

Abstract
The low molecular weight protein tyrosine phosphatase (LMPTP), encoded by the ACP1 gene, is a ubiquitously expressed phosphatase whose in vivo function in the heart and in cardiac diseases remains unknown. To investigate the in vivo role of LMPTP in cardiac function, we generated mice with genetic inactivation of the Acp1 locus and studied their response to long‐term pressure overload. Acp1−/− mice develop normally and ageing mice do not show pathology in major tissues under basal conditions. However, Acp1−/− mice are strikingly resistant to pressure overload hypertrophy and heart failure. Lmptp expression is high in the embryonic mouse heart, decreased in the postnatal stage, and increased in the adult mouse failing heart. We also show that LMPTP expression increases in end‐stage heart failure in humans. Consistent with their protected phenotype, Acp1−/− mice subjected to pressure overload hypertrophy have attenuated fibrosis and decreased expression of fibrotic genes. Transcriptional profiling and analysis of molecular signalling show that the resistance of Acp1−/− mice to pathological cardiac stress correlates with marginal re‐expression of fetal cardiac genes, increased insulin receptor beta phosphorylation, as well as PKA and ephrin receptor expression, and inactivation of the CaMKIIδ pathway. Our data show that ablation of Lmptp inhibits pathological cardiac remodelling and suggest that inhibition of LMPTP may be of therapeutic relevance for the treatment of human heart failure. © 2015 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: low molecular weight protein tyrosine phosphatase, ACP1, protein tyrosine phosphatase, cardiac hypertrophy, heart failure
Introduction
Cardiovascular diseases remain the leading cause of mortality worldwide, with heart failure being the fastest growing condition in the past decade. Heart failure complicates a number of common conditions (ie hypertension, myocardial infarction, and cardiomyopathies) and is associated with a phase of cardiac hypertrophy that is initially beneficial, but that often progresses to heart failure if the stress stimulus is not alleviated 1.
Numerous studies have implicated protein kinases in the pathogenesis of heart failure 1. On the other hand, fewer studies have addressed the role of protein phosphatases in cardiac pathologies. While several serine/threonine phosphatases play a role in heart diseases 2, 3, 4, 5, only three members of the protein tyrosine phosphatases (PTPs) 6 (ie the protein tyrosine phosphatase, non‐receptor type 11, PTPN11; the phosphatase and tensin homologue, PTEN; and the protein tyrosine phosphatase 1B, PTP1B) have been implicated in cardiac abnormalities 7, 8, 9. The low molecular weight protein tyrosine phosphatase (LMPTP) is another PTP highly expressed in the heart 10. However, its function in heart pathophysiology remains unknown. Encoded by the ACP1 gene, LMPTP is an 18 kDa cytosolic enzyme with a wide tissue distribution 11. As a result of alternative mRNA splicing, two active isoforms are generated, called ACP1‐F or ‐S and LMPTP‐A or ‐B in humans and IF1 and IF2 in rodents 12, 13, 14. A second IF2 isoform called IF2p not encoded by Acp1 and derived from a pseudogene has also been reported, although its molecular function remains undetermined 11.
Genetic association studies have suggested a role for ACP1 in human cardiovascular disorders 12, 13, 14, 15. LMPTP appears to modulate signalling through several tyrosine kinase receptors 16, 17, 18, 19, 20. Analysis of liver and fat tissue from mice injected with anti‐Lmptp antisense oligonucleotides suggested that Lmptp acts as a negative regulator of insulin signalling 21. However, a mouse model of Lmptp deficiency is still lacking and the functional role of Lmptp in cardiac diseases remains unknown.
In the present study, we show that LMPTP expression increases in end‐stage heart failure in mice and in humans and we addressed the function of Lmptp in heart failure by generating and characterizing the first mouse model carrying inactivation of the Acp1 gene. Acp1−/− mice subjected to chronic cardiac stress display strikingly preserved contractile function and attenuated histological and molecular evidence of cardiac fibrosis. This phenotype correlates with limited reactivation of the fetal gene programme. In addition, hearts from Acp1−/− mice show increased phosphorylation of the insulin receptor (IR), and enhanced expression of molecules involved in protective signalling and attenuation of signalling pathways with deleterious effects.
Materials and methods
Details of the methods and antibodies are provided in the Supplementary materials and methods.
Generation of Acp1 knockout mice
Acp1 gene‐trapped mice were generated according to Stryke et al 22. Acp1 knockout (KO) mice were backcrossed for at least ten generations onto the Balb/c background. Male Balb/c and Acp1 KO mice 13–15 weeks old were subject to trans‐aortic constriction (TAC) as previously described 23. Sham operations were performed by opening and closing the chest. The Vevo 770 (VisualSonics, Toronto, Canada) and Vivid E9 (GE Healthcare, Little Chalfont, Bucks, UK) high‐resolution In‐Vivo Imaging systems were used to measure cardiac dimension and function in mice. Before assessing cardiac functional parameters, mice were subjected to 1–2% isoflurane. At the study end‐point, hearts were collected after opening the pericardial cavity and immediately placed in liquid nitrogen or retro‐perfused to generate paraffin‐embedded sections.
Immunofluorescence
At the study end‐point, paraffin‐embedded heart sections were deparaffinized and after antigen retrieval in citrate buffer and blocking, wheat germ agglutinin antibody was incubated overnight at 4 °C. After washing, secondary antibody incubation was performed for 2 h. Cells were then washed and slides were mounted and visualized using a 20× objective and a Zeiss (Axio Imager) microscope. Cell surface areas were measured using a tool in NIS Element software that created regions of interest. Surface areas were calculated from 100 cells from three separate areas in three animals from each group of mice.
Primary neonatal rat cardiomyocytes
Primary neonatal rat cardiomyocytes were isolated using the Worthington Neonatal Cardiomyocytes System (Worthington Biochemical Corp, Lakewood, NJ, USA) according to the manufacturer's instructions.
Protein extraction, immunoblotting, and co‐immunoprecipitation
Whole heart extracts resuspended in a high salt lysis buffer containing a cocktail of protease and phosphatase inhibitors were prepared. Forty micrograms of heart extract was analysed by immunoblot. Whole heart extracts were immunoprecipitated using anti‐insulin receptor β antibody according to the manufacturer's instructions (Pierce Classic IP kit; Pierce Thermoscientific, Grand Island, NY, USA). Immunoprecipitated samples (30 µl) were analysed by immunoblotting using anti‐pTyr antibody. The membrane was then stripped and incubated with anti‐IRβ antibody.
Q‐PCR
Total RNA was extracted from mouse hearts using RNAEasy (Qiagen, Maryland, USA) according to the manufacturer's protocol. cDNA was synthesized using Superscript III (Invitrogen) using random hexamers. PCR reactions were run using 10 ng of cDNA and primers were added in PCR buffer IQ SYBR Green Supermix (Bio‐Rad Laboratories, Hercules, CA, USA). The primers used for fetal cardiac genes, PLN, and SERCA2A are listed in the Supplementary materials and methods.
Array hybridization and microarray analysis
Total RNA was isolated from heart tissues using standard protocols. Sample handling, cDNA synthesis, cRNA labelling and synthesis, hybridization, washing, array (GeneChip® Mouse Genome 430 2.0 Array; Affymetrix Inc, Santa Clara, CA, USA) scanning, and all related quality controls were performed according to the manufacturer's instructions. The open source R/Bioconductor packages 24 were used for processing and analysis of microarray data. Significantly modulated genes were defined as those with absolute fold change (FC) greater than 1.5 and unadjusted p value less than 0.05. Statistical analyses were performed by using ANOVA, SAS 9.2 (SAS Institute, Cary, NC, USA), MATLAB software packages (Mathworks, Natick, MA, USA), and PARTEK Genomics Suite (Partek Inc, St Louis, MO, USA). Microarray data are provided in the Supporting information.
Human subjects
All procedures were performed under approved protocol (RAC# 2100 023) from the Institutional Review Board at King Faisal Specialist Hospital and Research Centre. Patients with end‐stage heart failure secondary to idiopathic dilated cardiomyopathy were followed at King Faisal Heart Centre and were enrolled in our study after informed consent. After transplantation, cardiomyopathic hearts were collected and stored at −80 °C for further biochemical analysis. Normal human hearts were obtained from donors who died from accidental death or from causes other than cardiac diseases and that could not be transplanted because of incompatibility with the recipient.
Dissection and histological analyses of human and mouse hearts
Samples from the same region of the left ventricle were dissected from normal and cardiomyopathic hearts. Total proteins were prepared after grinding of the tissue as described above. After fixation in formalin and embedding in paraffin, heart sections were stained with Masson's trichrome using standard procedures.
Statistical analysis
For echocardiographic data, statistical analysis was performed using Prism 5 software. Data were analysed using repeated measures two‐way ANOVA followed by a Bonferroni post‐hoc test. For all other data analyses, Student's t‐test was performed. Graphical data are represented as the mean ± SEM. p values less than 0.05 were considered statistically significant.
Results
Developmental regulation of Lmptp in rodent heart
LMPTP is a cytosolic enzyme initially purified as a major PTP in bovine heart 10. As expected, we found that Lmptp is highly expressed in adult BALB/c mouse heart and is detected as two bands of molecular mass near 18 kDa (Figure 1A). Immunofluorescence of adult rodent cardiac sections showed high levels of Lmptp in the sarcolemmal membrane and lower levels in the myocardium (Figure 1B and Supplementary Figure 1A). Lmptp was also detected in neonatal rat ventricular myocytes (Supplementary Figure 1B). Next, we investigated if Lmptp expression is developmentally regulated. Lmptp protein was high at embryonic days 11.5 and 15, declined between embryonic days 15 and 19, and remained similar between postnatal days 1 and 7 and in young adulthood (Figures 1C and 1D). These results show that Lmptp is highly expressed during early embryonic development and is expressed in the adult heart.
Figure 1.
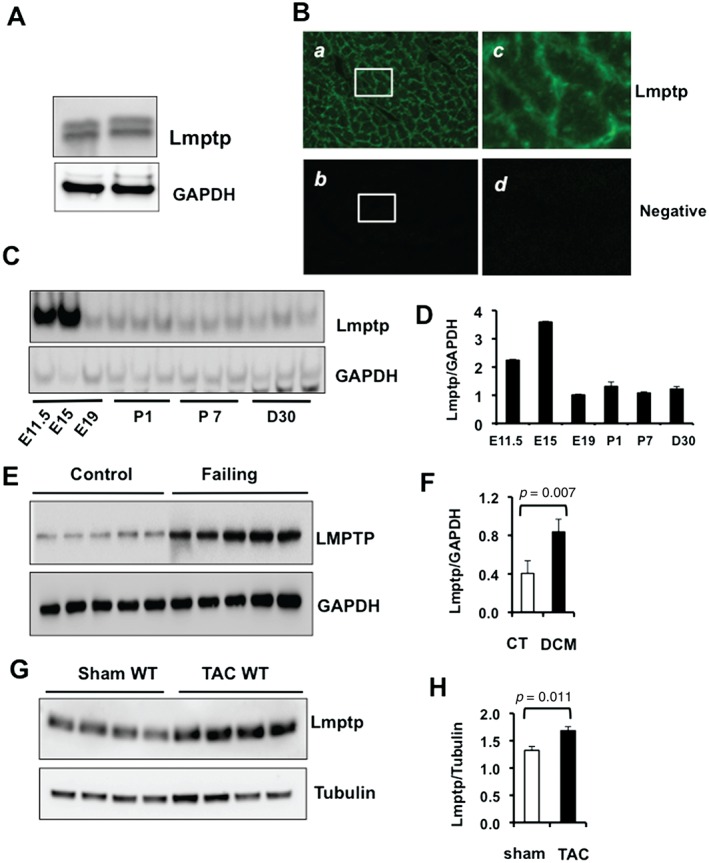
Expression of Lmptp during embryonic and postnatal cardiac development and after pathological stress. (A) Immunoblot of Lmptp in adult mouse heart. (B) Indirect immunofluorescence of adult mouse heart sections showing Lmptp expression in the sarcolemmal membrane and lower expression in the cytosol. Signals were visualized by confocal microscopy using a 20× objective. (a) Lmptp expression; (b) negative control; (c) higher magnification of a; (d) higher magnification of b. (C) Immunoblot showing the developmental time course of Lmptp expression in mouse heart at embryonic days E11.5, E15, and E19, and at postnatal days 1 (P1) and 7 (P7) and at day 30. (D) Quantitation of C. (E) LMPTP protein level in control and failing human hearts measured by immunoblot and (F) quantitative analysis (n = 5). (G) Immunoblot of Lmptp in the hearts of wild‐type (WT) mice subjected to sham operation or TAC for 10 weeks (n = 4) and (H) quantitative analysis. p values are indicated (Student's t‐test).
LMPTP is up‐regulated in failing human and rodent hearts
Many embryonic genes are up‐regulated in heart failure. Therefore, we investigated LMPTP expression in failing hearts from patients in end‐stage heart failure and in control hearts from subjects who suffered an unexpected accidental death. Failing hearts displayed compromised left ventricular (LV) function, with ejection fraction and fractional shortening below normal values (averaging 20% and 10%, respectively). LMPTP protein was significantly increased in failing human hearts compared with control hearts (Figures 1E and 1F). This was associated with enhanced fibrosis, which was prominent in failing human hearts but not in control hearts (Supplementary Figure 1C). Lmptp expression was also higher in BALB/c mouse hearts 10 weeks post‐TAC compared with sham controls (Figures 1G and 1H). This was associated with a significant increase in LV volume at diastole and systole, an increase of the LV mass over body weight ratio, and reduced ejection fraction and fractional shortening (Supplementary Figure 1D). Similar results were obtained when we analysed Lmptp expression after TAC in the C57BL/6 background (Supplementary Figure 2). We conclude that Lmptp expression is high in mouse heart during early embryonic development, decreases in the postnatal and adult stages, and is induced in response to pressure overload in humans and in mice.
Mice with Acp1 ablation do not have an overt phenotype under basal conditions
To investigate the role of Lmptp in vivo, we generated Lmptp KO mice by insertion of a gene trap in the Acp1 locus (Supplementary Figures 3A and 3B) 22. Acp1−/− mice expressed no Lmptp protein in the heart, whereas Acp1+/− mice expressed half the levels of Acp1+/+ (Supplementary Figure 3C). Lmptp expression was similarly undetectable by immunoblotting in lysates of liver, skeletal muscle, adipose tissue, and tails of Acp1−/− mice (data not shown), showing that our gene‐trapped mouse functions as a global KO. We did not detect expression of IF2p 11 in Acp1+/+ or Acp1−/− mice, suggesting that IF2p is a true pseudogene (Supplementary Figure 3D). Acp1+/− and Acp−/− mice were born at expected Mendelian ratios and developed normally. No gross difference in body anatomy was observed between Acp1+/+ and Acp−/− mice. Acp1−/− mice were slightly smaller than Acp+/+ mice at week 10; however, the difference was not significant at later times of development (Supplementary Figures 4A and 4B). Heart morphology (Supplementary Figures 4C and 4D) and resting cardiac function at 2 months (Supplementary Table 1) were similar in Acp1+/+ and Acp−/− mice. Mouse weights, as well as the weights of hearts, left and right atria, left and right ventricles, liver, and tibia lengths, were similar in Acp1+/+ and Acp−/− mice (Supplementary Table 2). Since Acp1‐null mice have not been reported previously, we performed a pathology survey of various tissues in aged Acp1+/+ and Acp−/− mice. Internal organs including the heart, brain, lungs, liver, kidney, small intestine, pancreas, skin, and reproductive organs from 16‐ to 21‐month‐old male and female mice did not display any obvious anatomical abnormalities or pathology (data not shown). We conclude that Acp1−/− mice are viable and fertile, do not develop spontaneous pathology, and appear to have a normal cardiac phenotype at baseline.
Mice with Acp1 ablation have preserved cardiac contractility, attenuated fibrosis, and reduced up‐regulation of heart failure markers after chronic pressure overload
Since Acp1−/− mice did not show an overt phenotype under basal conditions, we investigated their response to pressure overload hypertrophy induced by constriction of the aorta (TAC). No significant changes in LV dimension or cardiac function were observed in Acp1+/+ and Acp−/− mice over 10 weeks after sham operation (Figures 2A–2F). Acp1+/+ mice developed cardiac hypertrophy between weeks 4 and 7 post‐TAC, as evidenced by increased interventricular septum thickness (Figure 2A) and increased posterior wall thickness (Figure 2B and Supplementary Figure 5A). Starting at week 7, Acp1+/+ mice showed increased systolic volume and end‐systolic diameter after TAC (Figures 2C and 2D), whereas end‐diastolic diameter was not significantly affected (Supplementary Figure 5B). Also, starting 7 weeks post‐TAC, Acp1+/+ mice had reduced ejection fraction and fractional shortening (23.4% and 14.16%, respectively) (Figures 2E and 2F) indicative of severely compromised contractile function. In contrast, Acp−/− mice subjected to TAC displayed no signs of cardiac hypertrophy, no significant enlargement of the ventricular chambers (Figures 2C, 2D, and Supplementary Figure 5A), and no deterioration of haemodynamic function (Figures 2E and 2F). TAC‐induced increases in heart weight/body weight (HW/BW) ratio were significantly attenuated in Acp1−/− mice compared with Acp1+/+ after 10 weeks (Figures 3A and 3B). Consistent with these results, cell surface area as measured by wheat germ agglutinin staining showed no difference between Acp1+/+ and Acp−/− hearts at baseline. In contrast, pressure overload increased the cell surface area in Acp1+/+ hearts but not in Acp1−/− hearts (Figures 3C and 3D). Cardiac fibrosis was also markedly attenuated in Acp1−/− compared with Acp1+/+ mice 10 weeks post‐TAC (Figures 3E and 3F). Next, we measured fetal cardiac genes, which are down‐regulated after birth and re‐expressed in cardiac hypertrophy and heart failure. The results showed a significant up‐regulation of β‐myosin heavy chain (β‐Mhc) and atrial natriuretic peptide (Anp) mRNA in the hearts of Acp1+/+ mice after TAC. In contrast, Anp and β‐Mhc were not as strongly transcriptionally induced in Acp1−/− banded hearts (Figure 3G). These results show that ablation of Acp1 minimizes cardiac fibrosis and the genetic re‐programming of fetal cardiac genes in response to long‐term haemodynamic stress. Taken together, these results show that Acp1 deletion prevents the development of pathological cardiac hypertrophy and heart failure.
Figure 2.
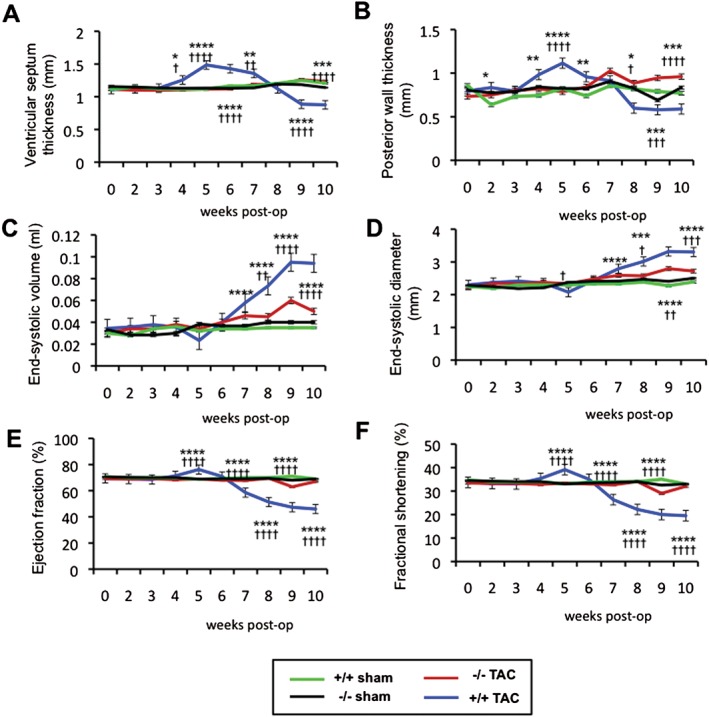
Acp1 −/− mice are resistant to pressure overload‐induced hypertrophy and heart failure. Longitudinal assessment of ventricular septum thickness and posterior wall thickness at systole (A, B), end‐systolic volume and end‐systolic diameter (C, D), ejection fraction (E), and fractional shortening (F) measured by echocardiography over 10 weeks after sham operation or TAC in Acp1+/+ or Acp1−/− mice. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.001 between Acp1+/+ TAC and Acp1−/− TAC animals. † p < 0.05, †† p < 0.01, ††† p < 0.001, and †††† p < 0.001 between Acp1+/+ TAC and Acp1+/+ sham animals (n = 9, Acp1+/+ sham; n = 8, Acp1+/+ TAC; n = 10, Acp1−/− sham; n = 10, Acp1−/− TAC). Statistical analysis was performed using ANOVA.
Figure 3.
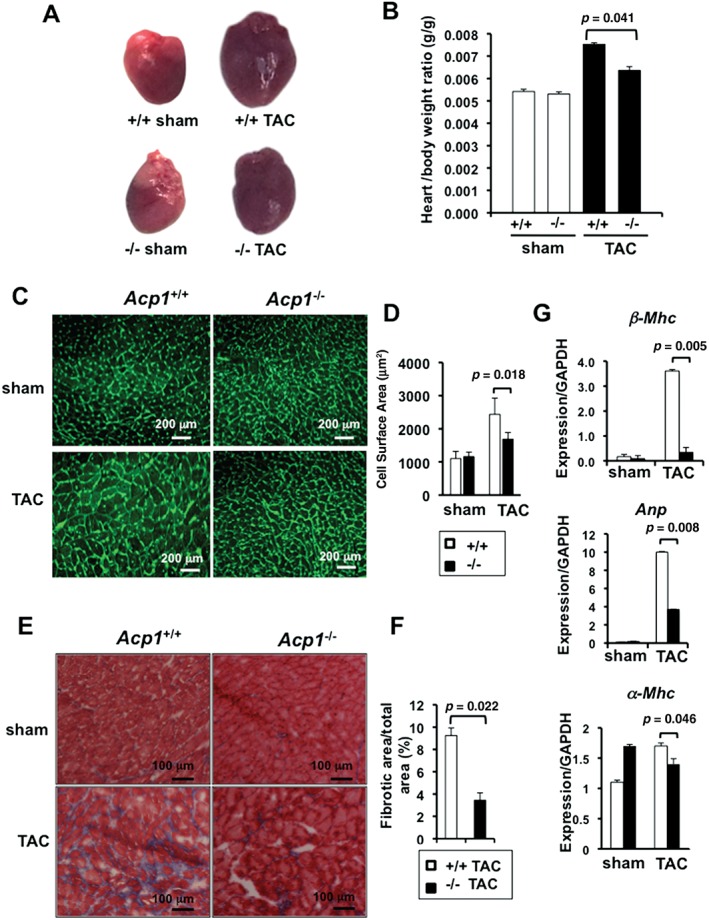
Acp1−/− mice are protected against fibrosis and heart failure. (A) Representative images of the hearts of Acp1+/+ and Acp−/− mice after 10 weeks of sham or TAC. (B) Heart/body weight ratio in Acp1+/+ and Acp−/− mice after 10 weeks of sham or TAC surgery. Acp1+/+ sham and Acp−/− sham n = 4, Acp1+/+ TAC n = 7, Acp1−/− TAC n = 4. (C) Wheat germ agglutinin staining in heart sections of Acp1+/+ and Acp1−/− mice 10 weeks after sham or TAC surgery and (D) quantitative measurement of the cell surface area from 100 cells in three different fields from three animals in each group of mice. (E) Masson's trichrome staining of heart sections of Acp1+/+ and Acp1−/− mice 10 weeks after sham or TAC surgery. (F) Significant reduction of cardiac fibrosis in Acp1−/− mice compared with Acp1+/+ mice after pressure overload, as evidenced by quantification of the fibrotic area over the total area (n = 5). (G) qPCR analysis for β‐Mhc, Anp, α‐Mhc, and Gapdh in whole heart lysates at 10 weeks post‐sham or TAC (n = 3). p values are shown on each graph (Student's t‐test).
Gene expression profiling in Acp1−/− mice after chronic pressure overload
To gain insight into the mechanism of the improved adaptation of Acp1−/− mice to pathological stress, we performed genome‐wide expression profiling in Acp1+/+ and Acp−/− hearts after 10 weeks of sham or TAC surgery. Microarray data represented as a ‘heat map’ showed global expression similarities between Acp1+/+ and Acp−/− hearts after sham operation (Figure 4A; see Supplementary Table 3 for a full list of the genes). Conversely, widespread expression differences were observed between Acp1+/+ sham hearts and Acp1+/+ TAC hearts (Figure 4A), with 828 differentially regulated genes (Supplementary Figure 6B), among which were heart failure marker genes such as Anp and β‐Mhc, and genes associated with fibrosis such as collagen type α (Supplementary Table 4; see Supplementary Table 5 for the full microarray data). Pressure overload altered the expression of 419 genes in Acp1−/− hearts compared with Acp1+/+ hearts (Supplementary Figure 6B; see Supplementary Table 6 for the full data). The DAVID functional clustering tool that groups genes according to their ontology (GO) 25, 26 revealed that genes encoding receptor and non‐receptor tyrosine kinases were among the most dysregulated (Supplementary Figure 7). Importantly, major diseases and functions to which these genes belonged were cardiac hypertrophy, dilation, and fibrosis (Figure 4B). Among the hypertrophy‐related genes, Camk2δ was expressed at a higher level in Acp1+/+ TAC hearts than in Acp−/− TAC hearts, which was validated by immunoblot analysis (Figures 4C and 4D). Also, Gαq/11 and PLCδ protein expression, which promotes pathological cardiac hypertrophy, was significantly higher in Acp1+/+ TAC hearts than in Acp1−/− TAC hearts (Figures 4C and 4D). Pro‐apoptotic protein Bax was strongly induced in Acp1+/+ TAC hearts but not in Acp1−/− TAC hearts, whereas anti‐apoptotic proteins Bcl2 and BclXL remained mostly unchanged (Figures 4E and 4F). Together, these results suggest that Acp1 deletion confers cardioprotection at least in part by enhanced expression of receptor and non‐receptor tyrosine kinases, blockade of the CaMKIIδ pathway, and reduced apoptosis.
Figure 4.
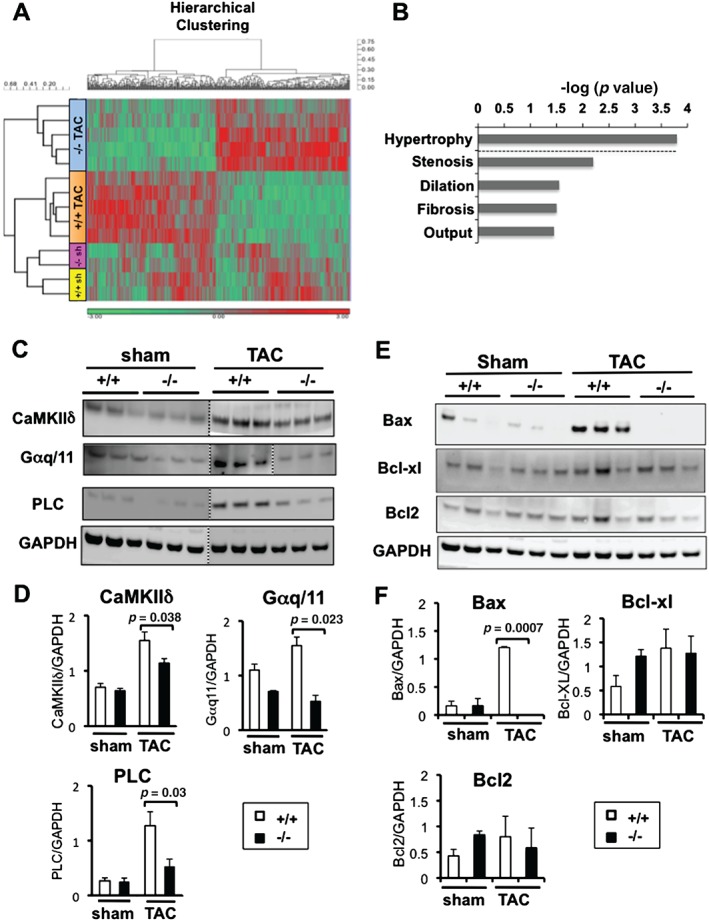
Microarray analysis of Acp1+/+ and Acp1−/− mouse hearts after sham or TAC surgery. (A) Heat map showing the clustering of significantly dysregulated genes in Acp1−/− and Acp1+/+ mice after sham surgery (n = 2) or TAC (n = 5) (p < 0.01, 428 probes). Red colour indicates genes significantly up‐regulated, while green indicates genes significantly down‐regulated. (B) Major diseases and functions connected to genes dysregulated in Acp1+/+ TAC hearts compared with Acp1−/− TAC hearts. (C) Immunoblot of CaMKIIδ, Gαq/11, and PLC protein expression in Acp1−/− TAC hearts compared with Acp1+/+ TAC hearts. (D) Quantitative analysis of C over GAPDH control (n = 3–5). (E) Immunoblot of anti‐ and pro‐apoptotic proteins in Acp1−/− TAC hearts compared with Acp+/+ TAC hearts. (F) Quantitative analysis of E over GAPDH control (n = 3–5). Significant p values are shown (Student's t‐test).
Mice with Acp1 deletion have enhanced IRβ phosphorylation, PKA, ephrin receptor, and reduced PLCβ/CaMKIIδ phosphorylation after short‐term pressure overload
Our transcriptional analysis suggested that increased signalling through the insulin receptor (IR); increased expression of PKA, NFκB, and ephrin receptor (Figure 5A); and inhibition of CaMKIIδ may confer cardioprotection in Acp1−/− mice after long‐term chronic stress (Figures 4B and 4C). To confirm that these pathways are implicated in early cardioprotection, we performed short‐term TAC (10 min and 24 h) and assessed these signalling pathways in the four groups of mice. Since Lmptp regulates IR phosphorylation in the liver 21, we first measured IRβ phosphorylation. IRβ phosphorylation increased in Acp1−/− hearts after 10 min of TAC compared with Acp1+/+ TAC hearts, with a further increase 24 h post‐TAC (Figures 5B and 5C). PKA and ephrin receptor were also significantly increased in Acp1−/− hearts after 10 min of TAC and this effect was augmented 24 h post‐TAC (Figures 5D and 5E). NFκB increased after 24 h of TAC in Acp1−/− hearts; however, the difference did not reach statistical significance. Next, we assessed the Akt pathway downstream of the IR 27, which has a protective role in the heart. Unexpectedly, phosphorylation of Akt increased in Acp1+/+ hearts after 24 h of TAC, with no further increase in Acp1−/− TAC hearts (Supplementary Figure 8). Assessment of the Erk and p38 pathways, which are also activated downstream of the IR, showed similar levels of phosphorylation in Acp1+/+ and Acp1−/− mice after 10 min or 24 h of TAC, suggesting that these pathways do not contribute to the protective phenotype of Acp1−/− mice (Supplementary Figure 8).
Figure 5.
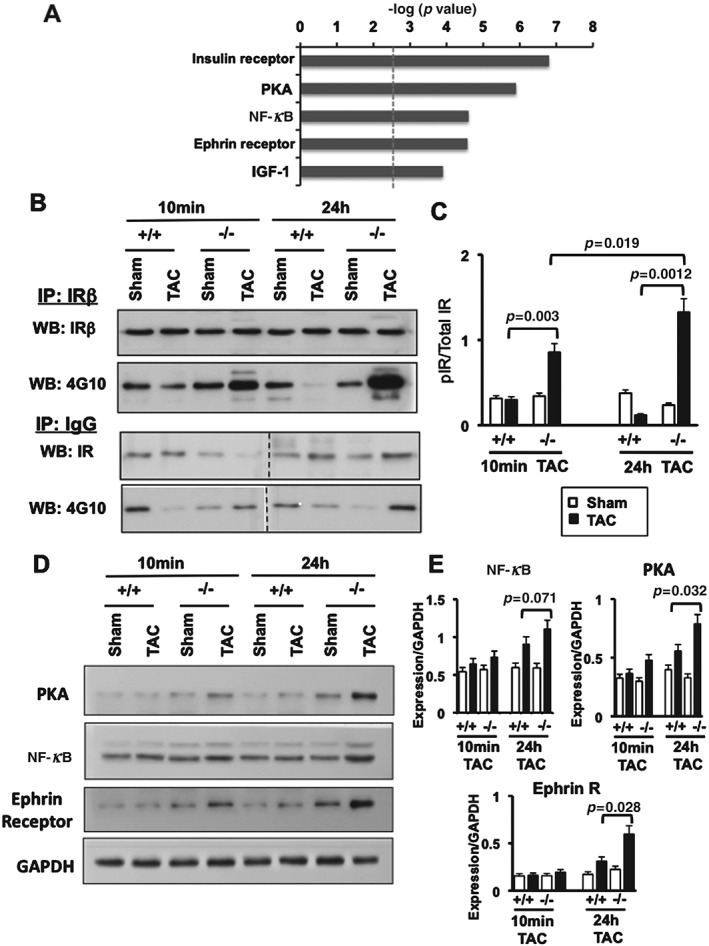
Increased IR phosphorylation, PKA, and ephrin receptor expression in Acp1−/− hearts subjected to TAC compared with Acp1+/+ TAC hearts. (A) Ingenuity pathway analysis showing the most significantly increased canonical pathways in Acp1−/− TAC hearts compared with Acp1+/+ TAC hearts after 10 weeks of TAC or sham surgery. (B) Immunoprecipitation from Acp1+/+ and Acp1−/− mouse hearts after 10 min and 24 h of sham or TAC using anti‐IRβ antibody followed by immunoblotting using anti‐4G10 antibody. (C) Quantitation of B; n = 3 in each group of mice. (D) Immunoblot for PKA, NFκB, and ephrin receptor in Acp1+/+ and Acp1−/− mouse hearts after 10 min and 24 h of sham or TAC. (E) Quantitation of D from three animals in each group. p values between Acp1+/+ TAC and Acp1−/− TAC hearts after Student's t‐test are shown.
Since our transcriptional analysis showed reduced CaMKIIδ expression in Acp1−/− hearts after chronic stress, we assessed this pathway after short‐term pressure overload. Strikingly, Gαq/11 and phosphorylation of PLCβ and CaMKIIδ, which have known deleterious effects in the heart, were increased after short‐term pressure overload in Acp1+/+ hearts but not in Acp1−/− hearts, indicating that CaMKIIδ inhibition likely provides protection in Acp1−/− hearts (Figures 6A and 6B). Next, we assessed whether deletion of Acp1 is associated with altered expression of calcium‐regulated proteins. Sarcoplasmic reticulum calcium ATPase (Serca2a) mRNA did not change in Acp1−/− and Acp1+/+ hearts after pressure overload (Supplementary Figure 8). Phospholamban (Pln), which regulates Serca2a and is phosphorylated by PKA and CaMKIIδ, was also assessed in the four groups of animals. Pln mRNA levels were increased in both Acp1+/+ and Acp−/− hearts after TAC. However, the difference was not statistically significant. Immunoblot analysis showed that Pln was mainly non‐phosphorylated at baseline in Acp1+/+ hearts and its phosphorylated form increased after 24 h of TAC. In contrast, total Pln was relatively low in Acp1−/− hearts at baseline and after TAC, while phosphorylated Pln was high in Acp−/− hearts regardless of the cardiac stress (Figures 6A and 6B). Together, these data suggest that Acp1−/− mice are protected against pathological stress due to alteration of several signalling pathways, including activation of protective pathways (ie IRβ, PKA, ephrin receptor) and inhibition of deleterious pathways such as CaMKIIδ/PLC signalling.
Figure 6.
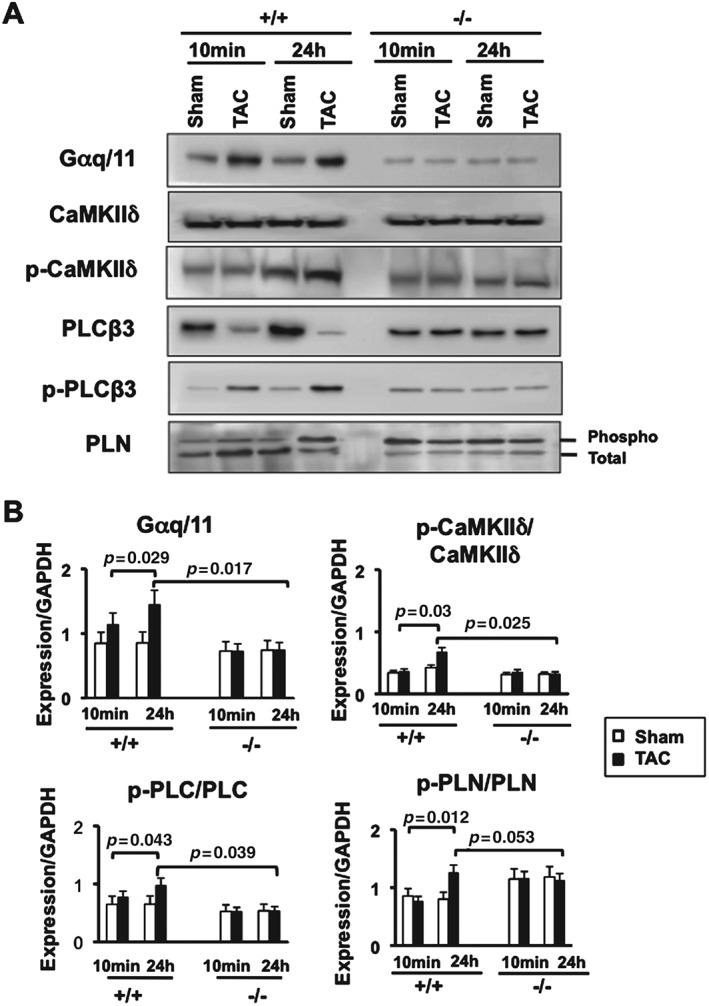
Reduced activation of CaMKIIδ and PLC pathways in Acp1−/−hearts subjected to TAC compared with Acp1+/+ TAC hearts. (A) Immunoblot showing Gαq/11, total and phosphorylated CaMKIIδ, PLCβ, and PLB in Acp1+/+ or Acp1−/−mouse hearts after 10 min and 24 h of TAC or sham surgery. (B) Quantitative analysis of A from three animals in each group. p values obtained after Student's t‐test are shown.
Discussion
LMPTP is a small PTP that defines a separate class of PTPs 6 whose function in heart muscle remains unknown. In the present study, we generated mice with global deletion of Lmptp (Acp1) and show that Lmptp deficiency protects against pathological cardiac hypertrophy and cardiac dysfunction, and attenuates cardiac fibrosis and apoptosis in response to long‐term stress. The protective mechanism involves limited genetic fetal reprogramming, activation of receptors and molecules with cardioprotective functions, and attenuation of deleterious pathways.
Acp1‐null mice showed normal heart development and function, and no organ pathology under basal conditions. Rather, deletion of Lmptp blocked cardiac hypertrophy, significantly attenuated fibrosis, and improved haemodynamic function in a pressure overload model of heart failure. Cardiac hypertrophy and heart failure are characterized by transcriptional reprogramming of genes expressed during fetal cardiac development, which are silenced after birth and induced in pathological conditions. Reactivation of fetal cardiac genes is considered a hallmark of cardiac hypertophy and failure. Likewise, we evaluated fetal genes in the four groups of mice. A strong induction of β‐MHC and ANF occurred in wild‐type mice after stress, while minimal expression was observed in Acp1‐null mice after pressure overload. These observations, together with the developmental regulation of Lmptp and up‐regulation of Lmptp in the adult heart after pathological stress, suggest that maintenance of the adult cardiac gene programme is a likely proximal protective mechanism. However, the recent report that pressure overload induces β‐MHC expression in a restricted number of small non‐hypertrophic cardiomyocytes raises the intriguing possibility, yet to be demonstrated, that fetal genes may be protective 28. Whether reactivation of the fetal gene programme has a protective or deleterious effect is difficult to address, as this process involves complex transcriptional, post‐transcriptional, and epigenetic alterations.
Several signalling pathways appear to be implicated in the protective mechanism of Lmptp ablation. Our transcriptional analysis showed increased expression of IR‐induced genes in Acp1−/−mice after long‐term stress. The IR is currently one of the most validated substrates of the LMPTP, based on in vitro and in vivo studies conducted using antisense oligonucleotides 16, 17, 18, 19, 20. On this basis, and since activation of physiological signalling pathways downstream of the IR is beneficial in the setting of cardiac failure 29, 30, 31, we hypothesized that activation of the IR/Akt pathway may confer cardioprotection. Our molecular analysis performed after short‐term stress showed a strong increase in IRβ phosphorylation in Acp1−/− mouse hearts, which was not associated with enhanced Akt activity. These results suggest that the IR/Akt pathway is not the primary pathway conferring cardioprotection after pathological cardiac stress. ERK and MAPK pathways are known to be activated by receptor tyrosine kinases. However, our results showed similar levels of ERK and MAPK activation in Acp1+/+ and Acp1−/− mice after sham and short‐term TAC. We attribute this unexpected observation to the BALB/c background of the mice. Relatedly, cardiac hypertrophy is usually a quick response following pressure overload; however, cardiac hypertrophy was observed between weeks 4 and 7 post‐TAC in Acp1+/+ mice, suggesting a delayed response to haemodynamic stress in BALB/c mice. PKA and ephrin receptor expression was increased in Acp1−/− mice after stress compared with Acp1+/+ TAC mice. Since there is evidence of a protective role of these effectors in the heart 32, 33, 34, 35, it is conceivable that they account for part of the cardioprotective effect of Lmptp deletion. Evaluation of the downstream effectors of these molecules will be required to ascertain their protective role in Acp1‐deficient hearts.
No activation of CaMKIIδ and PLCβ occurred in Acp1−/− mice after short‐term and prolonged pressure overload. Since PLC and CaMKIIδ are important mediators of pathological cardiac remodelling 36, 37, 38, 39, 40, it is highly likely that the resistance of Acp1−/− mice to pathological stress is mediated at least in part by blockade of PLC/CaMKIIδ pathways and possibly maintenance of calcium homeostasis. Serca2a mRNA levels, however, remained similar under basal conditions and after short‐term TAC in Acp1 +/+ and Acp1−/− mice. Since cardiac hypertrophy and heart failure are known to reduce Serca2a expression, our data suggest that the calcium pump is not regulated by short‐term stress. Phosphorylated Pln, however, appeared to be higher in Acp1‐deficient hearts at baseline and remained high after short‐term stress, suggesting that maintenance of calcium release from the sarcoplasmic reticulum is critical to preserve cardiac contractility in Acp1‐null mice.
In summary, our study reports for the first time that Acp1 deletion in mice prevents cardiac hypertrophy, fibrosis, ventricular chamber dilation, and cardiac dysfunction associated with pathological cardiac remodelling. Repression of the fetal cardiac gene programme is a likely proximal event in the protective mechanism, but additional mechanisms contribute to the preserved phenotype caused by Acp1 deficiency. IR phosphorylation, activation of PKA, ephrin receptors, and inhibition of Gαq11/PLC/CaMKIIδ are early responses following stress challenge, suggesting a role for these pathways in the cardioprotective function of Acp1 deletion. Detailed investigations of the effect of Lmptp in isolated cardiac myocytes, as well as the generation of models carrying conditional deletion of Lmptp in cardiac muscle, will be critical to complete the picture of the role of this phosphatase in the heart. Since global Acp1 KO has a benign phenotype, development of LMPTP‐targeted chemical inhibitors is warranted to further assess its role in cardiac failure and validate LMPTP as a possible target for therapy for cardiac diseases.
Author contribution statement
FW performed biochemical experiments, collected data, and participated in manuscript writing. SMA, MK, SMS, and YL performed biochemical experiments from mouse heart tissues. PQ, KA‐H, HT, and MCJ performed echocardiography in Acp1 mice, cardiac surgeries, and collected the data. RA and FA‐M were responsible for mice breeding, genotyping, and collection of embryos. QM performed all statistical analyses for the echocardiographic data and performed some of the biochemical experiments. DC analysed the microarray data and provided pathway and function analyses. KB performed biochemical experiments. EZ performed qPCR assays. KPR, AA, and MS provided intellectual discussions. WA‐H performed dissection of the human hearts after cardiac transplantation. NB and CP designed the project conceptually and wrote the manuscript.
SUPPORTING INFORMATION ON THE INTERNET.
The following supporting information may be found in the online version of this article:
Supplementary materials and methods.
Table S1. Resting cardiac dimensions and function evaluated in 2‐month‐old mice by echocardiography.
Table S2. Weights of the mice, hearts, left atria, right atria, left ventricle, right ventricle, and liver, and tibia lengths.
Table S3. 146 differentially expressed genes in Acp1 −/− sham hearts versus Acp1 +/+ sham hearts.
Table S4. Representative up‐regulated genes in Acp1 +/+ TAC hearts versus Acp1 +/+ sham hearts and their known biological process.
Table S5. 828 genes differentially regulated in Acp1 +/+ TAC hearts versus Acp1 +/+ sham hearts.
Table S6. 419 genes differentially regulated in Acp1 −/− TAC hearts versus Acp1 +/+ TAC hearts.
Figure S1. Lmptp immunofluorescence, fibrosis, and echocardiography.
Figure S2. Increased Lmptp levels in C57BL/6 mice subjected to TAC.
Figure S3. Targeting of mouse Acp1.
Figure S4. Characterization of Acp1 −/− mice at baseline.
Figure S5. Posterior wall thickness and end‐diastolic diameters after sham or TAC.
Figure S6. Principal component analysis and numbers of changed genes by microarray.
Figure S7. Gene ontology analysis of microarray data.
Figure S8. Immunoblot and RT‐PCR early after TAC.
Supporting information
Appendix S1. Supplementary materials and methods
Lmptp immunofluorescence, fibrosis, and echocardiography.
Increased Lmptp levels in C57BL/6 mice subjected to TAC.
Targeting of mouse Acp1.
Characterization of Acp1 −/− mice at baseline.
Posterior wall thickness and end‐diastolic diameters after sham or TAC.
Principal component analysis and numbers of changed genes by microarray.
Gene ontology analysis of microarray data.
Immunoblot and RT‐PCR early after TAC.
Resting cardiac dimensions and function evaluated in 2‐month‐old mice by echocardiography. The data show that the measured chamber dimensions and functional parameters were similar in rested Acp1 +/+ and ‐/‐ mice (n=5). VST: ventriculum septum thickness; EDD: end diastolic diameter; PWT: posterior wall thickness; ESD: end systolic diameter, HR: heart rate; LV FS: left ventricular fractional shortening; Vcf: velocity of circumferential fiber shortening; LV EF: left ventricular ejection fraction; LV mass: left ventricular mass; BW: body weight.
Weights of the mice (BW), hearts, left atria (LA), right atria (RA), left ventricle (LV), right ventricle (RV), liver, and tibia length are similar in Acp1 +/+ and ‐/‐ mice at 8 weeks of age under basal condition (n=4).
Microarray data showing 146 differentially expressed genes (90 were down‐regulated and 56 were up‐regulated) in Acp1 −/− sham hearts versus Acp1 +/+ sham hearts (n = 2). Only genes expression with fold changes ≥1.5 (up‐regulated) or ≤ ‐1.5 (down‐regulated) and p‐value <0.05 were considered statistically significant.
List of representative up‐regulated genes in Acp1 +/+ TAC hearts compared with Acp1 +/+ sham hearts and their known biological process. Only gene expression with fold changes ≥1.5 (up‐regulated) or ≤ ‐1.5 (down‐regulated) and p‐value <0.05 was considered statistically significant (Acp1 +/+ Sham, n=2; Acp1 +/+ TAC, n=5). The last column in right indicates the validated genes using qPCR and immunoblot analysis, (n=3, p<0.05).
Full microarray data showing the 828 genes significantly regulated (340 were down‐regulated and 488 were up‐regulated) in Acp1 +/+ after TAC vs. Acp1 +/+ after sham. Only genes expression with fold changes ≥1.5 (up‐regulated) or ≤ ‐1.5 (down‐regulated) and p‐value <0.05 were considered statistically significant (Acp1 +/+ Sham, n=2; Acp1 +/+ TAC, n=5).
Microarray data showing the 419 genes differentially regulated (205 were down‐regulated and 214 were up‐regulated) in Acp1 ‐/‐ TAC hearts vs. Acp1 +/+ TAC hearts. Only genes expression with fold changes ≥1.5 (up‐regulated) or ≤ ‐1.5 (down‐regulated) and p‐value <0.05 were considered statistically significant (n=5).
Acknowledgments
We thank Mohammad Pashmforoush (USC Keck School of Medicine, Los Angeles) for providing heart samples from C57BL/6 mice subjected to sham or TAC. CP thanks all the members of her laboratory for their hard work. We thank Haytham El Zein, Ahmed Abusaleem, Khalid Al‐Khatib, and other members of the Heart Transplant team at KFSHRC, and the Comparative Medicine Department staff for their help in various aspects of this project. This work was supported by funds from King Abdullaziz City for Science and Technology (KACST 10‐BIO 1347–20) awarded to CP and by USC and LJI Institutional funds awarded to NB.
No conflicts of interest were declared.
References
Note: References 41\endash 51 are cited in the Supporting information to this article.
- 1. Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nature Rev Mol Cell Biol 2006; 7 : 589–600. [DOI] [PubMed] [Google Scholar]
- 2. Schwartzbauer G, Robbins J. The tumor suppressor gene PTEN can regulate cardiac hypertrophy and survival. J Biol Chem 2001; 276 : 35786–35793. [DOI] [PubMed] [Google Scholar]
- 3. Carr AN, Schmidt AG, Suzuki Y, et al. Type 1 phosphatase, a negative regulator of cardiac function. Mol Cell Biol 2002; 22 : 4124–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shiraishi I, Melendez J, Ahn Y, et al. Nuclear targeting of Akt enhances kinase activity and survival of cardiomyocytes. Circ Res 2004; 94 : 884–891. [DOI] [PubMed] [Google Scholar]
- 5. Miyamoto S, Purcell NH, Smith JM, et al. PHLPP‐1 negatively regulates Akt activity and survival in the heart. Circ Res 2010; 107 : 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alonso A, Sasin J, Bottini N, et al. Protein tyrosine phosphatases in the human genome. Cell 2004; 117 : 699–711. [DOI] [PubMed] [Google Scholar]
- 7. Kontaridis MI, Yang W, Bence KK, et al. Deletion of Ptpn11 (Shp2) in cardiomyocytes causes dilated cardiomyopathy via effects on the extracellular signal‐regulated kinase/mitogen‐activated protein kinase and RhoA signaling pathways. Circulation 2008; 117 : 1423–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ruan H, Li J, Ren S, et al. Inducible and cardiac specific PTEN inactivation protects ischemia/reperfusion injury. J Mol Cell Cardiol 2009; 46 : 193–200. [DOI] [PubMed] [Google Scholar]
- 9. Gomez E, Vercauteren M, Kurtz B, et al. Reduction of heart failure by pharmacological inhibition or gene deletion of protein tyrosine phosphatase 1B. J Mol Cell Cardiol 2012; 52 : 1257–1264. [DOI] [PubMed] [Google Scholar]
- 10. Chernoff J, Li HC. A major phosphotyrosyl‐protein phosphatase from bovine heart is associated with a low‐molecular‐weight acid phosphatase. Arch Biochem Biophys 1985; 240 : 135–145. [DOI] [PubMed] [Google Scholar]
- 11. Magherini F, Giannoni E, Raugei G, et al. Cloning of murine low molecular weight phosphotyrosine protein phosphatase cDNA: identification of a new isoform. FEBS Lett 1998; 437 : 263–266. [DOI] [PubMed] [Google Scholar]
- 12. Bottini N, Bottini E, Gloria‐Bottini F, et al. Low‐molecular‐weight protein tyrosine phosphatase and human disease: in search of biochemical mechanisms. Arch Immunol Ther Exp (Warsz) 2002; 50 : 95–104. [PubMed] [Google Scholar]
- 13. Lazaruk KD, Dissing J, Sensabaugh GF. Exon structure at the human ACP1 locus supports alternative splicing model for f and s isozyme generation. Biochem Biophys Res Commun 1993; 196 : 440–446. [DOI] [PubMed] [Google Scholar]
- 14. Cirri P, Fiaschi T, Chiarugi P, et al. The molecular basis of the differing kinetic behavior of the two low molecular mass phosphotyrosine protein phosphatase isoforms. J Biol Chem 1996; 271 : 2604–2607. [DOI] [PubMed] [Google Scholar]
- 15. Rodriguez‐Rodriguez L, Lopez‐Mejias R, Garcia‐Bermudez M, et al. Genetic markers of cardiovascular disease in rheumatoid arthritis. Mediators Inflamm 2012; 2012 : 574817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Park EK, Warner N, Mood K, et al. Low‐molecular‐weight protein tyrosine phosphatase is a positive component of the fibroblast growth factor receptor signaling pathway. Mol Cell Biol 2002; 22 : 3404–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chiarugi P, Taddei ML, Schiavone N, et al. LMW‐PTP is a positive regulator of tumor onset and growth. Oncogene 2004; 23 : 3905–3914. [DOI] [PubMed] [Google Scholar]
- 18. Chiarugi P, Cirri P, Marra F, et al. LMW‐PTP is a negative regulator of insulin‐mediated mitotic and metabolic signalling. Biochem Biophys Res Commun 1997; 238 : 676–682. [DOI] [PubMed] [Google Scholar]
- 19. Stein E, Lane AA, Cerretti DP, et al. Eph receptors discriminate specific ligand oligomers to determine alternative signaling complexes, attachment, and assembly responses. Genes Dev 1998; 12 : 667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chiarugi P, Cirri P, Taddei L, et al. The low M(r) protein‐tyrosine phosphatase is involved in Rho‐mediated cytoskeleton rearrangement after integrin and platelet‐derived growth factor stimulation. J Biol Chem 2000; 275 : 4640–4646. [DOI] [PubMed] [Google Scholar]
- 21. Pandey SK, Yu XX, Watts LM, et al. Reduction of low‐molecular‐weight protein tyrosine phosphatase expression improves hyperglycemia and insulin sensitivity in obese mice. J Biol Chem 2007; 282 : 14291–14299. [DOI] [PubMed] [Google Scholar]
- 22. Stryke D, Kawamoto M, Huang CC, et al. BayGenomics: a resource of insertional mutations in mouse embryonic stem cells. Nucleic Acids Res 2003; 31 : 278–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Awad S, Al‐Haffar KM, Marashly Q, et al. Control of histone H3 phosphorylation by CaMKIIdelta in response to haemodynamic cardiac stress. J Pathol 2015; 235 : 606–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004; 5 : R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protoc 2009; 4 : 44–57. [DOI] [PubMed] [Google Scholar]
- 26. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37 : 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ibarra C, Estrada M, Carrasco L, et al. Insulin‐like growth factor‐1 induces an inositol 1,4,5‐trisphosphate‐dependent increase in nuclear and cytosolic calcium in cultured rat cardiac myocytes. J Biol Chem 2004; 279 : 7554–7565. [DOI] [PubMed] [Google Scholar]
- 28. Lopez JE, Myagmar BE, Swigart PM, et al. β‐myosin heavy chain is induced by pressure overload in a minor subpopulation of smaller mouse cardiac myocytes. Circ Res 2011; 109 : 629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li B, Setoguchi M, Wang X, et al. Insulin‐like growth factor‐1 attenuates the detrimental impact of nonocclusive coronary artery constriction on the heart. Circ Res 1999; 84 : 1007–1019. [DOI] [PubMed] [Google Scholar]
- 30. McMullen JR, Shioi T, Huang WY, et al. The insulin‐like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3‐kinase(p110alpha) pathway. J Biol Chem 2004; 279 : 4782–4793. [DOI] [PubMed] [Google Scholar]
- 31. Bernardo BC, Weeks KL, Pretorius L, et al. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther 2010; 128 : 191–227. [DOI] [PubMed] [Google Scholar]
- 32. Sanada S, Asanuma H, Tsukamoto O, et al. Protein kinase A as another mediator of ischemic preconditioning independent of protein kinase C. Circulation 2004; 110 : 51–57. [DOI] [PubMed] [Google Scholar]
- 33. Chung YW, Lagranha C, Chen Y, et al. Targeted disruption of PDE3B, but not PDE3A, protects murine heart from ischemia/reperfusion injury. Proc Natl Acad Sci U S A 2015; 112 : E2253–E2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dries JL, Kent SD, Virag JA. Intramyocardial administration of chimeric ephrinA1‐Fc promotes tissue salvage following myocardial infarction in mice. J Physiol 2011; 589 : 1725–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Goichberg P, Bai Y, D'Amario D, et al. The ephrin A1–EphA2 system promotes cardiac stem cell migration after infarction. Circ Res 2011; 108 : 1071–1083. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36. D'Angelo DD, Sakata Y, Lorenz JN, et al. Transgenic Gαq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci U S A 1997; 94 : 8121–8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mende U, Kagen A, Cohen A, et al. Transient cardiac expression of constitutively active Gαq leads to hypertrophy and dilated cardiomyopathy by calcineurin‐dependent and independent pathways. Proc Natl Acad Sci U S A 1998; 95 : 13893–13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Backs J, Backs T, Neef S, et al. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci U S A 2009; 106 : 2342–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ling H, Gray CB, Zambon AC, et al. CaMKIIδ mediates myocardial ischemia/reperfusion injury through NF‐κB. Circ Res 2013; 112 : 935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang R, Khoo MS, Wu Y, et al. Calmodulin kinase II inhibition protects against structural heart disease. Nature Med 2005; 11 : 409–417. [DOI] [PubMed] [Google Scholar]
- 41. Tailor P, Gilman J, Williams S, et al. Regulation of the low molecular weight phosphotyrosine phosphatase by phosphorylation at tyrosines 131 and 132. J Biol Chem 1997; 272 : 5371–5374. [DOI] [PubMed] [Google Scholar]
- 42. Reiner A, Yekutieli D, Benjamini Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 2003; 19 : 368–375. [DOI] [PubMed] [Google Scholar]
- 43. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 2001; 25 : 402–408. [DOI] [PubMed] [Google Scholar]
- 44. Li C, Wong WH. Model‐based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci U S A 2001; 98 : 31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Saeed AI, Bhagabati NK, Braisted JC, et al. TM4 microarray software suite. Methods Enzymol 2006; 411 : 134–193. [DOI] [PubMed] [Google Scholar]
- 46. Saeed AI, Sharov V, White J, et al. TM4: a free, open‐source system for microarray data management and analysis. Biotechniques 2003; 34 : 374–378. [DOI] [PubMed] [Google Scholar]
- 47. Wu Z, Irizarry RA. Stochastic models inspired by hybridization theory for short oligonucleotide arrays. J Comput Biol 2005; 12 : 882–893. [DOI] [PubMed] [Google Scholar]
- 48. Wu Z, Irizarry RA, Gentleman R, et al. A model based background adjustment for oligonucleotide expression arrays. J Am Stat Assoc 2004; 99 : 909–917. [Google Scholar]
- 49. Dennis G Jr, Sherman BT, Hosack DA, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 2003; 4 : P3. [PubMed] [Google Scholar]
- 50. Colak D, Chishti MA, Al‐Bakheet AB, et al. Integrative and comparative genomics analysis of early hepatocellular carcinoma differentiated from liver regeneration in young and old. Mol Cancer 2010; 9 : 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Colak D, Nofal A, Albakheet A, et al. Age‐specific gene expression signatures for breast tumors and cross‐species conserved potential cancer progression markers in young women. PLoS One 2013; 8 : e63204. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supplementary materials and methods
Lmptp immunofluorescence, fibrosis, and echocardiography.
Increased Lmptp levels in C57BL/6 mice subjected to TAC.
Targeting of mouse Acp1.
Characterization of Acp1 −/− mice at baseline.
Posterior wall thickness and end‐diastolic diameters after sham or TAC.
Principal component analysis and numbers of changed genes by microarray.
Gene ontology analysis of microarray data.
Immunoblot and RT‐PCR early after TAC.
Resting cardiac dimensions and function evaluated in 2‐month‐old mice by echocardiography. The data show that the measured chamber dimensions and functional parameters were similar in rested Acp1 +/+ and ‐/‐ mice (n=5). VST: ventriculum septum thickness; EDD: end diastolic diameter; PWT: posterior wall thickness; ESD: end systolic diameter, HR: heart rate; LV FS: left ventricular fractional shortening; Vcf: velocity of circumferential fiber shortening; LV EF: left ventricular ejection fraction; LV mass: left ventricular mass; BW: body weight.
Weights of the mice (BW), hearts, left atria (LA), right atria (RA), left ventricle (LV), right ventricle (RV), liver, and tibia length are similar in Acp1 +/+ and ‐/‐ mice at 8 weeks of age under basal condition (n=4).
Microarray data showing 146 differentially expressed genes (90 were down‐regulated and 56 were up‐regulated) in Acp1 −/− sham hearts versus Acp1 +/+ sham hearts (n = 2). Only genes expression with fold changes ≥1.5 (up‐regulated) or ≤ ‐1.5 (down‐regulated) and p‐value <0.05 were considered statistically significant.
List of representative up‐regulated genes in Acp1 +/+ TAC hearts compared with Acp1 +/+ sham hearts and their known biological process. Only gene expression with fold changes ≥1.5 (up‐regulated) or ≤ ‐1.5 (down‐regulated) and p‐value <0.05 was considered statistically significant (Acp1 +/+ Sham, n=2; Acp1 +/+ TAC, n=5). The last column in right indicates the validated genes using qPCR and immunoblot analysis, (n=3, p<0.05).
Full microarray data showing the 828 genes significantly regulated (340 were down‐regulated and 488 were up‐regulated) in Acp1 +/+ after TAC vs. Acp1 +/+ after sham. Only genes expression with fold changes ≥1.5 (up‐regulated) or ≤ ‐1.5 (down‐regulated) and p‐value <0.05 were considered statistically significant (Acp1 +/+ Sham, n=2; Acp1 +/+ TAC, n=5).
Microarray data showing the 419 genes differentially regulated (205 were down‐regulated and 214 were up‐regulated) in Acp1 ‐/‐ TAC hearts vs. Acp1 +/+ TAC hearts. Only genes expression with fold changes ≥1.5 (up‐regulated) or ≤ ‐1.5 (down‐regulated) and p‐value <0.05 were considered statistically significant (n=5).