

Abstract
The colibactins are hybrid polyketide–nonribosomal peptide natural products produced by certain strains of commensal and extraintestinal pathogenic Escherichia coli. The metabolites are encoded by the clb gene cluster as prodrugs termed precolibactins. clb+ E. coli induce DNA double-strand breaks in mammalian cells in vitro and in vivo and are found in 55–67% of colorectal cancer patients, suggesting that mature colibactins could initiate tumorigenesis. However, elucidation of their structures has been an arduous task as the metabolites are obtained in vanishingly small quantities (μg/L) from bacterial cultures and are believed to be unstable. Herein we describe a flexible and convergent synthetic route to prepare advanced precolibactins and derivatives. The synthesis proceeds by late-stage union of two complex precursors (e.g., 28 + 17 → 29a, 90%) followed by a base-induced double dehydrative cascade reaction to form two rings of the targets (e.g., 29a → 30a, 79%). The sequence has provided quantities of advanced candidate precolibactins that exceed those obtained by fermentation, and is envisioned to be readily scaled. These studies have guided a structural revision of the predicted metabolite precolibactin A (from 5a or 5b to 7) and have confirmed the structures of the isolated metabolites precolibactins B (3) and C (6). Synthetic precolibactin C (6) was converted to N-myristoyl-D-asparagine and its corresponding colibactin by colibactin peptidase ClbP. The synthetic strategy outlined herein will facilitate mechanism of action and structure–function studies of these fascinating metabolites, and is envisioned to accommodate the synthesis of additional (pre)colibactins as they are isolated.
Graphical abstract
INTRODUCTION
Bacteria residing in and on humans (the human microbiota) play an integral role in regulating physiology and disease.1 The intestinal tract has been estimated to contain 500–1000 species of bacteria constituting ~1.5 kg of biomass.2 Certain strains of gut commensal and extraintestinal pathogenic Escherichia coli harbor a gene cluster (clb or “pks”) that encodes a group of molecules termed precolibactins.3 Precolibactins are substrates for colibactin peptidase ClpP, a protease encoded within the clb gene cluster. ClbP is anchored within the inner periplasmic membrane of the bacteria4 and removes an N-acyl-D-asparagine side chain from the precolibactins. This cleavage step converts precolibactins to cytotoxic colibactins and likely constitutes a prodrug resistance mechanism in the bacteria.5 clb+ E. coli induce DNA double-strand breaks (DSBs) in mammalian cells in vitro3a and in vivo.6 Host inflammation promotes proliferation of E. coli7 and expression of clb,8 the clb pathway promotes colorectal cancer in colitis-susceptible mice treated with azoxymethane,7 and two studies revealed the presence of clb+ E. coli in 55–67% of colorectal cancer patients.7,9 Collectively, these data suggest that colibactins initiate tumorigenesis by a mechanism involving induction of DNA DSBs.
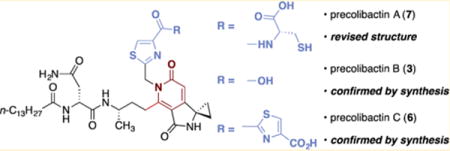
Fully elaborated (pre)colibactins have been difficult to isolate in homogeneous form, and the definitive structures of the most active metabolite(s) are not known. This has been attributed to the low levels of natural production of the metabolites, their instability under fermentation conditions, and the inflammation-dependent up-regulation of the native clb gene cluster. The metabolites 1,5c 2,10 3 (referred to hereafter as “precolibactin B”),11 and 411 were obtained in vanishingly small quantities (2.5–55 μg/L for 2–4) from the fermentation broth of genetically engineered clb+ E. coli and implicated as shunt metabolites and/or degradation products in the colibactin biosynthetic pathway (Figure 1). Using the isolation of 2, as well as HRMS analysis, isotope labeling, and bioinformatics based on established biosynthetic logic, the structure of precolibactin A was predicted as 5a or 5b.10a Key elements within the proposed structures include a hydrophobic N-terminal fragment, a spirocyclic aminocyclopropane, and (read from left to right) a thiazoline–thiazole chain. As the presence of the thiazoline–thiazole fragment was inferred by bioinformatic analysis,10a 5a and 5b could not be unequivocally distinguished at that time, and the absolute stereochemistry of the putative thiazoline ring was not determined. A compound with an exact mass corresponding to 5a was observed in unpurified extracts, but all efforts to isolate this structure were hampered by its low levels of production and instability.10a The pyridone structure 6 (referred to hereafter as “precolibactin C”) was recently proposed as a candidate precolibactin on the basis of biosynthetic considerations, isolation of precolibactin B (3), and HRMS analysis,11 and during the preparation of this paper, Balskus and co-workers reported the isolation of precolibactin C (6) from a mutant strain (0.5 mg of 6 was obtained from an optimized 48 L fermentation).12 Although one can envision cyclodehydration of 5a or 5b to form pyridones resembling 6, the biosynthetic relationship between these structures had not been established. 2 was shown to weakly cross-link DNA in vitro,10a suggesting that the colibactins may damage DNA by induction of replication-dependent DSBs.13 Detailed structure–function analyses of the colibactins have been impossible to conduct owing to their low yields of natural production and the absence of a synthetic route to the targets. However, the aminocyclopropane fragments within 2–6 are reminiscent of yatakemycin, CC-1065, and the duocarmycins, which have been shown to alkylate DNA via nucleophilic ring-opening,14 and the biheterocyclic fragment may serve as a DNA intercalation motif.15
Figure 1.
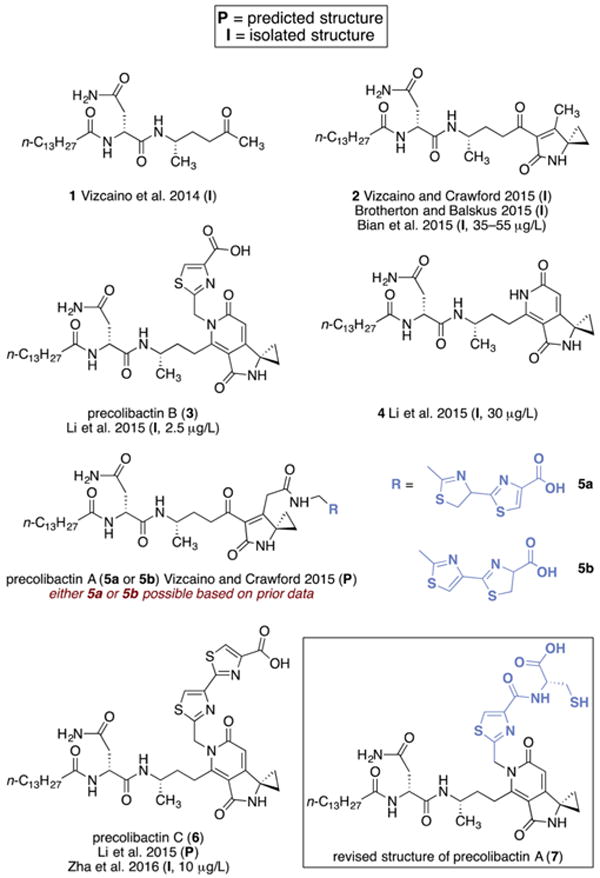
Structures of isolated, predicted, and synthesized candidate precolibactins. Fermentation yields (micrograms of product per liter of fermentation broth) are shown in parentheses, where available.
In light of the immense difficulties associated with isolating natural precolibactins, chemical synthesis provides an attractive avenue to resolve the ambiguities surrounding the composition of the active metabolite(s) and to enable mechanism of action and structure–function studies. Studies indicate the presence of an aminomalonyl unit in the biosynthetic pathway,12,16 suggesting additional colibactins are formed, but no evidence relevant to the structures of these metabolites exists, to our knowledge. Consequently, we initially focused on the synthesis of the predicted structures of precolibactin A (5a and 5b) and precolibactin C (6), as these represent the most advanced precolibactins for which structural data had been presented. Herein we report a convergent and high-yielding synthesis of structures 5a and 5b by cyclization of a fully linear precursor, establish that these materials are distinct from natural precolibactin A, propose and validate by synthesis a revised structure for precolibactin A (as 7), demonstrate that acyclic precolibactins undergo cyclodehydration to form the pyridone residues found in precolibactin B (3), 4, and precolibactin C (6) under mild conditions, and confirm the structures of precolibactins B (3) and C (6) by total synthesis. We anticipate that this route will be amenable to synthesis of more advanced structures, including those containing aminomalonyl units, as they are proposed.
RESULTS
As shown in Figure 1, at the time we began our studies, the structure of precolibactin A had been predicted as 5a or 5b. Consequently, we designed our synthetic route to accommodate either heterocyclic sequence and to provide access to the bithiazole found in precolibactin C (6). The synthesis of the common left-hand fragment began with Nα-(tert-butoxycarbonyl)-D-asparagine, which was coupled with (S)-hex-5-en-2-amine (8; prepared in three steps, 96% yield, and 88% ee from pent-4-en-al)17 using N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl) (Scheme 1). Cleavage of the tert-butoxycarbonyl protective group (hydrochloric acid) provided the amine hydrochloride 9 (84%, two steps). Acylation of the amine 9 with myristoyl chloride, followed by oxidative cleavage of the alkene (ruthenium chloride, sodium periodate) generated the carboxylic acid 10 (78%, two steps).
Scheme 1. Synthesis of the Carboxylic Acid 10a.
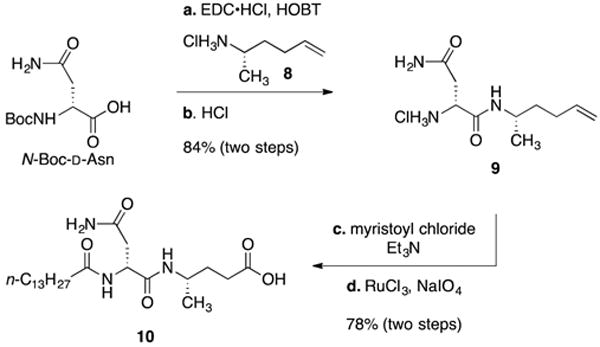
aReagents and conditions: (a) (S)-hex-5-en-2-amine hydrogen chloride (8), 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC·HCl), hydroxybenzotriazole (HOBT), N,N-diisopropylethylamine (DIPEA), DMF, 23 °C, 84%; (b) HCl, CH2Cl2–1,4-dioxane (7:1), 23 °C, >99%; (c) myristoyl chloride, triethylamine (Et3N), DMF, 23 °C, 82%; (d) RuCl3, NaIO4, H2O–EtOAc–CH3CN (3:2:2), 50 °C, 95%.
The isomeric thiazoline–thiazole and the related bithiazole fragments were prepared by the sequences shown in Scheme 2. Deuterated cysteine labeling experiments18 supported preservation of the L-amino acid configuration in precolibactin A,10a so we selected L-cysteine as the building block for the thiazoline ring.
Scheme 2. Synthesis of (A) the Thiazolin–Thiazole 17, (B) the Thiazole–Thiazoline 23, and (C) the Bithiazole 27a.
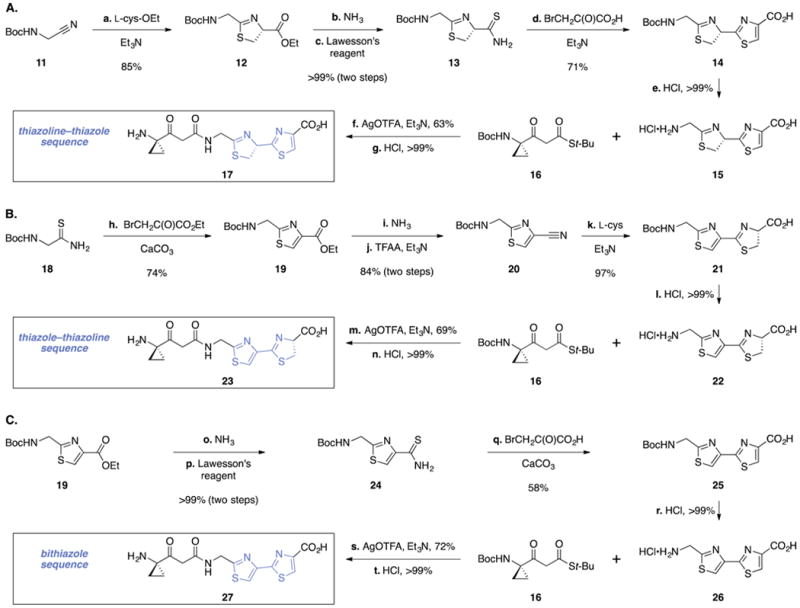
aReagents and conditions: (a) L-(+)-cysteine ethyl ester hydrochloride, Et3N, CH3OH, 23 °C, 85%; (b) NH3, CH3OH–H2O (2:1), 23 °C, >99%; (c) Lawesson’s reagent, CH2Cl2, 23 °C, >99%; (d) bromopyruvic acid, Et3N, CH3OH, reflux, 71%; (e) HCl, CH2Cl2–1,4-dioxane (4:1), 23 °C, >99%; (f) silver trifluoroacetate (AgO(TFA)), Et3N, DMF, 0 °C, 63%; (g) HCl, CH2Cl2–1,4-dioxane (4:1), 23 °C, >99%; (h) ethyl bromopyruvate, CaCO3, EtOH, 23 °C, 74%; (i) NH3, CH3OH–H2O (2:1), 23 °C, >99%; (j) trifluoroacetic anhydride (TFAA), Et3N, CH2Cl2, 0 → 23 °C, 84%; (k) L-(+)-cysteine, Et3N, CH3OH, reflux, 97%; (l) HCl, CH2Cl2–1,4-dioxane (8:1), 23 °C, >99%; (m) AgO(TFA), Et3N, DMF, 0 °C, 69%; (n) HCl, CH2Cl2–1,4-dioxane (4:1), 23 °C, >99%; (o) NH3, CH3OH–H2O (2:1), 23 °C, >99%; (p) Lawesson’s reagent, CH2Cl2, 23 °C, >99%; (q) bromopyruvic acid, CaCO3, EtOH, 23 °C, 58%; (r) HCl, CH2Cl2–1,4-dioxane (4:1), 23 °C, >99%; (s) AgO(TFA), Et3N, DMF, 0 °C, 72%; (t) HCl, CH2Cl2–1,4-dioxane (4:1), 23 °C, >99%.
Treatment of N-(tert-butoxycarbonyl)aminoacetonitrile (11) with L-cysteine ethyl ester provided the thiazoline 12 (85%, Scheme 2A). Aminolysis of the ester, followed by stirring with Lawesson’s reagent, generated the thioamide 13 (>99%, two steps). Exposure of 13 to bromopyruvic acid in the presence of triethylamine formed the thiazoline–thiazole 14 (71%). The thiazoline–thiazole and subsequent intermediates were found to be exceedingly unstable toward hydrolytic ring-opening and, to a lesser and variable extent, oxidation to a bithiazole. Accordingly, the identification of conditions to isolate and purify these intermediates without exposure to water was essential to the success of the route. Cleavage of the tert-butoxycarbonyl protective group (hydrochloric acid, >99%) generated the amine 15. Coupling (silver trifluoroacetate, triethylamine) of the amine 15 with the β-ketothioester 16 (prepared in one step and 56% yield from N-(tert-butoxycarbonyl)-1-aminocyclopropane-1-carboxylate)17 followed by carbamate cleavage (hydrochloric acid, >99%) furnished the thiazoline–thiazole fragment 17.
The isomeric thiazole–thiazoline fragment was prepared by a modified sequence (Scheme 2B). Treatment of N-(tert-butoxycarbonyl)-2-aminoethanethioamide (18) with ethyl bromopyruvate formed the thiazole 19 (74%). Aminolysis of 19 followed by dehydration of the resulting primary amide (trifluoroacetic anhydride, triethylamine) generated the nitrile 20 (84%, two steps). Coupling of 20 with L-cysteine formed the thiazole–thiazoline 21 (97%). In contrast to the isomeric intermediate 14, 21 was found to be stable toward aqueous workup and atmospheric oxygen. A three-step sequence analogous to that described above provided the thiazole–thiazoline 23 (69% overall).
The bithiazole fragment was prepared by the sequence shown in Scheme 2C. Aminolysis of the ester 19 followed by stirring with Lawesson’s reagent formed the thioamide 24 (>99%, two steps). Treatment of the thioamide 24 with bromopyruvic acid in the presence of calcium carbonate formed the bithiazole 25 (58%). Prior efforts to prepare and isolate 25 were impeded by its instability;19 we found that rigorous exclusion of water during workup and purification facilitated the isolation of 25 and subsequent intermediates in homogeneous form. A three-step sequence analogous to that used to prepare 17 and 23 then generated the bithiazole fragment 27 (72% overall).
To complete the synthesis of the precolibactin A skeleton, the carboxylic acid 10 was first converted to the β-ketothioester 28 by activation with carbonyldiimidazole followed by the addition of 3-(tert-butylthio)-3-oxopropanoic acid and magnesium ethoxide (Scheme 3).20 Silver-mediated coupling of 28 with the heterocyclic fragment 17, 23, or 27 then formed the penultimate intermediates 29a–c (90%, 87%, and 86% for 29a, 29b, and 29c, respectively). The stabilities of the fully linear precursors 29a–c paralleled those of 17, 23, or 27; the thiazole–thiazoline 29b was stable toward aqueous workup, while the thiazoline–thiazole 29a and the bithiazole 29c were unstable toward aqueous conditions.
Scheme 3. Synthesis of the Acyclic Advanced Precursors 29a–ca.
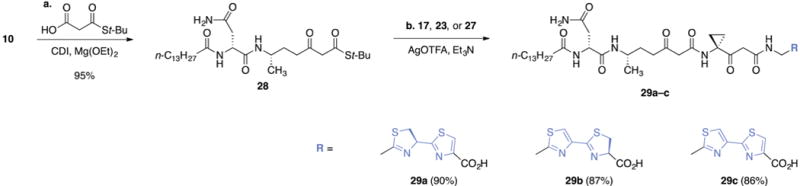
aReagents and conditions: (a) carbonyldiimidazole (CDI), 4 Å molecular sieves, DMF, then malonic acid half-thioester, magnesium ethoxide (Mg(OEt)2), 23 °C, 95%; (b) 17, 23, or 27, AgO(TFA), Et3N, DMF, 0 °C, 90% (29a), 87% (29b), 86% (29c).
Given the higher stability of the thiazole–thiazoline 29b, this compound was used to develop conditions to effect the key cyclization reaction (to 5b). Surprisingly, we found that in preparative experiments treatment of 29b with potassium carbonate in methanol at 0 °C resulted in formation of the pyridone 30b (80%, Scheme 4A). Similar outcomes were obtained on exposure of 29b to ammonium carbonate in ethanol or aqueous sodium hydroxide. Under these conditions, accumulation of the putative monocyclized intermediate 5b was not observed (LC/MS analysis). The pyridone 30b was fully characterized, and spectroscopic data for this compound were in good agreement with the isolated metabolite precolibactin B (3; Table 1). In particular, H-2 and H-4 of 30b resonated at 6.16 and 5.59/5.50 ppm, respectively; these values are nearly identical to those recorded for natural precolibactin B (3; 6.16 and 5.61/5.48 ppm, respectively).11 A plausible mechanism for the formation of the pyridone 30b involves cyclodehydration to 5b, 1,2-addition of the primary amide to the adjacent carbonyl, and aromatization. The facile formation of 30b from 29b provides evidence that the putative and isolated colibactin metabolites 3, 4, and 6 may derive from related acyclic precursors, although the timing of cyclizations in the modular biosynthetic pathway remains unknown. The cyclization of the thiazoline–thiazole derivative 29a and the bithiazole derivative 29c proceeded in a strictly analogous manner to provide the fully cyclized derivative 30a or precolibactin C (6), respectively.17 The mass spectroscopic fragmentation data and 1H NMR data for synthetic precolibactin C (6), as well as LC/MS co-injection with metabolite extracts, matched those of natural material (Figure S1 and Tables S1 and S5).11,12 In addition, synthetic precolibactin C (6) was converted to N-myristoyl-D-asparagine and its corresponding colibactin in a ClbP-dependent manner, indicating that precolibactin C (6) represents a suitable substrate for ClbP (Figure S3). This sequence provided multimilligram quantities of 30a, 30b, and precolibactin C (6), and is envisioned to be easily scalable.
Scheme 4. (A) Cyclodehydration of the Linear Precursors 29a–ca and (B) UV Trace (254 nm) of the Cyclization of 29b Using Potassium Carbonate in Dimethyl Sulfoxide at 24 °C.
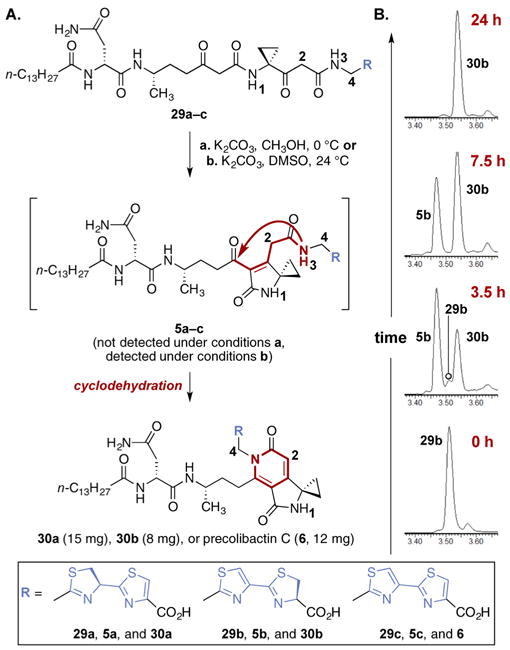
aReagents and conditions: (a) K2CO3, CH3OH, 0 °C, 79% (30a), 80% (30b), 83% (30c); (b) K2CO3, dimethyl sulfoxide, 24 °C (for 29b).
Table 1.
Selected 1H Chemical Shifts and LC/MS Data for 29b, 5b, 30b, and Precolibactin B (3)a
| position | 1 | 2 | 3 | 4 | tr (min) |
|---|---|---|---|---|---|
| 29b | 8.83 | 3.60 | 8.94 | 4.56 | 3.52 |
| 5b | 8.78 | b | 8.65 | b | 3.48 |
| 30b | 8.46 | 6.16 | 5.59, 5.50 | 3.55 | |
| precolibactin B (3)11 | 8.44 | 6.16 | 5.61, 5.48 |
1H spectroscopic data recorded in DMSO-d6 (500 MHz, 23 °C). Complete 1H NMR spectra (Figure S2) and LC/MS conditions are presented in the Supporting Information.
Not resolved.
We ultimately found that treatment of 29b with potassium carbonate (~3.0 equiv) in dimethyl sulfoxide at 24 °C proceeded more slowly and allowed for detection of 5b in the reaction mixture (LC/MS analysis, Scheme 4B). By conducting the reaction in dimethyl sulfoxide-d6, signals consistent with the monocyclized intermediate 5b could also be observed by 1H NMR analysis (Table 1).
Mass-selective LC/HRMS-QTOF analysis was conducted to determine if either 5a or 5b corresponded to the structure of natural precolibactin A. As shown in Figure 2, the concentrated ethyl acetate extracts of clb+ E. coli ΔclbP10a displayed a single prominent peak of m/z = 816.3788, which corresponds to [M + H]+ for the proposed structure of precolibactin A. However, the retention times of synthetic 5a and 5b were distinct, and the signals did not coalesce upon co-injection with the natural sample (parts A and B, respectively, of Figure 2). The retention times of 5a and 5b were nearly identical (tr = 15.70 and 15.80 min, respectively), as expected, and their differences with respect to natural precolibactin A (tr = 16.56 min) suggested a significant discrepancy in structure.
Figure 2.
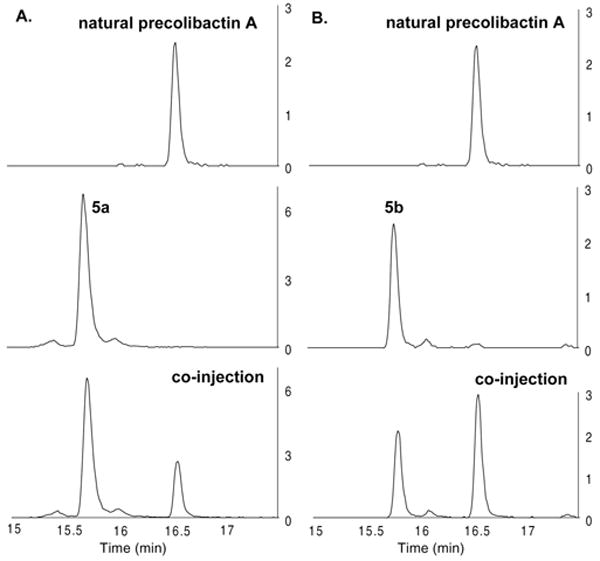
(A) Mass-selective (m/z = 816.3788) LC/HRMS-QTOF analysis of the ethyl acetate extracts of clb+ E. coli ΔclbP (top), synthetic 5a (middle), and co-injection (bottom). (B) Mass-selective (m/z = 816.3788) LC/HRMS-QTOF analysis of the ethyl acetate extracts of clb+ E. coli ΔclbP (top), synthetic 5b (middle), and co-injection (bottom). The y axis corresponds to the relative ion intensity (×104).
In light of the facile cyclization of our synthetic intermediates to the pyridone residues found in precolibactins B (3) and C (6), we reasoned that precolibactin A may also incorporate this substructure. The original isotope labeling, HRMS, and MSMS data for natural precolibactin A10a could not exclude this assignment. In addition, careful inspection of the initial report revealed that precolibactin A production was optimized by increasing the concentration of L-cysteine in the media 5-fold (to 1 g/L). Walsh and co-workers have previously reported the formation of cysteine derailment products in the biosynthesis of yersiniabactin.21 Accordingly, we hypothesized that the structure of natural precolibactin A may comprise the pyridone found in precolibactins B (3) and C (6) and a terminal cysteine residue appended to a single thiazole ring (Scheme 5A). This structure (7) possesses an exact mass that is identical to that of the originally predicted structures 5a or 5b and would similarly match the reported amino acid isotope labeling studies. Such a change in the terminal heterocyclic fragment would also be consistent with the large differences in retention times between 5a or 5b and natural precolibactin A. The synthesis of the revised precolibactin A structure 7 was readily accomplished using our synthetic strategy (Scheme 5B). Treatment of N-(tert-butoxycarbonyl)-2-aminoethanethioamide (18) with bromopyruvic acid22 followed by removal of the tert-butoxycarbonyl protective group generated the thiazole 31 (74%, two steps). Silver trifluoroacetate-mediated coupling of 31 and the thioester 16, followed by carbamate cleavage, formed the amine 32 (55%, two steps). Coupling of the amine 32 with the thioester 28 (Scheme 3, silver trifluoroacetate, triethylamine), followed by double cyclization (potassium carbonate, methanol), generated precolibactin B (3; 67% over two steps, 36 mg). NMR spectroscopic data for synthetic precolibactin B (3) and LC/MS co-injection with metabolite extracts matched those of the natural material (Figure S1 and Tables S3 and S4),11,12 thereby confirming the structure of the natural product.17 Finally, coupling of precolibactin B (3) with L-cysteine mediated by N-hydroxysuccinimide (NHS) and EDC· HCl generated 7 (89%). Mass-selective LC/HRMS-QTOF analysis against the concentrated ethyl acetate extracts of clb+ E. coli ΔclbP10a revealed that 7 corresponded exactly to natural material (Figure 3). In addition, both synthetic 7 and natural precolibactin A displayed identical mass spectral fragmentation patterns, providing further confirmation of the structure (Table S2). As natural precolibactin A is not isolable in amounts sufficient for NMR analysis,10a a direct comparison of the NMR spectra of synthetic and natural precolibactin A is not possible at this time.23
Scheme 5. (A) Originally Predicted (5a) and Revised (7) Structures of Precolibactin A and (B) Synthesis of the Revised Structure of Precolibactin A (7)a.
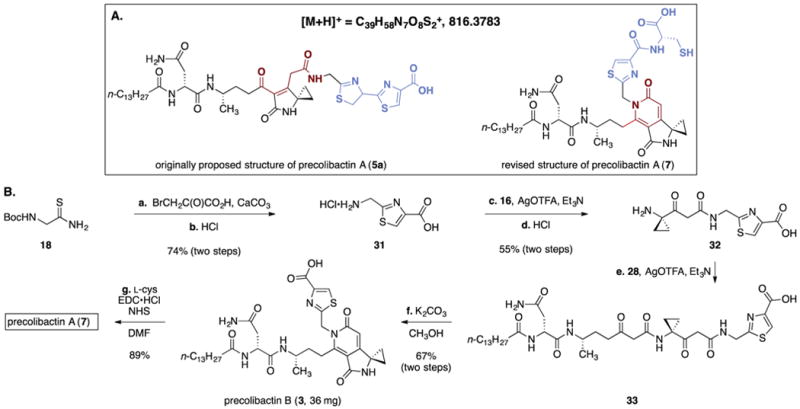
aReagents and conditions: (a) bromopyruvic acid, CaCO3, EtOH, 23 °C, 74%; (b) HCl, CH2Cl2–dioxane (3:1), 23 °C, >99%; (c) 16, AgO(TFA), Et3N, DMF, 0 °C, 55%; (d) HCl, CH2Cl2–1,4-dioxane (3:1), 23 °C, >99%; (e) 28, AgO(TFA), Et3N, DMF, 0 °C; (f) K2CO3, CH3OH, 0 °C, 67% (two steps); (g) L-cysteine, N-hydroxysuccinimide (NHS), EDC·HCl, Et3N, DMF, 0 → 23 °C, 89%.
Figure 3.
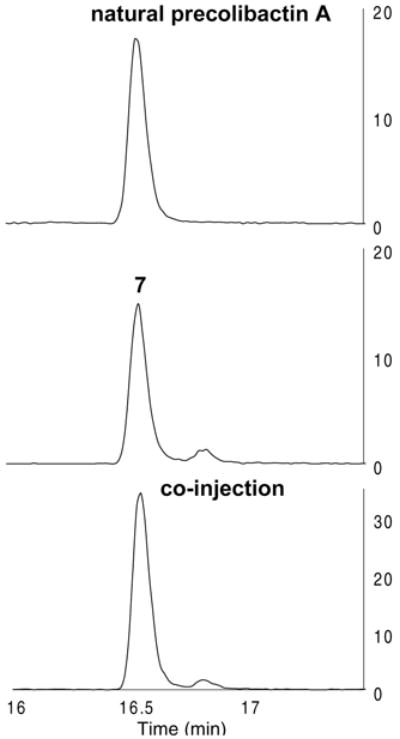
Mass-selective (m/z = 816.3788) LC/HRMS-QTOF analysis of the ethyl acetate extracts of clb+ E. coli ΔclbP (top), synthetic 7 (middle), and co-injection (bottom).
DISCUSSION AND CONCLUSION
The colibactins are a fascinating family of natural products that are produced by certain strains of commensal and extraintestinal E. coli, and the pathway has been implicated in the progression of colorectal cancer.7,8 Despite over a decade of intensive research, their complete structures and mechanism of action have remained unresolved. As highlighted in Figure 1, many of these compounds have been isolated in astoundingly low yields (μg/L) by painstaking fermentation experiments. By bringing the power of modern bioinformatics, enzymology, and mass spectrometry to bear on this problem, the structures of additional precolibactins, which are recalcitrant to isolation, have been predicted.
At the time we began our work, 3–6 represented the most complex precolibactin structures in the literature. Precolibactin B (3) and 4 were fully characterized by isolation,10 while 5a10a and precolibactin C (6)11 were predicted. While this paper was in preparation, Balskus and co-workers12 reported the isolation of precolibactin C (6; 0.5 mg from a 48 L fermentation) from a mutant strain. Biosynthetic studies now suggest that additional precolibactins incorporating an aminomalonyl unit exist,12,16 but no evidence for their structures has been presented, to our knowledge.
We have developed a high-yielding and modular synthetic route to the most advanced known precolibactin structures. The left-hand fragment 10, which is common to all of the precolibactins, is prepared in six steps and 63% overall yield from pent-4-en-al (Scheme 1). We have executed the synthesis of four distinct heterocyclic side chain fragments, in 4–7 steps and 37–41% overall yield (Schemes 2 and 5). Finally, these intermediates are elaborated to advanced precolibactins in three steps and ~50% overall yield (Schemes 3–5). We have confirmed the structures of the isolated precolibactins B (3) and C (6) by chemical synthesis, and revised the structure of precolibactin A, from 5a or 5b to 7. This structural revision also supports an unexpected biosynthetic route to colibactin bithiazole formation, in which biosynthesis of the first thiazole ring may precede heterocyclization and oxidation of the C-terminal L-cysteine moiety. This is in contrast to bioinformatic proposals for bleomycin bithiazole biosynthesis.24 Our synthetic studies also provide insights into the reactivities of these structures, and the facile cyclization of the linear precursors 29a–c to pyridones suggests this element as a common substructure. We envision that the synthetic strategy we have presented will be amenable to the synthesis of precolibactins incorporating an aminomalonyl substituent or other modifications, as their structures are proposed.
The synthetic route outlined herein provides a means to procure sufficient quantities of material to study the cellular responses to isolated colibactins and elucidate their mechanism of action for the first time. As noted in the Introduction, mammalian cells were shown to accumulate DNA DSBs when cocultured with pks+ E. coli cells,3a but no essential follow-up studies employing single metabolites derived from the clb pathway have been reported, to our knowledge. We expect that our modular synthetic strategy will finally open the door to examining these types of questions regarding colibactin’s mode of action with molecular-level resolution.25
Supplementary Material
Acknowledgments
Financial support from the National Institutes of Health (Grants R01GM110506 to S.B.H. and 1DP2-CA186575 to J.M.C.) is gratefully acknowledged. We thank Mr. Herman Nikolayevskiy and Mr. Steven Swick for assistance with the DFT calculations.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b02276.
Supplementary Figures S1–S6, Supplementary Tables S1–S5, Supplementary Scheme S1, general experimental procedures, and detailed experimental procedures and characterization data for all new compounds (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Donia MS, Fischbach MA. Science. 2015;349:395. doi: 10.1126/science.1254766. For a review, see. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu J, Gordon JI. Proc Natl Acad Sci U S A. 2003;100:10452. doi: 10.1073/pnas.1734063100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Nougayrède J-P, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, Buchrieser C, Hacker J, Dobrindt U, Oswald E. Science. 2006;313:848. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]; (b) Balskus EP. Nat Prod Rep. 2015;32:1534. doi: 10.1039/c5np00091b. For recent reviews, see. [DOI] [PubMed] [Google Scholar]; (c) Trautman EP, Crawford JM. Curr Top Med Chem. 2015;16:1. doi: 10.2174/1568026616666151012111046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dubois D, Baron O, Cougnoux A, Delmas J, Pradel N, Boury M, Bouchon B, Bringer MA, Nougayrede JP, Oswald E, Bonnet R. J Biol Chem. 2011;286:35562. doi: 10.1074/jbc.M111.221960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Brotherton CA, Balskus EP. J Am Chem Soc. 2013;135:3359. doi: 10.1021/ja312154m. [DOI] [PubMed] [Google Scholar]; (b) Bian X, Fu J, Plaza A, Herrmann J, Pistorius D, Stewart AF, Zhang Y, Muller R. ChemBioChem. 2013;14:1194. doi: 10.1002/cbic.201300208. [DOI] [PubMed] [Google Scholar]; (c) Vizcaino MI, Engel P, Trautman E, Crawford JM. J Am Chem Soc. 2014;136:9244. doi: 10.1021/ja503450q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrède J-P. Proc Natl Acad Sci U S A. 2010;107:11537. doi: 10.1073/pnas.1001261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan T-J, Campbell BJ, Abujamel T, Dogan B, Rogers AB, Rhodes JM, Stintzi A, Simpson KW, Hansen JJ, Keku TO, Fodor AA, Jobin C. Science. 2012;338:120. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arthur JC, Gharaibeh RZ, Mühlbauer M, Perez-Chanona E, Uronis JM, McCafferty J, Fodor AA, Jobin C. Nat Commun. 2014;5:4724. doi: 10.1038/ncomms5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buc E, Dubois D, Sauvanet P, Raisch J, Delmas J, Darfeuille-Michaud A, Pezet D, Bonnet R. PLoS One. 2013;8:e56964. doi: 10.1371/journal.pone.0056964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Vizcaino MI, Crawford JM. Nat Chem. 2015;7:411. doi: 10.1038/nchem.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bian X, Plaza A, Zhang Y, Müller R. Chem Sci. 2015;6:3154. doi: 10.1039/c5sc00101c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Brotherton CA, Wilson M, Byrd G, Balskus EP. Org Lett. 2015;17:1545. doi: 10.1021/acs.orglett.5b00432. [DOI] [PubMed] [Google Scholar]
- 11.Li ZR, Li Y, Lai JY, Tang J, Wang B, Lu L, Zhu G, Wu X, Xu Y, Qian PY. ChemBioChem. 2015;16:1715. doi: 10.1002/cbic.201500239. [DOI] [PubMed] [Google Scholar]
- 12.Zha L, Wilson MR, Brotherton CA, Balskus EP. ACS Chem Biol. doi: 10.1021/acschembio.6b00014. [Online early access] Published Online: Feb 18, 2016 http://pubs.acs.org/doi/abs/10.1021/acschembio.6b00014p (accessed March 28, 2016) [DOI] [PubMed]
- 13.Zhang J, Walter JC. DNA Repair. 2014;19:135. doi: 10.1016/j.dnarep.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tichenor MS, Boger DL. Nat Prod Rep. 2008;25:220. doi: 10.1039/b705665f. For a review, see. [DOI] [PubMed] [Google Scholar]
- 15.Wu W, Vanderwall DE, Turner CJ, Hoehn S, Chen J, Kozarich JW, Stubbe J. Nucleic Acids Res. 2002;30:4881. doi: 10.1093/nar/gkf608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brachmann AO, Garcie C, Wu V, Martin P, Ueoka R, Oswald E, Piel J. Chem Commun. 2015;51:13138. doi: 10.1039/c5cc02718g. [DOI] [PubMed] [Google Scholar]
- 17.See the Supporting Information
- 18.Bode HB, Reimer D, Fuchs SW, Kirchner F, Dauth C, Kegler C, Lorenzen W, Brachmann AO, Grün P. Chem - Eur J. 2012;18:2342. doi: 10.1002/chem.201103479. [DOI] [PubMed] [Google Scholar]
- 19.Souto JA, Vaz E, Lepore I, Pöppler A-C, Franci G, Álvarez R, Altucci L, de Lera ÁR. J Med Chem. 2010;53:4654. doi: 10.1021/jm100244y. [DOI] [PubMed] [Google Scholar]
- 20.Sodeoka M, Sampe R, Kojima S, Baba Y, Usui T, Ueda K, Osada H. J Med Chem. 2001;44:3216. doi: 10.1021/jm0100741. [DOI] [PubMed] [Google Scholar]
- 21.Gehring AM, Mori I, Perry RD, Walsh CT. Biochemistry. 1998;37:11637. doi: 10.1021/bi9812571. [DOI] [PubMed] [Google Scholar]
- 22.Videnov G, Kaiser D, Kempter C, Jung G. Angew Chem, Int Ed Engl. 1996;35:1503. [Google Scholar]
- 23.Synthetic precolibactin A (7) was found to readily undergo disulfide bond formation, which may explain its instability in complex cellular organic extracts
- 24.Galm U, Wendt-Pienkowski E, Wang L, Huang S-X, Unsin C, Tao M, Coughlin JM, Shen B. J Nat Prod. 2011;74:526. doi: 10.1021/np1008152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Our data suggest that prior proposals of the colibactin mechanism of action may require revision. See the Supporting Information for a discussion
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.