

Abstract
We are living through exciting times during which we are able to unravel the “microbial dark matter” in and around us through the application of high‐resolution “meta‐omics”. Metaproteomics offers the ability to resolve the major catalytic units of microbial populations and thereby allows the establishment of genotype‐phenotype linkages from in situ samples. A decade has passed since the term “metaproteomics” was first coined and corresponding analyses were carried out on mixed microbial communities. Since then metaproteomics has yielded many important insights into microbial ecosystem function in the various environmental settings where it has been applied. Although initial progress in analytical capacities and resulting numbers of proteins identified was extremely fast, this trend slowed rapidly. Here, we discuss several representative metaproteomic investigations of activated sludge, acid mine drainage biofilms, freshwater and seawater microbial communities, soil, and human gut microbiota. By using these case studies, we highlight current challenges and possible solutions for metaproteomics to realize its full potential, i.e. to enable conclusive links between microbial community composition, physiology, function, interactions, ecology, and evolution in situ.
Keywords: Integrated omics, Metagenomics, Metaproteomics, Microbial community, Microbial systems ecology, Microbiology
Abbreviations
- EBPR
enhanced biological phosphorus removal
- n‐damo
nitrite‐dependent anaerobic methane oxidation
1. Introduction
High‐throughput “meta‐omic” approaches have found widespread application in microbial ecology as they allow unprecedented insights into the organismal and functional make‐up of natural consortia in situ. By bridging genetic potential and final phenotype, metaproteomics (also often referred to as community proteogenomics 1) offers the ability to resolve the main functional components driving microbial ecosystem function. We originally defined metaproteomics as “the large‐scale characterization of the entire protein complement of environmental microbiota at a given point in time” 2 and, in our opinion, this definition remains pertinent. Through the application of metaproteomics to different microbial consortia over the past decade, we have learnt much about key functional traits in the various environmental settings where they occur. Initial progress in analytical capabilities was extremely rapid. Though we were initially able to identify three proteins excised from 2D‐PAGE by de novo peptide sequencing using LC‐MS/MS in 2004 3, this number was already superseded a year later when Jill Banfield and colleagues were able to identify over 2000 proteins from a microbial consortium using a shotgun LC‐MS/MS approach and by searching the resulting data against a tailored metagenomic database 3. Here, we discuss several representative studies and conclude by highlighting the key challenges and possible solutions to bring metaproteomics on a par with other community omic approaches, in particular metagenomics and metatranscriptomics, to allow metaproteomics to fulfil its keystone role in microbial systems ecology in the future.
Correspondence concerning this and other Viewpoint articles can be accessed on the journals' home page at: http://viewpoint.proteomics‐journal.de
Correspondence for posting on these pages is welcome and can also be submitted at this site.
2. Activated sludge
Activated sludge is the most common form of wastewater treatment employed in the industrialised world and optimization of the process requires improved understanding of these relatively complex microbial ecosystems 4. Metaproteomics has its origins in activated sludge research and initial studies were conducted on laboratory‐scale systems performing enhanced biological phosphorus removal (EBPR) 1, 5. These studies relied on 2D‐PAGE protein separation and identification of excised proteins by LC‐MS/MS and de novo peptide sequencing. Subsequent metaproteomic studies of EBPR were more insightful as metagenomes of these ecosystems became available 6 and the analyses became far more automated through use of nano‐LC for peptide separation rather than 2D‐PAGE 7. By focusing on the dominant bacterium involved in EBPR, key functions relevant to the performance were revealed 7. Additionally, detection of enzyme variants indicated that some degree of genetic diversity is important for stable EBPR performance 7. These studies also revealed that only minor differences in the metaproteome are detected between the rapidly alternating anaerobic and aerobic phases of EBPR 1, 7. Consequently, radiolabeled 35S‐methionine was used for the detection of only newly synthesized proteins within the EBPR phases 8. Findings suggest that the glyoxylate cycle is important in the aerobic phase, which contrasted with previous speculations of EBPR metabolism. In another study, rapid changes (within 15 min) in activated sludge proteomes could be detected, in response to a previously unseen environmental stimulant of Cd exposure 9. Additionally, the sensitivity of metaproteomics has been examined in activated sludge, such that proteins from an added strain could be detected when this was at around one thousandth of the biomass 10. Finally, the metaproteome of extracellular polymeric substances, which are integral to the system, has been studied in detail revealing a number of cytoplasmic proteins, which may play differing roles in activated sludge biomass 11, 12, 13, 14.
Metaproteomic studies of activated sludge have revealed important details of metabolism, interactions, and physiology, which overall have significantly enhanced our understanding of the microbial communities underpinning the process. So far, public sequence databases have mostly been used, and improved identifications will be obtainable in the future by use of sample‐derived metagenomic sequences. Additionally, very few studies have so far used a quantitative approach. Such information would however allow refinement of metabolic models and permit more detailed comparative investigations for example between contrasting aerobic and anaerobic phases.
3. Acid mine drainage biofilms
From all the different microbial communities to which metaproteomics has been applied, acidophilic biofilms from the Richmond Mine in Northern California represent by far the most deeply sampled. A first shotgun proteomic analysis resulted in the identification of over 2000 proteins with high protein coverage (48%) obtained for the dominant Leptospirillum rubarum 2. One highly abundant protein of unknown function was further investigated and found to be an iron‐oxidizing cytochrome (Cyt579), a key component of energy generation in the biofilms 2. Thus, the proteomic results were instrumental in guiding the ensuing detailed biochemical investigations 15. The use of high mass accuracy data further allowed the differentiation between peptides originating from discrete strains of L. rubarum and subsequently allowed a mapping of intraspecies recombination 16. This approach was expanded to conduct extensive proteomics‐based genotype surveys of distinct biofilm samples and indicated which genes are involved in niche partitioning across different biofilm development stages 17, 18. Based on detectable amino acid modifications due to hydrolysis, a distinction between proteins derived from active community members and lysed cells was also achievable 19. Other important physiological insights revealed through metaproteomics include species‐specific hydrogen isotope fractionation in proteins 20, nitrogen flow patterns through these communities 21, and diverse posttranslational modifications specific to distinct biofilm developmental stages 22. Encouragingly for metaproteomic validation studies on other microbial communities, in situ protein expression was largely mirrored in dedicated laboratory‐based bioreactor experiments 23 and this experimental approach has allowed the assessment of the impact of specific perturbations, i.e. an increase in temperature, on the metaproteome 24. While the metaproteome of these biofilms arguably remains the most explored, recent taxonomic survey data from such biofilms 25 suggest that even with the most advanced proteomic technologies, only around 1% of the expected protein complement is currently resolvable (Table 1).
Table 1.
Estimated microbial species and protein richness as well as numbers and examples of proteins identified
| Ecosystem | Estimated number of taxaa | Estimated number of unique proteinsb | Number of identified proteinsc | Examples of signature proteins and potential biomarkers |
|---|---|---|---|---|
| Acid mine drainage biofilm | 159 25 | 477 000 | 4259 22 | Specific cytochromes involved in iron oxidation 2, 15. |
| Activated sludge | 1000 73 | 3 000 000 | 5000 69 | Proteins constituting exopolymeric substances 12, 13, 14 |
| Ocean water | 160 74 | 480 000 | 5600 28 | Proteorhodopsins 30, 31, transport proteins 30, 31 |
| Surface freshwater | 20 000 75 | 60 000 000 | 1800 37 | N and P cycling 41, anthropogenic contaminant degradation 45 |
| Soil | 50 000 76–8 000 000 50 | 150 000 000–24 000 000 000 | 7000 56 | Saprophytic enzymes 53, 54, Nif proteins 55, methane monooxygenase 55 |
| Human‐associated | ||||
| Saliva | >5400 77 | 16 200 000 | >2000 78 | Glycoproteinolytic enzymes 78 |
| Feces | >21 000 77 | >63 000 000 | >2900 62 | Carbohydrate active enzymes 62 |
As defined by author(s) of referenced work.
Estimated number of unique proteins based on average environmental microbial genome size of 3 Mbp and 1 kbp of sequence coding for one gene. Uniqueness does not include strain‐level variation. Numbers do not reflect intra‐ and intertaxon protein abundance differences.
As defined by author(s) of referenced work.
4. Marine systems
A study of Chesapeake Bay microbial communities kicked off metaproteomic investigations of marine systems 26. These are now numerous and include diverse marine environments including coastal systems 27, 28, 29, surface waters 30, 31, low oxygen waters 32, ship hull biofilms 33, animal gut symbionts 34, 35, and sediments 36, 37.
Surface waters differ substantially with regards to nutrients, light, and microbial activities. Metaproteomics has been conducted on a range of different samples: small bacterioplankton (<1.2 μm) in nutrient‐rich waters (Oregon coast) 29; membrane‐enriched fractions of small cells (<0.8 μm) from coastal waters and nutrient‐depleted waters from the open ocean (South Atlantic) 30; bacterial populations during a phytoplankton bloom (North Sea) 31; and seasonally sampled cold coastal waters of the West Antarctic Peninsular 38. Surface water proteins are often dominated by those from the SAR11, Roseobacter, and the oligotrophic marine gammaproteobacteria clades [29, 30, 31, 38] whereas cyanobacterial proteins are more prominent in nutrient‐depleted ocean water samples 30. On a functional level, metaproteomics has contributed important knowledge regarding carbon and nutrient cycling in surface waters, with notable functionalities including proteorhodopsin‐mediated light‐driven proton pumps and methylotrophy 30, 31.
From diffuse hydrothermal venting sediments of the Norwegian‐Greenland Sea (at 90°C and 564 m deep), covered with white microbial mats, metaproteomics has allowed assignment of major metabolic pathways to the dominant taxa 36. The metaproteome reflected autotrophic sulfide oxidation, sulphate reduction coupled to CO2 fixation, and aerobic methane oxidation. This study used a combined sequence database of the metagenomic and metatranscriptomic data to improve the identification of proteins. Additionally, a large degree of overlap between the metatranscriptome and metaproteome was apparent 36. In cold seep sediments, also of the Norwegian Sea (746 m deep), anaerobic oxidation of methane by anaerobic methanotrophic archaea (ANME) was found to be coupled to bacterial sulphate reduction. In a study of the sediments, genes and proteins from anaerobic methanotrophic archaea dominated the metagenomic and metaproteomic data 37. Notably, this study revealed that the protein MetF, expressed in the sediment, may replace its structural analogue Mer in the reverse methanogenesis pathway. However, this metaproteomic discovery requires detailed future biochemical validation but, in analogy to the discovery of Cyt579 in acid mine drainage biofilms, highlights the power of metaproteomics to inform future work on previously elusive biochemical transformations.
Symbiosis with deep‐sea marine animals enables microbial communities to survive in nutrient‐poor environments. A combined metaproteomic and meta‐metabolomic investigation was conducted on a gutless marine worm, which hosts a stable community of five bacterial endosymbionts 34. Some of the more remarkable discoveries made on the symbionts include: the potential to use CO oxidation coupled to sulphate reduction for energy generation; a pathway for utilization of the host's waste fermentation products; the preponderance of high‐affinity uptake transporters for various organic molecules; and a mechanism for rapid evolution of the symbionts. These discoveries provide new insights on the basis of the symbiosis and have thereby significantly advanced our understanding of the oligotrophic marine environment.
5. Surface freshwater and aquifer systems
Early metaproteomic studies of surface freshwater and aquifer systems tested methods of cell concentration 39 and protein extraction 39, 40 as sample‐related challenges here include low biomass yields and interfering substances such as humic acids. In a study that included two freshwater lakes (NY, USA), proteins from Betaproteobacteria and Cyanobacteria dominated 41. The study provided evidence of nutrient cycling as well as details on photosynthesis and electron transport 41. Interestingly, in contrast to marine systems, the lakes contained relatively low levels of transport proteins suggesting that nutrients are less limiting in these environments compared to marine systems.
Ace Lake, an Antarctic meromictic lake has been subjected to metagenomic and metaproteomic analyses 42, 43. The lake is brackish above the chemocline and is increasingly saline and anoxic toward the bottom with a green sulfur bacterial population dominating dense communities at the chemocline 42, 43. Metaproteomics highlighted the importance of bacteriochlorophylls adapted to low light intensities, membrane fluidity, and syntrophic sulfur transformations in this bacterial population thereby describing essential physiological and metabolic traits contributing to life in this cold oligotrophic environment 42, 43.
Studies of groundwater systems are currently allowing significant insight into microbial activities in situ. Nitrite‐dependent anaerobic methane oxidation (n‐damo) is potentially an important sink for methane in freshwaters. Evidence of n‐damo activity was investigated in a coal tar contaminated aquifer 44. The metaproteome indicated the presence of the organism proposed to be responsible for methane oxidation Candidatus Methylomirabilis oxyfera, and the near complete n‐damo pathway was detected directly in the aquifer 44. In another contaminated aquifer, the metaproteome, obtained after protein stable isotope probing, revealed the presence of key bacterial populations and the degradation of contaminating polycyclic aromatic hydrocarbons 45. Importantly, in both these studies, the roles of particular bacteria with specific functions were detected to prove their activities directly in these contaminated field sites rather than in “artificial” laboratory experiments thereby underpinning the importance of in situ metaproteomic investigations.
6. Soil
Soil is among the most challenging microbial ecosystems to study 46, 47, not least for metaproteomics. This is due to several reasons: interference of soil components with the analyses (e.g. humic acids), seasonal variability, spatial complexity, and nestedness, as well as the presence of different macroorganisms including invertebrates and plants. Soil heterogeneity leads to high diversity even though the latter has been subject of considerable debate 48, 49 since Gans et al. 50 published an estimate of 8 × 106 different taxa per gram of soil. More recent estimates put the number at around 50 000 76. Nonetheless, given the diversity and overall complexity, the need for dedicated protein extraction methods has been highlighted by recent studies, in which protein yield and measurable proteomic diversity could be increased markedly 51, 52.
Despite the difficulties mentioned above, soil particles and forest soil water were among the first environmental samples studied using a metaproteomic approach 53. In the latter study, differences in contribution of distinct taxa to the metaproteome were associated with seasonal changes and the state and nature of plant cover on the forest groundwater proteome. In addition, several enzyme classes involved in the decomposition of plant material in soil particles were identified. Litter degradation has also been the focus of a more recent metaproteomic study, which provided deep insights into succession during litter decomposition and used advanced bioinformatic analyses to resolve taxa contributing the most active litter decomposing enzymes 54.
In addition to the decomposition of plant material, the soil microbiome is also closely associated with living plant roots and thereby contributes greatly to their productivity. Plant‐microbe interactions have been the subject of metaproteomic investigations. In particular, Bao et al. recently combined the analytical power of metaproteomics and spatial resolution of CARD‐FISH to elegantly link nitrogen fixation and methane oxidation to bacteria of the family Methylocystaceae, which inhabit vascular bundles and epidermal cells of rice roots 55.
Permafrost soil, the subject of a recent comprehensive study integrating metagenomic, metatranscriptomic, and metaproteomic data, is a unique habitat, which hosts a surprising diversity of psychrophilic microorganisms 56. This study identified proteins expected to be highly expressed in the permafrost microbiota, for example cold shock proteins, as well as more surprising functions including proteins linked to organismal motility. However, the unique preservative nature of this environment may limit the usual proteomic paradigm that presence of proteins indicates their activity. Such studies would therefore benefit from in situ stable isotope labeling approaches, which would allow the subsequent identification of labeled proteins and their linkage to community members with actual activity.
7. Human gastrointestinal microbiota
Human gastrointestinal microbiota arguably represents the best‐studied host‐associated microbiome. The human gastrointestinal microbiome has been mostly studied using fecal samples as these represent an easily accessible, noninvasive proxy. However, metaproteomic approaches have also been developed for studying the microbial mucosa‐lumen interface of different intestinal sites 57 and have been applied in a cohort study of inflammatory bowel disease 58, which, despite limited resolution of the metaproteome, demonstrated strong differences between inflamed and healthy mucosa linked to the resident microbiome.
The first in‐depth metaproteomic study of the human fecal microbiome was conducted by Verberkmoes et al. in 2008 59 and this study already allowed identification of up to 1340 nonredundant proteins per sample. This study also established that fecal metaproteomics are impacted extensively by host proteins with 30% of the measured spectra being matched to a human protein database. Therefore, depletion of host cells prior to measurements can dramatically increase the depth of coverage of the microbial proteome 60. However, these human proteome measurements can also be very informative. A comparison of metaproteomes of fecal samples from individuals with Crohn's disease and healthy individuals mirrored the previous findings from metagenomic studies including a reduced functional richness in the fecal microbiota of individuals with Crohn's disease 61, while differential abundances of proteins relating to carbohydrate degradation and human recognition of bacteria were also detected 62. In addition, human proteins involved in intestinal epithelial barrier function were found to vary significantly between individuals with Crohn's disease and healthy individuals.
The comparison between metagenomic and metaproteomic data has highlighted a congruency in the abundance of different taxa and the abundance of identified proteins from the same organismal groups, with a few notable exceptions such as the highly active, but lowly abundant Bifidobacteria 63. The same study described temporal stability of a core metaproteome similar to previous metagenomic observations. However, disturbance of such a stable state was observed in a multi‐omic time series study of the effect of antibiotic treatment 64. These studies demonstrate the ability of metaproteomics to resolve clinically relevant activities of gut‐associated microbiota, which along with other signature proteins from other environments (Table 1) may be exploited as biomarkers in the coming years.
8. Current challenges and future prospects
Given the complexities of metaproteomes, the vast dynamic ranges of protein abundances (driven by intra‐ and interpopulation abundance differences) and current analytical limitations, metaproteomic analyses face multiple challenges at the different stages of the analytical workflow (Fig. 1). Given the multitude of metaproteomic studies of different microbial consortia carried out so far, essential considerations for metaproteomic analyses according to our assessment include: (i) sample‐specific comprehensive cell lysis and protein extraction procedures, (ii) standardized subcellular proteome fractionation procedures (soluble, membrane, extracellular fractions), (iii) proteome fractionation based on physicochemical properties prior to LC‐MS/MS, (iv) quantification methods that do not require in situ metabolic labeling, (v) enhanced liquid chromatographic separation, (vi) use of mass spectrometers with fast scan speeds and high mass accuracies, (vii) mass spectral search databases based on high‐quality metagenomic and metatranscriptomic data, (viii) detection of posttranslational modifications, (ix) means of integrating the metaproteomic data with other meta‐omic data and their visualization, and (x) targeted validation of signature proteins (biomarkers) including those identified in earlier metaproteomic studies. Although specific solutions are already available for the highlighted challenges, their deployment has arguably been rather unmethodical and their systematic application will, in our opinion, allow the field to advance in the coming decade. More specifically, existing and emerging solutions include: (i) comprehensive cell lysis and protein extraction procedures, which can be applied to even the most challenging sample matrices, e.g. soil 51, 52, 56. (ii) Subcellular proteome fractionation procedures were developed early on 2 but should to be more routinely applied in metaproteomic studies on all samples. (iii) Proteome fractionation based on liquid IEF (off‐gel) has been used prior to 2D‐PAGE 5 or LC 65 and in both cases has led to a marked increase in numbers of proteins identified. Furthermore, 1D‐PAGE is now routinely applied by many groups prior to LC and does significantly improve metaproteome coverage. Therefore, combined fractionation procedures exploiting both molecular weight and charge prior to LC will allow much deeper metaproteome coverage. (iv) Given the challenges of in situ metabolic labeling, methods that allow direct quantitation of peptides without the need for incubation experiments involving labeled substrates are highly desirable. In this context and according to our assessment, an important recent development in proteomic technologies is SWATH‐MS 66. SWATH‐MS represents a data‐independent acquisition method, which combines the advantages of shotgun proteomics, i.e. high‐throughput, and selected reaction monitoring, i.e. high reproducibility and consistency. As both characteristics represent essential requirements for metaproteomics, we foresee that SWATH‐MS will find wide application in microbial community proteomics, not least enabling the immediate investigation of certain proteins of interest using a hypothesis‐driven approach. (v) New chip‐based approaches for multidimensional chromatographic peptide fractionation allow the identification of higher numbers of peptides, consume less sample, improve the lower limits of quantitation, and exhibit improved reproducibility 67. Due to their modularity, these procedures offer exciting opportunities for enhanced LC‐based metaproteomics. (vi) Given the need for precise peptide identification and sample complexity, high‐mass accuracy and fast scan‐speed mass spectrometers are already commonplace in metaproteomics. The development of mass spectrometers with enhanced characteristics will continue to benefit the field in the coming decade. (vii) The availability of high‐quality metagenomic and metatranscriptomic data from the same samples, e.g. 56, 68, 69, allows tailored search databases to be constructed, which in turn leads to marked improvement in the numbers of proteins that can be identified. As costs of metagenomic and metatranscriptomic analyses decrease, these will be routinely carried out in parallel to metaproteomic analyses. (viii) Advanced computational, e.g. 22, and MS, e.g. the Thermo Scientific Orbitrap Fusion Lumos instrument, methods already exist for the comprehensive detection of posttranslational modifications in metaproteomic data. We foresee that these will become more widely accessible with reductions in the cost of computation and instrumentation. (ix) As already highlighted in point VII above, we project that metagenomic and metatranscriptomic data will in future be routinely generated alongside metaproteomic analyses. Approaches for integrating the resulting meta‐omic data already exist and include population‐centric 68 and community‐wide 69 approaches. However, given the high‐dimensionality of the data, advanced data integration, analysis and visualization (e.g. 70, 71) methods will need to be further developed. (x) Protein signatures identified will require independent validation including using targeted MS analyses or classical immunoblots, and may be used as biomarkers for the different environmental settings in the future. Given that solutions already exist for the identified challenges and very few hurdles exist limiting their implementation, we postulate that metaproteomics will flourish in the coming decade.
Figure 1.
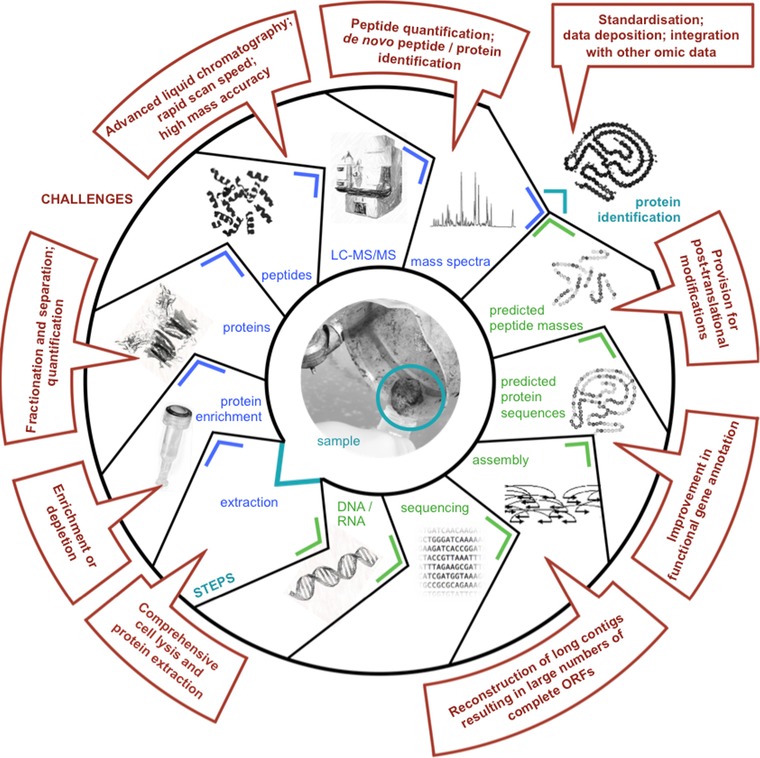
A typical metaproteomic analytical workflow with associated challenges. Given that metaproteomic analyses rely on comprehensive mass spectral search databases (ideally sample specific), specific challenges associated with concomitant metagenomic/metranscriptomic analyses have also been included. LC‐MS/MS: liquid chromatography coupled to tandem mass spectrometry; ORF: open reading frame.
Given the central role of the metaproteome in governing microbial community function, metaproteomics will be keystone to ecosystematic studies of microbial communities in the future. Integrated omics including metaproteomic analyses have already provided important fundamental ecological insight into microbial consortia including revelations that the divergence of microbial species is reflected in functional differentiation in situ 72, that microbial generalists fine‐tune their gene expression according to substrate availability 68, and that key functional traits are encoded by keystone species 69. However, for metaproteomics to fulfill its full potential in more conclusively informing our understanding of community function, great strides still need to be made in resolving the vast dynamic ranges of the different metaproteomes (Table 1) as well as in the routine detection of protein modifications, highlighting the need for the dedicated and deep multi‐omic characterization of different microbial communities. We foresee that these two aspects will become the major foci of the field in the coming decade.
Through the relatively recent advent of inexpensive high‐throughput sequencing, the application of metagenomics and metatranscriptomics is now commonplace. In comparison, metaproteomics remains somewhat of a niche discipline restricted to a few well‐established laboratories worldwide. In our opinion, metaproteomics will however gain more traction if the major challenges described above are addressed in the coming years. Apart from the highlighted challenges, we are also of the opinion that metaproteomics has to be more actively promoted by the community to occupy its rightful place among the other meta‐omics, especially in the context of future integrated omic analyses of microbial consortia. Finally, reduced instrumentation costs similar to what has been seen for DNA sequencing would also allow metaproteomics to find wider application. For this, more active collaborations between instrument manufacturers and academic partners with interests in metaproteomics will be fruitful.
The authors have declared no conflict of interest.
9 References
- 1. Armengaud, J. , Marie Hartmann, E. , Bland, C. , Proteogenomics for environmental microbiology. Proteomics 2013, 13, 2731–2742. [DOI] [PubMed] [Google Scholar]
- 2. Wilmes, P. , Bond, P. L. , The application of two‐dimensional polyacrylamide gel electrophoresis and downstream analyses to a mixed community of prokaryotic microorganisms. Environ. Microbiol. 2004, 6, 911–920. [DOI] [PubMed] [Google Scholar]
- 3. Ram, R. J. , Verberkmoes, N. C. , Thelen, M. P. , Tyson, G. W. et al., Community proteomics of a natural microbial biofilm. Science 2005, 308, 1915–1920. [PubMed] [Google Scholar]
- 4. Daims, H. , Taylor, M. W. , Wagner, M. , Wastewater treatment: a model system for microbial ecology. Trends Biotechnol. 2006, 24, 483–489. [DOI] [PubMed] [Google Scholar]
- 5. Wilmes, P. , Wexler, M. , Bond, P. L. , Metaproteomics provides functional insight into activated sludge wastewater treatment. PLoS ONE 2008, 3, e1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Martin, H. G. , Ivanova, N. , Kunin, V. , Warnecke, F. et al., Metagenomic analysis of two enhanced biological phosphorus removal (EBPR) sludge communities. Nat. Biotechnol. 2006, 24, 1263–1269. [DOI] [PubMed] [Google Scholar]
- 7. Wilmes, P. , Andersson, A. F. , Lefsrud, M. G. , Wexler, M. et al., Community proteogenomics highlights microbial strain‐variant protein expression within activated sludge performing enhanced biological phosphorus removal. ISME J. 2008, 2, 853–864. [DOI] [PubMed] [Google Scholar]
- 8. Wexler, M. , Richardson, D. J. , Bond, P. L. , Radiolabelled proteomics to determine differential functioning of Accumulibacter during the anaerobic and aerobic phases of a bioreactor operating for enhanced biological phosphorus removal. Environ. Microbiol. 2009, 11, 3029–3044. [DOI] [PubMed] [Google Scholar]
- 9. Lacerda, C. M. R. , Choe, L. H. , Reardon, K. F. , Metaproteomic analysis of a bacterial community response to cadmium exposure. J. Proteome Res. 2007, 6, 1145–1152. [DOI] [PubMed] [Google Scholar]
- 10. Collado, N. , Buttiglieri, G. , Kolvenbach, B. A. , Comas, J. et al., Exploring the potential of applying proteomics for tracking bisphenol A and nonylphenol degradation in activated sludge. Chemosphere 2013, 90, 2309–2314. [DOI] [PubMed] [Google Scholar]
- 11. Hansen, S. H. , Stensballe, A. , Nielsen, P. H. , Herbst, F.‐A. , Metaproteomics: evaluation of protein extraction from activated sludge. Proteomics 2014, 14, 2535–2539. [DOI] [PubMed] [Google Scholar]
- 12. Albertsen, M. , Stensballe, A. , Nielsen, K. L. , Nielsen, P. H. , Digging into the extracellular matrix of a complex microbial community using a combined metagenomic and metaproteomic approach. Water Sci. Technol. 2013, 67, 1650–1656. [DOI] [PubMed] [Google Scholar]
- 13. Park, C. , Novak, J. T. , Helm, R. F. , Ahn, Y.‐O. , Esen, A. , Evaluation of the extracellular proteins in full‐scale activated sludges. Water Res. 2008, 42, 3879–3889. [DOI] [PubMed] [Google Scholar]
- 14. Silva, A. F. , Carvalho, G. , Soares, R. , Coelho, A. V. , Barreto Crespo, M. T. , Step‐by‐step strategy for protein enrichment and proteome characterisation of extracellular polymeric substances in wastewater treatment systems. Appl. Microbiol. Biotechnol. 2012, 95, 767–776. [DOI] [PubMed] [Google Scholar]
- 15. Jeans, C. , Singer, S. W. , Chan, C. S. , Verberkmoes, N. C. et al., Cytochrome 572 is a conspicuous membrane protein with iron oxidation activity purified directly from a natural acidophilic microbial community. ISME J. 2008, 2, 542–550. [DOI] [PubMed] [Google Scholar]
- 16. Lo, I. , Denef, V. J. , Verberkmoes, N. C. , Shah, M. B. et al., Strain‐resolved community proteomics reveals recombining genomes of acidophilic bacteria. Nature 2007, 446, 537–541. [DOI] [PubMed] [Google Scholar]
- 17. Denef, V. J. , Kalnejais, L. H. , Mueller, R. S. , Wilmes, P. et al., Proteogenomic basis for ecological divergence of closely related bacteria in natural acidophilic microbial communities. Proc. Natl. Acad. Sci. USA 2010, 107, 2383–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mueller, R. S. , Denef, V. J. , Kalnejais, L. H. , Suttle, K. B. et al., Ecological distribution and population physiology defined by proteomics in a natural microbial community. Mol. Syst. Biol. 2010, 6, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Justice, N. B. , Pan, C. , Mueller, R. , Spaulding, S. E. et al., Heterotrophic Archaea Contribute to Carbon Cycling in Low‐pH, Suboxic Biofilm Communities. Appl. Environ. Microbiol. 2012, 78, 8321–8330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fischer, C. R. , Bowen, B. P. , Pan, C. , Northen, T. R. , Banfield, J. F. , Stable‐isotope probing reveals that hydrogen isotope fractionation in proteins and lipids in a microbial community are different and species‐specific. ACS Chem. Biol. 2013, 8, 1755–1763. [DOI] [PubMed] [Google Scholar]
- 21. Justice, N. B. , Li, Z. , Wang, Y. , Spaudling, S. E. et al., 15N‐ and 2H proteomic stable isotope probing links nitrogen flow to archaeal heterotrophic activity. Environ. Microbiol. 2014, 16, 3224–3237. [DOI] [PubMed] [Google Scholar]
- 22. Li, Z. , Wang, Y. , Yao, Q. , Justice, N. B. et al., Diverse and divergent protein post‐translational modifications in two growth stages of a natural microbial community. Nat. Commun. 2014, 5, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Belnap, C. P. , Pan, C. , Verberkmoes, N. C. , Power, M. E. et al., Cultivation and quantitative proteomic analyses of acidophilic microbial communities. ISME J. 2009, 4, 520–530. [DOI] [PubMed] [Google Scholar]
- 24. Mosier, A. C. , Li, Z. , Thomas, B. C. , Hettich, R. L. et al., Elevated temperature alters proteomic responses of individual organisms within a biofilm community. ISME J. 2014, 9, 180–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aliaga Goltsman, D. S. , Comolli, L. R. , Thomas, B. C. , Banfield, J.F. , Community transcriptomics reveals unexpected high microbial diversity in acidophilic biofilm communities. ISME J. 2015, 9, 1014–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kan, J. , Hanson, T. E. , Ginter, J. M. , Wang, K. , Chen, F. , Metaproteomic analysis of Chesapeake Bay microbial communities. Saline Systems 2005, 1, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dong, H.‐P. , Hong, Y.‐G. , Lu, S. , Xie, L.‐Y. , Metaproteomics reveals the major microbial players and their biogeochemical functions in a productive coastal system in the northern South China Sea. Environ. Microbiol. Rep. 2014, 6, 683–695. [DOI] [PubMed] [Google Scholar]
- 28. Georges, A. A. , El‐Swais, H. , Craig, S. E. , Li, W. K. W. , Walsh, D.A. , Metaproteomic analysis of a winter to spring succession in coastal northwest Atlantic Ocean microbial plankton. ISME J. 2014, 8, 1301–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sowell, S. M. , Abraham, P. E. , Shah, M. , Verberkmoes, N. C. et al., Environmental proteomics of microbial plankton in a highly productive coastal upwelling system. ISME J 2011, 5, 856–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Morris, R. M. , Nunn, B. L. , Frazar, C. , Goodlett, D. R. et al., Comparative metaproteomics reveals ocean‐scale shifts in microbial nutrient utilization and energy transduction. ISME J. 2010, 4, 673–685. [DOI] [PubMed] [Google Scholar]
- 31. Teeling, H. , Fuchs, B. M. , Becher, D. , Klockow, C. et al., Substrate‐Controlled Succession of Marine Bacterioplankton Populations Induced by a Phytoplankton Bloom. Science 2012, 336, 608–611. [DOI] [PubMed] [Google Scholar]
- 32. Hawley, A. K. , Brewer, H. M. , Norbeck, A. D. , Pasa‐Tolic, L. , Hallam, S. J. , Metaproteomics reveals differential modes of metabolic coupling among ubiquitous oxygen minimum zone microbes. Proc. Natl. Acad. Sci. USA 2014, 111, 11395–11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Leary, D. H. , Li, R. W. , Hamdan, L. J. , Hervey, W. J. et al., Integrated metagenomic and metaproteomic analyses of marine biofilm communities. Biofouling 2014, 30, 1211–1223. [DOI] [PubMed] [Google Scholar]
- 34. Kleiner, M. , Wentrup, C. , Lott, C. , Teeling, H. et al., Metaproteomics of a gutless marine worm and its symbiotic microbial community reveal unusual pathways for carbon and energy use. Proc. Natl. Acad. Sci. USA 2012, 109, E1173–E1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Markert, S. , Gardebrecht, A. , Felbeck, H. , Sievert, S. M. et al., Status quo in physiological proteomics of the uncultured Riftia pachyptila endosymbiont. Proteomics 2011, 11, 3106–3117. [DOI] [PubMed] [Google Scholar]
- 36. Urich, T. , Lanzén, A. , Stokke, R. , Pedersen, R. B. et al., Microbial community structure and functioning in marine sediments associated with diffuse hydrothermal venting assessed by integrated meta‐omics. Environ. Microbiol. 2014, 16, 2699–2710. [DOI] [PubMed] [Google Scholar]
- 37. Stokke, R. , Roalkvam, I. , Lanzén, A. , Haflidason, H. , Steen, I.H. , Integrated metagenomic and metaproteomic analyses of an ANME‐1‐dominated community in marine cold seep sediments. Environ. Microbiol. 2012, 14, 1333–1346. [DOI] [PubMed] [Google Scholar]
- 38. Williams, T. J. , Long, E. , Evans, F. , DeMaere, M. Z. et al., A metaproteomic assessment of winter and summer bacterioplankton from Antarctic Peninsula coastal surface waters. ISME J. 2012, 6, 1883–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pierre‐Alain, M. , Christophe, M. , Severine, S. , Houria, A. et al., Protein extraction and fingerprinting optimization of bacterial communities in natural environment. Microb. Ecol. 2007, 53, 426–434. [DOI] [PubMed] [Google Scholar]
- 40. Benndorf, D. , Balcke, G. U. , Harms, H. , von Bergen, M. , Functional metaproteome analysis of protein extracts from contaminated soil and groundwater. ISME J. 2007, 1, 224–234. [DOI] [PubMed] [Google Scholar]
- 41. Hanson, B. T. , Hewson, I. , Madsen, E. L. , Metaproteomic survey of six aquatic habitats: discovering the identities of microbial populations active in biogeochemical cycling. Microb. Ecol. 2014, 67, 520–539. [DOI] [PubMed] [Google Scholar]
- 42. Lauro, F. M. , DeMaere, M. Z. , Yau, S. , Brown, M. V. et al., An integrative study of a meromictic lake ecosystem in Antarctica. ISME J. 2011, 5, 879–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ng, C. , DeMaere, M. Z. , Williams, T. J. , Lauro, F. M. et al., Metaproteogenomic analysis of a dominant green sulfur bacterium from Ace Lake, Antarctica. ISME J. 2010, 4, 1002–1019. [DOI] [PubMed] [Google Scholar]
- 44. Hanson, B. T. , Madsen, E. L. , In situ expression of nitrite‐dependent anaerobic methane oxidation proteins by Candidatus Methylomirabilis oxyfera co‐occurring with expressed anammox proteins in a contaminated aquifer. Environ. Microbiol. Rep. 2015, 7, 252–264. [DOI] [PubMed] [Google Scholar]
- 45. Herbst, F.‐A. , Bahr, A. , Duarte, M. , Pieper, D. H. et al., Elucidation of in situ polycyclic aromatic hydrocarbon degradation by functional metaproteomics (protein‐SIP). Proteomics 2013, 13, 2910–2920. [DOI] [PubMed] [Google Scholar]
- 46. Mocali, S. , Benedetti, A. , Exploring research frontiers in microbiology: the challenge of metagenomics in soil microbiology. Res. Microbiol. 2010, 161, 497–505. [DOI] [PubMed] [Google Scholar]
- 47. Lombard, N. , Prestat, E. , van Elsas, J. D. , Simonet, P. , Soil‐specific limitations for access and analysis of soil microbial communities by metagenomics. FEMS Microbiol. Ecol. 2011, 78, 31–49. [DOI] [PubMed] [Google Scholar]
- 48. Bunge, J. , Comment on “computational improvements reveal great bacterial diversity and high metal toxicity in soil.” Science 2006, 313, 918c–918c. [DOI] [PubMed] [Google Scholar]
- 49. Volkov, I. , Comment on “computational improvements reveal great bacterial diversity and high metal toxicity in soil.” Science 2006, 313, 918a. [DOI] [PubMed] [Google Scholar]
- 50. Gans, J. , Wolinsky, M. , Dunbar, J. , Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science 2005, 309, 1387–1390. [DOI] [PubMed] [Google Scholar]
- 51. Bastida, F. , Hernández, T. , García, C. , Metaproteomics of soils from semiarid environment: functional and phylogenetic information obtained with different protein extraction methods. J. Proteomics 2014, 101, 31–42. [DOI] [PubMed] [Google Scholar]
- 52. Nicora, C. D. , Anderson, B. J. , Callister, S. J. , Norbeck, A. D. et al., Amino acid treatment enhances protein recovery from sediment and soils for metaproteomic studies. Proteomics 2013, 13, 2776–2785. [DOI] [PubMed] [Google Scholar]
- 53. Schulze, W. X. , Gleixner, G. , Kaiser, K. , Guggenberger, G. et al., A proteomic fingerprint of dissolved organic carbon and of soil particles. Oecologia 2004, 142, 335–343. [DOI] [PubMed] [Google Scholar]
- 54. Schneider, T. , Keiblinger, K. M. , Schmid, E. , Sterflinger‐Gleixner, K. et al., Who is who in litter decomposition? Metaproteomics reveals major microbial players and their biogeochemical functions 2012, 6, 1749–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bao, Z. , Okubo, T. , Kubota, K. , Kasahara, Y. et al., Metaproteomic identification of diazotrophic methanotrophs and their localization in root tissues of field‐grown rice plants. Appl. Environ. Microbiol. 2014, 80, 5043–5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hultman, J. , Waldrop, M. P. , Mackelprang, R. , David, M. M. et al., Multi‐omics of permafrost, active layer and thermokarst bog soil microbiomes. Nature 2015, 521, 208–212. [DOI] [PubMed] [Google Scholar]
- 57. Li, X. , LeBlanc, J. , Truong, A. , Vuthoori, R. et al., A Metaproteomic Approach to Study Human‐Microbial Ecosystems at the Mucosal Luminal Interface. PLoS ONE 2011, 6, e26542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Presley, L. L. , Ye, J. , Li, X. , LeBlanc, J. et al., Host–microbe relationships in inflammatory bowel disease detected by bacterial and metaproteomic analysis of the mucosal–luminal interface. Inflamm. Bowel Dis. 2012, 18, 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Verberkmoes, N. C. , Russell, A. L. , Shah, M. , Godzik, A. et al., Shotgun metaproteomics of the human distal gut microbiota. ISME J. 2008, 3, 179–189. [DOI] [PubMed] [Google Scholar]
- 60. Xiong, W. , Giannone, R. J. , Morowitz, M. J. , Banfield, J. F. , Hettich, R. L. , Development of an enhanced metaproteomic approach for deepening the microbiome characterization of the human infant gut. J. Proteome Res. 2015, 14, 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Manichanh, C. , Reduced diversity of faecal microbiota in Crohn's disease revealed by a metagenomic approach. Gut 2006, 55, 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Erickson, A. R. , Cantarel, B. L. , Lamendella, R. , Darzi, Y. et al., Integrated Metagenomics/Metaproteomics Reveals Human Host‐Microbiota Signatures of Crohn's Disease. PLoS ONE 2012, 7, e49138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kolmeder, C. A. , de Been, M. , Nikkilä, J. , Ritamo, I. et al., Comparative metaproteomics and diversity analysis of human intestinal microbiota testifies for its temporal stability and expression of core functions. PLoS ONE 2012, 7, e29913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pérez‐Cobas, A. E. , Gosalbes, M. J. , Friedrichs, A. , Knecht, H. et al., Gut microbiota disturbance during antibiotic therapy: a multi‐omic approach. Gut 2013, 62, 1591–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kohrs, F. , Heyer, R. , Magnussen, A. , Benndorf, D. et al., Sample prefractionation with liquid isoelectric focusing enables in depth microbial metaproteome analysis of mesophilic and thermophilic biogas plants. Anaerobe 2014, 29, 59–67. [DOI] [PubMed] [Google Scholar]
- 66. Gillet, L.C. , Navarro, P. , Tate, S. , Roest, H. et al., Targeted data extraction of the MS/MS spectra generated by data‐independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 2012, O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Krisp, C. , Yang, H. , van Soest, R. , Molloy, M.P. , Online peptide fractionation using a multiphasic microfluidic liquid chromatography chip improves reproducibility and detection limits for quantitation in discovery and targeted proteomics. Mol. Cell. Proteomics 2015, 14, 1708–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Muller, E. E. L. , Pinel, N. , Laczny, C. C. , Hoopmann, M. R. et al., Community‐integrated omics links dominance of a microbial generalist to fine‐tuned resource usage. Nat. Commun. 2014, 5, 5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Roume, H. , Heintz‐Buschart, A. , Muller, E. E. L. , May, P. et al., Comparative integrated omics: identification of key functionalities in microbial community‐wide metabolic networks. npj Biofilms Microbiomes 2015, 1, 15007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Muth, T. , Behne, A. , Heyer, R. , Kohrs, F. et al., The MetaProteomeAnalyzer: a powerful open‐source software suite for metaproteomics data analysis and interpretation. J. Proteome Res. 2015, 14, 1557–1565. [DOI] [PubMed] [Google Scholar]
- 71. Penzlin, A. , Lindner, M. S. , Doellinger, J. , Dabrowski, P. W. et al., Pipasic: similarity and expression correction for strain‐level identification and quantification in metaproteomics. Bioinformatics 2014, 30, i149–i156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wilmes, P. , Bowen, B. P. , Thomas, B. C. , Mueller, R. S. et al., Metabolome‐proteome differentiation coupled to microbial divergence. MBio 2010, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang, T. , Shao, M.‐F. , Ye, L. , 454 Pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. ISME J. 2011, 6, 1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Curtis, T. P. , Sloan, W. T. , Scannell, J. W. , Estimating prokaryotic diversity and its limits. Proc. Natl. Acad. Sci. USA 2002, 99, 10494–10499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Palmer, M. A. , Covich, A. P. , Lake, S. , Biro, P. et al., Linkages between aquatic sediment biota and life above sediments as potential drivers of biodiversity and ecological processes. Bioscience 2000, 50, 1062–1075. [Google Scholar]
- 76. Roesch, L. F. W. , Fulthorpe, R. R. , Riva, A. , Casella, G. et al., Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 2007, 1, 283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Huse, S. M. , Ye, Y. , Zhou, Y. , Fodor, A. A. , A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS ONE 2012, 7, e34242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Rudney, J. D. , Xie, H. , Rhodus, N. L. , Ondrey, F. G. , Griffin, T. J. , A metaproteomic analysis of the human salivary microbiota by three‐dimensional peptide fractionation and tandem mass spectrometry. Mol. Oral Microbiol. 2010, 25, 38–49. [DOI] [PMC free article] [PubMed] [Google Scholar]