

Abstract
Single-walled carbon nanotubes (SWNTs) are used in the near infrared (NIR)-mediated thermal ablation of tumor cells because they efficiently convert absorbed NIR light into heat. Despite the therapeutic potential of SWNTs, there have been no published studies that directly quantify how many SWNTs need be associated with a cell to achieve a desired efficiency of killing, or what is the most efficient subcellular location of SWNTs for killing cells. Herein we measured dose response curves for the efficiency of killing correlated to the measured amounts of folate-targeted SWNTs that were either on the surface or within the vacuolar compartment of normal rat kidney (NRK) cells. Folate-targeted SWNTs on the cell surface were measured after different concentrations of SWNTs in medium were incubated with cells for 30 min at 4 °C. Folate-targeted SWNTs within the vacuolar compartments were measured after cells were incubated with different concentrations of SWNTs in medium for 6 h at 37 °C. It was observed that a SWNT load of ~13 pg/cell when internalized was sufficient to kill 90% of the cells under standard conditions of NIR light irradiation. When ~3.5 pg/cell of SWNTs were internalized within the endosomal/lysosomal compartments, ~50% of the cells were killed, but when ~3.5 pg/cell of SWNTs were confined to the cell surface only ~5% of the cells were killed under the same NIR irradiation condition. The SWNT subcellular locations were verified using Raman imaging of SWNTs merged with fluorescence images of known subcellular markers. To our knowledge, this is the first time that SWNT amounts at known subcellular locations have been correlated with a dose-normalized efficacy of thermal ablation and the results support the idea that SWNTs confined to the plasma membrane are not as effective in NIR-mediated cell killing as an equivalent amount of SWNTs when internalized within the endosomal/lysosomal vesicles.
Keywords: carbon nanotubes, near infrared, photo-thermal ablation, cancer therapy
1. INTRODUCTION
Single-walled carbon nanotubes (SWNTs) absorb near infrared (NIR) radiation and efficiently transduce the energy to heat. This phenomenon has been explored to thermally ablate tumor cells in vitro [1–7] and in vivo [8–13]. There are two general approaches to SWNT-mediated NIR thermal ablation. One is to introduce non-targeted SWNTs to cells in vitro or in vivo where they may be taken up by fluid-phase endocytosis or other mechanisms that do not require receptors [9, 10, 12–17], followed by exposure to NIR light. The second and more specific approach is to use SWNTs targeted to tumor cells by ligand-receptor interactions in vitro [1, 3–5, 7] or in vivo [8, 11, 18]. In this scenario, the SWNT-ligand combination would accumulate in the tumor cells bearing the specific receptor for the ligand with the intent to target only the specific cells of interest.
The use of targeted approaches with ligands or monoclonal antibodies (MAbs) has several potential advantages over a non-targeted approach. One is specificity since cells containing the receptor for the ligand should accumulate a higher load of SWNTs than cells lacking the receptor. A second is that lower SWNT concentrations may be used because of the high affinity of the ligand for receptors. A potential third advantage is that it should be possible, with judicial choice of ligands, to target SWNTs not only to specific receptors on cells, but to specific subcellular compartments (such as lysosomes) where concentrated local heating may be more effective in cell killing. The complexity of ligand-targeted approaches, however, raises several basic questions about the ablation process that are not well understood.
One question that has not been answered is what quantity of SWNTs need to be on or in a cell to achieve a given ablation efficacy. Numerous published papers report thermal ablation of cancer cells using targeted killing in vitro [1, 3–7] and in vivo [8, 11] but the actual dose of SWNTs that need to be on or within a cell to achieve effective ablation is not well formulated. A main reason for this information gap is the difficulty in quantifying the small amount of SWNTs associated with cells [19]. In previous work, we developed a method for quantifying cell-associated SWNTs that exploits sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) of SWNTs extracted from cells and organisms [20–22]. Here, the method was used to quantify carboxylated SWNTs functionalized to contain folic acid (FA) and targeted to folate receptors (FRs) on normal rat kidney (NRK) cells made to over-express FRs. Cell killing under standard NIR ablation conditions was then measured, which resulted in dose response curves that correlated the extent of ablation with the actual SWNT load per cell.
Another question is whether the efficiency of cell-associated SWNTs in mediating cell killing is affected by the subcellular locations of the SWNTs. For example, are SWNTs on the cell surface more or less efficient in cell killing than SWNTs in endosomal/lysosomal vesicles? There have been approaches to answer this question in the literature. Xiao et al. demonstrated that SWNTs targeted and confined to the surface of HER-2 positive breast tumor cells could kill the cells upon exposure to NIR light, but they did not compare the killing efficacy of surface-bound SWNTs with the same material internalized within the cells [6]. Work of Marches et al. compared the efficacy of surface-bound SWNTs versus internalized SWNTs, also on HER-2 positive breast tumor cells, and suggested that internalized material was more potent in cell killing [7]. However, approaches to this question should normalize the efficiency of ablation to the actual dose of SWNTs per cell, either on the cell surface or within the cell, to control for differences in local dose that could affect the efficiency of killing, and this has not yet been done. In the present paper, different doses of SWNTs were targeted to FRs overexpressed in NRK cells and were either confined to the cell surface by binding at 4 °C where internalization does not occur, or were allowed to populate the endosomal/lysosomal compartment upon internalization at 37 °C. The extent of NIR-mediated cell killing was then assessed as a function of the cell-associated SWNT doses at different subcellular locations. The results presented here support the idea that SWNTs confined on the plasma membrane are not as effective in ablating cells as an equivalent amount of SWNTs accumulated within endosomal/lysosomal compartments.
2. MATERIALS AND METHODS
2.1 Materials and chemicals
The SWNTs (Product no. P3, batch no. 03-A010) produced by a Ni/Y-catalyzed electric arc discharge method and functionalized with nitric acid to contain 1.0–3.0 atomic % of carboxylic groups were purchased from Carbon Solutions, Inc. (Riverside, CA). Caution: a particulate respirator should be worn when handling dry SWNT powders. According to the manufacturer specification, the carbonaceous purity of the SWNT powder was >90% and the individual SWNTs were 0.5–3.0 μm in length and approximately 1.4 nm in diameter. Phospholipid-polyethylene glycol-amine (PL-PEG-NH2) (product no. 880128P) was purchased from Avanti Polar Lipids (Alabaster, AL) and stored at −24 °C. Sodium phosphate buffer stock (0.1 M, pH 7.4) was sterilized by autoclaving at 121 °C for 45 min. FA (lot no. SLBC2647V) was purchased from Sigma Aldrich (St. Louis, MO) and was stored in the dark.
2.2 Tissue culture materials
Normal rat kidney (NRK) epithelial cells (ATCC number CRL-6509) were obtained from the American Type Culture Collection (Manassas, VA). Dulbecco’s modified Eagle medium (DMEM) and trypsin were purchased from Gibco (Grand Island, NY). The standard cell culture medium contains DMEM supplemented with 10 mM HEPES, 3.7 g/L sodium bicarbonate, 4.5 g/L D-glucose, 0.584 g/L L-glutamine, and 10% (v/v) fetal bovine serum (FBS, purchased from Hyclone, Logan, UT). Standard incubation conditions were at 37 °C in a humidified 10% CO2 incubator. DMEM with similar media constituents but without FA was purchased from Sigma Aldrich (Lot no. RNBD3838). Phosphate buffered saline (PBS; 0.8 mM phosphate, 150 mM NaCl, pH 7.4) was sterilized by autoclaving at 121 °C for 45 min. Deionized water (18.3 MΩ-cm) was obtained using a Milli-Q Integral water purification system (Billerica, MA). Penicillin at 10,000 U/mL/Streptomycin at 10 mg/mL (P/S) and Amphotericin B (Amp B) at 12.5 mg/mL were purchased from Sigma Aldrich and used only in terminal cultures at 1/100 dilutions.
2.3 Preparation of phospholipid-polyethylene glycol-folic acid (PL-PEG-FA)
PL-PEG-FA was prepared by modification of the method described by Kam et al.[1] FA (3.5 mM) was first dissolved in just enough 0.1 M NaOH to completely dissolve the FA because excessive NaOH resulted in precipitation of the construct. PL-PEG-NH2 (0.35 mM) was dissolved in 10 mM sodium phosphate buffer and 5 mM 1-ethyl-3-(3-dimethylamine-propyl) carbodiimide (EDC) was added. To this mixture, 3.5 mM FA was added and reacted for 3 h with gentle rocking. After the reaction, the solution was dialyzed against 10 mM sodium phosphate buffer using a dialysis membrane (Spectrum Labs, Rancho Dominguez, CA) with a molecular weight cutoff of 6000–8000 Da to remove unreacted FA and EDC. The dialysis was carried out for 3 d with frequent replacement of the buffer.
2.4 Spectrophotometric analysis and SDS-PAGE for detection of conjugation and the presence of free FA
To determine the presence and the amount of FA in the final PL-PEG-FA construct, a UV-Vis absorption spectrum was acquired from 200 to 450 nm to detect the characteristic peak of FA at 360 nm. A standard curve was plotted for FA and PL-PEG-NH2 and from the known concentrations of the standards, the extinction coefficient of each at 360 nm was calculated. Using the extinction coefficients for the FA and PL-PEG-NH2 it was determined that they were in a 1:1 molar ratio in the PL-PEG-FA construct.
To validate the removal of free FA following dialysis and to determine whether PL-PEG was present in the PL-PEG-FA conjugate, ten microliters of FA, PL-PEG-NH2, and PL-PEG-FA before and after dialysis were analyzed in a 4% stacking and 15% resolving SDS polyacrylamide gel. The samples were electrophoresed at 100 V for 90 min and fixed in the gel with a solution of 50% methanol and 10% acetic acid for 5 min at room temperature. The gel was soaked in 5% BaCl2 for 10 min and the excess BaCl2 was removed. The gel was then soaked in 0.1 N iodine for 5 min and destained with water to remove the background staining [23, 24]. The BaI2 stains the PEG component of the PL-PEG construct and the FA could be viewed directly in the gel due to its distinct yellow color. The gels were scanned and digitized with a HP Scanjet G3110 flatbed scanner.
2.5 Non-covalent functionalization of SWNTs with PL-PEG-FA
SWNT powder (5 mg) was weighed into a glass vial and 10 mL of dialyzed PL-PEG-FA in 10 mM sodium phosphate solution was added, followed by sonication using an Elmasonic P30H Ultrasonic Cleaner bath sonicator (Singen, Germany) in a 4 °C cold room. The vial was placed in a central location in the sonication bath filled with 1400 mL of cold DI water and sonicated at 37 kHz and 120 W for 20 min. The temperature of the bath was controlled with a cooling coil that circulated water at 2 °C through the bath. During sonication the temperature never exceeded 18 °C. It was noted that the power delivered to the sonicator was ~80 W for our sonication system [24]. Following sonication, a two-step centrifugation procedure was done to minimize impurities and to enrich for individually dispersed SWNT-PL-PEG-FA. The SWNT-PL-PEG-FA suspension was dispensed into 1 mL aliquots in centrifuge tubes and centrifuged at 20,000 g for 5 min. 900 μL of each collected supernatant was then transferred to another centrifuge tube and centrifuged at 20,000 g for 30 min. 800 μL of the supernatant was collected from each tube and pooled. The SWNT concentration in the final SWNT-PL-PEG-FA stock solution was quantified based on its absorbance at 500 nm. Briefly, an aliquot of the sonicated but not centrifuged sample was used as the standard with a known SWNT concentration of 0.5 mg/mL. A serial dilution of the standard provided a set of samples to construct a calibration curve that correlated the absorbance measured at 500 nm to known SWNT concentrations ranged from 50 to 300 μg/mL. The prepared SWNT-PL-PEG-FA stock solution with unknown concentration was diluted to 12.5%, 25%, and 50% and the absorbance measured in triplicate. The SWNT concentration of the stock solution was determined using the linear regression of the standard calibration curve.
The particle size distribution (PSD) of the SWNT-PL-PEG-FA construct was measured by dynamic light scattering (DLS) using a 633 nm laser source at a fixed angle of 173° (Zetasizer Nano-ZS 3600, Malvern Instrument, Worcestershire, UK). The SWNT-PL-PEG-FA stock solution was diluted to a SWNTs concentration of 20 μg/mL in PBS prior to analysis. 500 μL of the sample was then placed in a disposable cuvette and 10 consecutive 30 second runs were taken per measurement at 25 °C. Three independent DLS measurements were acquired and averaged to determine the relative homogeneity of the SWNT-PL-PEG-FA particles in suspension expressed in terms of the mean hydrodynamic diameter (HD) and polydispersiblity index (PDI) values.
2.6 FR overexpression and measurement in NRK cells
To maximize the number of folate receptors expressed in NRK cells, the cells were deprived of folate in the culture medium to induce FR expression [1, 8, 25]. NRK cells were cultured in DMEM without FA for at least 4 rounds of sub-culturing to maximize FR overexpression. Cells with overexpressed FRs were termed FR(+) and those without induced overexpression of the FRs were termed FR(−). The overexpression of FRs was monitored by immunofluorescence using a rabbit polyclonal IgG anti-folate receptor primary antibody (Santa Cruz Biotech, Dallas, TX, lot no. K2911) and an Alexa Fluor® 488 goat anti-rabbit secondary antibody (Invitrogen Molecular Probes, Grand Island, NY, lot no. 1073082). FR(−) and FR(+) NRK cells were grown on glass cover slips (Ted Pella, Inc.; Prod No. 26023) in DMEM with or without FA respectively and supplemented with 10% FBS. The cells were washed thrice using PBS and fixed with 4% (w/v) paraformaldehyde (PFA) in PBS followed by three more washes with PBS. The cells were incubated with anti-folate receptor primary antibody at a concentration of 4 μg/mL under moist conditions for 1 h. The cells were washed thrice with PBS at 10 min intervals. The cells were then incubated with goat anti-rabbit secondary antibody labeled with Alexa Fluor® 488 at a concentration of 1.25 μg/mL for 20 min under moist and dark conditions. Cover slips were mounted on a glass slide and viewed using a Nikon Eclipse TE 300 fluorescence microscope with an excitation wavelength of 495 nm and emission wavelength at 525 nm. Cells that were not treated with primary or secondary antibodies and cells treated with only secondary antibodies were used as negative controls. The relative levels of FR expression in FR(+) and FR(−) cells were determined based on the FR specific fluorescence intensity acquired from respective fluorescence images of the cells. The fluorescence images were analyzed and the average fluorescence intensity per cell was quantified using ImageJ software.
2.7 Targeted binding of SWNT-PL-PEG-FA to NRK cells at 4 °C as a function of SWNT concentration in media
FR(−) and FR(+) NRK cells were plated on a 6-well plate at a density of ~2 × 105 cells per well in 2 mL of DMEM-based growth media. FR(−) cells were grown in regular media with FA while FR(+) cells were grown in media that contained no FA. For SWNT uptake experiments with FR(+) cells, 2X concentrated DMEM media supplemented with P/S and Amp B but without FA or FBS were mixed with an equal volume of 10 mM sodium phosphate buffer (control) or with an equal volume of SWNT-PL-PEG-FA dispersion (experimental) so that the final concentrations of DMEM in control or experimental media were the same as in standard growth medium. The final concentration of the sodium phosphate buffer solution was 5 mM and the concentrations of the SWNT-PL-PEG-FA in the media were also halved. In preparation for a surface binding experiment, cells were first grown at 37 °C for 24 h after which the growth media was replaced with fresh control media that contained no SWNT-PL-PEG-FA or experimental media with SWNT-PL-PEG-FA at final concentrations of 10, 25, 50, or 90 μg/mL in the media. The cells were incubated at 4 °C for 30 min with SWNTs to allow binding to cell surface receptors without internalization, which is blocked at 4 °C. Following incubation, the cells were washed thrice with ice cold DMEM and PBS and lysed in 1% SDS, 1 mM CaCl2, and 1 mM MgCl2. A cell scraper was used to help lift the cells from the bottom of the well and to mix with the lysis buffer to ensure complete lysis of the cells. Subsequently, the cell lysate was treated with DNase I (0.1 μg/μL) and incubated for 6 h at 37 °C to degrade released DNA and reduce the viscosity of the lysate. A assay procedure using a BCA protein assay kit (Pierce Chemical, Rockford, IL) was adopted from Wang et al. [20] for the quantitation of protein content in cell lysate samples. The average cellular protein content for FR (+) and FR (−) NRK cell was calculated through BCA assay. In FR (+) NRK cells the cellular protein content is 3.84 × 10−4 μg per cell, and in FR (−) cell the cellular protein content is 1.29 × 10−4 μg per cell. The protein content of the cell lysate sample was used to calculate the number of cells the sample was prepared from. The amount of SWNTs in the cell lysate samples was then determined using the SDS-PAGE method, described next and in the Supplemental Information section (Figure S2). Finally, the average cell-associated SWNTs amount, expressed as pg SWNTs per cell, was derived from the amount of SWNTs measured in a cell lysate and the known number of cells from which the lysate was prepared.
2.8 Uptake of SWNT-PL-PEG-FA by FR(+) NRK cells at 37 °C
For SWNT accumulation experiments, similar to SWNT surface binding experiments described in the previous section, FR(+) NRK cells at ~2 × 105 cells/well were grown in a 6 well plate in DMEM-based growth media without FA. After a 24 h incubation at 37 °C, the growth media was replaced with fresh control media that contained no SWNT-PL-PEG-FA or experimental media that contained SWNT-PL-PEG-FA at final SWNTs concentrations of 1, 5, or 10 μg/mL in the media. Cells were incubated at 37 °C for 6 h to allow internalization and accumulation of SWNTs. Following incubation, the cells were washed thrice with DMEM and PBS and lysed. The total cellular protein content was then measured with the BCA assay as described above and the cell-associated SWNT content in the lysate was quantified with the SDS-PAGE method described next. The uptake of SWNT-PL-PEG-FA was then presented as the average pg SWNTs per cell for an indicated control or experimental population of FA(+) cells.
2.9 Quantification of SWNTs in cell lysates by SDS-PAGE
The amount of SWNTs in cell lysates was determined using a modification of the SDS-PAGE method as described in our previous work [19–21]. A Hoefer mini vertical gel caster for 10 cm × 8 cm plates with 1.5 mm thick spacers and 10 well combs were used. 10 % polyacrylamide gels were prepared from a 40% stock solution that had a ratio of acrylamide to bisacrylamide of 29:1. Samples were mixed with 2X SDS sample loading buffer at a 1:1 ratio to a final concentration of 2% SDS and 5% 2-mercaptoethanol, boiled for 3 min to reduce the disulfide bonds in proteins, and loaded into the wells. SWNTs of known amounts from 50 to 200 ng were loaded as standards. Cell lysates with unknown SWNT amounts were loaded as a function of increasing protein content that had been determined using the BCA assay. An electric current was applied on the SDS-PAGE system at 100 V for 2 h. Following electrophoresis, images of the SWNT bands in the gels were acquired using a HP Scanjet G 3110 flatbed scanner with the resolution set at 1200 dpi. Quantification of the digitized bands was performed using ImageJ software. All sample gel band pixel intensities were background subtracted using the intensities from a control gel band that did not contain SWNTs. The pixel intensities of SWNTs that were loaded in known amounts were quantified and a calibration curve of pixel intensities-versus-the known amount of SWNTs was plotted. This linear calibration curve was then used to estimate the unknown amount of SWNTs in the cell lysates and expressed as SWNTs/known amount of cellular protein. From the amount of SWNTs in a lysate sample containing a known amount of protein and the amount of protein/cell for NRK cells [20], the amount of SWNTs per cell was then determined. A detailed example of the calculation is presented in the Supplemental Information section (Figure S2). However, it should be recognized that the stated amount of SWNTs/cell is an average for the cell population from which the lysate was prepared.
2.10 Determining intracellular localizations of SWNTs by merging Raman and immunofluorescence images
To determine the intracellular localization of SWNTs in cells, FR(+) NRK wells were seeded on gridded glass cover slips (stock no. 1916-91818, Bellco Biotechnology, Vineland, NJ). Cells were either incubated at 37 °C for 6 h with 10 μg/mL SWNT-PL-PEG-FA or were incubated at 4 °C for 30 min with 90 μg/mL SWNT-PL-PEG-FA in the media. Post incubation, the cells were washed thrice with DMEM and PBS, fixed with 4% PFA for 20 min, washed thrice with PBS to remove PFA, permeabilized with 0.4% (v/v) Tween 20 for 15 min, and incubated with 1% BSA for 1 h at room temperature. 50 μL of a rabbit polyclonal primary antibody against lysosome associated membrane protein-1 (LAMP-1) (Abcam, Cambridge, MA) was applied on the cells at a concentration of 2 μg/mL for 1 h. The cells were washed thrice with PBS at 10 min intervals to remove unbound excess LAMP-1 antibody. 50 μL of a goat anti-rabbit IgG secondary antibody with Alexa Fluor® 488 (from Abcam, Cambridge, MA) was applied on the cells at a concentration of 2 μg/mL for 1 h. Again, the cells were washed thrice with PBS at 5 min intervals to remove excess unbound secondary antibody. The nuclei of the cells were stained with DAPI at a concentration of 2 μg/mL for 5 min and the cells were washed again, thrice with PBS, and thrice with water. The coverslips were mounted on glass slides using fluormount and immunofluorescence microscopy was performed as described next.
All immunofluorescence images were acquired using a Nikon Eclipse TE 300 microscope equipped with a 60X oil immersion lens. Images were acquired with an excitation wavelength of 495 nm, an emission wavelength of 525 nm, and an exposure time of 500 ms. DAPI fluorescence images were acquired with an excitation wavelength of 350 nm, an emission wavelength of 470 nm, and an exposure time of 800 ms. Phase contrast images were acquired using an exposure time of 300 ms. Following the acquisition of fluorescence images, the glass slide was placed under regular fluorescent room light for 72 h to photobleach the Alexa Fluor® 488 and DAPI fluorescence. The completion of the photo-bleaching was verified by fluorescence microscopy. The coverslips mounted on glass slides were washed gently with water, peeled gently off the glass slide, washed again gently with water to remove all the fluoromount and air dried for Raman microscopy analysis, as detailed in the next section. Control images of cells without SWNT-PL-PEG-FA and antibodies, without SWNT-PL-PEG-FA but with antibodies, or with SWNT-PL-PEG-FA but without antibodies were also acquired.
All Raman spectra and images were acquired using a WITec alpha 300R confocal Raman microscope system equipped with a 532 nm laser. Wavenumber calibration was performed using the 520.5 cm−1 line of a silicon wafer with a spectral resolution of ~1 cm−1. The power density of the laser was measured using a Newport model-1918-C power meter with an 818-SL photodetector. Raman spectra were acquired from the same cells on which fluorescence images were acquired previously by locating the cells using their known position on a coverslip grid. The laser was adjusted to a power density of 10 mW/cm2 and focused using a 100X objective lens. The typical integration time was 0.1 s with 20 accumulation cycles. The fluorescence and Raman images were merged using Adobe Photoshop CS6 software to correlate the intracellular location of lysosomes that had been identified by immunofluorescence microscopy with the location of SWNTs that had been identified by their signature Raman G-band signal.
Additional control experiments were performed to avoid potential false signals resulting from fluorescence of SWNTs in immunofluorescence experiments or laser excitation of the fluorophores during Raman scanning. The fluorescent images of FR(+) cells incubated with SWNTs, but not with LAMP-1 primary or Alexa Fluor® 488-tagged secondary antibodies, revealed negligible background fluorescence using an excitation wavelength of 495 nm and emission at 525 nm optimized for detecting Alexa Fluor® 488 (data not shown). This indicated that the SWNTs themselves did not contribute detectable fluorescence during LAMP-1 immunofluorescence microscopy. Correspondingly, reciprocal control experiments were performed with FR(+) cells not incubated with SWNTs but labeled with both primary antibodies to LAMP-1 and Alexa Fluor® 488-tagged secondary antibodies. Under these conditions the LAMP-1 fluorescence signal was evident by fluorescence microscopy, but after the photo bleaching step no SWNT Raman peaks were observed during subsequent Raman imaging (data not shown). This indicated that after photo-bleaching the Alexa Fluor® 488-tagged secondary antibody was not excited by the 532 nm laser that might be registered as a Raman SWNT signal.
2.11 NIR laser irradiation of cells and viability assays
In a typical NIR laser ablation experiment, ~20,000 FR(+) NRK cells were seeded per well in a 96-well plate in DMEM media without FA and incubated for 24 h under standard cell culture conditions at 37 °C with 10% CO2. To minimize unintended NIR laser exposure to neighboring wells during irradiation, only 4–8 wells in a 96-well plate were seeded with cells and they were spaced so that surrounding wells were left blank. The cells were then exposed to various experimental or control conditions, washed thrice with 4 °C PBS and another three times with 4 °C DMEM without FA and 50 μL of irradiation media containing P/S, Amp B, and FBS but without FA was added at 4 °C. Wells were irradiated one at a time in sequence. Just before irradiation, the media was replaced with fresh 4 °C irradiation media and ice cold water was added to the surrounding empty wells to help dissipate heat generated during laser irradiation. All NIR laser irradiations were performed using an Integra MP 808 nm laser system (Spectra Physics) operated in continuous mode and controlled by Integra Soft Version 3.0.4 software. The laser spot area was adjusted to 1.76 cm2 and the actual power density at the irradiated spot was measured using a Newport power meter (model 1928-C).
Following irradiation, the cells were incubated for another 24 h under standard culture conditions in 200 μL of fresh DMEM containing P/S, Amp B, and FBS but without FA to allow viable cells to multiply. The cells were then inspected by microscopy where it was evident that dead or dying cells affected by NIR light were rounded up and detached from the plate. The number of cells remaining after irradiation were assessed with a crystal violet (CV) staining assay as previously described [21, 26]. The wells were washed thrice with cold PBS to remove dead cells and the nucleic acids in the surviving cells were stained with 200 μL of 0.1% (w/v) CV in 10% ethanol for 20 min. Excess CV was washed away with water, and the bound CV was extracted with 200 μL of 10% acetic acid. 100 μL of the extract from each well was transferred to a corresponding well in a new 96-well plate where the CV dye absorbance at 590 was acquired with a microplate reader (BioTek SynergyMx). The background absorbance at 700 nm was subtracted from the CV peak absorbance at 590 nm. It has been recognized that the CV absorbance intensity measured in this assay is directly proportional to the number of cells in the well. Furthermore, this correlation has been validated in our previous work by a direct comparison of the CV assay results with corresponding cell numbers counted using a Coulter counter [21, 26]. Each data point is the average from 4 replica wells and the viability of experimental cells is expressed as a percent of the viability of the control (i.e., cells not incubated with SWNTs nor irradiated) that was set to 100%.
2.12 The effect of laser power density, laser exposure time, and SWNT-PL-PEG-FA concentration on cell viability after exposure to NIR light
To expose cells to SWNT-PL-PEG-FA, the growth media was replaced with SWNT-containing media prepared by mixing 2X concentrated DMEM lacking FA and FBS but containing P/S and Amp B with a SWNT-PL-PEG-FA solution in a 1:1 ratio. The concentration of the SWNT-PL-PEG-FA solution was adjusted to be 2X the desired final SWNT-PL-PEG-FA concentration in the media. Three types of controls were typically performed in these experiments: 1) control cells were incubated with the same concentrations of SWNT-PL-PEG-FA as the testing group but were not irradiated, 2) control cells were not incubated with SWNT-PL-PEG-FA but irradiated either as a function of NIR laser power density or NIR exposure time, and 3) control cells were neither incubated with SWNTs nor irradiated. To assess the effect of laser power density, cells were incubated with media that contained SWNT-PL-PEG-FA at a concentration of 10 μg/mL in the media for 6 h at 37 °C. The cells were prepared for NIR irradiation as described and irradiated for 0, 2, 4, 6, 7, 8, and 9 W/cm2 for a fixed 5 min exposure time. To assess the effect of laser exposure time, cells were incubated with 10 μg/mL SWNT-PL-PEG-FA in media for 6 h at 37 °C as before but irradiated at a constant power density of 8 W/cm2 for 0, 1, 2, 3, 4, and 5 min. To assess the effect of surface–bound SWNT-PL-PEG-FA on cell killing as a function of SWNT concentration in media, cells were incubated with media that contained SWNT-PL-PEG-FA at 0, 10, 25, 50, and 90 μg/mL at 4 °C for 30 min, washed, and irradiated at 8 W/cm2 for 5 min. The effect of internalized SWNT-PL-PEG-FA on cell viability was assessed by incubating cells with SWNT-PL-PEG-FA in media at 0, 1, 5, 10, 15, and 20 μg/mL at 37 °C for 6 h. The cells were then washed, and irradiated also at 8 W/cm2 for 5 min. Cell viability was measured after a 24-h rest at 37 °C post irradiation, as described in the preceding section.
3. RESULTS
3.1 Preparation and characterization of the SWNT-phospholipid-polyethylene glycol-folic acid (SWNT-PL-PEG-FA) construct
The general approach in this work was to non-covalently functionalize carboxylated SWNTs with PL-PEG-FA to target FRs that are often over-expressed on tumor cells. Mildly carboxylated SWNTs were chosen because this type of SWNT can be degraded both in vitro and in vivo by myeloperoxidase present in neutrophils and by a superoxide/peroxynitrite oxidative pathway in macrophages, providing a way to clear the SWNTs from an intact organism [27–30]. FA was non-covalently attached to SWNTs using PL-PEG-NH2 as generally described in the literature [1]. It has been noted that there is very little dissociation of PL-PEG from the SWNTs in full mouse serum, attesting to the stability of the non-covalently functionalized material [31]. In brief, FA was conjugated to PL-PEG-NH2 and dialyzed to remove unbound FA. To verify the attachment of FA to the PL-PEG, UV-Vis spectra from 200 to 450 nm for PL-PEG-NH2, PL-PEG-FA, and non-conjugated FA were compared. As expected, PL-PEG-NH2 had little absorbance in this region, whereas the spectrum for dialyzed PL-PEG-FA had peaks similar to non-conjugated free FA (Figure 1). To determine whether the FA was covalently or non-covalently associated with PL-PEG-NH2, PL-PEG-FA samples before and after dialysis were subjected to SDS-PAGE analysis side-by-side with non-conjugated free FA and PL-PEG-NH2 as reference materials. SDS binds PL-PEG polymer, imparts a negative charge, and the constructs migrate to the anode, as previously noted [24]. The presence of free FA in the gel is indicated by the yellow color in the dye front that is present in both FA and PL-PEG-FA prior to dialysis (Figure 2). This band is absent in lanes containing PL-PEG-NH2 and PL-PEG-FA sample after dialysis, evidence of removal of free FA from the PL-PEG-FA construct. In addition, the gel was stained with barium iodine (BaI2) to detect the PEG portion of PL-PEG-NH2 and PL-PEG-FA [23, 24] as described in the Materials and Methods section. It is apparent in Figure 2 that free FA and PL-PEG-NH2 have distinct electrophoretic mobilities of their own while the PL-PEG-FA bands are slightly shifted up and have tails, evidence that the FA is covalently attached to the PL-PEG-NH2 in the PL-PEG-FA construct. Based on the maximum absorbance of FA that peaks at 360 nm, as shown in Figure 1, a standard curve was prepared with known amounts of FA and the stoichiometry of FA with respect to the amount of PL-PEG-FA was estimated to be a molar ratio of 1:1.
Figure 1. Absorption spectra of free and PL-PEG-bound folate.
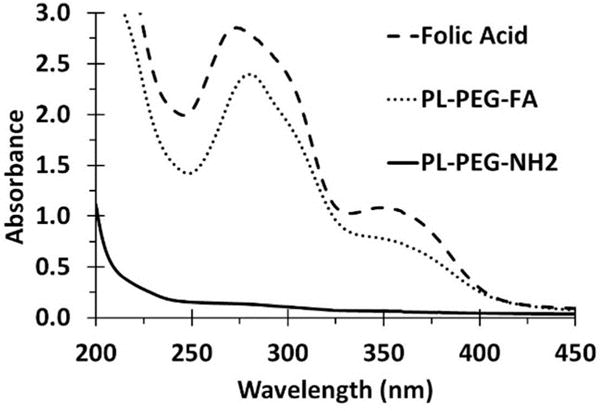
Representative absorption spectra of free folic acid (FA), PL-PEG-NH2, and dialyzed PL-PEG-FA.
Figure 2. SDS-PAGE analysis of PL-PEG-FA before and after dialysis to remove unbound FA.
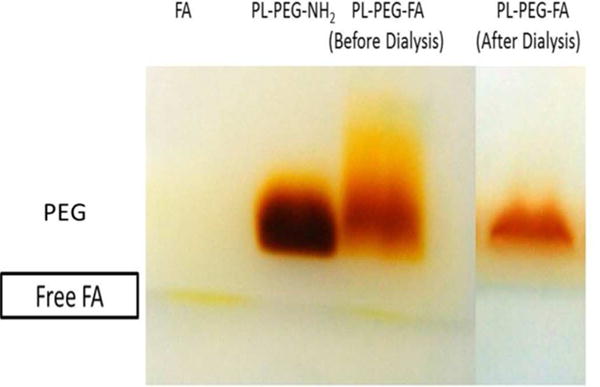
PL-PEG-NH2 was conjugated with FA and the excess, unbound free FA was removed by dialysis for 3 d using a 6000–8000 Da dialysis membrane. To verify the complete removal of unbound FA by dialysis, FA, PL-PEG-NH2, and PL-PEG-FA before and after dialysis were resolved on SDS-PAGE followed by BaI2 staining for PL-PEG.
SWNTs were non-covalently functionalized with PL-PEG-FA by a sonication method that has been widely used in the literature [1, 8]. However, a caveat with the sonication approach is that materials containing PEG are very susceptible to sonolytic degradation [32], which not only destroys the PEG, but may generate toxic byproducts [26]. The conditions of sonication used here were carefully chosen to avoid breakdown of PL-PEG-FA [24] and the integrity of the PEG in the final SWNT-PL-PEG-FA construct was verified using the SDS-PAGE method followed by the BaI2 staining (data not shown), as we previously described [23, 24]. The particle size distribution and aggregation states of the SWNT-PL-PEG-FA construct in PBS were analyzed by DLS (Figure S1). The average HD was found to be 101.75 ± 4.69 nm and the PDI was 0.24 ± 0.04. The relatively small mean hydrodynamic partial sizes near 100 nm in diameter accompanied by a low PDI suggests that the SWNT-PL-PEG-FA constructs were not extensively aggregated in physiological solutions.
3.2 Overexpression of FRs on NRK cells and binding of SWNT-PL-PEG-FA to cells at 4 °C
NRK cells were chosen as the model cell line because they can be made to over-express FRs by culturing them in medium with low FA and they have a flat morphology conducive to imaging experiments [26]. NRK cells were cultured in DMEM without FA for at least 4 passages to maximize FR overexpression. Cells with overexpressed FRs were termed FR(+) and those without overexpression are termed FR(−). To assess overexpression, cells were grown on glass cover slips, fixed, and incubated with an anti-folate receptor primary antibody and an Alexa Fluor® 488-tagged secondary antibody followed by immunofluorescence imaging. Negative control FR(+) and FR(−) cells that received neither primary nor secondary antibody and cells that received only the secondary antibody displayed negligible fluorescence (Figure 3A, top and middle). FR(+) NRK cells incubated with both primary and secondary antibody showed strong fluorescence whereas FR(−) cells were weakly fluorescent (Figure 3A, bottom). The relative increase in fluorescence intensity in FR(+) cells was determined using ImageJ software and there was a 20-fold increase in the fluorescence of FR(+) cells compared to FR(−) NRK cells (Figure 3B) indicating high expression of FR in FR(+) cells.
Figure 3. Overexpression of FRs on NRK cells.
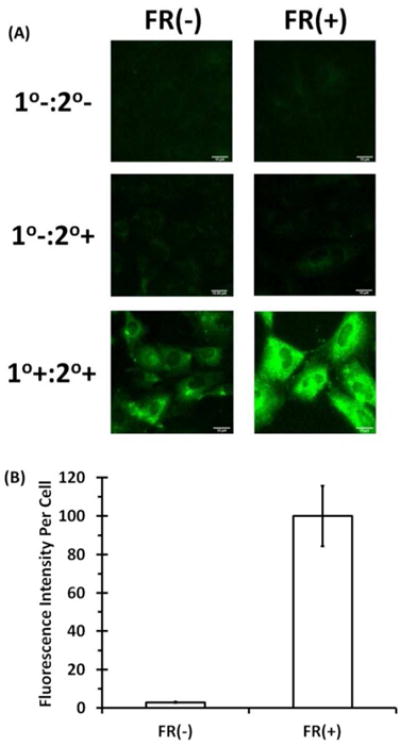
To maximize the number of FRs on NRK cells, the cells were incubated in media without FA. (A) Immunofluorescence images of FR(−) and FR(+) cells stained with rabbit anti-folate receptor primary antibody followed by Alexa fluor 488 goat anti-rabbit secondary antibody. (1°−:2° −) no primary or secondary antibody; (1°−:2°+) primary antibody absent, secondary antibody present; (1°+:2°+) both primary and secondary antibodies present. (B) The fluorescence intensity for each cell line was then quantified using Image J. Immunofluorescence was performed on three independent coverslips with at least 20 images acquired per coverslip. The bars are the mean fluorescence intensities per cell ± the standard error of the mean (SEM). The mean fluorescence intensity per cell of the FR(+) cells treated with both the primary and secondary antibodies was set as 100%.
To test the targeting of SWNT-PL-PEG-FA to FR(+) and FR(−) cells, binding assays were performed at 4 °C to minimize energy-dependent internalization and the amount of SWNTs associated with cells as a function of SWNT-PL-PEG-FA concentration in media was determined. FR(+) and FR(−) NRK cells were incubated with media that contained SWNT-PL-PEG-FA at concentrations of 0, 10, 25, 50, and 90 μg/mL at 4 °C for 30 min. The cells were washed, lysed, and the amounts of cell-associated SWNTs were measured by SDS-PAGE as described in the Materials and Methods section. Additional details regarding this procedure and a representative SWNT concentration calibration curve used to estimate the SWNT content in a cell lysate sample are provided in Supplemental Information (Figure S2). With FR(+) cells, binding increased as a function of SWNT concentration in media and a near plateau level of binding was detected with a SWNT concentration of ~50 μg/mL, suggesting an approach to binding saturation (Figure 4B). FR(−) NRK cells showed a significantly lower binding level, evidence that SWNT-PL-PEG-FA binding was specific for FRs on overexpressing NRK cells. It is interesting to note that FR(+) cells expressed ~20 times more FRs than FR(−) cells, demonstrated by immunofluorescence analysis with anti-FR antibodies (Figure 3), but they did not bind 20 times as much SWNT-PL-PEG-FA, instead, a 3–4 times greater binding of SWNT-PL-PEG-FA to FR(+) than to FR(−) cells was observed. One explanation for this is that SWNT-PL-PEG-FA is likely to be multivalent (i.e., a single SWNT particle will have multiple PL-PEG-FA moieties associated with it) and thus is able to bind multiple FRs on FR(+) cells where the receptor density is much higher than on FR(−) cells. Hence, a higher ratio of FRs to SWNT-PL-PEG-FA in FR(+) cells may result in a less than expected total amount of SWNT-PL-PEG-FA bound, nevertheless, still significantly greater than the amount bound to FR(−) cells (Figure 4B).
Figure 4. Binding of SWNT-PL-PEG-FA to cells at 4 °C.
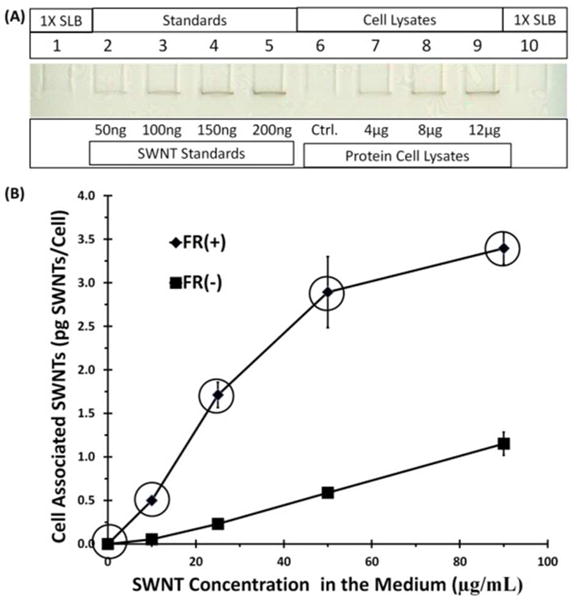
FR(+) and FR(−) NRK cells were incubated with SWNT-PL-PEG-FA at concentrations of 0, 10, 25, 50, and 90 μg/mL for 30 min at 4 °C. The cells were lysed and extracted SWNTs were quantified by SDS-PAGE. (A) Image of a representative gel used for quantification with sample loading buffer (SLB) loaded on lanes 1 and 10 as blank background controls, lanes 2–5 loaded with increasing known amounts of SWNTs in ng per lane, lane 6 (Ctrl.) was a cell lysate background control loaded with 12 μg of protein cell lysate from FR(+)/FR(−) cells not incubated with SWNT-PL-PEG-FA, and lanes 7–9 were loaded with cell lysates in μg protein per lane prepared from FR(+) cells that had been incubated with 50 μg/mL of SWNT-PL-PEG-FA. (B) Plots of SWNT concentrations in the medium versus the amount of SWNTs bound at 4 °C. Data points are the average of three independent experiments, with quadruplicates of each data point in each experiment, ± SEM. The circled data points were used in Figure 8 (see text).
3.3 Accumulation of SWNT-PL-PEG-FA by FR(+) NRK cells at 37 °C
The results in the previous section suggested that the binding of SWNT-PL-PEG-FA to FR(+) cells was a saturable and specific event that occurs at 4 °C where internalization is inhibited. To determine whether internalization of SWNT-PL-PEG-FA would occur at 37 °C, the accumulation of SWNT-PL-PEG-FA by FR(+) cells was measured also as a function of SWNT-PL-PEG-FA concentration in media after a 6 h continuous exposure at 37 °C. FR(+) NRK cells were incubated in media that contained SWNT-PL-PEG-FA at sub-saturating concentrations of 0, 1, 5, and 10 μg/mL for 6 h at 37 °C and the amount of cell-associated SWNTs were measured by SDS-PAGE as described in the Materials and Methods section. The observation that the amount of cell-associated SWNTs after incubation at 37 °C was much greater (~13 pg SWNTs/cell after incubation with 10 μg/mL SWNTs in the medium) than that after surface binding at 4 °C (~3.5 pg SWNTs/cell after incubation with 90 μg/mL SWNTs in the medium), which indicates that internalization and accumulation of SWNT-PL-PEG-FA was robust at physiological temperature (Figure 5B). It was also notable that a plateau level of SWNT accumulation was detected with a SWNT-PL-PEG-FA concentration of ~5 μg/mL in the media. One possible explanation for the plateau is that the internalized SWNTs are degraded with time such that the amount of SWNTs entering the cell equals the amount degraded, resulting in a constant amount of SWNTs in the cells; however, the enzymes known to degrade SWNTs are present primarily in neutrophils and macrophages [27–29, 33, 34]. Another explanation is that the internalized SWNTs are subsequently exocytosed such that rates of uptake and exocytosis are equal, resulting in a steady state where the cell associated SWNT level does not change [15]. Alternatively, all the available FRs in FR(+) cells were occupied upon continuous incubation with excess SWNT-PL-PEG-FA in the media so that no further binding of FA-targeted SWNTs occurs when higher concentrations of SWNT-PL-PEG-FA were present in the media. The latter explanation is the most likely one, but further work is necessary to distinguish these possibilities.
Figure 5. Accumulation of SWNT-PL-PEG-FA by cells at 37 °C as a function of SWNT concentration in the medium.
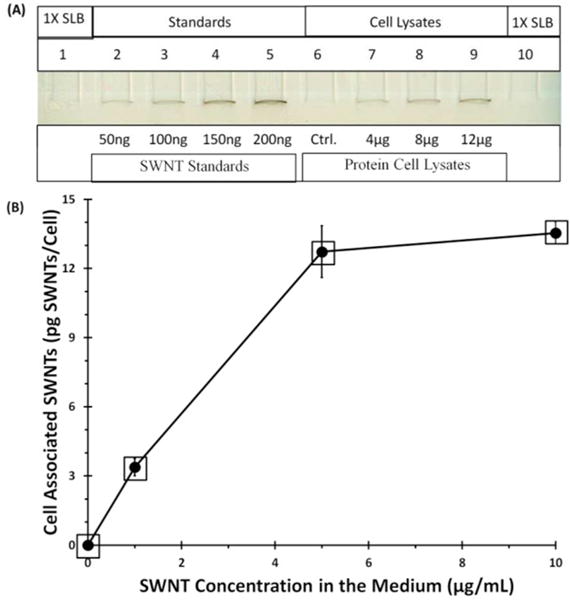
FR(+) NRK cells were incubated with SWNT-PL-PEG-FA at 0, 1, 5, and 10 μg/mL for 6 h at 37 °C. Cell associated SWNTs were extracted and quantified as in Figure 4. (A) Image of a representative gel used for quantification with sample loading buffer (SLB) loaded on lanes 1 and 10 as blank background controls, lanes 2–5 loaded with increasing known amounts of SWNTs in ng per lane, lane 6 (Ctrl.) was a cell lysate background control loaded with 12 μg of protein cell lysate from FR(+) cells not incubated with SWNT-PL-PEG-FA, and lanes 7–9 were loaded with cell lysates in μg protein per lane prepared from FR(+) cells that had been incubated with 10 μg/mL of SWNT-PL-PEG-FA. (B) Plots of SWNT concentration in the medium versus cell associated SWNTs. Data points are the average of three independent experiments, with quadruplicates of each data point in each experiment, ± SEM. The squared data points were used in Figure 8 (see text).
3.4 Assessing the subcellular location of SWNTs by combining Raman and immunofluorescence imaging
To assess the subcellular location of SWNT-PL-PEG-FA in FR(+) NRK cells, a combination of Raman and immunofluorescence imaging techniques was performed on the same cells. FR(+) NRK cells were seeded on gridded cover slips and incubated either with 10 μg/mL of SWNT-PL-PEG-FA in media at 37 °C for 6 h, a condition that permit SWNT-PL-PEG-FA internalization, or with 90 μg/mL of SWNT-PL-PEG-FA in media at 4 °C for 30 min, a condition that confine SWNT-PL-PEG-FA bound to the cell surface. Lysosomes in cells were identified by immunofluorescence using a primary antibody to LAMP-1 and an Alexa Fluor® 488-tagged secondary antibody. After fluorescence images were acquired, the samples were prepared for Raman imaging. The coverslips were gently removed from the glass slides, washed to remove the fluoromount, photo bleached to diminish Alexa Fluor® 488 fluorescence, and air dried. Cells whose lysosomes had been previously identified by LAMP-1 immunofluorescence were located by their grid positions and subjected to laser scanning confocal Raman microscopy. The subcellular locations of SWNTs in the cells were identified by the presence of the SWNT signature Raman G-band (~1590 cm−1) upon laser excitation at 532 nm.
Figure 6A shows a representative immunofluorescence image of a FR(+) cell, after incubation with SWNT-PL-PEG-FA at 37 °C for 6 h, where the locations of the nucleus (blue, DAPI) and lysosomes (green, Alexa Fluor® 488) were identified. The same cell was subsequently imaged by laser scanning confocal Raman microscopy to locate the characteristic SWNT G-band Raman signal and the resulting Raman scan image was merged with the fluorescence image (Figure 6B). Figure 6C shows the bright field microscopy image of a single cell overlaid with both the fluorescence and Raman images. It is evident from Figures 6A, B, and C that some of the SWNTs co-localized with lysosomes (an example is noted with the white arrow) near the nuclear region, whereas others are in LAMP-1 negative punctate structures, most likely endosomes. There is no significant localization of SWNTs with the nucleus. The corresponding phase contrast, fluorescence, and Raman scan images of a representative FR(+) cells with internalized SWNTs are shown individually in the Supplemental Information section (Figure S3, panels A1–A6).
Figure 6. Assessing the subcellular location of SWNTs by combining Raman and immunofluorescence imaging.
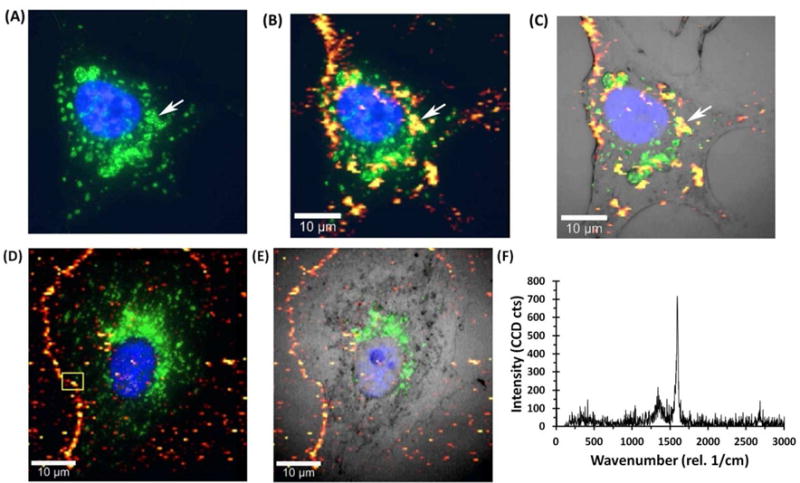
Raman and immunofluorescence images were merged to locate SWNTs with respect to the outer membrane, nucleus, and lysosomes. For SWNTs internalized (A, B, and C), FR(+) NRK cells were incubated with 10 μg/mL SWNT-PL-PEG-FA for 6 h at 37 °C. For SWNTs localized to the cell surface (D and E), FR(+) NRK cells were incubated with 90 μg/mL SWNT-PL-PEG-FA for 30 min at 4 °C. Immunofluorescence was first performed using anti-LAMP-1 antibody to locate lysosomes. The nucleus was stained with the fluorescent dye DAPI. After immunofluorescence, the cells were bleached by placing the samples under standard room light for 72 h to reduce the fluorescent signal and SWNTs were imaged by confocal laser scanning Raman microscopy of the same cells. (A) Alexa Fluor® 488 tagged lysosomal (green) and nuclear (blue) staining with DAPI. The arrow indicates a representative lysosome. (B) Merging of the Raman image intensity of the G-band (on a scale from red to yellow with yellow being the most intense) and the immunofluorescence image. The white arrow indicates an example of co-localization of LAMP-1 and SWNTs in the same structures. (C) Merging of Raman and immunofluorescence images with an optical light image of the same cell. (D) Merging of the Raman image for surface bound SWNTs and the immunofluorescence image for lysosomal staining. (E) Merging of the Raman image for surface bound SWNTs and immunofluorescence for lysosomal staining with a light microscope image of the cell. (F) Representative Raman spectrum acquired from the boxed area in D where the two characteristic SWNT Raman shifts (i.e., the D-band at ~1350 cm−1 and G-band at ~1590 cm−1) are evident.
Figure 6D shows the merged fluorescence and Raman images of a representative FR(+) cell incubated with SWNT-PL-PEG-FA at 4 °C and Figure 6E shows the bright field microscopy image of the same cell. Only a background level of SWNT Raman signal was detected within the cell boundary at this condition where endocytosis is inhibited by the low temperature. Most importantly, there is a Raman G-band signal detected along the perimeter of the cell suggesting the presence of SWNTs on the cell surface. Figure 6F shows a typical Raman spectrum acquired from a pixel in Figure 6D, indicated by the yellow square box, where the characteristic SWNT D-band (~1350 cm−1) and G-band (~1590 cm−1) Raman shifts were evident. In addition, corresponding phase contrast, fluorescence, and Raman scan images of a representative FR(+) cells with surface bound SWNTs are shown individually in the Supplemental Information section (Figure S3, panels B1–B6). Altogether, the results shown in Figures 6 and S3 suggest that SWNT-PL-PEG-FA are on the cell surface and not internalized at 4 °C, but at 37 °C, they are internalized and accumulate in the vacuolar apparatus that includes LAMP-1 positive lysosomes and possibly LAMP-1 negative endosomes.
3.5 Optimizing NIR laser conditions for targeted ablation of FR(+) NRK cells using SWNT-PL-PEG-FA
Optimum laser conditions were identified prior to assessing the efficacy of NIR-mediated thermal ablation as a function of SWNT concentration and whether the SWNT-PL-PEG-FA was internalized or bound to the cell surface. To optimize the NIR laser power, FR(+) NRK cells were incubated either with no SWNTs, or with 10 μg/mL SWNT-PL-PEG-FA in the media at 37 °C for 6 h, washed, and irradiated at various NIR power densities of 0, 2, 4, 6, 7, 8, and 9 W/cm2 with a fixed 5 min exposure time (Figure S4A). Similarly, experiments were performed to assess the effect of NIR exposure time where FR(+) NRK cells were incubated with 10 μg/mL SWNT-PL-PEG-FA in the media at 37 °C for 6 h as before but irradiated for 0, 1, 2, 3, 4, and 5 min at a constant power density of 8 W/cm2 (Figure S4B). The temperatures of the media of the control cells were measured immediately after irradiation and were found to be less than 35 °C. Post irradiation, fresh media was added and control and experimental cells were incubated at 37 °C for an additional 24 h. Cell survival was measured with a CV assay as described in Materials and Methods [21, 26]. Results shown in Figures S4A and S4B demonstrate that lethality increased as a function of both NIR laser power density and NIR exposure time. Control cells not incubated with SWNTs but irradiated with NIR were unaffected under the same NIR laser conditions. Thus, the optimum NIR laser conditions were determined to be 8 W/cm2 and 5 min continuous irradiation to achieve near complete ablation of FR(+) cells that accumulated SWNTs and assert no detectable adverse effects on control cells.
3.6 Ablation of FR(+) NRK cells as a function of SWNT-PL-PEG-FA concentration in the medium
Using the optimized NIR laser condition described in the previous section, the effect of the applied SWNT concentration on ablation efficiency was assessed by incubating FR(+) NRK cells with media that contained SWNT-PL-PEG-FA at various concentrations at either 4 °C to confine the SWNTs to the cell surface or at 37 °C to allow SWNTs to be internalized. For the cell surface-bound scenario, FR(+) NRK cells were incubated with SWNT-PL-PEG-FA at 0, 10, 25, 50, and 90 μg/mL in the media at 4 °C for 30 min. For the internalized scenario, FR(+) NRK cells were incubated with SWNT-PL-PEG-FA construct in the media at concentrations (0, 1, 5, 10, 15, and 20 μg/mL) at 37 °C for 6 h. The cells were washed and irradiated at 8 W/cm2 for 5 min, fresh medium was added and cell survival was assessed after 24 h incubation under standard growth conditions. For cell surface-bound SWNT-PL-PEG-FA, there was no significant cell death observed (Figure 7A), even for cells incubated at 4 °C with 90 μg/mL SWNT-PL-PEG-FA in the media, the highest concentration tested at which a plateau of binding was demonstrated (see Figure 4). In contrast, there was more than 90% cell death observed for cells incubated at 37 °C with only 5 μg/mL of SWNT-PL-PEG-FA in the media, when the SWNT-PL-PEG-FA was internalized at physiological temperature (Figure 7B). In addition, no cell death was detected in control cells that were allowed to internalize SWNT-PL-PEG-FA under the same incubation condition but not subjected to NIR laser irradiation (Figure 7B), indicating that the observed 90% cell death was a combined effort that require the presence of internalized SWNT-PL-PEG-FA in these cells accompanied by proper NIR laser irradiation. All together, these results support the idea that differences in subcellular locations (i.e., SWNTs either internalized or cell surface bound) impact the efficacy of NIR irradiation on cell killing.
Figure 7. Targeted ablation of FR(+) NRK cells using SWNT-PL-PEG-FA as a function of incubated SWNT concentration.
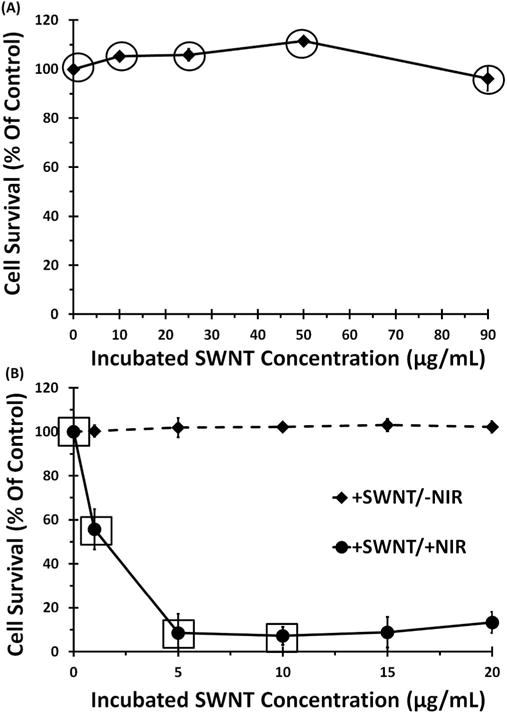
FR(+) NRK cells were incubated with SWNT-PL-PEG-FA (A) at 4 °C for 30 min to confine binding to the cell surface or (B) at 37 °C for 6 h to allow internalization. Cells were irradiated under identical conditions at a laser power density of 8 W/cm2 for 5 min. Controls were as described in Figure S4. Cell viability after irradiation was determined using CV assay, as described in Materials and Methods. Data points are the average of four independent experiments, with quadruplicates repeats in each experiment, ± SEM. The circled data points from (A) and squared data points from (B) were used in Figure 8.
3.7 Correlating cell death with the amount of internalized and/or surface-bound SWNTs
Data in the previous sections provided information on 1) the amounts of cell-associated SWNTs as a function of SWNT-PL-PEG-FA concentration in media and 2) the extent of NIR mediated cell killing also as a function of SWNT-PL-PEG-FA concentration in media. Together, these two sets of data can be combined to correlate the amount of cell-associated SWNTs with the extent of NIR-mediated cell killing. For SWNTs on the surface of FR(+) cells, the circled data points in Figure 4B give the average amounts of SWNTs on the cell surface corresponding to the concentrations in the incubation medium of 0, 10, 25, 50, and 90 μg/mL. The effects of NIR irradiation on cells incubated with these same concentrations of SWNT-PL-PEG-FA in media are shown as circled data points in Figure 7A. The relationship between the dose of cell surface-bound SWNTs and the NIR ablation efficacy is shown in Figure 8A and revealed that there was little NIR-mediated killing even at the highest cell surface-bound dose tested, ~3.5 pg SWNTs/cell (Figure 8A). Similarly, for cells that had internalized SWNTs at 37 °C for 6 h, the squared data points in Figure 5B show the amounts of SWNTs accumulated as a function of SWNT-PL-PEG-FA concentration in the media and the squared data points in Figure 7B show the corresponding effect of NIR irradiation on cells as a function of SWNT-PL-PEG-FA concentration in the media. This information is combined in Figure 8B to correlate the amounts of internalized SWNTs dose with the effects of NIR irradiation on the cells. Cell viability declined as a function of accumulated SWNT dose and survival was less than 10% at ~13.5 pg SWNTs/cell. Most interesting, however, was the direct comparison of the effects of NIR irradiation on cells that contain the same SWNT dose of ~3.5 pg/cell but differed only in the subcellular locations of the SWNT: NIR-irradiation had little effect on cells with the SWNTs bound to the cell surface whereas viability was reduced to ~50% when the SWNTs were accumulated in the endosomal/lysosomal compartments (Figure 8B inset, p value of ≤ 0.005).
Figure 8. Correlation between NIR-mediated cell death and the quantities of surface-bound or internalized SWNTs.
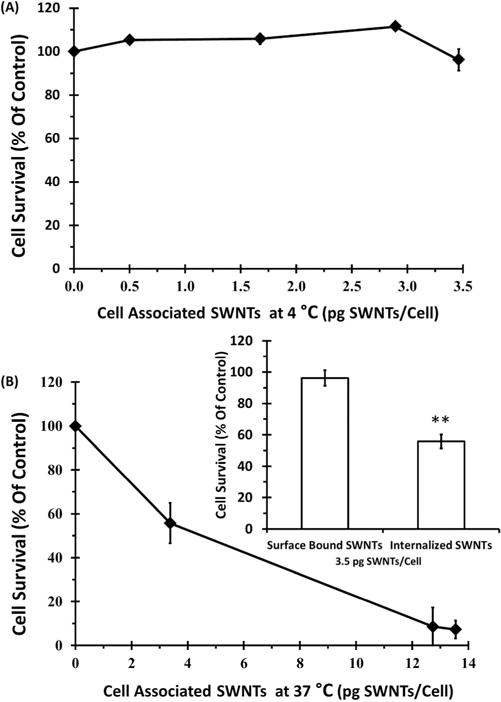
The efficacies of NIR ablation indicated by the decrease in cell viability after laser irradiation were plotted as a function of cell-associated SWNT doses measured in the internalized or the cell surface bound systems. (A) SWNTs confined on cell surface system: cell surface bound SWNTs doses (circled data points in Figure 4B) were correlated to the extent of cell killing (circled data points in Figure 7A). (B) SWNTs internalized system: doses of internalized SWNTs (squared data points in Figure 5B) were correlated to ablation (squared data points in Figure 7B). (B, inset) The cell viability after NIR irradiation at equivalent ~3.5 pg/cell SWNTs under internalized or cell surface bound conditions are compared and a p value of ≤ 0.005 was acquired by 2 tail t-test with equal variances (**).
4. DISCUSSION
An important question in the field of photo-thermal cell ablation mediated by nanoparticle absorption of NIR light is what quantity of nanoparticles need to be associated with cells to efficiently kill them upon exposure to NIR light. This issue has been experimentally addressed for metal or metal-containing nanoparticles using inductively coupled plasma (ICP)-mass spectrometry (MS) and ICP-optical emission spectroscopy (OES) to quantify metal nanoparticle dose in the cell. For example, Au et al. studied gold nanocages (AuNCs) conjugated with anti-HER-2 MAb to target SK-BR-3 breast cancer cells bearing the HER-2 marker [35]. The total number of AuNCs per cell was roughly 400 ± 90 as determined by flow cytometry and ICP-MS. At this dose per cell, the AuNCs responded to NIR laser irradiation (1.6 W/cm2) and caused cell damage. A later study quantifying Au nanorods (AuNRs) and Au nanoshells (AuNSs) was carried out by Wu et al. in A549 and HeLa cells [36]. The authors measured EGF-targeted metal nanoparticles by ICP-MS and demonstrated that the minimum number of particles required for effective ablation of cells depended on the type of nanoparticle. The minimum numbers of Her-2 targeted AuNRs and AuNSs were 1.1 × 105 and 4.1 × 102 particles per cell, respectively. The minimum numbers of EGFR targeted AuNRs and AuNSs were at 1.9 × 105 and 5.4 × 102 particles per cell. This type of quantitative information collected with cells in vitro provides guidelines for dosing tumor targets with intact animals. However, similar data has not been available for SWNTs.
Several groups have described the use of folate to target SWNTs to folate receptors in cell studies [1, 8, 37]; however, none have addressed the question of what SWNT load per cell is needed to kill cells, nor compared whether SWNTs on the cell surface are as potent as SWNTs inside the cells in mediating cell death. In the present work, the quantity of folate-targeted SWNTs confined to the plasma membrane at 4 °C were measured and correlated with the efficiency of cell killing upon exposure to NIR light as a function of the dose of cell surface-bound SWNTs. The approach was extended to quantify targeted SWNTs that had populated endosomal/lysosomal vesicles after incubation at 37 °C and the corresponding efficiency of cell killing acquired under identical laser ablation conditions. Acquisition of both data sets allowed a comparison of NIR-mediated cell killing efficacies derived from cell surface-bound SWNTs versus internalized SWNTs, normalized to equivalent SWNTs dose per cell at either location, information not previously available.
The binding of folate-targeted SWNTs to the surface of FR(+) NRK cells at low temperature showed binding as a function of dose that approached saturation at ~3.5 pg SWNTs/cell, indicating a limit on the dose per cell under conditions where internalization could not occur. The SWNTs were primarily on the plasma membrane and not within the cell, based on direct detection of the SWNT G-band by scanning confocal Raman imaging. Viability measurements at various SWNT doses in the binding isotherm upon exposure to NIR light revealed very little effect on cells, even at a dose near saturation. On the contrary, Xiao et al. reported that multi-wall CNTs bound to HER-2 receptors on the plasma membrane of SK-BR-3 cells were lethal upon exposure to NIR light [6]. A greater cell killing efficacy using our SWNT-PL-PEG-FA construct would have been possible if there were a higher number of FA receptors on the NRK cell membrane. Nevertheless, a dose of ~3.5 pg/cell of SWNTs on the cell surface provided a benchmark dose where NIR exposure was not lethal.
When FR(+) NRK cells were incubated for 6 h with folate-targeted SWNTs at 37 °C to permit receptor-mediated endocytosis, SWNTs accumulated as a function of SWNT-PL-PEG-FA concentration in the medium and the accumulation levels approached a plateau at SWNT-PL-PEG-FA concentrations of 10 μg/mL or higher in the media. A combination of laser-scanning confocal Raman imaging and fluorescence microscopy lead to the conclusion that most of the SWNTs were accumulated within the cell after a 6 h incubation at 37 °C. Some SWNTs co-localized with the lysosomal marker LAMP-1, while LAMP-1 negative structures that contained SWNTs were probably endosomes. No significant accumulation of the SWNTs within the nucleus was observed.
The data relating the efficiency of NIR-mediated cell killing to either the amount of SWNTs bound to the cell surface or the amount of SWNTs internalized supports the conclusion that SWNTs inside the cell are more potent than those on the cell surface. To our knowledge there was not sufficient data in the current literature to adequately address this question. For example, AuNRs were targeted to folate receptors and the material on the plasma membrane was reported to be more effective than internalized material for photothermal ablation [38]. Results of studies that failed to consider the possible differences in the doses of the surface-bound and the internalized AuNRs should be taken with caution. For SWNTs targeted by HER-2 antibodies to the surface of SK-BR-3 breast tumor cells, Xiao et al. reported that surface bound SWNTs can be lethal, but provided no information on the efficacy of intracellular SWNTs [6]. Additionally, Marches et al. compared HER-2-targeted SWNTs located either on BT-474 cell surfaces or within cells for NIR-mediated cell killing and observed that internalized material was more lethal; however, these data were also not normalized to the amounts of SWNTs in either of the two locations [7].
There are several potential explanations for why SWNTs within the vacuolar compartment are more effective in cell killing. One is that damaging lysosomes by local heating upon NIR irradiation releases lysosomal hydrolases to the cytoplasm that enhance cell killing by digesting cytoplasmic material. Another possibility is that SWNTs could be more concentrated locally in the vacuolar membrane after endocytosis than in the plasma membrane, which might result in more efficient membrane damage and higher lethality on exposure to NIR light. In addition, lysosomal membranes could be inherently more sensitive to local thermal damage than the plasma membrane. Nevertheless, understanding that internalized SWNTs are more effective in killing cells in this in vitro model suggests that application of the approach to intact animals would benefit from strategies that optimize the amount of material internalized by target cells; for example, by targeting receptors that are efficiently endocytosed and by using sufficient incubation times to support significant internalization. Photo-thermal ablation approaches using other nanoparticles might also be aided by strategies that maximize nanoparticle internalization.
5. CONCLUSION
This paper addresses whether SWNTs on the cell surface or within the endosomal/lysosomal compartments are optimal for the NIR-mediated photo-thermal ablation of cells by folate-targeted SWNTs. Cells were incubated with the folate-targeted SWNT-PL-PEG-FA at 4 °C to confine the SWNTs to the cell surface, or were incubated with the folate-targeted SWNT-PL-PEG-FA at 37 °C to allow the construct to populate the endosomal/lysosomal compartments. The location of the SWNTs was verified by a combination of laser scanning confocal Raman microscopy to locate SWNTs merged with fluorescence images that identified lysosomes and the nucleus. The amount of the SWNTs either on the cell surface or within the endosomal/lysosomal vacuolar compartments was quantitated and the extent of cell killing after NIR irradiation was normalized to a equivalent quantity of SWNTs in the two locations. The results revealed that ~3.5 pg of SWNTs/cell on the cell surface killed less than ~5% of the cells whereas the same amount of SWNTs internalized within the vacuolar compartment killed ~50% of the cells. When the amount of internalized SWNTs reached ~13 pg/cell of SWNTs, greater than 90% of the cells were killed upon exposure to NIR light. These results support the conclusion that internalized SWNTs are more efficient in NIR-mediated photo-thermal cell killing than SWNTs on the cell surface.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Cancer Institute under award number R15-CA152917 and the National Institute for Environmental Health Sciences under award number R15-ES023666.
Footnotes
Conflict of Interest: The authors declare no competing financial interest.
Disclaimer: The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the National Institute for Environmental Health Sciences.
References
- 1.Kam NWS, O’Connell M, Wisdom JA, Dai H. Carbon Nanotubes as Multifunctional Biological Transporters and Near-Infrared Agents for Selective Cancer Cell Destruction. Proc Natl Acad Sci USA. 2005;102:11600–5. doi: 10.1073/pnas.0502680102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Panchapakesan B, Lu S, Sivakumar K, Taker K, Cesarone G, Wickstrom E. Single-Wall Carbon Nanotube Nanobomb Agents for Killing Breast Cancer Cells. NanoBiotechnology. 2005;1:133–9. [Google Scholar]
- 3.Shao N, Shaoxin L, Eric W, Balaji P. Integrated Molecular Targeting of IGF1R and HER2 Surface Receptors and Destruction of Breast Cancer Cells using Single Wall Carbon Nanotubes. Nanotechnology. 2007;18:315101. [Google Scholar]
- 4.Chakravarty P, Marches R, Zimmerman NS, Swafford ADE, Bajaj P, Musselman IH, Pantano P, Draper RK, Vitetta ES. Thermal Ablation of Tumor Cells with Antibody-Functionalized Single-Walled Carbon Nanotubes. Proc Natl Acad Sci USA. 2008;105:8697–702. doi: 10.1073/pnas.0803557105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marches R, Chakravarty P, Musselman IH, Bajaj P, Azad RN, Pantano P, Draper RK, Vitetta ES. Specific Thermal Ablation of Tumor Cells using Single-Walled Carbon Nanotubes Targeted by Covalently-Coupled Monoclonal Antibodies. Intl J Cancer. 2009;125:2970–7. doi: 10.1002/ijc.24659. [DOI] [PubMed] [Google Scholar]
- 6.Xiao Y, Gao X, Taratula O, Treado S, Urbas A, Holbrook RD, Cavicchi R, Avedisian CT, Mitra S, Savla R, Wagner P, Srivastava S, He H. Anti-HER2 IgY Antibody-Functionalized Single-Walled Carbon Nanotubes for Detection and Selective Destruction of Breast Cancer Cells. BMC Cancer. 2009;9:351–62. doi: 10.1186/1471-2407-9-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marches R, Mikoryak C, Wang R, Pantano P, Draper RK, Vitetta ES. The Importance of Cellular Internalization of Antibody-Targeted Carbon Nanotubes in the Photothermal Ablation of Breast Cancer Cells. Nanotechnology. 2011;22:095101. doi: 10.1088/0957-4484/22/9/095101. [DOI] [PubMed] [Google Scholar]
- 8.Zhou F, Resasco DE, Chen WR, Xing D, Ou Z, Wu B. Cancer Photothermal Therapy in the Near-Infrared Region by Using Single-Walled Carbon Nanotubes. J Biomed Opt. 2009;14:0210091–7. doi: 10.1117/1.3078803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moon HK, Lee SH, Choi HC. In Vivo Near-Infrared Mediated Tumor Destruction by Photothermal Effect of Carbon Nanotubes. ACS Nano. 2009;3:3707–13. doi: 10.1021/nn900904h. [DOI] [PubMed] [Google Scholar]
- 10.Liu X, Tao H, Yang K, Zhang S, Lee S-T, Liu Z. Optimization of Surface Chemistry on Single-Walled Carbon Nanotubes for in vivo Photothermal Ablation of Tumors. Biomaterials. 2010;32:144–51. doi: 10.1016/j.biomaterials.2010.08.096. [DOI] [PubMed] [Google Scholar]
- 11.Neves FFL, John JK, Brent DVR, Rajagopal R, Daniel ER, Roger GH. Targeting Single-Walled Carbon Nanotubes for the Treatment of Breast Cancer Using Photothermal Therapy. Nanotechnology. 2013;24:375104. doi: 10.1088/0957-4484/24/37/375104. [DOI] [PubMed] [Google Scholar]
- 12.Antaris AL, Robinson JT, Yaghi OK, Hong G, Diao S, Luong R, Dai H. Ultra- Low Doses of Chirality Sorted (6,5) Carbon Nanotubes for Simultaneous Tumor Imaging and Photothermal Therapy. ACS Nano. 2013;7:3644–52. doi: 10.1021/nn4006472. [DOI] [PubMed] [Google Scholar]
- 13.Liang C, Diao S, Wang C, Gong H, Liu T, Hong G, Shi X, Dai H, Liu Z. Tumor Metastasis Inhibition by Imaging-Guided Photothermal Therapy with Single-Walled Carbon Nanotubes. Adv Mater. 2014;26:5646–52. doi: 10.1002/adma.201401825. [DOI] [PubMed] [Google Scholar]
- 14.Robinson J, Welsher K, Tabakman SM, Sherlock S, Wang H, Luong R, Dai H. High Performance In Vivo Near-IR (>1 um) Imaging and Photothermal Cancer Therapy with Carbon Nanotubes. Nano Res. 2010;3:779–93. doi: 10.1007/s12274-010-0045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin H, Heller DA, Strano MS. Single-Particle Tracking of Endocytosis and Exocytosis of Single-Walled Carbon Nanotubes in NIH-3T3 Cells. Nano Lett. 2008;8:1577–85. doi: 10.1021/nl072969s. [DOI] [PubMed] [Google Scholar]
- 16.Jin H, Heller DA, Sharma R, Strano MS. Size-Dependent Cellular Uptake and Expulsion of Single-Walled Carbon Nanotubes: Single Particle Tracking and a Generic Uptake Model for Nanoparticles. ACS Nano. 2009;3:149–58. doi: 10.1021/nn800532m. [DOI] [PubMed] [Google Scholar]
- 17.Lacerda L, Russier J, Pastorin G, Herrero MA, Venturelli E, Dumortier H, Al-Jamal KT, Prato M, Kostarelos K, Bianco A. Translocation Mechanisms of Chemically Functionalised Carbon Nanotubes Across Plasma Membranes. Biomaterials. 2012;33:3334–43. doi: 10.1016/j.biomaterials.2012.01.024. [DOI] [PubMed] [Google Scholar]
- 18.Hashida Y, Tanaka H, Zhou S, Kawakami S, Yamashita F, Murakami T, Umeyama T, Imahori H, Hashida M. Photothermal Ablation of Tumor Cells Using a Single-Walled Carbon Nanotube–Peptide Composite. J of Contrld Rel. 2014;173:59–66. doi: 10.1016/j.jconrel.2013.10.039. [DOI] [PubMed] [Google Scholar]
- 19.Pantano P, Draper R, Mikoryak C, Wang R. Electrophotreic Methods to Quantify Carbon Nanotubes in Biological Cells in Handbook of Carbon Nanomaterials. Wrld Sci Pub. 2012;3:83–106. [Google Scholar]
- 20.Wang R, Mikoryak C, Chen E, Li S, Pantano P, Draper RK. Gel Electrophoresis Method to Measure the Concentration of Single-Walled Carbon Nanotubes Extracted from Biological Tissue. Anal Chem. 2009;81:2944–52. doi: 10.1021/ac802485n. [DOI] [PubMed] [Google Scholar]
- 21.Wang R, Mikoryak C, Li S, Bushdiecker D, Musselman IH, Pantano P, Draper RK. Cytotoxicity Screening of Single-Walled Carbon Nanotubes: Detection and Removal of Cytotoxic Contaminants from Carboxylated Carbon Nanotubes. Mol Pharm. 2011;8:1351–61. doi: 10.1021/mp2001439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang R, NMeredith A, Lee M, Deutsch D, Miadzvedskaya L, Braun E, Pantano P, Harper S, Draper R. Toxicity Assessment and Bioaccumulation in Zebrafish Embryos Exposed to Carbon Nanotubes Suspended in Pluronic® F-108. Nanotoxicology. 2015 doi: 10.3109/17435390.2015.1107147. “In Press”. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kurfurst MM. Detection and Molecular Weight Determination of Polyethylene Glycol-Modified Hirudin by Staining after Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis. Anal Biochem. 1992;200:244–8. doi: 10.1016/0003-2697(92)90460-o. [DOI] [PubMed] [Google Scholar]
- 24.Murali VS, Wang R, Mikoryak CA, Pantano P, Draper R. Rapid Detection of Polyethylene Glycol Sonolysis Upon Functionalization of Carbon Nanomaterials. Exp Biol Med. 2015;240:1147–51. doi: 10.1177/1535370214567615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antony AC. Folate Receptors. Annu Rev Nutr. 1996;16:501–21. doi: 10.1146/annurev.nu.16.070196.002441. [DOI] [PubMed] [Google Scholar]
- 26.Wang R, Hughes T, Beck S, Vakil S, Li S, Pantano P, Draper RK. Generation of Toxic Degradation Products by Sonication of Puronic® Dispersants: Implications for Nanotoxicity Testing. Nanotoxicology. 2012;7:1272–81. doi: 10.3109/17435390.2012.736547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kagan VE, Konduru VN, Fend W, Allen LB, Conroy J, Volkov Y, Vlasova II, Belikova AN, Yanamala N, LKapralov A, Tyurina YY, Shi J, Kisin RE, Murray RA, Franks J, Stolz D, Gou P, Klein-Seetharaman J, Fadeel B, Star A, Shvedova A. Carbon Nanotubes Degraded by Neutrophil Myeloperoxidase Induce Less Pulmonary Inflammation. Nat Nanotech. 2010;6:354–59. doi: 10.1038/nnano.2010.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kagan VE, Kapralov AA, St Croix CM, Watkins SC, Kisin ER, Kotchey GP, Balasubramanian K, Vlasova II, Yu J, Kim K, Seo W, Mallampalli RK, Star A, Shvedova AA. Lung Macrophages “Digest” Carbon Nanotubes Using a Superoxide/Peroxynitrite Oxidative Pathway. ACS Nano. 2014;8:5610–21. doi: 10.1021/nn406484b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kotchey GP, Zhao Y, Kagan VE, Star A. Peroxidase-Mediated Biodegradation of Carbon Nanotubes In Vitro and In Vivo. Adv Drug Del Rev. 2013;65:1921–32. doi: 10.1016/j.addr.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andón FT, Kapralov AA, Yanamala N, Feng W, Baygan A, Chambers BJ, Hultenby K, Ye F, Toprak MS, Brandner BD, Fornara A, Klein-Seetharaman J, Kotchey GP, Star A, Shvedova AA, Fadeel B, Kagan VE. Biodegradation of Single-Walled Carbon Nanotubes by Eosinophil Peroxidase. Small. 2013;9:2721–9. doi: 10.1002/smll.201202508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Z, Cai W, He L, Nakayama N, Chen K, Sun X, Chen X, Dai H. In Vivo Biodistribution and Highly Efficient Tumour Targeting of Carbon Nanotubes in Mice. Nat Nano. 2007;2:47–52. doi: 10.1038/nnano.2006.170. [DOI] [PubMed] [Google Scholar]
- 32.Zeineldin R, Al-Haik M, Hudson LG. Role of Polyethylene Glycol Integrity in Specific Receptor Targeting of Carbon Nanotubes to Cancer Cells. Nano Lett. 2009;9:751–7. doi: 10.1021/nl8033174. [DOI] [PubMed] [Google Scholar]
- 33.Bianco A, Kostarelos K, Prato M. Making Carbon Nanotubes Biocompatible and Biodegradable. Chem Commun. 2011;47:10182–8. doi: 10.1039/c1cc13011k. [DOI] [PubMed] [Google Scholar]
- 34.Kunzmann A, Andersson B, Thurnherr T, Krug H, Scheynius A, Fadeel B. Toxicology of Engineered Nanomaterials: Focus on Biocompatibility, Biodistribution and Biodegradation. Biochim Biophys Acta (BBA) 2011;1810:361–73. doi: 10.1016/j.bbagen.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 35.Au L, Zheng D, Zhou F, Li Z-Y, Li X, Xia Y. A Quantitative Study on the Photothermal Effect of Immuno Gold Nanocages Targeted to Breast Cancer Cells. ACS Nano. 2008;2:1645–52. doi: 10.1021/nn800370j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu P-C, Shieh D-B, Cheng F-Y. Nanomaterial-Mediated Photothermal Cancer Treatment: The Pivotal Role of Cellular Uptake on Photothermal Therapeutic Efficacy. RSC Adv. 2014;4:53297–306. [Google Scholar]
- 37.Yang X, Zhang Z, Liu Z, Ma Y, Yang R, Chen Y. Multi-Functionalized Single- Walled Carbon Nanotubes as Tumor Cell Targeting Biological Transporters. J of Nano Res. 2008;10:815–22. [Google Scholar]
- 38.Tong L, Zhao Y, Huff TB, Hansen MN, Wei A, Cheng J-X. Gold Nanorods Mediate Tumor Cell Death by Compromising Membrane Integrity. Adv Mater. 2007;19:3136–41. doi: 10.1002/adma.200701974. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.