

Abstract
Sleep is critical to health and functionality, and several studies have investigated the inherited component of insomnia and other sleep disorders using genome‐wide association studies (GWAS). However, genome‐wide studies focused on sleep duration are less common. Here, we used data from participants in the Coriell Personalized Medicine Collaborative (CPMC) (n = 4,401) to examine putative associations between self‐reported sleep duration, demographic and lifestyle variables, and genome‐wide single nucleotide polymorphism (SNP) data to better understand genetic contributions to variation in sleep duration. We employed stepwise ordered logistic regression to select our model and retained the following predictive variables: age, gender, weight, physical activity, physical activity at work, smoking status, alcohol consumption, ethnicity, and ancestry (as measured by principal components analysis) in our association testing. Several of our strongest candidate genes were previously identified in GWAS related to sleep duration (TSHZ2, ABCC9, FBXO15) and narcolepsy (NFATC2, SALL4). In addition, we have identified novel candidate genes for involvement in sleep duration including SORCS1 and ELOVL2. Our results demonstrate that the self‐reported data collected through the CPMC are robust, and our genome‐wide association analysis has identified novel candidate genes involved in sleep duration. More generally, this study contributes to a better understanding of the complexity of human sleep. © 2015 The Authors. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics Published by Wiley Periodicals, Inc.
Keywords: genomic, genetic, self‐reported, ordered logistic regression
Abbreviations
- AIC
akaike information criterion
- CLIA
Clinical Laboratory Improvement Amendments
- CPMC
Coriell Personalized Medicine Collaborative
- DNA
deoxyribonucleic acid
- GWAS
genome‐wide association studies
- kb
kilobase
- PCA
principle components analysis
- PC
principal component
- PTSD
post traumatic stress disorder
- SNP
single nucleotide polymorphism
- QC
quality control
INTRODUCTION
Sleep is a complex and critical biological process that is impacted by both genetic and non‐genetic factors in humans. Inadequate sleep can lead to several health issues such as impaired immune function [Aldabal and Bahammam, 2011], increased risk for type II diabetes and obesity [Knutson et al., 2007], and cognitive impairment [Van Dongen et al., 2003; Durmer and Dinges, 2005]. Furthermore, sleep deprivation is associated with common psychiatric conditions such as anxiety and depression [van Mill et al., 2010]. While it is unclear to what extent sleep deprivation may be contributing to these conditions as opposed to resulting from them [van Mill et al., 2010], one study of military personnel has shown that individuals reporting symptoms of pre‐deployment insomnia or short sleep (<6 hr of sleep a night) are more likely to suffer from new‐onset post‐deployment post‐traumatic stress disorder (PTSD) Gehrman et al., 2013]. Thus, lifestyle changes and medical interventions that improve sleep quantity may lead to improved physical and emotional health.
Here, we have interrogated self‐reported sleep data, demographic and lifestyle data, as well as genome‐wide single nucleotide polymorphism (SNP) data collected through the Coriell Personalized Medicine Collaborative (CPMC) to identify genetic variants that contribute to variation in sleep duration. The CPMC is a prospective study designed to evaluate the utility of genomics in clinical decision‐making and health management [Keller et al., 2010]. Participants self‐report information related to family history, demographics, and lifestyle. They then receive personalized reports that incorporate their genetic and non‐genetic risk factors [Stack et al., 2011]. We believe that a better understanding of both genetic and non‐genetic factors involved in sleep duration will improve our ability to identify individuals that will most benefit from lifestyle changes and/or medical management to improve sleep quantity.
To date, several studies have identified non‐genetic and genetic risk factors for inadequate sleep duration and insomnia. One of the more recent studies included more than 100,000 individuals and identified age, ethnicity, smoking status, alcohol consumption, education, socio‐economic status, marital status, weight, and activity level among other non‐genetic variables as associated with sleep duration [Krueger and Friedman, 2009]. Several candidate genes for involvement in sleep duration, a sub‐set of which are involved in related biological mechanisms and have supporting functional data [Allebrandt et al., 2013; Byrne et al., 2013], have also been identified in previous genome‐wide association studies (GWAS) [Gottlieb et al., 2014, 2007; Ollila et al., 2014]; however, due to limitations in sample size, many of the reported candidate variants have neither reached genome‐wide significance nor been replicated in independent analyses. Results from our current analyses lend further support to several previously identified candidate genes involved in sleep duration: ABCC9 [Allebrandt et al., 2013; Ollila et al., 2014], TSHZ2 [Gottlieb et al., 2007], and FBXO15 [Byrne et al., 2013]. Moreover, we have identified several novel sleep duration candidate genes, including SORCS1 and ELOVL2.
MATERIALS AND METHODS
Samples
The CPMC is a prospective study comprised of several cohorts included in the current study [Keller et al., 2010; Stack et al., 2011]: a CPMC community cohort recruited from the general population (n = 2,686), a cancer (breast and prostate) cohort recruited through oncologists at Fox Chase Cancer Center (n = 74), a chronic disease (congestive heart failure and hypertension) cohort recruited through primary care physicians or cardiologists at Ohio State University Medical Center (n = 191), a community cohort recruited through Ohio State University (n = 188), and an Air Force Medical Service cohort recruited through the United States Air Force (n = 1,262). All participants are adults (at least 18 years old) that have given written informed consent to enroll in the study. No participants were excluded based on comorbidities including any health conditions related to heart disease, stroke, or sleep apnea. In total, information from 4,401 participants was included in the current study. The Coriell Institute Institutional Review Board (IRB) has reviewed and approved protocols for each of the abovementioned cohorts. In addition, the Institutional Review Boards of Fox Chase Cancer Center, Virtua Health System, Ohio State University Medical Center, and the United States Air Force have approved their respective cohort‐specific protocols.
Genotyping
Each participant has provided a saliva sample to the study from which DNA was extracted using the Oragene method (DNA Genotek, Inc., Ottawa, Ontario, Canada). Coriell's in‐house Clinical Laboratory Improvement Amendments (CLIA) certified Genotyping and Microarray Center [Keller et al., 2010; Stack et al., 2011] used the Affymetrix 6.0 GeneChip to genotype 909,622 SNPs. In total, 901,083 SNPs passed our research quality control (QC) filters (no more than 10% missing data for any given marker) and were retained for further analyses. All individual samples with genetic data (n = 3,948) had at least 97% complete SNP data and were retained for downstream analyses.
Non‐Genetic Data Collection
CPMC participants use a secure web‐based portal [Keller et al., 2010; Stack et al., 2011] to provide information related to medical history, family history, lifestyle, and demographics, including the information used in the current study (average amount of sleep per night, gender, age, weight, physical activity, smoking status, alcohol intake, and ethnicity).
The physical activity question includes the following options: none, recent, occasional, want to start, or regularly for at least 6 months. However, anyone who responded “want to start” was re‐coded as “none” for the current analysis. The amount of physical activity at work question includes the following options: sedentary, standing, physical, heavy, or unemployed. However, anyone who responded as unemployed was re‐coded as “sedentary” for the current analysis. Smoking was coded as currently smoking or not currently smoking. Alcohol use was coded as consumed in the past month or not consumed in the past month (Table I).
Table I.
Participant Characteristics
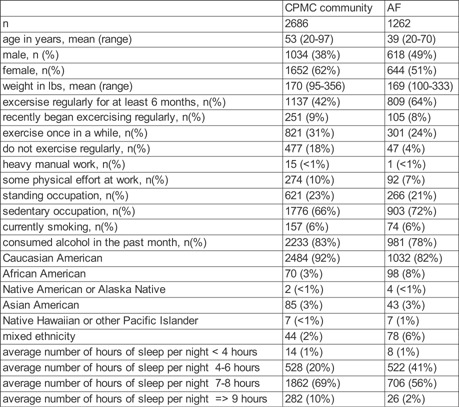
In addition, since the inception of the project in 2007, the participant questionnaire was updated such that the sleep question changed from “indicate the average number of hours of sleep you get a night” (less than 4 hr, 4–6 hr, 6–8 hr, 8 or more hours) to “During the past 30 days, indicate the average number of hours of sleep you get a night” (less than 4 hr, 4–6 hr, 7–8 hr, more than 8 hr). The entire Air Force cohort (n = 1262) and 214 (8%) of the CPMC cohort (n = 2686) answered the later version of the question. We considered the defined categories as equivalent for the current study.
Principle Components Analysis
Given that our cohorts include individuals with diverse genetic backgrounds, we used principle components analysis (PCA) to correct for any potential population structure in our statistical modeling (described further below). We used PLINK [Purcell et al., 2007] to generate a pruned set of relatively independent genome‐wide SNPs (R2 <0.2), and we used a custom R script (available upon request) and the svd function [Team, 2014] to calculate the eigenvalues and eigenvectors of the covariance matrix of normalized genotype data. We visualized principal components (PCs) 1–20, but by PC7, the distribution of individuals appeared to be all noise, so we retained PCs 1–6 for downstream analyses (Fig. S1a–g).
Statistical Modeling
The sleep duration variable is collected as an ordered category that we have imposed over an assumed underlying latent variable (sleep duration) with a continuous distribution. We, therefore, employed ordinal logistic regression with the polr function in the MASS library [Ripley, 2002] in R [Team, 2014] to test for associations between sleep duration and genetic and non‐genetic variables.
Since over‐parameterizing regression models can lead to a better fit simply due to the number of parameters, we conducted a step‐wise analysis of all non‐genetic factors and calculated akaike information criterion (AIC) for each model using the polr function in the MASS R package [Ripley, 2002]. We retained the model with the lowest AIC value. For the initial analysis of all participants (from all five cohorts, n = 4401), we used the following model: sleep hours ∼ age + gender + weight + physical activity + physical activity at work + smoking status + alcohol intake + ethnicity + cohort. After identifying participant cohort as a significant variable contributing to sleep duration (P = 2.6 × 10−56, Wilcoxon rank sum test, also see Table II), we designed a two phase genome‐wide analysis in which only the CPMC community cohort (n = 2,152) was used in phase I, and only the Air Force cohort (n = 1,262) was used in phase II. This study design, therefore, reduced the risk that we would identify false positives due to differential environmental or lifestyle factors related to either cohort.
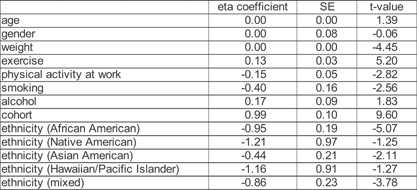
Table II.
Regression Results for Demographic and Lifestyle Variables
Taken together, we included genetic data from 3,414 total participants. For the phase I analysis, we used the following model in the CPMC community cohort for all genome‐wide SNPs: sleep hours ∼ age + gender + weight + physical activity + physical activity at work + smoking status + alcohol intake + ethnicity + PC1 + PC2 + PC3 + PC4 + PC5 + PC6 + SNP genotype. Given the modest sample size available for the current study, we prioritized retaining individuals and not variants for the genome‐wide association testing, and retained 169,252 SNPs with complete genotyping data in phase I. We then tested only the most significant SNPs (n = 173) in the phase II Air Force cohort with the same model. In addition, we went back to the top candidate regions and performed association tests of all of the genetic variants with at least 90% complete data within 500 kb of a given candidate variant in the CPMC cohort. SNP genotypes were coded as 0/1/2, where 0 corresponds to two copies of the reference allele, 1 corresponds to one copy of each allele, and 2 corresponds to two copies of the non‐reference allele. Finally, we imputed missing genotypes using Beagle Version 4.0 [Browning and Browning, 2007] with the following parameters: niterations = 10 and nsamples = 4. We excluded monomorphic SNPs, singleton SNPs, and SNPs with allelic R2 < 0.7. We retained 870,428 SNPs with imputed data for an exploratory genome‐wide association analysis in the CPMC community cohort (n = 2,152).
Assigning Statistical Significance
We used ordered logistic regression to maximize statistical power for the genome‐wide analysis of the community cohort and used the polr function in R to implement the model. The output of polr includes the eta coefficient, the standard error, and the affiliated t‐value (the coefficient divided by the standard error). The eta coefficients are the linear predictors of the explanatory variables, which do not follow a standard t‐distribution. Therefore, we were not able to analytically assign P‐values to our t‐values. Given our sample size for the genome‐wide analysis of the community cohort (n = 2,152), we instead chose an absolute t‐value cutoff of 10. This cutoff roughly corresponds to the 0.1% of the empirical tails of the t‐value distribution (estimated from 169,252 SNPs with no missing genotyping data). Assuming a normal distribution of t‐values, we estimate that an absolute t‐value cutoff of 10 approximately corresponds to a P‐value of 10−6. We note, however, that our t‐values are asymptotically distributed, and therefore this P‐value estimate is only provided as a very rough estimate.
Variants meeting the absolute t‐value cutoff of 10 (173 SNPs) were then tested in the Air Force cohort. From this set, 37 variants had an absolute t‐value of 2 or higher in the Air Force cohort (n = 1,262), which roughly corresponds to the top 1% empirical tail of the absolute t‐value distribution. We calculated bootstrap P‐values with 1,000 bootstraps for each of the 37 variants that met the above criteria in both cohorts.
Pathway Enrichment Analysis
We used the GREAT resource [McLean et al., 2010] to test for molecular function, biological process, and pathway enrichment in our set of 37 candidate loci. In particular, we included GO molecular functions, GO biological processes, PANTHER and MSigDB, and chose the hypergeometric gene enrichment test. We limited our enrichment testing to categories containing at least two candidate loci for sleep duration.
RESULTS
Non‐Genetic Factors
Consistent with previously reported analyses [Krueger and Friedman, 2009], we identified several non‐genetic factors that are associated with the average amount of sleep reported by CPMC participants. For example, reported non‐smokers get significantly more sleep than reported smokers (W = 552,877, P = 0.0032; Wilcoxon rank sum test; Table SI), and women get significantly less sleep than men (W = 2,430,874, P = 0.0485, Wilcoxon rank sum test; Table SI). We also discovered that cohort membership has a significant association with reported sleep (X2 = 261.86, P < 2.20 × 10−16, Kruskall–Wallis rank sum test; Table SI). Furthermore, this result was driven by two of the cohorts. In particular, individuals recruited into the Air Force cohort report significantly less sleep than individuals recruited into the CPMC community cohort (W = 1,251,280, P = 2.5666 × 10−56; Wilcoxon rank sum test; Fig. S2). We additionally modeled the combined set of non‐genetic factors using ordered logistic regression (Table II).
Genetic Candidates
Given the striking impact of cohort membership, we designed a two‐stage analysis in which we tested a set of genome‐wide SNPs in the larger CPMC community cohort (n = 2,152), and tested the subset of most associated variants (absolute(t‐value) >10) in the Air Force cohort. The 173 SNPs (Table SII) that met the phase I threshold have t‐values that roughly correspond to the most extreme 0.1% of the empirical distribution. Table III displays the subset of 37 variants that met our filter (absolute(t‐value) >2) in phase II, which roughly corresponds to the most extreme 1% of the empirical distribution; however, none of the tested genetic variants reached genome‐wide significance after correcting for multiple testing. We note that for a subset of these candidate variants, the direction of association (positive vs. negative eta value) is not consistent between the CPMC and Air Force cohorts. We tested for pathway enrichment using the 2 kb region surrounding each of the 37 variants listed in Table III and did not find any significant enrichment after correction for multiple testing (Tables SIII, SIV, and SV). However, we did find suggestive associations with two GO molecular functions (GO:0008188, neuropeptide receptor activity, uncorrected P = 0.00038; GO:0042923, neuropeptide binding, uncorrected P= 0.00049, also see Table SIV) and one GO biological process (GO:0007218, neuropeptide signaling pathway, P = 0.000052, also see Table SV) related to neuropeptides, which may be of general relevance to sleep duration [Steiger and Holsboer, 1997; Prospero‐Garcia and Mendez‐Diaz, 2004].
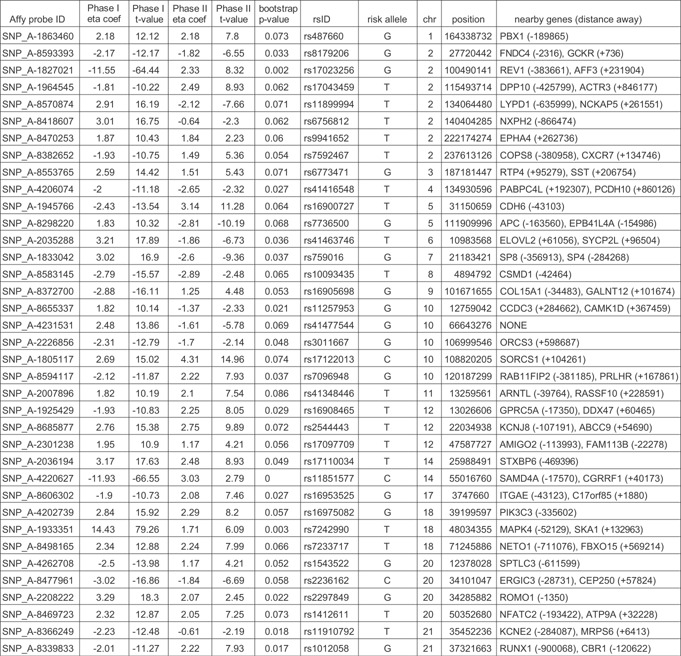
Table III.
Candidate Genetic Variants
Identification of Previous Candidate Genes
While we were not able to replicate any previously identified genetic variants, Table III includes several variants present in or near previously implicated candidate genes for involvement in sleep. Rs2544443, an intronic SNP located within the ATP‐binding cassette sub‐family C member 9 (ABCC9) gene, is one of the 37 variants that passed our filters in both phases of the analysis (Fig. 1). The ATP binding cassette transporter pathway was previously found to be enriched in a pathway analysis for sleep duration [Ollila et al., 2014], and ABCC9 has previously been implicated in a meta‐analysis of GWAS of sleep duration [Allebrandt et al., 2013]. In particular, an intronic variant (rs11046205) reached genome‐wide statistical significance. This variant was unfortunately not present in our dataset. However, we did test rs11046211 (eta = −0.17, SE = 0.14, t‐value = −1.18), which is also evaluated by Allebrandt et al. [2013] (beta = 0.20, P = 9.9 × 10−6) and did not find an especially strong association signal. The closest SNP (within 50 kb) with the strongest signal in our analysis is rs704191 (∼23 kb away, eta = −5.37, t‐value = −11.80). One possibility for this discrepancy is that there is more than one signal of association in the same genomic region. Although, perhaps a simpler explanation is that the patterns of linkage disequilibrium between the presumed underlying functional variant and the evaluated SNPs are not consistent across study population samples. Indeed, the allele frequency range across population samples included in Allebrandt et al.'s meta‐analysis [Allebrandt et al., 2013] for rs11046211 is 0.042–0.119. In addition, the correlation (as measured with R2) between rs704191 and rs11046205 in the 1000 Genomes EUR population sample is only 0.10 [Genomes Project et al., 2012].
Figure 1.
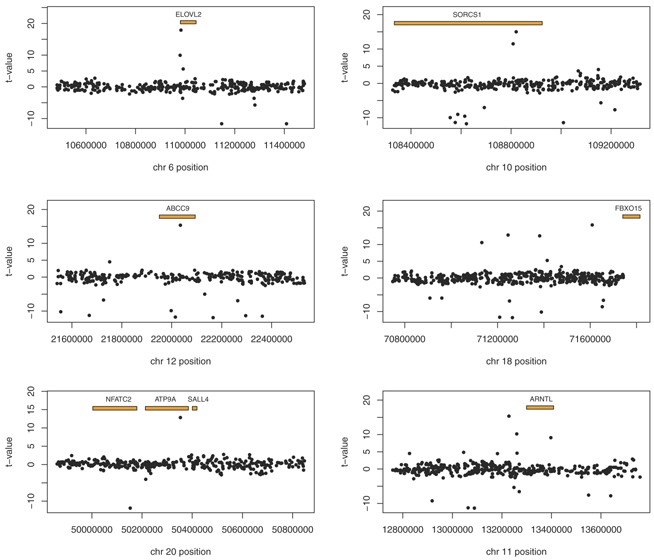
Top candidate gene regions. [Color figure can be seen in the online version of this article, available at http://wileyonlinelibrary.com/journal/ajmgb]
Several other genic regions identified in the current analysis have been previously implicated but did not originally reach genome‐wide significance. Rs7233717 lies ∼500 kb upstream of FBXO15 (Fig. 1), which was identified in a GWAS of sleep duration but did not reach genome‐wide significance in the original analysis [Byrne et al., 2013]. The SNP identified by Byrne et al. [2013], rs2278331 was not associated with sleep duration in our analysis (eta = −0.06, t‐value = −0.90). In our targeted analysis of additional variants in the region (Fig. 1), we identified rs17088578 (27∼kb away from rs2278331, eta = 2.88, t‐value = 15.88), which is located ∼200 kb upstream of FBXO15. Again, one possibility for this discrepancy is that there is more than one signal of association in the same genomic region. Although, in this case, rs2278331 was imputed by Byrne et al. [2013], and an alternate explanation is that there is a biological signal in the region that is being ‘tagged’ by different variants in the two datasets due to differences in the way that the alleles were collected.
In addition, we identified rs41348446 (eta = 1.82, t‐value = 10.19), located on chromosome 11 approximately 40 kb upstream of ARNTL (Fig. 1). ARNTL is a circadian rhythm gene that has been associated with later sleep and wake times in an elderly cohort [Evans et al., 2013] as well as with seasonal affective disorder [Partonen et al., 2007]. We additionally identified a stronger signal of association with rs931186 (∼71 kb upstream of ARNTL, eta = 2.76, t‐value = 15.35).
We also identified a region on chromosome 20 that contains four genes with suggestive relationships to sleep (Fig. 1). Rs2256551, the SNP we identified in the GWAS is located in the intronic region of ATPase, class II, type 9A (ATP9A), which is upstream to spalt‐like transcription factor 4 (SALL4). SALL4 mutations cause Duane radial ray syndrome (Okihiro syndrome), which is associated with narcolepsy [Butterworth and Shneerson, 2014]. ATP9A is also located 1.2 Mb upstream of teashirt zinc finger homeobox 2 (TSHZ2), a gene that was implicated in sleep duration in a GWAS conducted by Gottlieb et al. [2007] but did not reach genome‐wide significance in the original analysis. Finally, ATP9A lies downstream of nuclear factor of activated T‐cells, cytoplasmic, calcineurin‐dependent 2 (NFATC2), which is associated with narcolepsy [Shimada et al., 2010]. Therefore, it appears that this region may contain multiple loci involved in sleep and sleep‐related disorders.
Two more recent studies identified several additional candidate variants associated with sleep duration. Ollila et al., [2014] identified a SNP on chromosome 5 (rs114725) that is less than 2 Mb away from one of our identified candidate SNPs (rs16900727, Table III), and a SNP on chromosome 10 (rs10886445) that is less than 1 Mb away from another one of our identified candidate SNPs (rs7096948, Table III). Gottlieb et al. [2014] identified a region on chromosome 2 (position 113,785‐491‐113,811,454) that is not in the tails of our empirical distribution (absolute t‐values range from 0.40–0.91). We did, however, identify a nearby region (position 115,493,714; rs17043459) that is less than 2 Mb away. However, given the distances between the previously reported signals of association and the current study, it is difficult to determine whether these represent one or more suggestive signals of association.
To increase our genomic coverage, we additionally performed an exploratory association analysis with an expanded set of 870,428 SNPs that included imputed genotypes for individuals with missing data. The top 0.1% of the empirical distribution of absolute t‐values are included in Table SVI; however, we were not able to replicate any previously reported genetic candidate variants for sleep duration with the expanded association analysis.
Novel Candidate Genes
We have identified two novel genes that have been implicated in sleep in other mammals. Rs17122013 is located in the intronic region of Sortilin‐related VPS10 domain containing receptor 1 (SORCS1) (Fig. 1) which is deleted in an inbred short sleep mouse strain [Dumas et al., 2014]. Rs41463746 is located in the 3′ UTR of ELOVL fatty acid elongase 2 (ELOVL2) (Fig. 1), which is up‐regulated in liver tissue from hibernating winter bears [Fedorov et al., 2009]. It is worth noting that the direction of the effect of rs41463746 on sleep is not consistent across the CPMC and Air Force cohorts (eta = 3.21 and −1.86, respectively). However, as displayed in Figure 1, there are two additional SNPs (rs6919269, eta = −5.37, t‐value = −11.67; rs4713206, eta = −5.35, t‐value = −11.66) that are consistent across both cohorts.
DISCUSSION
Here, we leverage the data collected from the CPMC research study to investigate putative candidate genes involved in sleep duration. We have taken an approach that is different and complimentary to previously reported genome‐wide association studies of sleep. In particular, we have incorporated an expanded set of lifestyle and demographic predictor variables in our statistical modeling.
While we have not replicated any previously identified candidate variants for sleep duration, our results provide independent support for previously identified candidate genes that have (ABCC9) and have not yet (FBOX15, TSHZ2) reached genome‐wide significance in previously reported GWAS of sleep duration [Allebrandt et al., 2013; Byrne et al., 2013]. In addition, Allebrandt et al. [2013] conducted a functional experiment in Drosophila melanogaster of ABCC9 and demonstrated that flies with non‐functional transcripts were sleepless for 3 hr on average more per night compared to wild‐type flies. Thus, the self‐reported data collected through the CPMC related to sleep duration as well as the lifestyle and demographic variables included in our statistical model appear to be robust, and our analytical approach appears to have identified biologically meaningful associations with sleep duration.
We also find it intriguing that one of the regions identified in the current analysis on chromosome 20 contains multiple genes with putative biological relationships to sleep and sleep disorders. TSHZ2 was identified as a suggestive candidate gene for sleep duration [Gottlieb et al., 2007], and SALL4 and NFATC2 are associated with narcolepsy [Shimada et al., 2010; Butterworth and Shneerson, 2014]. This phenomenon is not without precedent in that previous work has identified physical clusters of genes with related biological functions (e.g., [Scheinfeldt et al., 2011]). While ATP9A itself has not been previously identified as a candidate gene for involvement in sleep duration, work in rats shows that expression levels of ATP9A in liver change as a function of time of day, and ATP9A is involved in ion transport. Some have speculated that ATP9A and other similar genes may be involved in pharmacokinetics and pharmacodynamics [Nainwal et al., 2011], which raises the possibility that ATP9A may mediate the effects of sleep medications.
Moreover, we found compelling evidence in the non‐human literature for a relationship between two of our strongest candidate genes and sleep. Work in lab strains of inbred short and long sleep mice demonstrates that SORCS1 is deleted in the short sleep strain relative to the long sleep strain suggesting that the absence of this gene may contribute to shorter sleep duration [Dumas et al., 2014]. Perhaps even more intriguing is the finding that ELOVL2 is up‐regulated in the livers of hibernating bears [Fedorov et al., 2009]. In humans, ELOVL2 is associated with aging [Garagnani et al., 2012], and omega‐3 fatty acid levels were associated with sleep in an epidemiological study of children in the United Kingdom [Montgomery et al., 2014]. These results suggest that ELOVL2 may be important not just during hibernation, but for routine daily sleep in humans as well.
Taken together, six of out 37 candidate gene regions have been previously implicated in human and/or mammalian research. As a set, however, there is no significant enrichment of any one biological pathway, function or process. This negative result is consistent with the inherited component of sleep involving more than one biological mechanism, and more work needs to be done to identify and test what we assume are underlying functional variants contributing to sleep.
There are several limitations to the current study, the most important being that our sample size was too modest to identify any genome‐wide significant candidate genes. Indeed, the direction of association is not always consistent between the CPMC and Air Force cohorts, potentially due to the limited sample sizes included in each analysis. Furthermore, we were limited by the included variants in the genome‐wide array, especially in that the vast majority of them are not functional variants, and instead act as proxies for what we assume are underlying functional variants. Therefore, resequencing studies will be necessary to identify any putative functional variants contributing to sleep variation. Moreover, there are regions of the genome that are not well covered with the variants included in the current study. In addition, there are lifestyle and demographic variables of interest that we either do not currently collect through the CPMC (e.g., do you live with young children?) or cannot incorporate into the model due to incomplete data (e.g., caffeine and stimulant intake, sleep medication usage, and co‐morbidities, which are missing for the vast majority of study participants). Error may also be introduced when participants do not correctly report their average sleep duration. Finally, we used a categorical self‐reported measure of sleep duration, and it will be useful to collect continuous measures of sleep duration in the future, which may be more powerful data for identifying signals of association.
In summary, the CPMC research study uses both genetic and non‐genetic information to customize individual risk reports for complex diseases, and here we have leveraged those data to better understand the role of common genetic variation in sleep duration. Not only have we identified several candidate genes previously implicated in sleep duration in human studies, but we have also identified several novel putative candidate genes involved in human sleep duration. Our approach demonstrates that using the CPMC participant self‐reported data in combination with genome‐wide genetic data is robust. Future work that explores functional variants related to sleep duration will contribute to our ability to identify individuals at increased risk of sleep problems and associated health disorders and tailor recommended lifestyle changes and/or medications to improve sleep quantity and overall general health.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supporting Information Figure S1.
Supporting Information Figure S2.
Supporting Information Table S1.
Supporting Information Table S2.
Supporting Information Table S3‐S5.
Supporting Information Table S6.
ACKNOWLEDGMENTS
This work was supported by grants from the RNR Foundation and the United States Air Force. In addition, the authors would like to thank Daniel Lynch and Andrew Brangan for helpful discussions. Finally, we are grateful to all of the participants of the Coriell Personalized Medicine Collaborative.
Scheinfeldt LB, Gharani N, Kasper RS, Schmidlen TJ, Gordon ES, Jarvis JP, Delaney S, Kronenthal CJ, Gerry NP, Christman MF. 2015. Using the Coriell Personalized Medicine Collaborative Data to Conduct a Genome‐Wide Association Study of Sleep Duration. Am J Med Genet Part B 168B:697–705.
REFERENCES
- Aldabal L, Bahammam AS. 2011. Metabolic, endocrine, and immune consequences of sleep deprivation. Open Respir Med J 5:31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allebrandt KV, Amin N, Muller‐Myhsok B, Esko T, Teder‐Laving M, Azevedo RV, Hayward C, van Mill J, Vogelzangs N, Green EW, Melville SA, Lichtner P, Wichmann HE, Oostra BA, Janssens AC, Campbell H, Wilson JF, Hicks AA, Pramstaller PP, Dogas Z, Rudan I, Merrow M, Penninx B, Kyriacou CP, Metspalu A, van Duijn CM, Meitinger T, Roenneberg T. 2013. A K(ATP) channel gene effect on sleep duration: From genome‐wide association studies to function in Drosophila . Mol Psychiatry 18(1):122–132. [DOI] [PubMed] [Google Scholar]
- Browning SR, Browning BL. 2007. Rapid and accurate haplotype phasing and missing‐data inference for whole‐genome association studies by use of localized haplotype clustering. Am J Hum Genet 81(5):1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterworth JW, Shneerson JM. 2014. Narcolepsy associated with Duane's syndrome. Clin Med Insights Case Rep 7:1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne EM, Gehrman PR, Medland SE, Nyholt DR, Heath AC, Madden PA, Hickie IB, Van Duijn CM, Henders AK, Montgomery GW, Martin NG, Wray NR, Chronogen C. 2013. A genome‐wide association study of sleep habits and insomnia. Am J Med Genet Part B Neuropsychiatr Genet 162B(5):439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas L, Dickens CM, Anderson N, Davis J, Bennett B, Radcliffe RA, Sikela JM. 2014. Exome sequencing and arrayCGH detection of gene sequence and copy number variation between ILS and ISS mouse strains. Mamm Genome 25(5–6):235–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durmer JS, Dinges DF. 2005. Neurocognitive consequences of sleep deprivation. Semin Neurol 25(1):117–129. [DOI] [PubMed] [Google Scholar]
- Evans DS, Parimi N, Nievergelt CM, Blackwell T, Redline S, Ancoli‐Israel S, Orwoll ES, Cummings SR, Stone KL, Tranah GJ. Study of Osteoporotic F, Osteoporotic Fractures in Men Study G. 2013. Common genetic variants in ARNTL and NPAS2 and at chromosome 12p13 are associated with objectively measured sleep traits in the elderly. Sleep 36(3):431–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov VB, Goropashnaya AV, Toien O, Stewart NC, Gracey AY, Chang C, Qin S, Pertea G, Quackenbush J, Showe LC, Showe MK, Boyer BB, Barnes BM. 2009. Elevated expression of protein biosynthesis genes in liver and muscle of hibernating black bears (Ursus americanus). Physiol Genomics 37(2):108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garagnani P, Bacalini MG, Pirazzini C, Gori D, Giuliani C, Mari D, Di Blasio AM, Gentilini D, Vitale G, Collino S, Rezzi S, Castellani G, Capri M, Salvioli S, Franceschi C. 2012. Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell 11(6):1132–1134. [DOI] [PubMed] [Google Scholar]
- Gehrman P, Seelig AD, Jacobson IG, Boyko EJ, Hooper TI, Gackstetter GD, Ulmer CS, Smith TC. 2013. Predeployment sleep duration and insomnia symptoms as risk factors for new‐onset mental health disorders following military deployment. Sleep 36(7):1009–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project C, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491(7422):56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb DJ, Hek K, Chen TH, Watson NF, Eiriksdottir G, Byrne EM, Cornelis M, Warby SC, Bandinelli S, Cherkas L, Evans DS, Grabe HJ, Lahti J, Li M, Lehtimaki T, Lumley T, Marciante KD, Perusse L, Psaty BM, Robbins J, Tranah GJ, Vink JM, Wilk JB, Stafford JM, Bellis C, Biffar R, Bouchard C, Cade B, Curhan GC, Eriksson JG, Ewert R, Ferrucci L, Fulop T, Gehrman PR, Goodloe R, Harris TB, Heath AC, Hernandez D, Hofman A, Hottenga JJ, Hunter DJ, Jensen MK, Johnson AD, Kahonen M, Kao L, Kraft P, Larkin EK, Lauderdale DS, Luik AI, Medici M, Montgomery GW, Palotie A, Patel SR, Pistis G, Porcu E, Quaye L, Raitakari O, Redline S, Rimm EB, Rotter JI, Smith AV, Spector TD, Teumer A, Uitterlinden AG, Vohl MC, Widen E, Willemsen G, Young T, Zhang X, Liu Y, Blangero J, Boomsma DI, Gudnason V, Hu F, Mangino M, Martin NG, O'Connor GT, Stone KL, Tanaka T, Viikari J, Gharib SA, Punjabi NM, Raikkonen K, Volzke H, Mignot E, Tiemeier H. 2014. Novel loci associated with usual sleep duration: The CHARGE Consortium Genome‐Wide Association Study. Mol Psychiatry. [Epub ahead of print] doi:10.1038/mp.2014.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb DJ, O'Connor GT, Wilk JB. 2007. Genome‐wide association of sleep and circadian phenotypes. BMC Med Genet 8(Suppl 1):S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller MA, Gordon ES, Stack CB, Gharani N, Sill CJ, Schmidlen TJ, Joseph M, Pallies J, Gerry NP, Christman MF. 2010. Coriell Personalized Medicine Collaborative®: A prospective study of the utility of personalized medicine. Pers Med 7(3):301–317. [DOI] [PubMed] [Google Scholar]
- Knutson KL, Spiegel K, Penev P, Van Cauter E. 2007. The metabolic consequences of sleep deprivation. Sleep Med Rev 11(3):163–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger PM, Friedman EM. 2009. Sleep duration in the United States: A cross‐sectional population‐based study. Am J Epidemiol 169(9):1052–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. 2010. GREAT improves functional interpretation of cis‐regulatory regions. Nat Biotechnol 28(5):495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery P, Burton JR, Sewell RP, Spreckelsen TF, Richardson AJ. 2014. Fatty acids and sleep in UK children: Subjective and pilot objective sleep results from the DOLAB study‐a randomized controlled trial. J Sleep Res 23(4):364–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nainwal R, Nanda D, Rana V. 2011. Fundamentals of chronopharmacology and chronopharmacotherapy. J Pharm Res 4(8):2692–2695. [Google Scholar]
- Ollila HM, Kettunen J, Pietilainen O, Aho V, Silander K, Kronholm E, Perola M, Lahti J, Raikkonen K, Widen E, Palotie A, Eriksson JG, Partonen T, Kaprio J, Salomaa V, Raitakari O, Lehtimaki T, Sallinen M, Harma M, Porkka‐Heiskanen T, Paunio T. 2014. Genome‐wide association study of sleep duration in the Finnish population. J Sleep Res 23(6):609–618. [DOI] [PubMed] [Google Scholar]
- Partonen T, Treutlein J, Alpman A, Frank J, Johansson C, Depner M, Aron L, Rietschel M, Wellek S, Soronen P, Paunio T, Koch A, Chen P, Lathrop M, Adolfsson R, Persson ML, Kasper S, Schalling M, Peltonen L, Schumann G. 2007. Three circadian clock genes Per2, Arntl, and Npas2 contribute to winter depression. Ann Med 39(3):229–238. [DOI] [PubMed] [Google Scholar]
- Prospero‐Garcia O, Mendez‐Diaz M. 2004. The role of neuropeptides in sleep modulation. Drug News Perspect 17(8):518–522. [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd‐Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. 2007. PLINK: A tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet 81(3):559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripley WNVaBD. 2002. Modern applied statistics with S. New York: Springer. [Google Scholar]
- Scheinfeldt LB, Biswas S, Madeoy J, Connelly CF, Akey JM. 2011. Clusters of adaptive evolution in the human genome. Front Genet 2:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada M, Miyagawa T, Kawashima M, Tanaka S, Honda Y, Honda M, Tokunaga K. 2010. An approach based on a genome‐wide association study reveals candidate loci for narcolepsy. Hum Genet 128(4):433–441. [DOI] [PubMed] [Google Scholar]
- Stack CB, Gharani N, Gordon ES, Schmidlen T, Christman MF, Keller MA. 2011. Genetic risk estimation in the Coriell Personalized Medicine Collaborative. Genet Med 13(2):131–139. [DOI] [PubMed] [Google Scholar]
- Steiger A, Holsboer F. 1997. Neuropeptides and human sleep. Sleep 20(11):1038–1052. [PubMed] [Google Scholar]
- Team RC. 2014. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Van Dongen HP, Maislin G, Mullington JM, Dinges DF. 2003. The cumulative cost of additional wakefulness: Dose‐response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivation. Sleep 26(2):117–126. [DOI] [PubMed] [Google Scholar]
- van Mill JG, Hoogendijk WJ, Vogelzangs N, van Dyck R, Penninx BW. 2010. Insomnia and sleep duration in a large cohort of patients with major depressive disorder and anxiety disorders. J Clin Psychiatry 71(3):239–246. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supporting Information Figure S1.
Supporting Information Figure S2.
Supporting Information Table S1.
Supporting Information Table S2.
Supporting Information Table S3‐S5.
Supporting Information Table S6.