

Summary
The Chinese indigenous pig breeds in the Taihu Lake region are the most prolific pig breeds in the world. In this study, we investigated the genetic diversity and population structure of six breeds, including Meishan, Erhualian, Mi, Fengjing, Shawutou and Jiaxing Black, in this region using whole‐genome SNP data. A high SNP with proportions of polymorphic markers ranging from 0.925 to 0.995 was exhibited by the Chinese indigenous pigs in the Taihu Lake region. The allelic richness and expected heterozygosity also were calculated and indicated that the genetic diversity of the Meishan breed was the greatest, whereas that of the Fengjing breed was the lowest. The genetic differentiation, as indicated by the fixation index, exhibited an overall mean of 0.149. Both neighbor‐joining tree and principal components analysis were able to distinguish the breeds from each other, but structure analysis indicated that the Mi and Erhualian breeds exhibited similar major signals of admixture. With this genome‐wide comprehensive survey of the genetic diversity and population structure of the indigenous Chinese pigs in the Taihu Lake region, we confirmed the rationality of the current breed classification of the pigs in this region.
Keywords: genetic distance, genetic variation, Next‐generation sequencing, pig genome
The Chinese indigenous pigs in the Taihu Lake region are the most prolific breeds in the world. These pigs are the domestic breeds of China and inhabit a narrow region with a mild subtropical climate around the Taihu Lake region in the lower Yangtze River Valley. The first comprehensive survey of pig resources in China regarded these animals as a single breed termed Taihu pigs (Zhang 1986). However, according to the latest classification of Chinese indigenous pig breeds, these pigs are now divided to six breeds (i.e., the Meishan, Erhualian, Mi, Fengjing, Shawutou and Jiaxing Black breeds) (China National Commission of Animal Genetic Resources 2011). The Meishan breed can also be subdivided into two types, that is the Small Meishan and the Middle Meishan (Zhang 1991; China National Commission of Animal Genetic Resources 2011). The classification of these breeds is based primarily on their phenotypic characteristics (such as body and ear size) or their performances (i.e., reproductive and meat quality traits) (China National Commission of Animal Genetic Resources 2011) as well as their locations.
Fan et al. (2002) performed a genetic variation analysis using 27 microsatellites markers to evaluate the levels of diversity of four breeds in the Taihu Lake region. These authors found that the Small Meishan exhibited the least genetic variability, and the studied population exhibited an average fixation index (F ST) of 0.18. Recently, several studies have also extensively evaluated the genetic variations of some Chinese pigs using high‐density SNP markers (Ai et al. 2013, 2014) or whole‐genome sequencing data (Ai et al. 2015). These studies confirmed the divergent evolutions and distinct population structures of Chinese and Western pigs and revealed the adaptive evolutionary history of the pig. However, to our knowledge, an original survey of the genome‐wide genetic markers of all six Chinese indigenous pig breeds in the Taihu Lake region has not yet been conducted. The full characterization of their genetic diversity and population structure based on genome‐wide markers also remains unexplored. Therefore, in this study, we applied the genotyping by genome reducing and sequencing (GGRS) approach (Chen et al. 2013), which is a next‐generation sequencing technology that is capable of identifying and genotyping hundreds of thousands of markers across the genome in a cost‐effective and highly reproducible manner (especially for outbred species with large genomes), to assess genetic variation and population structure of the six breeds in the Taihu Lake region.
A total of 252 unrelated or distantly related pigs from the six Chinese indigenous breeds in the Taihu Lake region (Table 1) were selected based on pedigree records. The DNA samples were genotyped according to the GGRS protocol (http://klab.sjtu.edu.cn/GGRS/; Chen et al. 2013). Briefly, high molecular weight genomic DNA samples were extracted from ear tissue, digested with AvaII and then ligated with a unique adapter barcode. Next, the samples were pooled and enriched to construct a sequencing library. Finally, the sequence libraries (fragments ranging from 300 to 400 bp, including the adapter barcode sequence) were sequenced on an Illumina Hiseq2000 (the sequencing process is given in detail by the manufacturer, Illumina) instrument with a paired‐end (2 × 100 bp) pattern. The SNPs were identified and genotyped using samtools (Li et al. 2009), and these variants were retained for further analysis according to the following criteria: more than 75 (30%) genotyped samples and sequencing depth greater than fivefold on average. The imputation analysis for the missing genotypes described below was performed with iblup (http://klab.sjtu.edu.cn/iBLUP/), which accounts for both the identity‐by‐descent and linkage disequilibrium information and can impute missing genotypes with greater accuracy than can other imputation methods, such as beagle (Yang et al. 2014). A total of 105 550 high‐confidence SNPs with minor allele frequencies (MAFs) ≥ 0.05 were obtained. Among these SNPs, 29 575 (28%; submitted to dbSNP of Genbank) were novel based on comparisons with a database of known SNPs that contained 28 722 391 SNPs, downloaded from http://hgdownload.soe.ucsc.edu/goldenPath/susScr3/database/ (updated on March 2, 2014). According to the Ensembl pig gene annotation set (Ensembl release 78, ftp://ftp.ensembl.org/pub/release-78/gtf/sus_scrofa/; Flicek et al. 2013), 37 484 SNPs were mapped to gene regions, of which four were mapped to regions of start or stop codons, 2400 were mapped as exons, 20 249 were mapped as introns and 14 831 were mapped as UTRs. The SNP data can be downloaded from our website (http://klab.sjtu.edu.cn/iBLUP/genotype_taihu_pigs.zip). The SNP positions within the chromosomes were based on the pig reference genome (SGSC Sscrofa10.2) (Groenen et al. 2012).
Table 1.
Sample sizes and genetic diversities of the seven pig populations in the Taihu Lake region
| Population | Origin | Sample size | Indices of genetic diversity | NSNP | ||
|---|---|---|---|---|---|---|
| AR | PN | HE | ||||
| Meishan (Middle) | Jiading, Shanghai | 50 | 1.983 | 0.993 | 0.375 | 25 724 |
| Meishan (Small) |
Jurong, Jiangsu (n = 33) Taicang, Jiangsu (n = 36) |
69 | 1.981 | 0.989 | 0.382 | 26 202 |
| Fengjing | Jinshan, Shanghai | 16 | 1.861 | 0.925 | 0.315 | 19 625 |
| Shawutou | Chongming, Shanghai | 21 | 1.962 | 0.987 | 0.371 | 24 588 |
| Erhualian | Changshu, Jiangsu | 31 | 1.983 | 0.995 | 0.378 | 25 125 |
| Mi | Jintan, Jiangsu | 36 | 1.982 | 0.991 | 0.382 | 25 694 |
| Jiaxing Black | Jiaxing, Zhejiang | 29 | 1.933 | 0.959 | 0.350 | 23 047 |
AR, allelic richness; PN, proportion of SNPs that displayed polymorphisms among the 34 789 SNPs selected from the set of 105 550; HE, expected heterozygosity; NSNP, number of SNPs in the 34 789 SNP subset with MAF > 0.2.
Intrapopulation genetic variability
The allelic richness (AR), proportion of polymorphic markers (PN) and expected heterozygosity (HE) were used to investigate the genome‐wide genetic variability within these six breeds. AR estimation was conducted using adze v1.0 (Szpiech et al. 2008). PN and HE were calculated using plink v1.07 (Purcell et al. 2007). To reduce the ascertainment bias that was likely caused by low frequency SNPs, we used a subset of 34 789 SNPs with MAFs ≥ 0.2 to calculate these three measures. Overall, the Chinese indigenous pigs in the Taihu Lake region displayed a comparably high level of SNP with PN values that ranged from 0.925 to 0.995 (Table 1). We found that each breed sustained high levels of genetic variability, which was likely due to the less intensive selection and greater genetic diversity of their wild ancestors compared with Western pigs. These results are consistent with previous reports (Fan et al. 2002; Yang et al. 2003; Ai et al. 2013). Specifically, the Middle Meishan breed exhibited a higher level of polymorphisms than did the Small Meishan breed, implying that the Small Meishan breed might have a smaller effective population or might have experienced intensive selection that decreased the extent of its polymorphisms. This observation agrees with the history of the breeding programs of the Small Meishan breed, which experienced a high level of inbreeding in the mid‐1980s (Fan et al. 2002). Although imputation‐based estimates of diversity might be biased (Fu 2014), such bias might be largely reduced by the high level of accuracy of the imputation method used in our study. We should note that we used only a common SNP set (MAF > 0.2) in the above analysis, which might have resulted in an underestimation of diversity; thus, our results most likely reflect only the relative values of three measures of studied breeds. In addition, linkage disequilibrium values (r 2 0.3, predicted as described by Ai et al. 2013) ranged from 15 kb (Middle Meishan) to 34 kb (Fengjing), and the interpopulation linkage disequilibrium extent across the six Chinese pigs breeds was only 9 kb (Fig. S1), which is close to the estimation of Ai et al. (2013).
Interpopulation genetic variability
Genetic distance
All 105 550 SNPs were used to estimate the genetic distances between the populations. The average proportions of alleles shared were calculated as the D st with plink v1.07 (Purcell et al. 2007). The genetic distances (D) between all of the pair‐wise combinations of individuals were calculated as follows: D = 1 − D st. Neighbor‐joining (NJ) trees for the relationships between the individuals were constructed with mega v4 (Tamura et al. 2007). The average genetic distance between individuals was 0.23 ± 0.03 across all six breeds. For the individual populations, these values ranged from 0.16 ± 0.01 (Small Meishan) to 0.22 ± 0.01 (Erhualian). Although the Small Meishan pigs came from two different farms, some Jurong individuals migrated from Taicang, and extensive gene exchange occurred (Fan et al. 2002). Moreover, as mentioned above, the Small Meishan pigs had experienced a high level of inbreeding (Fan et al. 2002). Artificial selection, small sample size and inbreeding might explain the finding that the genetic distance of this breed was shorter than those of the other populations. The NJ tree revealed that all individuals from the same populations clustered together. The Middle Meishan and Small Meishan pigs were clustered into two separate clades, whereas the Fengjing and Jiaxing Black pigs were grouped into a single clade (Fig. S2).
Determination of genetic differentiation estimates
To investigate the extent of population differentiation among the six Chinese indigenous pig breeds in the Taihu Lake region, unbiased genetic differentiation estimates of F ST (Weir & Cockerham 1984) were calculated (as described in Akey et al. 2002) using the entire SNP dataset (except the SNPs on the sex chromosome). Because the range of F ST was originally defined as 0 to 1 (Wright 1951), we set the negative F ST values (i.e., those without a biological interpretation) to 0. The criteria for interpreting the F ST values were those of Li et al. (2014). For the entire population, the F ST value exhibited an overall mean of 0.149 and a standard deviation of 0.124. The F ST values for all of the pair‐wise population comparisons are shown in Table S1. The Fengjing breed exhibited the greatest F ST value among all of the populations (with the exception of the Shawutou breed) and the greatest divergence from the Small Meishan breed. The Mi and Erhualian displayed the least divergence among all of the pair‐wise population comparisons. The Middle Meishan breed exhibited the least divergence with the Small Meishan among all other populations.
Analysis of the population structure
An analysis of the population structure was performed using structure (run with 10 000 iterations using the correlated allele model) (Hubisz et al. 2009). The number of markers was reduced to 34 789 by filtering those with MAFs < 0.2 from the above‐mentioned 105 550 SNPs to minimize the SNP ascertainment bias among the different breeds and to save running time. distruct (Rosenberg 2004) was employed to plot the results (K = 2 to 6 are shown, and K = 6 had the highest likelihood value). Principal components analysis (PCA) was conducted using gcta (ver 1.24) (Yang et al. 2011). structure analysis revealed that the Jiaxing Black and Small Meishan breed formed the first two independent populations (K = 2) followed by the Middle Meishan (K = 3), Fengjing (K = 5) and Shawutou (K = 6) breeds (Fig. 1a). However, the Erhualian and Mi breeds appeared to share a common ancestry. These observations are reasonable considering the assumption that the breeds with geographically close origins likely shared common ancestors and crossbred with each other. A previous study also reported that the Mi breed was formed by crossbreeding the Dahualian (extinct) with the Huaizhu breeds and was backcrossed with the Dahualian breed to produce the Erhualian breed (Fan et al. 2002). In addition, there was a time when the Mi breed was nearly extinct, and it was regarded as the same as the Erhualian breed for conservation; thus, crossing might have occurred between these breeds. These factors might explain why the Mi and Erhualian pigs are closely related and exhibited similar major signals of admixture. PCA revealed that the first two PC axes could clearly distinguish the pigs from each breed (including the two subdivisions of the Meishan, i.e., the Small Meishan and Middle Meishan) (Fig. 1b).The Erhualian, Mi, Fengjing and Jiaxing Black breeds were closely clustered. This finding agreed well with the NJ tree and population structure mentioned above. The phylogenic relationships among the six breeds might be altered considering all the local Chinese pig breeds. Thus, further investigations should be conducted to clarify this issue.
Figure 1.
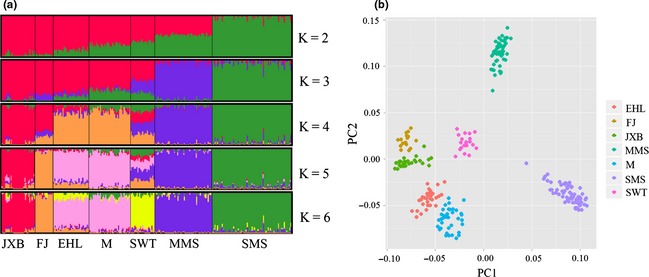
Genetic diversity and structure of Chinese indigenous pigs in the Taihu Lake region. (a) Population structure of the Chinese indigenous pigs in the Taihu Lake region revealed with structure software. (b) Population structures of Chinese indigenous pigs in the Taihu Lake region revealed by principal components analysis. MMS, Middle Meishan; SWT, Shawutou; EHL, Erhualian; M, Mi; FJ, Fengjing; JXB, Jiaxing Black; SMS, Small Meishan.
Regarding the Middle Meishan and Small Meishan breeds, the PCA and NJ analyses revealed that they could be classified into two different groups. Moreover, these two breeds exhibited a moderate level of genetic differentiation (F ST = 0.081) that was greater than that of the Erhualian and Mi breeds (F ST = 0.061), which are now regarded as two breeds (China National Commission of Animal Genetic Resources 2011). The number of putatively breed‐specific SNPs of these four breeds was further calculated to compare genetic difference among them. We found that the number of putatively breed‐specific SNPs between the Middle Meishan and Small Meishan breeds (19 928, Fig. S3) was greater than that between the Erhualian and Mi breeds (15 407, Fig. S3), which indicates a greater genetic difference between the Middle Meishan and Small Meishan breeds than between the Erhualian and Mi breeds. Therefore, it might be reasonable to separate the Middle Meishan and Small Meishan into two different breeds rather than two types of a single breed for conservation to enhance the genetic diversity of indigenous Chinese pigs.
In conclusion, we confirmed the rationality of the current breed classification of the Chinese indigenous pigs in the Taihu Lake region, and we suggest that the Middle Meishan and Small Meishan were separated into two breeds for conservation. We conducted, for the first time, a comprehensive survey of the nucleotide variability of six Chinese indigenous pigs in the Taihu Lake region on a genome‐wide scale.
Conflict of interest
The authors declare no conflict of interest.
Authors′ contributions
Y. P. designed the study. Y. P. and Q. W. supervised the study. Z.W. analyzed the data. Z.W. wrote the manuscript. Y. Y. implemented the method in the iblup software package with the help of J. Z. and Z. Z. Q. C. developed the GGRS approach for outbred populations with the help of Z. C, R. L and X. Z., and M. X, H. Y and Y. Z assisted pig sample collection. All authors have read and edited the manuscript.
Supporting information
Figure S1 Extents of LD (predicated as r 2) within and across the six breeds. 1000 Genome Project Data Processing Subgroup.
Figure S2 The neighbor‐joining tree of the pigs from the six breeds based on the genome‐wide SNPs.
Figure S3 The numbers of SNPs that were breed‐specific among the Erhualian, Mi, Middle Meishan and Small Meishan breeds. A SNP was labeled as breed specific when the allele was present only in one of the populations and not detected in any of the others.
Table S1 Genetic differentiations (F ST values) between the seven pig populations.
Acknowledgements
This study was supported by the 2011–2015 Animal Germplasm Resources Conservation Project from the Ministry of Agriculture of China, the National Natural Science Foundation of China (grant no 31472069, U1402266, 31370043, 31272414) and the National 948 Project of China (2012‐Z26, 2011‐G2A).
The copyright line for this article was changed on 8 October after original online publication.
References
- Ai H., Huang L. & Ren J. (2013) Genetic diversity, linkage disequilibrium and selection signatures in Chinese and Western pigs revealed by genome‐wide SNP markers. PLoS One 8, e56001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai H., Yang B., Li J., Xie X., Chen H. & Ren J. (2014) Population history and genomic signatures for high‐altitude adaptation in Tibetan pigs. BMC Genomics 15, 834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai H., Fang X., Yang B. et al (2015) Adaptation and possible ancient interspecies introgression in pigs identified by whole‐genome sequencing. Nature Genetics 47, 217–25. [DOI] [PubMed] [Google Scholar]
- Akey J.M., Zhang G., Zhang K., Jin L. & Shriver M.D. (2002) Interrogating a high‐density SNP map for signatures of natural selection. Genome Research 12, 1805–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q., Ma Y., Yang Y. et al (2013) Genotyping by genome reducing and sequencing for outbred animals. PLoS One 8, e67500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- China National Commission of Animal Genetic Resources (2011) Animal Genetic Resources in China Pigs. China Agriculture Press, Beijing. [Google Scholar]
- Fan B., Wang Z.G., Li Y.J. et al (2002) Genetic variation analysis within and among Chinese indigenous swine populations using microsatellite markers. Animal Genetics 33, 422–7. [DOI] [PubMed] [Google Scholar]
- Flicek P., Ahmed I., Amode M.R. et al (2013) Ensembl 2013. Nucleic Acids Research 41, D48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.B. (2014) Genetic diversity analysis of highly incomplete SNP genotype data with imputations: an empirical assessment. G3 (Bethesda) 4, 891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenen M.A., Archibald A.L., Uenishi H. et al (2012) Analyses of pig genomes provide insight into porcine demography and evolution. Nature 491, 393–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubisz M.J., Falush D., Stephens M. & Pritchard J.K. (2009) Inferring weak population structure with the assistance of sample group information. Molecular Ecology Resources 9, 1322–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. & 1000 Genome Project Data Processing Subgroup. (2009) The Sequence Alignment/Map format and samtools . Bioinformatics 25, 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Yang S., Tang Z., Li K., Rothschild M.F., Liu B. & Fan B. (2014) Genome‐wide scans to detect positive selection in Large White and Tongcheng pigs. Animal Genetics 45, 329–39. [DOI] [PubMed] [Google Scholar]
- Purcell S., Neale B., Todd‐Brown K. et al (2007) plink: a tool set for whole‐genome association and population‐based linkage analyses. American Journal of Human Genetics 81, 559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg N.A. (2004) distruct: a program for the graphical display of population structure. Molecular Ecology Notes 4, 137–8. [Google Scholar]
- Szpiech Z.A., Jakobsson M. & Rosenberg N.A. (2008) adze: a rarefaction approach for counting alleles private to combinations of populations. Bioinformatics 24, 2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Dudley J., Nei M. & Kumar S. (2007) mega4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Molecular Biology and Evolution 24, 1596–9. [DOI] [PubMed] [Google Scholar]
- Weir B.S. & Cockerham C.C. (1984) Estimating F‐statistics for the analysis of population structure. Evolution 38, 1358–70. [DOI] [PubMed] [Google Scholar]
- Wright S. (1951) The genetical structure of populations. Annals of Eugenics 15, 323–54. [DOI] [PubMed] [Google Scholar]
- Yang S.L., Wang Z.G., Liu B., Zhang G.X., Zhao S.H., Yu M., Fan B., Li M.H., Xiong T.A. & Li K. (2003) Genetic variation and relationships of eighteen Chinese indigenous pig breeds. Genetics Selection Evolution 35, 657–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Lee S.H., Goddard M.E. & Visscher P.M. (2011) gcta: a tool for genome‐wide complex trait analysis. American Journal of Human Genetics 88, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Wang Q., Chen Q., Liao R., Zhang X., Yang H., Zheng Y., Zhang Z. & Pan Y. (2014) A new genotype imputation method with tolerance to high missing rate and rare variants. PLoS One 9, e101025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. (1986) Chinese Pig Breed Records. Shanghai Science and Technology Press, Shanghai, China. [Google Scholar]
- Zhang Z. (1991) Chinese Taihu Pig. Shanghai Scientific and Technical Publishers, Shanghai. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Extents of LD (predicated as r 2) within and across the six breeds. 1000 Genome Project Data Processing Subgroup.
Figure S2 The neighbor‐joining tree of the pigs from the six breeds based on the genome‐wide SNPs.
Figure S3 The numbers of SNPs that were breed‐specific among the Erhualian, Mi, Middle Meishan and Small Meishan breeds. A SNP was labeled as breed specific when the allele was present only in one of the populations and not detected in any of the others.
Table S1 Genetic differentiations (F ST values) between the seven pig populations.