

Abstract
Although the BRAF V600E base substitution is an approved target for the BRAF inhibitors in melanoma, BRAF gene fusions have not been investigated as anticancer drug targets. In our study, a wide variety of tumors underwent comprehensive genomic profiling for hundreds of known cancer genes using the FoundationOne™ or FoundationOne Heme™ comprehensive genomic profiling assays. BRAF fusions involving the intact in‐frame BRAF kinase domain were observed in 55 (0.3%) of 20,573 tumors, across 12 distinct tumor types, including 20 novel BRAF fusions. These comprised 29 unique 5′ fusion partners, of which 31% (9) were known and 69% (20) were novel. BRAF fusions included 3% (14/531) of melanomas; 2% (15/701) of gliomas; 1.0% (3/294) of thyroid cancers; 0.3% (3/1,062) pancreatic carcinomas; 0.2% (8/4,013) nonsmall‐cell lung cancers and 0.2% (4/2,154) of colorectal cancers, and were enriched in pilocytic (30%) vs. nonpilocytic gliomas (1%; p < 0.0001), Spitzoid (75%) vs. nonSpitzoid melanomas (1%; p = 0.0001), acinar (67%) vs. nonacinar pancreatic cancers (<1%; p < 0.0001) and papillary (3%) vs. nonpapillary thyroid cancers (0%; p < 0.03). Clinical responses to trametinib and sorafenib are presented. In conclusion, BRAF fusions are rare driver alterations in a wide variety of malignant neoplasms, but enriched in Spitzoid melanoma, pilocytic astrocytomas, pancreatic acinar and papillary thyroid cancers.
Keywords: cancer, solid tumors, BRAF fusions, pilocytic astrocytoma, pancreatic acinar carcinoma, Sptizoid melanoma, comprehensive genomic profiling, NGS, targeted therapy
Short abstract
What's new?
New results may help target a rare genetic alteration that promotes cancer. Activation of the BRAF gene is already known to spur tumor growth, and usually that activation results from a single amino acid substitution. BRAF‐inhibiting treatments, then, target that mutation. However, in some cases, BRAF gets revved up by a gene fusion. In our study, the authors tested 20,000 tumors and identified 55 BRAF gene fusions in 12 different tumor types. They found the gene fusions occurred more frequently in certain histologic subtypes, information which will help guide treatment strategies for patients with these tumor subtypes.
BRAF encodes a RAF kinase, which signal downstream of RAS and activate the MAPK pathway, and has emerged as a major oncogenic driver and a potential therapy target in a wide variety of solid tumors and hematological malignancies.1, 2, 3, 4 BRAF signaling is critical for cell division and differentiation and activating BRAF mutations result in uncontrolled growth and tumorigenesis.2, 3, 4 Over 90% of activating BRAF mutations in cancer cells occur within the kinase domain at amino acid V600, most commonly resulting in V600E, which is an approved target for the inhibitors dabrafenib and vemurafenib in the treatment of metastatic malignant melanoma.5, 6, 7 Melanomas with other BRAF mutations have been shown to respond to BRAF inhibitors, although generally to a lesser extent than tumors harboring V600E.5, 6, 7 Some nonmelanoma malignancies with activating BRAF alterations such as V600E have responded to BRAF‐targeted therapy,8, 9, 10, 11 whereas others have not.12
BRAF gene fusions represent a different mechanism of BRAF activation and have been described in several solid tumor types.13 However, reports describing the use of anti‐BRAF therapies for tumors with BRAF fusion alterations have been limited to date. Moreover, first‐generation BRAF inhibitors such as sorafenib may not only be ineffective in BRAF fusion‐driven malignancies, tumor progression may actually be promoted by the use of these agents; thus, drug sensitivity of BRAF fusions remains unclear and controversial.14 In the following study, genomic profiling of >20,500 malignancies identified BRAF gene fusions is in a panorama of tumor types, revealing enrichment in certain histologic subtypes and providing additional examples of response to therapies targeting activated BRAF fusions.
Material and Methods
A database of 20,573 consecutive clinical samples of primarily relapsed and refractory solid tumors and hematologic malignancies was evaluated retrospectively to search for BRAF gene fusions. Local site permissions to use clinical samples were obtained for our study. Comprehensive genomic profiling (CGP)was performed on all formalin fixed paraffin embedded tissues using a hybrid capture‐based next generation sequencing platform (FoundationOneTM) on the Illumina HiSeq2500 instrument.15 Extracted DNA was adaptor‐ligated and capture was performed for all coding exons of 182 cancer‐related genes and 37 introns of 14 genes frequently rearranged in cancer (earlier version of the test) or all coding exons from 236 cancer‐related and 47 introns of 19 genes frequently rearranged in cancer (current version of the test). Captured libraries were sequenced to a median exon coverage depth of >600×, and resultant sequences were analyzed for base substitutions, insertions, deletions, copy number alterations (focal amplifications and homozygous deletions) and gene fusions, as previously described.15 The sequence analysis methods and validation of the comprehensive genomic profiling platform used in our study included extensive comparisons to orthogonal methodologies.14 Base substitution detection is performed using a Bayesian methodology, which allows detection of novel somatic mutations at low mutant allele frequency (MAF) and increased sensitivity for mutations at hotspot sites through the incorporation of tissue‐specific prior expectations.15 Reads with mapping quality <25 are discarded, as are base calls with quality ≤2. Final calls are made at MAF ≥5% (MAF ≥1% at hotspots) after filtering for strand bias (Fisher's test, p < 1e–6), read location bias (KS test, p <1e–6), and presence in two or more normal controls. To detect indels, de novo local assembly in each targeted exon is performed using the de‐Bruijn approach.16, 17 After read pairs are collected and decomposed, the statistical support for competing haplotypes is evaluated and candidate indels are aligned against the reference genome. Filtering of indel candidates is carried out as described for base substitutions. Gene amplifications and homozygous deletions are detected by comparing complete chromosomal copy number maps to reference process‐matched normal control samples. Finally, gene fusions and rearrangements are detected by analysis of chimeric read pairs as follows.15 Genomic rearrangements are identified by analyzing chimeric read pairs (read pairs for which reads map to separate chromosomes, or at a distance of over 10 kbp). Pairs are clustered by genomic coordinate of the pairs, and clusters containing at least five chimeric pairs (three for known fusions) are identified as rearrangement candidates. Filtering of candidates is performed by mapping quality (MQ >30) and distribution of alignment positions (standard deviation >10). Rearrangements are annotated for predicted function (e.g. creation of fusion gene).
Clinically relevant alterations were defined as those that could be targeted using anticancer therapies currently on the market for any tumor type with known primary site or alterations required for entry in a mechanism‐driven registered clinical trial. Local site permissions to utilize clinical samples and approval by the Albany Medical College IRB to analyze and report patient data were obtained for our study. The frequencies of BRAF fusions in the various tumor types were evaluated for significance using the Fisher's exact test.
Results
BRAF fusions containing the intact BRAF kinase domain were observed in 55 (0.3%) of the 20,573 tumors (Table 1, Supporting Information Table 1 and Fig. 1), including 20 novel BRAF fusions. These comprised 29 unique 5′ fusion partners, of which 31% (9) were known and 69% (20) were novel. The median age of the 55 patients whose tumors harbored BRAF fusions was 56 years with a range of 1–83 years, with 29 (53%) of those patients female and 26 (47%) male. The primary tumor was sequenced in 33 (60%) of the cases and a metastasis biopsy was sequenced in 22 (40%). Of the 430 distinct tumor types evaluated in our study (Supporting Information Table 2), BRAF fusions were distributed across 12 (3%) tumors including melanoma (3%; 14/531); glioma (2%; 15/701); thyroid cancers (1.0%; 3/294); pancreatic carcinoma (0.3%; 3/1,062); nonsmall‐cell lung cancer (0.2%; 8/4,013) and colorectal cancers (0.2%; 4/2,154). Additional tumor types for which multiple samples were found with BRAF fusions included breast carcinomas and unknown primary carcinomas. Tumor types featuring only a single case included in data analysis with BRAF fusion included esophageal, prostatic carcinoma, head and neck carcinoma and soft tissue sarcoma. Four additional BRAF fusions were more recently identified each in a single tumor type as follows: rectal adenocarcinoma, uterine endometrial adenocarcinoma, ovarian serous carcinoma and pleural malignant mesothelioma. These four cases, numbers 56 through 59 are included in Table 1 and Supporting Information Table 2, but are not included in the statistical frequency and data analysis due to the fact that these fusions have not been fully characterized.
Table 1.
Fifty‐five cases of solid tumors with BRAF gene fusions
| Case number | Tumor group | Histologic diagnosis | Gender | Age | Sample source | Fusion |
|---|---|---|---|---|---|---|
| 1 | Breast carcinoma | Breast invasive ductal carcinoma (IDC) | F | 62 | Metastasis | KIAA1549‐BRAF |
| 5 | Breast carcinoma | Breast carcinoma (NOS) | F | 61 | Metastasis | KIAA1549‐BRAF |
| 4 | Colorectal carcinoma | Colon adenocarcinoma (CRC) | M | 56 | Primary | MKRN1‐BRAF |
| 2 | Colorectal carcinoma | Colon adenocarcinoma (CRC) | F | 71 | Metastasis | TRIM24‐BRAF |
| 6 | Colorectal carcinoma | Colon adenocarcinoma (CRC) | F | 52 | Metastasis | TRIM24‐BRAF |
| 3 | Colorectal carcinoma | Colon adenocarcinoma (CRC) | F | 59 | Primary | AGAP3‐BRAF |
| 7 | Esophageal carcinoma | Esophagus adenocarcinoma | M | 61 | Primary | ZC3HAV1‐BRAF |
| 18 | Glioma | Brain desmoplastic infantile ganglioglioma | F | 5 | Primary | KIAA1549‐BRAF |
| 12 | Glioma | Brain pilocytic astrocytoma | M | 17 | Primary | KIAA1549‐BRAF |
| 19 | Glioma | Brain pleomorphic xanthoastrocytoma | F | 64 | Primary | KIAA1549‐BRAF |
| 20 | Glioma | Spinal cord low‐grade glioma (NOS) | M | 4 | Primary | KIAA1549‐BRAF |
| 14 | Glioma | Brain pilocytic astrocytoma | M | 31 | Primary | AKAP9‐BRAF |
| 8 | Glioma | Brain pleomorphic xanthoastrocytoma | M | 2 | Primary | CCDC6‐BRAF |
| 17 | Glioma | Brain pilocytic astrocytoma | F | 2 | Primary | KIAA1549‐BRAF |
| 21 | Glioma | Spinal cord low‐grade glioma (NOS) | M | 8 | Primary | KIAA1549‐BRAF |
| 11 | Glioma | Brain pilocytic astrocytoma | M | 6 | Primary | KIAA1549‐BRAF |
| 15 | Glioma | Brain pilocytic astrocytoma | M | 8 | Primary | KIAA1549‐BRAF |
| 13 | Glioma | Brain pleomorphic xanthoastrocytoma | M | 21 | Primary | AGK‐BRAF |
| 9 | Glioma | Not pilocytic. Anaplastic oligodendroglioma | M | 20 | Primary | AGK‐BRAF |
| 16 | Glioma | Brain pilocytic astrocytoma | F | 2 | Primary | KIAA1549‐BRAF |
| 43 | Glioma | Brain pilocytic astrocytoma | M | 1 | Primary | KIAA1549‐BRAF |
| 10 | Glioma | Not pilocytic. Anaplastic ganglioglioma | F | 47 | Primary | KIAA1549‐BRAF |
| 22 | Head & Neck Carcinoma | Head and neck neuroendocrine carcinoma | F | 53 | Primary | MKRN1‐BRAF |
| 23 | Lung Carcinoma | Lung adenocarcinoma | F | 60 | Metastasis | EPS15‐BRAF |
| 29 | Lung Carcinoma | Lung nonsmall‐cell lung carcinoma (NOS) | M | 69 | Primary | NUP214‐BRAF |
| 26 | Lung Carcinoma | Lung adenocarcinoma | F | 69 | Primary | ARMC10‐BRAF |
| 28 | Lung Carcinoma | Lung adenocarcinoma | M | 70 | Primary | BTF3L4‐BRAF |
| 27 | Lung Carcinoma | Lung adenocarcinoma | F | 83 | Primary | AGK‐BRAF |
| 24 | Lung Carcinoma | Lung adenocarcinoma | M | 68 | Metastasis | GHR‐BRAF |
| 25 | Lung Carcinoma | Lung adenocarcinoma | F | 66 | Primary | ZC3HAV1‐BRAF |
| 30 | Lung Carcinoma | Lung nonsmall‐cell lung carcinoma (NOS) | M | 73 | Primary | TRIM24‐BRAF |
| 35 | Melanoma | Cutaneous melanoma Spitzoid | F | 62 | Primary | TRIM24‐BRAF |
| 39 | Melanoma | Mucosal melanoma non‐Spitzoid | F | 56 | Metastasis | ZNF767‐BRAF |
| 49 | Melanoma | Cutaneous melanoma non‐Spitzoid | M | 63 | Metastasis | CCDC91‐BRAF |
| 34 | Melanoma | Cutaneous melanoma Spitzoid | F | 25 | Primary | DYNC1I2‐BRAF |
| 32 | Melanoma | Cutaneous melanoma Spitzoid | F | 60 | Metastasis | AKAP9‐BRAF |
| 38 | Melanoma | Cutaneous melanoma Spitzoid | F | 46 | Metastasis | ZKSCAN1‐BRAF |
| 51 | Melanoma | Unknown primary melanoma | M | N/A | Metastasis | GTF2I‐BRAF |
| 42 | Melanoma | Cutaneous melanoma non‐Sptizoid | M | 54 | Metastasis | AGAP3‐BRAF |
| 37 | Melanoma | Cutaneous melanoma Spitzoid | F | 44 | Metastasis | AGK‐BRAF |
| 41 | Melanoma | Cutaneous melanoma Spitzoid | M | 27 | Metastasis | MZT1‐BRAF |
| 31 | Melanoma | Cutaneous melanoma Spitzoid | F | 52 | Metastasis | AGK‐BRAF |
| 33 | Melanoma | Cutaneous melanoma non‐Spitzoid | F | 1 | Primary | RAD18‐BRAF |
| 40 | Melanoma | Cutaneous melanoma Spitzoid | F | 60 | Metastasis | CUX1‐BRAF |
| 36 | Melanoma | Cutaneous melanoma Spitzoid | F | 30 | Metastasis | SLC12A7‐BRAF |
| 47 | Pancreatic carcinoma | Pancreas ductal adenocarcinoma | M | 63 | Primary | MYRIP‐BRAF |
| 46 | Pancreatic carcinoma | Pancreas acinar cell carcinoma | F | 75 | Primary | SND1‐BRAF |
| 45 | Pancreatic carcinoma | Pancreas acinar cell carcinoma | M | 67 | Metastasis | SND1‐BRAF |
| 48 | Prostatic carcinoma | Prostate acinar adenocarcinoma | M | 57 | Metastasis | NUB1‐BRAF |
| 50 | Sarcoma | Malignant solid fibrous tumor | F | 56 | Primary | KIAA1549‐BRAF |
| 53 | Thyroid carcinoma | Thyroid papillary carcinoma | M | 61 | Primary | KLHL7‐BRAF |
| 54 | Thyroid carcinoma | Thyroid papillary carcinoma | M | 67 | Primary | TANK‐BRAF |
| 52 | Thyroid carcinoma | Thyroid papillary carcinoma | F | 64 | Metastasis | RBMS3‐BRAF |
| 44 | Unknown primary carcinoma | Unknown primary, adenocarcinoma | F | N/A | Metastasis | STRN3‐BRAF |
| 55 | Unknown primary carcinoma | Unknown primary, carcinoma (NOS) | M | 65 | Metastasis | SND1‐BRAF |
| S1 | Pleura mesothelioma | Pleura mesothelioma | F | 48 | Primary | STK35‐BRAF |
| S2 | Rectum adenocarcinoma | Rectum adenocarcinoma | M | 56 | Metastasis | ETFA‐BRAF |
| S3 | Uterus endometrial carcinoma | Uterus endometrial adenocarcinoma (NOS) | F | 74 | Metastasis | SVOPL‐BRAF |
| S4 | Ovary serous carcinoma | Ovary serous carcinoma | F | 62 | Metastasis | JHDM1D‐BRAF |
Cases S1–S4 are supplemental, have not been fully characterized and were not included in the data analysis.
Melanomas
The 14 melanomas harbored BRAF fusions and 9 (64%) featured an epithelioid and spindle cell histology characteristic of the so‐called Spitzoid melanoma (Fig. 2 a). For the 531 melanomas evaluated, the enrichment of BRAF fusions in Spitzoid melanomas (9/12, 75%) compared to non‐Sptizoid tumors (5/519, 1%) was highly significant (p = 0.0001). BRAF base substitution alterations were identified in 191/531 (36%) melanomas analyzed.
Figure 2.
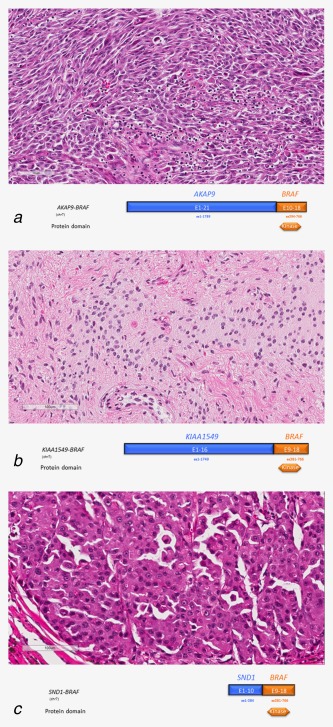
BRAF fusions in a variety of solid tumors. (a) (Case 32) Spitzoid metastatic malignant melanoma in a 60‐year‐old Caucasian female. Note the diffuse distribution of so‐called spindle or elongate cells and mixed with scattered round epithelioid cells with abundant pink cytoplasm and tumor‐infiltrating lymphocytes. Lymph node and cutaneous metastases present at the time of sequencing. The tumor features a fusion of AKAP9 (exons 1–21)–BRAF (exons 10–18) (hematoxylin and eosin 100×). (b) (Case 15) A pilocytic astrocytoma partially resected from the parietal lobe in an 8‐year‐old male with a KIAA1549 (exons 1–16)–BRAF (exons 9–18) fusion. Image shows a well‐differentiated low‐grade astrocytoma with widely separated oval to elongate tumor cell nuclei associated with tangles of eosinophilic fibrils (rosenthal fibers) in the lower right (hematoxylin and eosin 100×). (c) (Case 45) Pulmonary metastasis from a primary pancreatic acinar carcinoma in a 67‐year‐old Caucasian man. Sequencing revealed a rearrangement consistent with an inversion on chromosome 7, juxtaposing the 5′ region of SND1 to the complete kinase domain of BRAF, resulting in the generation of a predicted in‐frame SND1 (exons 1–10)–BRAF (exons 9–18) fusion protein (Hematoxylin and eosin X 100). In an expanded study of 44 pancreatic acinar carcinomas, we identified recurrent rearrangements involving BRAF and RAF1 (CRAF) in 23% of the tumors. The image shows solid nests of polygonal neoplastic cells with granular eosinophilic cytoplasm (hematoxylin and eosin 100×).
Gliomas
Of the 15 gliomas with BRAF fusions detected in our study, 7 (47%) were pilocytic astrocytomas (Fig. 2 b). Of the 701 gliomas analyzed, the enrichment of BRAF fusion in pilocytic astrocytomas (7/23; 30%) compared to the nonpilocytic gliomas (8/678; 1%) was highly significant (p < 0.0001). In addition, 3 (38%) of the 8 nonpilocytic gliomas harboring BRAF fusions featured high grade anaplastic astrocytoma histology with large histocytic‐like giant cells in the pattern of the pleomorphic xanthoastrocytoma. Of the entire set of gliomas evaluated, 28 (4%) featured base substitution alterations in BRAF.
Nonsmall‐cell lung carcinomas
BRAF fusions were identified in <1% of NSCLC samples. In contrast, 270/4,013 (7%) NSCLC harbored BRAF base substitution alterations. All NSCLC with BRAF fusions were adenocarcinomas or NSCLC with adenocarcinoma features. BRAF fusions were not seen in squamous or small cell lung cancers.
Colorectal carcinomas
Less than 1% of the 2,154 CRC tumors evaluated harbored BRAF fusions, in contrast to the 284 (13%) of the CRC that featured BRAF base substitution alterations. There were no distinctive morphologic features in the CRC tumors with BRAF fusions.
Pancreatic carcinomas
Of 1,062 pancreatic cancers, 3 featured BRAF fusions; this subset comprised 2 (67%) acinar carcinomas (Fig. 2 c) and 1 (33%) ductal adenocarcinoma. The cohort of pancreatic tumors analyzed featured only three acinar carcinomas, and the enrichment of BRAF fusions in acinar carcinomas (2/3; 67%) compared to nonacinar carcinomas (1; <0.1%) was significant (p < 0.0001).
Thyroid carcinomas
The three thyroid carcinomas with BRAF fusions identified in our study were papillary thyroid carcinomas (3/94; 3%), with no fusions identified in nonpapillary thyroid carcinomas (0/200; 0%) (p = 0.03). In contrast, BRAF base substitutions were found in 82 (28%) of the total thyroid tumors with 65 (79%) of these mutations identified in papillary thyroid carcinomas and 17 (21%) in nonpapillary thyroid tumors. Information pertaining to radiation exposure in the thyroid cancer patients was not available for our study.
Figure 1 summarizes the exon composition of the BRAF fusions identified in our study, all 55 of which preserved an intact BRAF kinase domain, encoded by exons 11–18, and are considered activating. Fusions between KIAA1549 and BRAF were the most frequent BRAF fusions identified in the study and involved 14 (25%) of the 55 BRAF fusion positive tumors. Eleven (20%) of the KIAA1549‐BRAF fusions were identified in brain tumors. The AGK‐BRAF, TRIM24‐BRAF and SND1‐BRAF fusions were the next most frequent, identified in 5, 4 and 3 tumors, respectively. A total of 20 novel fusion partners not previously reported in public databases (COSMIC and TCGA) or the published literature (PubMed) were identified across 20 samples (36%). The remaining 25 fusions have been previously reported (Table 1).18, 19, 20, 21, 22, 23, 24, 25, 26 All 55 BRAF fusions were in‐frame with breakpoints on the BRAF hotspot introns 7, 8, 9 and 10. One fusion MKRN1‐BRAF (Case 22) was found in a head and neck carcinoma with breakpoint on MKRN1 Exon 4 and BRAF intron 9, which is predicted as in‐frame with MKRN1 exons 1–3, partial exon4 and BRAF exons 11‐18. MKRN1‐BRAF was identified in another colorectal carcinoma with a known structure of MKRN1 (exons 1–4)–BRAF (exons 11–18).24 Most fusions retained BRAF exons 9–18 (24/55, 44%).
Figure 1.
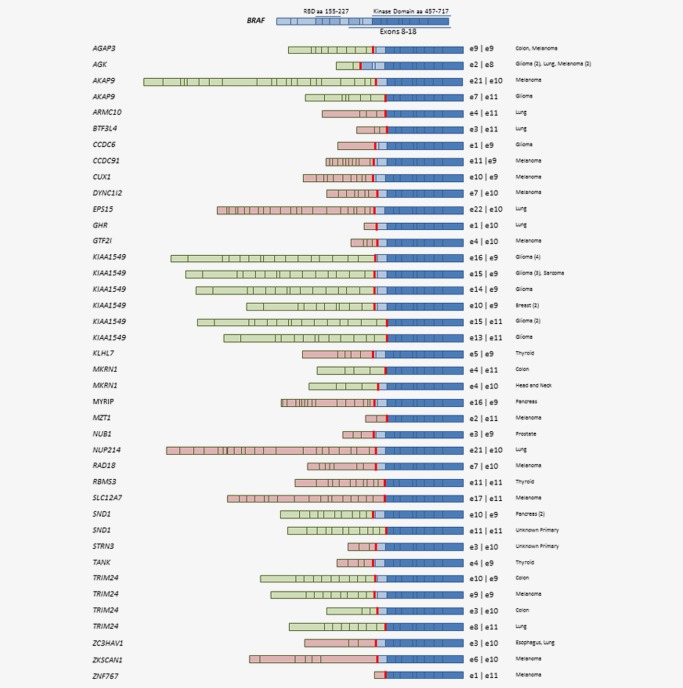
Structure of 55 BRAF fusions discovered from 20,573 solid tumors detected by comprehensive genomic profiling. Novel fusions were in pink, and known fusions were in green.
In the 55 tumors harboring BRAF fusions, 207 additional genomic alterations involving the targeted genes of the sequencing panel were identified in genes such as CDKN2A/B (29%), TP53 (22%), PTEN (11%), PIK3CA (9%), PBRM1, APC and EGFR (each at 7%). The long tail of additional alterations found in fewer than three tumors included clinically relevant alterations affecting, MET, PDGFRA, RET and TSC2 (Fig. 3). In 54/55 (98%), tumors the BRAF fusion was the only BRAF alteration identified, although a single case of metastatic non‐Spitzoid melanoma in a 54‐year‐old man (Case 42) featured both a BRAF V600E base substitution and an AGAP3‐BRAF fusion.
Figure 3.
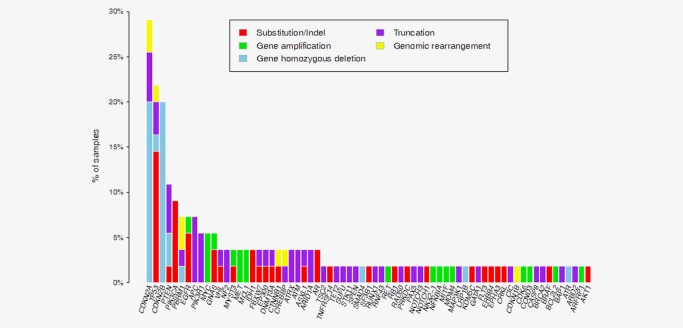
Distribution plot of additional genomic alterations identified in the targeted genes of the sequencing panel in 55 cases of BRAF fusion associated solid tumors.
Clinical outcomes are available for only two patients included in our study. A Spitzoid melanoma from a 46‐year‐old Caucasian woman that harbored a ZKSCAN1‐BRAF fusion responded to treatment with the MEK inhibitor trametinib given at full dose (2 mg/day orally) (Case 38) (Fig. 4). Subcutaneous tumor nodules exhibited overt clinical responses within 14 days of therapy, and her dominant bulky right lung metastases showed significant response by Day 45 such that she subsequently underwent robotic‐assisted lobectomy. This previously unresectable tumor was removed with clean surgical margins, and without any of the 16 recovered lymph nodes involved with melanoma. Similarly, in a recent study, significant clinical activity was demonstrated when trametinib was used in the treatment of a patient with metastatic melanoma harboring a BRAF fusion.27
Figure 4.
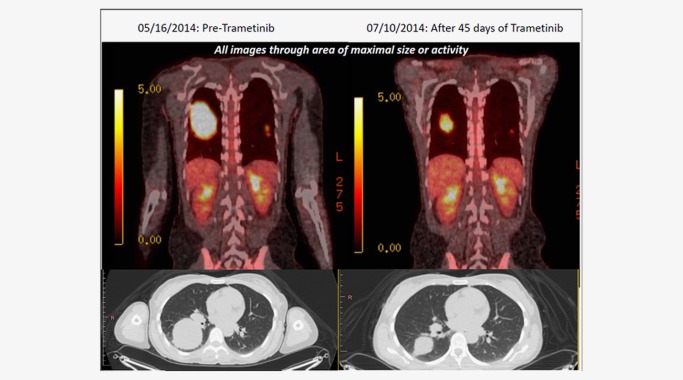
Fused PET/CT imaging results of trametinib therapy in a metastatic Spitzoid melanoma (Case 38) from a 46‐year‐old Caucasian woman that featured a ZKSCAN1‐BRAF fusion (ZKSCAN1 exons 1–5–BRAF exons 10–18) and responded to the MEK inhibitor trametinib. Subcutaneous tumor nodules exhibited overt clinical responses within 14 days of therapy, and her dominant bulky right lung metastases showed significant response by Day 45 such that she subsequently underwent robotic‐assisted lobectomy. The patient is currently alive with stable disease at 6 months post‐thoracic surgery.
A malignant spindle cell tumor of the chest wall treated as a soft tissue sarcoma featured a KIAA1549‐BRAF fusion (Fig. 5) and responded to treatment with the pan‐kinase inhibitor sorafenib in combination with bevacizumab and temsirolimus (Case 50).
Figure 5.
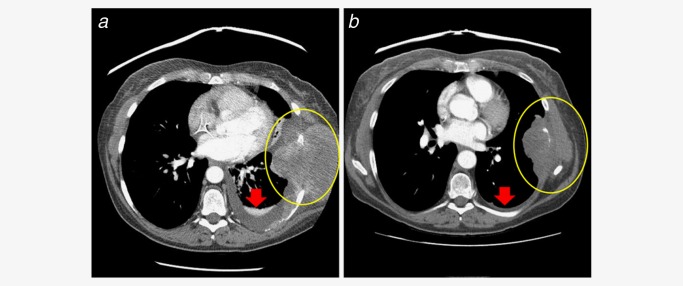
A malignant spindle cell tumor (Case 50) of the chest wall treated as a soft tissue sarcoma that featured a KIAA1549‐BRAF fusion (KIAA1549 exons 1–15–BRAF exons 9–18) showing pre‐ and post‐treatment CT scan images featuring tumor response to treatment with bevacizumab, temsirolimus and sorafenib.39 [Permission to re‐publish this figure provided by the publisher]. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Discussion
The above data represent the most diverse series of BRAF gene fusions described to date. Although BRAF fusions are infrequent in advanced solid tumors, both the present data and the published literature demonstrate enrichment in certain histologic subsets including pilocytic astrocytoma,14, 21, 28, 29, 30 Spitzoid melanoma,18, 20, 31, 32 pancreatic acinar carcinoma33 and papillary thyroid cancer.2 Other datasets including the COSMIC database accessed in December 2014 for our study report even fewer examples of tumors driven by BRAF fusions which are restricted to fewer tumor types.17 However, it should be noted that the public databases such as COSMIC likely include tumors that were evaluated for BRAF base substitutions only and may not have included a sequencing assay capable of detecting gene fusions. Thus, such discrepancies may be explained by the limitations of analyses not optimized or designed to identify gene fusions. For example, of 4,299 gliomas studied for BRAF sequence in COSMIC, 268 (6%) featured an alteration with a 106 (2%) incidence of BRAF fusions limited to pilocytic astrocytomas. Similarly, in the melanomas listed in COSMIC, 16,403 tumors included BRAF sequencing and 7,110 (43%) had BRAF alterations, but no BRAF fusions were listed in the entire group or in the 53 Spitzoid melanomas in the database. Of 2,533 pancreatic cancers sequenced for BRAF at COSMIC, 27 (1%) featured BRAF alterations with 0 BRAF fusions. Interestingly, in an expanded study of 44 pancreatic acinar carcinomas, we identified recurrent rearrangements involving BRAF and RAF1 (CRAF) in 23% of the tumors.33 Of the 46,463 thyroid tumors sequenced for BRAF at COSMIC, 19,297 (42%) had BRAF alterations with three (<0.1%) BRAF fusions identified all restricted to the papillary carcinoma subtype.
Of interest is the fact that BRAF fusions are similar to other kinase fusions in occurring in a mutually exclusive pattern with other activating mutations in the MAP kinase signaling pathway. Only one (2%) BRAF V600E base substitution was identified in the 55 cases of BRAF fusions which occurred in a case of cutaneous melanoma (Case 42). No KRAS mutations were identified in the 55 cases. In contrast, there were the alterations in GNAS (3 cases; 5%), IDH1 (2 cases; 4%) and EGFR (4 cases; 7%) in the 55 BRAF fusion‐positive tumors.
The greater frequency and wider tumor‐type distribution of BRAF fusions presented in the current study in comparison with COSMIC database is most likely the result of differing techniques used in the tumor analysis. The COSMIC database includes tumors sequenced by nonhybrid capture‐based technologies either not optimized to identify or unable to detect gene fusions. The current assay utilized a DNA bait set only.15 A small series of BRAF rearrangements was also uncovered in this cohort of >20,000 clinical tumor samples, but these alterations could not be completely characterized using DNA sequencing alone. It is possible that, with RNA sequencing, these rearrangements could be more precisely characterized as BRAF fusions.
Figure 1 shows the exon composition of the BRAF fusions identified in this cohort, which includes both a series of previously described fusions and a set of novel fusions described here for the first time. Although direct in vitro assays were not conducted as part of our study, based on the published studies for the known BRAF fusions and using published models for confirming activation and prediction of the protein amino acid sequences, we expect that the novel fusions identified to be similarly oncogenic. Several BRAF fusions, including many identified here, have been previously characterized as activating and oncogenic.18, 19, 20, 21, 22 Modeling and protein domain analysis shows that these fusions, as well as the 20 novel fusions described in Figure 1, all maintain the kinase domain of BRAF, suggesting a universal mechanism of BRAF activation, irrespective of the 5′ fusion partner. Previous studies have shown that loss of the autoinhibitory region upstream of the BRAF kinase domain, which is predicted for all of the fusions described here, leads to activation of BRAF signaling.34 Although the adverse prognostic significance of BRAF base substitution, such as V600E, is widely described for a variety of solid tumors,35, 36, 37 given their rarity, the significance of BRAF fusions for clinical outcome is unknown.
Evidence supporting the treatment of solid tumors harboring BRAF fusions with therapies targeting this kinase has started to emerge.18, 38, 39 As shown in Figures 4 and 5, tumor responses to kinase inhibitors in combination with nontargeted cytotoxic agents indicate that RAF kinases or downstream signaling pathways can be targeted when activated by BRAF fusion. Sorafenib, a multikinase inhibitor that inhibits RAF, has had limited efficacy as an anticancer drug in patients with BRAF activating point mutations.40 In Figure 5, sorafenib was used to treat the soft tissue sarcoma with a KIAA1549‐BRAF fusion, but the MTOR inhibitor temsirolimus and the antiangiogenic antibody therapeutic bevacizumab were also given to the patient, and these latter therapies may well have provided the primary tumor response shown in the tumor images.39 In a study of low‐grade astrocytomas, the impact of sorafenib therapy was mixed with both deleterious effects and stabilized disease seen.14 Studies on melanoma, in contrast, have shown evidence of significant benefit from sorafenib treatment.41 Thus, the sensitivity of BRAF fusion‐driven malignancies to sorafenib remains unclear and controversial. In addition, the major tumor response in the patient with the Spitzoid metastatic melanoma featuring a ZKSCAN1‐BRAF fusion shown in Figure 4 responded to the MEK inhibitor trametinib rather than to a RAF kinase inhibitor. Unfortunately, the extremely low frequency of BRAF fusions in solid tumors precludes a prospective randomized clinical trial evaluating the efficacy of treatment with RAF kinase and MEK inhibitors. However, the expanded clinical use of next‐generation DNA sequencing and comprehensive genomic profiling in oncology practice may provide data from Phase I trials and published case reports that will validate the use of agents targeting BRAF fusions and bring significant clinical improvement for patients with disease driven by this rare but distinctive genomic alteration.
Supporting information
Supporting Information
Supporting Information
COI Disclosure: J.S.R., K.W., J.C., L.G., A.J., J.C., R.Y., D.L., S.M.A., J.A.E., J.‐A.V., S.R., V.A.M. and P.J.S. all disclose that they have employment and equity positions in Foundation Medicine, Inc.
References
- 1. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–54. [DOI] [PubMed] [Google Scholar]
- 2. El‐Osta H, Falchook G, Tsimberidou A, et al. BRAF mutations in advanced cancers: clinical characteristics and outcomes. PLoS One 2011;6:e25806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pakneshan S, Salajegheh A, Smith RA, et al. Clinicopathological relevance of BRAF mutations in human cancer. Pathology 2013;45:346–56. [DOI] [PubMed] [Google Scholar]
- 4. Vultur A, Villanueva J, Herlyn M. Targeting BRAF in advanced melanoma: a first step toward manageable disease. Clin Cancer Res 2011;17:1658–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cantwell‐Dorris ER, O'Leary JJ, Sheils OM. BRAF(V600E): implications for carcinogenesis and molecular therapy. Mol Cancer Ther 2011;10:385–94. [DOI] [PubMed] [Google Scholar]
- 6. Holderfield M, Deuker MM, McCormick F, et al. Targeting RAF kinases for cancer therapy: BRAF‐mutated melanoma and beyond. Nat Rev Cancer 2014;14:455–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Montagut C, Settleman J. Targeting the RAF‐MEK‐ERK pathway in cancer therapy. Cancer Lett 2009;283:125–34. [DOI] [PubMed] [Google Scholar]
- 8. Dadu R, Shah K, Busaidy NL, et al. Efficacy and tolerability of vemurafenib in patients with BRAF(V600E)‐positive papillary thyroid cancer: MD Anderson Cancer Center Off Label Experience. J Clin Endocrinol Metab 2015;100:E77–E81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haroche J, Cohen‐Aubart F, Emile J‐F, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim‐Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood 2013;121:1495–500. [DOI] [PubMed] [Google Scholar]
- 10. Haroche J, Cohen‐Aubart F, Emile J‐F, et al. Reproducible and sustained efficacy of targeted therapy with vemurafenib in patients with BRAF(V600E)‐mutated Erdheim‐Chester disease. J Clin Oncol 2015;33:411–U52. [DOI] [PubMed] [Google Scholar]
- 11. Pettirossi V, Santi A, Imperi E, et al. BRAF inhibitors reverse the unique molecular signature and phenotype of hairy cell leukemia and exert potent antileukemic activity. Blood 2015;125:1207–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Corcoran RB, Ebi H, Turke AB, et al. EGFR‐mediated reactivation of MAPK signaling contributes to insensitivity of BRAF‐mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov 2012;2:227–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Palanisamy N, Ateeq B, Kalyana‐Sundaram S, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med 2010;16:793–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Karajannis MA, Legault G, Fisher MJ, et al. Phase II study of sorafenib in children with recurrent or progressive low‐grade astrocytomas. Neuro Oncol 2014;16:1408–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31:1023+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Compeau PEC, Pevzner PA, Tesler G. How to apply de Bruijn graphs to genome assembly. Nat Biotechnol 2011;29:987–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Forbes SA, Bindal N, Bamford S, et al. COSMIC: mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res 2011;39:D945–D50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Botton T, Yeh I, Nelson T, et al. Recurrent BRAF kinase fusions in melanocytic tumors offer an opportunity for targeted therapy. Pigment Cell Melanoma Res 2013;26:845–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ciampi R, Knauf JA, Kerler R, et al. Oncogenic AKAP9‐BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. J Clin Invest 2005;115:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hutchinson KE, Lipson D, Stephens PJ, et al. BRAF fusions define a distinct molecular subset of melanomas with potential sensitivity to MEK inhibition. Clin Cancer Res 2013;19:6696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jones DTW, Kocialkowski S, Liu L, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 2008;68:8673–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee NV, Lira ME, Pavlicek A, et al. A novel SND1‐BRAF fusion confers resistance to c‐Met inhibitor PF‐04217903 in GTL16 cells though MAPK activation. PLoS One 2012;7:e39653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stransky N, Cerami E, Schalm S, et al. The landscape of kinase fusions in cancer. Nat Commun 2014;5:4846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jones DT, Hutter B, Jäger N, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 2013;45:927–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim HS, Jang KW, Jung M, et al. Oncogenic BRAF fusion induces MAPK‐pathway activation targeted by MEK inhibitor and phosphatidylinositol 3‐kinase inhibitor combination treatment in mucosal melanoma AACR abstract. 2015;75(15 Suppl):Abstract 3942. [Google Scholar]
- 26. Yeh I, Botton T, Talevich E, et al. Activating MET kinase rearrangements in melanoma and Spitz tumours. Nat Commun 2015;6:7174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Menzies AM, Yeh I, Botton T, et al. Clinical activity of the MEK inhibitor trametinib in metastatic melanoma containing BRAF kinase fusion. Pigment Cell Melanoma Res 2015;28:607–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cohen AL, Colman H. Glioma biology and molecular markers. Cancer Treat Res 2015;163:15–30. [DOI] [PubMed] [Google Scholar]
- 29. Jones DTW, Kocialkowski S, Liu L, et al. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene 2009;28:2119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sadighi Z, Slopis J. Pilocytic astrocytoma: a disease with evolving molecular heterogeneity. J Child Neurol 2013;28:625–32. [DOI] [PubMed] [Google Scholar]
- 31. Goel VK, Lazar AJF, Warneke CL, et al. Examination of mutations in BRAF, NRAS, and PTEN in primary cutaneous melanoma. J Invest Dermatol 2006;126:154–60. [DOI] [PubMed] [Google Scholar]
- 32. Wiesner T, He J, Yelensky R, et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun 2014;5:3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chmielecki J, Hutchinson KE, Frampton GM, et al. Comprehensive genomic profiling of pancreatic acinar cell carcinomas identifies recurrent RAF fusions and frequent inactivation of DNA repair genes. Cancer Discov 2014;4:1398–405. [DOI] [PubMed] [Google Scholar]
- 34. Tran NH, Wu XC, Frost JA. BRaf and Raf‐1 are regulated by distinct autoregulatory mechanisms. J Biol Chem 2005;280:16244–53. [DOI] [PubMed] [Google Scholar]
- 35. Baitei EY, Zou M, Al‐Mohanna F, et al. Aberrant BRAF splicing as an alternative mechanism for oncogenic B‐Raf activation in thyroid carcinoma. J Pathol 2009;217:707–15. [DOI] [PubMed] [Google Scholar]
- 36. Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 2011;29:2046–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yokota T, Ura T, Shibata N, Takahari D, Shitara K, Nomura M, Kondo C, Mizota A, Utsunomiya S, Muro K, Yatabe Y. BRAF mutation is a powerful prognostic factor in advanced and recurrent colorectal cancer. Br J Cancer 2011;104:856–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Passeron T, Lacour J‐P, Allegra M, et al. Signalling and chemosensitivity assays in melanoma: is mutated status a prerequisite for targeted therapy? Exp Dermatol 2011;20:1030–2. [DOI] [PubMed] [Google Scholar]
- 39. Subbiah V, Westin SN, Wang K, et al. Targeted therapy by combined inhibition of the RAF and mTOR kinases in malignant spindle cell neoplasm harboring the KIAA1549‐BRAF fusion protein. J Hematol Oncol 2014;7:8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wilhelm S, Carter C, Lynch M, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov 2006;5:835–44. [DOI] [PubMed] [Google Scholar]
- 41. Passeron T, Lacour JP, Allegra M, et al. Signalling and chemosensitivity assays in melanoma: is mutated status a prerequisite for targeted therapy? Exp Dermatol. 2011;20:1030–2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information