler A. Toll-like receptor ligands cause proinflammatory and prodiabetic activation of adipocytes via phosphorylation of extracellular signal-regulated kinase and c-Jun N-terminal kinase but not interferon regulatory factor-3. Endocrinology. 2010;151:1097–1108. doi: 10.1210/en.2009-1140. [DOI] [PubMed] [Google Scholar]
- 144.Brenner C, Simmonds RE, Wood S, Rose V, Feldmann M, Turner J. TLR signalling and adapter utilization in primary human in vitro differentiated adipocytes. Scand J Immunol. 2012;76:359–370. doi: 10.1111/j.1365-3083.2012.02744.x. [DOI] [PubMed] [Google Scholar]
- 145.Straub RH. Insulin resistance, selfish brain, and selfish immune system: an evolutionarily positively selected program used in chronic inflammatory diseases. Arthritis Res Ther. 2014;16(Suppl 2):S4. doi: 10.1186/ar4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhou YJ, Liu C, Li CL, et al. Increased NOD1, but not NOD2, activity in subcutaneous adipose tissue from patients with metabolic syndrome. Obesity (Silver Spring) 2015;23:1394–1400. doi: 10.1002/oby.21113. [DOI] [PubMed] [Google Scholar]
- 147.Zhou YJ, Zhou H, Li Y, Song YL. NOD1 activation induces innate immune responses and insulin resistance in human adipocytes. Diabetes Metab. 2012;38:538–543. doi: 10.1016/j.diabet.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 148.Stienstra R, van Diepen JA, Tack CJ, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci USA. 2011;108:15324–15329. doi: 10.1073/pnas.1100255108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kim S, Joe Y, Jeong SO, et al. Endoplasmic reticulum stress is sufficient for the induction of IL-1beta production via activation of the NF-kappaB and inflammasome pathways. Innate Immun. 2014;20:799–815. doi: 10.1177/1753425913508593. [DOI] [PubMed] [Google Scholar]
- 150.Watanabe Y, Nagai Y, Honda H, et al. Isoliquiritigenin attenuates adipose tissue inflammation in vitro and adipose tissue fibrosis through inhibition of innate immune responses in mice. Sci Rep. 2016;6:23097. doi: 10.1038/srep23097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Alcorn JF, Kolls JK. Physiology. Killer fat. Science. 2015;347:26–27. doi: 10.1126/science.aaa4567. [DOI] [PubMed] [Google Scholar]
- 152.Wensveen FM, Valentić S, Šestan M, Turk Wensveen T, Polić B. The “Big Bang” in obese fat: Events initiating obesity-induced adipose tissue inflammation. Eur J Immunol. 2015;45:2446–2456. doi: 10.1002/eji.201545502. [DOI] [PubMed] [Google Scholar]
- 153.Hanses F, Kopp A, Bala M, et al. Intracellular survival of Staphylococcus aureus in adipocyte-like differentiated 3T3-L1 cells is glucose dependent and alters cytokine, chemokine, and adipokine secretion. Endocrinology. 2011;152:4148–4157. doi: 10.1210/en.2011-0103. [DOI] [PubMed] [Google Scholar]
- 154.Celinski K, Dworzanski T, Fornal R, et al. Comparison of anti-inflammatory properties of peroxisome proliferator-activated receptor gamma agonists rosiglitazone and troglitazone in prophylactic treatment of experimental colitis. J Physiol Pharmacol. 2013;64:587–595. [PubMed] [Google Scholar]
- 155.Celinski K, Dworzanski T, Fornal R, Korolczuk A, Madro A, Slomka M. Comparison of the anti-inflammatory and therapeutic actions of PPAR-gamma agonists rosiglitazone and troglitazone in experimental colitis. J Physiol Pharmacol. 2012;63:631–640. [PubMed] [Google Scholar]
- 156.Celinski K, Dworzanski T, Korolczuk A, et al. Effects of peroxisome proliferator-activated receptors-gamma ligands on dextran sodium sulphate-induced colitis in rats. J Physiol Pharmacol. 2011;62:347–356. [PubMed] [Google Scholar]
- 157.Dworzanski T, Celinski K, Korolczuk A, et al. Influence of the peroxisome proliferator-activated receptor gamma (PPAR-γ) agonist, rosiglitazone and antagonist, biphenol-A-diglicydyl ether (BADGE) on the course of inflammation in the experimental model of colitis in rats. J Physiol Pharmacol. 2010;61:683–693. [PubMed] [Google Scholar]
- 158.Sánchez-Hidalgo M, Martín AR, Villegas I, de la Lastra CA. Rosiglitazone, a PPARgamma ligand, modulates signal transduction pathways during the development of acute TNBS-induced colitis in rats. Eur J Pharmacol. 2007;562:247–258. doi: 10.1016/j.ejphar.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 159.Sánchez-Hidalgo M, Martín AR, Villegas I, Alarcón De La Lastra C. Rosiglitazone, an agonist of peroxisome proliferator-activated receptor gamma, reduces chronic colonic inflammation in rats. Biochem Pharmacol. 2005;69:1733–1744. doi: 10.1016/j.bcp.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 160.Ramakers JD, Verstege MI, Thuijls G, Te Velde AA, Mensink RP, Plat J. The PPARgamma agonist rosiglitazone impairs colonic inflammation in mice with experimental colitis. J Clin Immunol. 2007;27:275–283. doi: 10.1007/s10875-007-9074-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Adachi M, Kurotani R, Morimura K, et al. Peroxisome proliferator activated receptor gamma in colonic epithelial cells protects against experimental inflammatory bowel disease. Gut. 2006;55:1104–1113. doi: 10.1136/gut.2005.081745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Saubermann LJ, Nakajima A, Wada K, et al. Peroxisome proliferator-activated receptor gamma agonist ligands stimulate a Th2 cytokine response and prevent acute colitis. Inflamm Bowel Dis. 2002;8:330–339. doi: 10.1097/00054725-200209000-00004. [DOI] [PubMed] [Google Scholar]
- 163.Han X, Osuntokun B, Benight N, Loesch K, Frank SJ, Denson LA. Signal transducer and activator of transcription 5b promotes mucosal tolerance in pediatric Crohn’s disease and murine colitis. Am J Pathol. 2006;169:1999–2013. doi: 10.2353/ajpath.2006.060186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Desreumaux P, Dubuquoy L, Nutten S, et al. Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome proliferator-activated receptor gamma (PPARgamma) heterodimer. A basis for new therapeutic strategies. J Exp Med. 2001;193:827–838. doi: 10.1084/jem.193.7.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lytle C, Tod TJ, Vo KT, Lee JW, Atkinson RD, Straus DS. The peroxisome proliferator-activated receptor gamma ligand rosiglitazone delays the onset of inflammatory bowel disease in mice with interleukin 10 deficiency. Inflamm Bowel Dis. 2005;11:231–243. doi: 10.1097/01.mib.0000160805.46235.eb. [DOI] [PubMed] [Google Scholar]
- 166.Takaki K, Mitsuyama K, Tsuruta O, Toyonaga A, Sata M. Attenuation of experimental colonic injury by thiazolidinedione agents. Inflamm Res. 2006;55:10–15. doi: 10.1007/s00011-005-0002-8. [DOI] [PubMed] [Google Scholar]
- 167.Takagi T, Naito Y, Tomatsuri N, et al. Pioglitazone, a PPAR-gamma ligand, provides protection from dextran sulfate sodium-induced colitis in mice in association with inhibition of the NF-kappaB-cytokine cascade. Redox Rep. 2002;7:283–289. doi: 10.1179/135100002125000802. [DOI] [PubMed] [Google Scholar]
- 168.Tanaka T, Kohno H, Yoshitani S, et al. Ligands for peroxisome proliferator-activated receptors alpha and gamma inhibit chemically induced colitis and formation of aberrant crypt foci in rats. Cancer Res. 2001;61:2424–2428. [PubMed] [Google Scholar]
- 169.Siegmund B, Sennello JA, Jones-Carson J, et al. Leptin receptor expression on T lymphocytes modulates chronic intestinal inflammation in mice. Gut. 2004;53:965–972. doi: 10.1136/gut.2003.027136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ballak DB, van Asseldonk EJ, van Diepen JA, et al. TLR-3 is present in human adipocytes, but its signalling is not required for obesity-induced inflammation in adipose tissue in vivo. PLoS One. 2015;10:e0123152. doi: 10.1371/journal.pone.0123152. [DOI] [PMC free article] [PubMed] [Google Scholar]