

Abstract
Immunotherapeutic approaches have emerged as promising strategies to treat various cancers, including breast cancer. A single approach, however, is unlikely to effectively combat the complex, immune evasive strategies found within the tumor microenvironment, thus novel, effective combination treatments must be explored. In this study, we investigated the efficacy of a combination therapy consisting of PD-L1 immune checkpoint blockade and whole cell vaccination in a HER-2 positive mouse model of breast cancer. We demonstrate that tumorigenicity is completely abrogated when adjuvanted with immune stimulatory molecules (ISMs) B7-1 and a cell-surface anchored (GPI) form of IL-12 or GM-CSF. Irradiated cellular vaccines expressing the combination of adjuvants B7-1 and GPI-IL-12 completely inhibited tumor formation which was correlative with robust HER-2 specific CTL activity. However, in a therapeutic setting, both cellular vaccination and PD-L1 blockade induced only 10–20% tumor regression when administered alone but resulted in 50% tumor regression as a combination therapy. This protection was significantly hindered following CD4 or CD8 depletion indicating the essential role played by cellular immunity. Collectively, these pre-clinical studies provide a strong rationale for further investigation into the efficacy of combination therapy with tumor cell vaccines adjuvanted with membrane-anchored ISMs along with PD-L1 blockade for the treatment of breast cancer.
Keywords: breast cancer, GPI-anchored proteins, HER-2, immunotherapy, PD-L1 blockade, tumor cell vaccines
Abbreviations
- ISMs
immune stimulatory molecules
- GPI
glycophosphatidylinositol
- PD-L1
programmed death-ligand 1
- CTLA-4
cytotoxic T-lymphocyte-associated protein 4
- GM-CSF
granulocyte macrophage colony-stimulating factor
- HER-2
human epidermal growth factor receptor 2
Introduction
A primary benchmark of efficacy of many immunotherapies is the induction of robust cellular immune responses which mediate clinical benefit. This goal can potentially be achieved in multiple ways, most notably through active vaccination to increase the pool of tumor-specific effector cells or by the removal of checkpoints that limit the induction and magnitude of these antitumor responses. Active vaccination involving the use of whole tumor cells as the vaccine material, allows for a broad repertoire of tumor antigens to be presented and recognized by the host's immune system which is more likely to minimize tumor escape and reduce the likelihood of metastasis.1 Though capable of inducing antitumor immune responses in mouse models as well as in patients, the therapeutic efficacy of allogeneic cellular vaccination has been limited, likely due to its inability to overcome the layers of tumor-derived immune suppression.2 Whereas, blockade of inhibitory immune pathways such as CTLA-4 and PD1/PD-L1, holds the potential to “reactivate” immunological responses that are significantly suppressed by tumor derived factors. This knowledge provides a solid rationale for combining active and immunomodulatory strategies to fight cancer.
Immune checkpoint therapies such as Ipilimumab (a CTLA-4 blocking antibody),3 Pembrolizumab and Nivolumab (PD-1 blocking antibodies)4,5 have been proven efficacious against multiple tumor types, most notably melanoma, however, only a subset of patients (10–20%) are responsive to these therapies. Moreover, only a few studies have investigated the efficacy of PD1/PD-L1 blockade against breast cancer.6,7 Due to the up-regulation of PD-L1 among breast cancer tumors,8 combining this therapy with cellular vaccination is likely to further boost tumor specific cellular responses thus leading to more complete responses. As a result, we sought to investigate the potential “synergy” of these immunotherapies in a murine breast cancer model.
The D2F2/E2 model has been transfected to express the well-characterized, clinically relevant tumor antigen human HER-2.9 We have adjuvanted the tumor cells with membrane anchored immune stimulatory molecules (ISMs), B7-1, glycolipid (GPI) IL-12 and GPI-GM-CSF, which we have previously shown to effectively reduce local immune suppression and mediate tumor growth inhibition.10 Due to the induction of a robust, endogenous anti- HER-2 response, the D2F2/E2 model is considered to be representative of HER-2 tumors that are immunogenic yet remain resistant to HER-2 targeted therapies.11,12 Membrane anchored ISMs are likely to remain at the site of vaccination for extended periods of time which has the potential for a prolonged induction of local antitumor responses. Additionally, these ISMs may not cause the systemic toxicity that is typically seen with the administration of soluble cytokines, particularly IL-12.13
Herein, we demonstrate, for the first time, the enhanced efficacy of combining PD-L1 blockade with cellular vaccination which induced complete tumor regression in 50% of treated mice.
Results
Expression of membrane-anchored ISMs inhibits D2F2/E2 tumorigenicity and induces protective immunity against secondary tumor challenge
Previous studies have shown that progressive tumor growth is observed in the D2F2/E2 mammary model despite the induction of an endogenous HER-2 specific immune response, most notably HER-2 specific IgG and IFNγ production.11 Here we investigated whether tumor growth inhibition could be induced when the D2F2/E2 tumor microenvironment is altered by expressing membrane anchored B7-1, GPI-IL-12 or GPI-GM-CSF on the cell surface. D2F2/E2 cells were stably transfected with cDNA encoding each ISM and surface expression of the ISMs was verified by flow cytometry analysis (Fig. 1A). BALB/c mice were then challenged with the live, adjuvanted cells to directly assess the effect of each ISM on the tumorigenicity of D2F2/E2 cells (Fig. 1B). We observed that mice challenged with the adjuvanted tumor cells completely rejected these tumors while the wild-type D2F2/E2 tumors (WT) grew progressively (Fig. 1C). Mice bearing WT tumors had significantly higher HER-2 specific IgG responses, with a predominance of IgG1, relative to tumor-free mice receiving the live adjuvanted cells (Fig. 1D), which is consistent with a previous report.11 This indicates that the tumorigenicity of this HER-2 positive cell line can be abolished by the expression of membrane-anchored ISMs.
Figure 1.

ISM expression completely abolishes the tumorigenicity of D2F2/E2 cells and induce partial protective immune responses following secondary tumor challenge. (A) Surface expression of HER-2 and B7-1, GPI-IL-12, GPI-GM-CSF on D2F2/E2 cells, Gray histogram (isotype control). (B) Mice were directly challenged with 2 × 105 live D2F2/E2 wild-type (WT) cells or live cells adjuvanted with B7-1, GPI-IL-12 or GPI-GM-CSF. (C) Primary tumor growth curve and tumor free survival is shown (D) Serum collected prior to secondary challenge was diluted (1:250) and used to assess for HER-2 specific IgG antibodies using WT cells by a cell ELISA as described in Materials and Methods. (E) Tumor-free mice were subjected to a secondary challenge 5 weeks after primary challenge with 2 × 105 live WT cells on the contralateral flank. Tumor growth and tumor free survival was monitored. Mean ± SEM is plotted. Representative data from 2 individual experiments (n=5 per group) is shown. Significance relative to WT, *p < 0.05, **p < 0.01, ***p < 0.001, **** p < 0.0001
Tumor-free mice were then subjected to a secondary challenge 5 weeks later with WT cells on the contralateral flank to determine if protective, systemic immunity was induced. Mice previously challenged with B7-1 or GPI-IL-12 adjuvanted cells, developed tumors, on average, one week after the control mice, with several mice remaining tumor-free following secondary challenge (Fig. 1E). Tumor progression was significantly inhibited in mice given adjuvanted tumor cells. Mice previously challenged with GPI-GMCSF adjuvanted tumor cells were completely protected against secondary tumor challenge with WT cells. These observations indicate that the expression of GPI-ISMs inhibits D2F2/E2 tumor growth and induces long-term, protective immune responses.
Lastly, to determine whether the GPI-ISMs were being shed and entering the circulation, serum and plasma was collected from mice 24 hrs following challenge with 2×105 or 4×105 GPI-IL-12 or GPI-GM-CSF adjuvanted D2F2/E2 cells for cytokine analysis by ELISA. We observed no significant increase in IL-12 or GM-CSF levels in the serum or plasma relative to naïve and D2F2/E2 WT challenged mice (Fig. 2), which suggest minimal shedding of the GPI-ISMs in vivo and demonstrates that the GPI-ISMs remain locally at the vaccination site.
Figure 2.
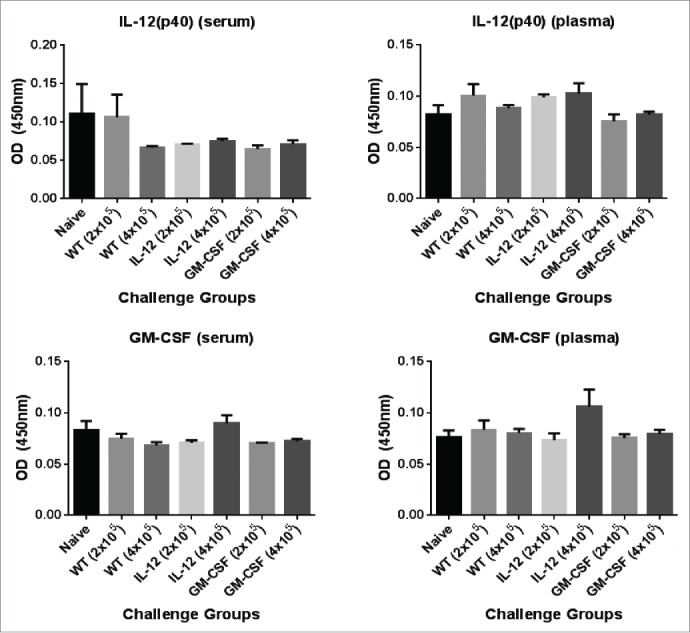
Analysis of circulating levels of IL-12 and GM-CSF in mice challenged with D2F2/E2 cells expressing GPI-GM-CSF and GPI-IL-12. Serum and plasma was collected from mice 24hrs post challenge with 2 × 105 or 4×105 D2F2/E2 live cells adjuvanted with GPI-IL-12 or GPI-GM-CSF. Cytokine levels were quantified by a sandwich ELISA. Mean ± SEM is plotted (n = 3–5 mice/group).
Prophylactic vaccination with irradiated B7-1 and GPI-IL-12 expressing tumor cells confers complete protection against subsequent tumor challenge
Due to the observed protection following live cell challenge, we investigated the efficacy of these adjuvanted cells in a prophylactic setting. In order to do so, we inactivated the tumor cells using gamma (γ) irradiation, which is a standard approach used in pre-clinical and clinical studies.14 Due to the established synergy of B7-1 with IL-12 or GM-CSF,15–17 we investigated the efficacy of co-administering these irradiated cellular vaccines. Two weeks following a single vaccination dose, mice were challenged with D2F2/E2 WT cells (Fig. 3A). We observed that mice vaccinated with irradiated B7-1 and GPI-IL-12 (Irr-B7-1/IL-12) expressing cellular vaccines were completely protected against WT tumor challenge (Fig. 3B), whereas other vaccinated groups developed tumors. Irr-B7-1 alone led to significant tumor inhibition. Tumor-free mice were then re-challenged with WT cells approximately 2 ½ months later (Fig. 3C). All vaccinated mice remained tumor-free for up to 40 d following re-challenge which indicates that irradiated cellular vaccines adjuvanted with GPI-ISMs can inhibit tumor formation as well as elicit sustainable immune responses. It remains unclear at this time as to why GPI-GM-CSF adjuvanted vaccines were completely protective as live “vaccines” (Fig. 1E) yet failed to induce protection when given as irradiated cellular vaccines in a prophylactic setting (Fig. 3B). While others have shown similar findings with other cytokines,18,19 it could be due to the differential recruitment and activation of myeloid cells by GPI-GMCSF in each treatment study and will require further investigation.
Figure 3.

Prophylactic vaccination with B7-1 and GPI-IL-12 adjuvanted cellular vaccines led to complete protection against subsequent D2F2/E2 tumor challenge. (A) Two weeks after vaccination (s.c.) with irradiated cellular vaccines expressing GPI-ISMs (2 × 105) mice were challenged (s.c.) with 2 × 105 live D2F2/E2 WT cells on the contralateral hind flank (s.c.). (B) Primary tumor growth and tumor free survival curves are shown. (C) Tumor-free mice were subjected to a secondary challenge 77 d post primary challenge. Secondary tumor growth and tumor free survival curves are shown. Mean ±SEM is plotted (n=5/group). Significance relative to PBS, *p< 0.05, **p < 0.01.
B7-1 and GPI-IL-12 expressing cellular vaccines induce minimal HER-2 specific humoral immunity but robust CTL activity
To gain insight into the protection conferred by Irr-B7-1/IL-12 vaccines, we assessed the humoral and cellular immune responses induced following vaccination. Serum was collected 2 weeks after vaccination. All cellular vaccines expressing ISMs induced highly significant IgG responses relative to control mice except mice receiving Irr-IL-12 or Irr-B7-1/IL-12 vaccines (Fig. 4A). Interestingly, although Irr-B7-1 significantly inhibited tumor growth (Fig. 3B) this group also had a high antibody response whereas when GPI-IL-12 was combined with the B7-1 expressing vaccines, protection was enhanced yet antibody responses were significantly inhibited suggesting that the expression of GPI-IL-12 is capable of modulating the induced humoral immune responses.
Figure 4.

Induction of significant HER-2 specific CTL response following prophylactic vaccination with B7-1 and GPI-IL-12 adjuvanted cellular vaccines. (A) Serum collected from mice 2 weeks after vaccination with cellular vaccines expressing GPI-ISMs (2 × 105) was diluted as indicated to assess for total IgG antibodies against D2F2/E2 WT cells by a cell ELISA as described in Materials and Methods. (B) Splenocytes were isolated from mice 2 weeks post vaccination and re-stimulated in vitro for 5 d with mitomycin-treated D2F2/E2 WT (5:1). CTLs were incubated with D2F2/E2 WT, D2F2 or 4TO7WT target cells for 4 h at indicated effector: target (E:T) ratios. Percentage of specific lysis was quantified by LDH release using the CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega). ANOVA analysis was used for serum and CTL analysis. Significance relative to PBS, *p < 0.05, **p < 0.01, ***p < 0.001, **** p < 0.0001.
Because our serum analysis indicated negligible induction of antibody responses following Irr-B7-1/IL-12 vaccination, we assessed the level of cytotoxic T lymphocyte (CTL) activity of splenocytes from vaccinated mice following 5-day in vitro stimulation with mitomycin-C treated D2F2/E2 WT cells. HER-2 specific cytotoxicity was determined after a 4 h incubation with D2F2/E2 WT cells. HER-2 negative mouse breast cancer cells, D2F2 and 4TO7, were used as specificity controls. Our data indicates that splenocytes from Irr-B7-1/IL-12 vaccinated mice are capable of killing approximately 50% of D2F2/E2 WT cells in a HER-2 specific manner (Fig. 4B), with minimal CTL activity against the HER-2 negative target cell lines, D2F2 and 4TO7. Splenocytes from Irr-WT vaccinated mice demonstrated minimal CTL activity against all target cell lines indicating that the adjuvants B7-1 and GPI-IL-12 expressed by the irradiated vaccines enhances HER-2 specific cellular immunity. These results clearly indicate that co-administration of B7-1 and GPI-IL-12 adjuvanted cellular vaccines is able to induce significant levels of HER2-specific CTLs capable of lysing D2F2/E2 tumor cells in an antigen-specific manner and is likely to mediate, at least in part, the conferred protection in vivo. Taken together, these data indicate that tumor inhibition is likely mediated by cellular immunity rather than humoral responses.
Cellular vaccines expressing B7-1 and GPI-IL-12 significantly reduced average tumor burden in a therapeutic setting
To determine whether cellular vaccines adjuvanted with ISMs could induce inhibition of established tumors, mice were vaccinated 7 d following tumor challenge (Fig. 5A). We observed that the co-injection of irradiated B7-1 and GPI-IL-12 expressing cellular vaccines led to a significant reduction in the average tumor burden of challenged mice from 178 mm2 to 48.6 mm2 and prolonged tumor-free survival (Fig. 5B).
Figure 5.

ISM expression by cellular vaccines significantly reduced average tumor burden and prolonged tumor-free survival. (A) Mice were challenged (s.c.) with 2 × 105 live D2F2/E2 WT cells. One week later, mice received one dose of 2 × 105 irradiated cellular vaccines on the contralateral flank (s.c.). (B) Tumor growth and tumor free survival was assessed. (C) D2F2/E2 cells cultured in vitro (Cultured Cells, solid black line) and freshly isolated tumors harvested from WT challenged mice (Tumor, dashed line) were assessed for the expression of PD-L1 by flow cytometry analysis (MFI-mean fluorescence intensity). (D) Splenocytes and tumor infiltrating lymphocytes (TILs) from tumor-bearing mice were isolated and analyzed for PD1 expression on CD8+ and CD4+ T cells by flow cytometry, mean fluorescence intensity (MFI). Data is representative of 2 individual experiments. Significance relative to PBS-treated. *p < 0.05, **p < 0.01, ***p ≤ 0.001.
Since the protection was not complete in the therapeutic setting, we sought to investigate whether combining our cellular vaccines with immune checkpoint inhibitors will augment the potency of our cellular vaccines. PD-L1 is known to be expressed by many tumors and is associated with a poor clinical prognosis among breast cancer patients20 and PD-1 expression by tumor infiltrating lymphocytes (TILs) has been reported to be an independent negative prognostic indicator for human breast cancer.21 Thus we assessed the tumor microenvironment for PD1 and PD-L1 expression among TILs and tumor tissue, respectively. We observed a significant upregulation of PD-L1 expression on freshly isolated tumors relative to in vitro cultured cells (Fig. 5C). Upon analysis of TILs we found that a significant percentage of CD4+ and CD8+ TILs expressed PD-1 relative to splenic T cells (Fig. 5D). Based on our findings and previous investigations that have demonstrated the adjuvant effect of PD-L1 immunological checkpoint blockade,22,23 we tested the therapeutic efficacy of PD-L1 blockade in this HER-2 breast cancer model.
Blockade of PD1/PD-L1 enhances therapeutic efficacy of cellular vaccines
Mice with early stage tumor growth (day 7) were co-administered B7-1 and GPI-IL-12 expressing irradiated cellular vaccines on the contralateral flank. An intraperitoneal injection of αPD-L1 (100µg) was given on days 7, 10 and 13 post tumor challenge (Fig. 6A). Our data indicates that αPD-L1 treatment enhanced the efficacy of the cellular vaccines as evident by significantly smaller average tumor burden by day 56 after tumor challenge relative to mice that received the cellular vaccine alone, 20.2 mm2 vs 88.6 mm2, respectively (Fig. 6B). Most importantly, 50% of mice receiving the combination therapy showed complete regression whereas only 10–20% of mice receiving single therapy demonstrated complete regression by the end of the study (Fig. 6B). Interestingly, mice given the combination therapy induced significantly less HER-2 specific IgG responses compared to mice given either therapy alone. This suggests that humoral immunity is likely not mediating the production conferred by the combination therapy and that cellular immunity is more dominant. Additionally, tumor-free mice were completely protected against secondary challenge for >60 d indicating the induction of prolonged and protective antitumor immunity for all treated mice (Fig. 6D).
Figure 6.

Enhanced therapeutic effect induced following cellular vaccination and PD-L1 blockade. (A) Mice were challenged (s.c.) with 2 × 105 live D2F2/E2 WT cells. One week later, mice received one dose of 2 × 105 irradiated cellular vaccines on the contralateral flank (s.c.). Additional groups of mice were treated with PD-L1 blockade (α-PD-L1: 100µg- i.p.) on days 7, 10 and 13 post tumor challenge either alone or in combination with the cellular vaccine (Combo). (B) Primary tumor growth and tumor incidence curves are shown. (C) Collected serum from treated mice were used to assess for HER-2 specific IgG. (D). Secondary tumor growth and tumor free survival curves are shown. Mean ±SEM is plotted. Representative of 2 individual experiments (n=5/group). Significance relative to PBS-treated mice based on ANOVA analysis. *p < 0.05, **p < 0.01.
Cellular immunity contributes significantly to the therapeutic efficacy of the combination therapy
Lastly, cellular depletion experiments were used to assess the relative roles of CD4+ or CD8+ cells during the priming phase of combination therapy treatment. Following establishment of D2F2/E2 tumors for 6 days, mice were depleted of CD4+ or CD8+ cells and then treated with the combination therapy (Fig. 7A). We observed that depletion of CD4+ or CD8+ cells significantly reversed the therapeutic effect observed following combination therapy (Fig. 7B). Interestingly, the overall tumor growth kinetics were comparable following CD4 or CD8 depletion with no significant difference observed relative to untreated mice. This study indicated that both CD4+ and CD8+ T cells were required for efficacy of the combination therapy, since depletion of either T-cell subset during the priming phase of treatment abrogated the development of systemic immunity.
Figure 7.
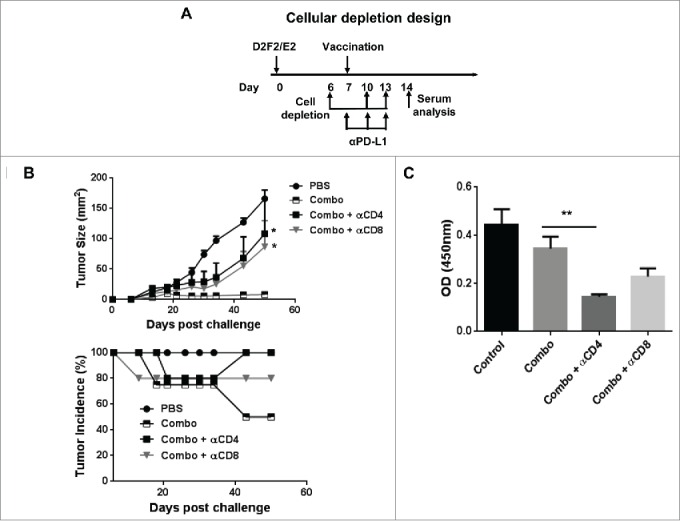
Both CD4+ and CD8+ (T)cells were required for efficacy of the combination therapy. (A) Mice were challenged (s.c.) with 2 × 105 live D2F2/E2 WT cells. One week later, mice received one dose of 2 × 105 irradiated cellular vaccines on the contralateral flank (s.c.) and PD-L1 blockade (100µg- i.p.) as indicated (combo). CD4+ and CD8+ cells were depleted by i.p. injection of 200µg of anti-CD4 (clone GK1.5) and anti-CD8 (clone 2.43) antibodies as indicated. (B) Tumor growth and tumor incidence curves following depletion are shown. (C) Serum collected after cellular depletion was used to assess for HER-2 specific IgG responses. Mean ±SEM is plotted. N=5/group. Significance relative to combination therapy (combo). *p < 0.05, **p< 0.01.
Upon analysis of humoral immunity following cellular depletion, we observed a significant decrease in HER-2 specific IgG following only CD4 depletion, which was expected due to the loss of CD4+ T cell help (Fig. 7B). Because CD8 depletion had minimal effect on humoral immune responses yet abrogated therapeutic efficacy similarly to CD4 depletion (which affected both cellular and humoral responses), it suggest a more critical role for cellular immune-mediated protection conferred by the combination therapy.
Discussion
Further development and investigation of combination therapies for effective cancer treatment are critically needed to inhibit multiple pathways of tumor growth and drug resistance.24,25 Standard combination strategies comprise of chemo/radiation therapy and surgery or the administration of multiple chemotherapeutic drugs, while the incorporation of various immunotherapies is emerging as viable options for many patients. Though successful at killing tumor cells, chemotherapy is known to induce a variety of harmful, off-target side effects which can be quite detrimental to patients. Thus the use of immunotherapeutic approaches, whether active, passive or immunodulatory, may serve as safer alternative strategies for cancer patients.
Many types of immunotherapy, which seek to harness and enhance the power and specificity of the immune system to effectively combat tumors, are currently being studied.26 Tumor cell vaccines, which allow for a wide repertoire of tumor antigens to be presented to the host, are often investigated clinically in a minimal, residual disease setting following tumor resection.27,28 The use of immunological adjuvants, such as soluble IL-12 and GM-CSF, have been used for over a decade to augment the quality of immune responses toward tumor antigens primarily through the enhancement in functionality and activation of antigen presenting cells and T cells.29,30 An emerging immunotherapeutic strategy is the use of immune checkpoint blockades, such as anti-CTLA-4 and anti-PD-1/PD-L1, which seek to amplify antitumor immune responses by blocking inhibitory pathways that regulate T cell signaling thus alleviating immune suppression and “reinvigorating” effector T cell responses.31 Our studies found that adjuvanted cellular vaccines expressing B7-1 and GPI-IL-12 (Irr-B7/IL-12) were capable of significantly reducing tumor burden when given during early tumor development, once palpable tumors were present. We also found that PD-L1, which is associated with poorer overall survival among patients with HER-2+ breast cancer,20 is up-regulated in vivo and isolated TILs expressed significant levels of PD-1 relative to splenic T cells. Thus we combined Irr-B7/IL-12 with PD-L1 blockade and observed a significantly augmented therapeutic effect leading to complete tumor regression in 50% of treated mice. Both subsets of T cells (CD4+ and CD8+) were found to be critical for this observed effect, with minimal contribution by antigen-specific antibody production. Further, based on our findings from our prophylactic studies, this enhanced therapeutic response is likely mediated through the rescue from exhaustion of pre-existing, antigen-specific CTLs by blockade of the PD-1/PD-L1 pathway.32,33
Despite the promising results of PD-1/PD-L1 blockade therapies, only a fraction of patients (10–30%) demonstrate clinical benefit following treatment.34 Additionally, as this checkpoint molecule plays a critical role in maintaining peripheral tolerance, its administration has led to varying degrees of immune-related adverse events in some patients.6,34 Because of this, combining this therapy with other strategies has been explored and proven efficacious against different tumors including melanoma35,36 yet had not been demonstrated in breast cancer models.
Irradiation is generally thought to enhance the immunogenicity of tumor cells,37 however GPI-GMCSF adjuvanted cellular vaccines were less effective following irradiation in a prophylactic vaccination setting relative to the live cell challenge studies. This finding was unexpected particularly due to the extensive studies done with GM-CSF secreting vaccines (GVAX) and its proven efficacy in multiple animal models and patients.38-40 A primary difference is that in our model, the GM-CSF is tethered to the tumor cell surface via a glycolipid anchor rather than being in secretory form which may lead to disparate results. While it remains unclear at this time the reason for the observed discrepancy in effectiveness between our 2 studies, others have reported similar findings while investigating other cytokines.18,19 It is important to note, however, that irradiated cellular vaccines expressing GPI-GM-CSF (Irr-GM-CSF) induced robust antibody responses whereas following live cell challenge, such levels of antibody responses were not detected. Consistently in our studies, it appears that HER-2 specific IgG responses are not sufficient to confer protection and mediate tumor rejection which may provide insight into the lack of prophylactic efficacy observed by the Irr-GM-CSF vaccine. On the other hand, the Irr-B7/IL-12 vaccine completely protected mice from primary and secondary tumor challenges following prophylactic vaccination. We, and others, have previously reported the synergy of these molecules in the induction of antitumor immune responses which has been shown to be mediated by a reduction in MDSCs and Tregs,10 the induction of CTL activity41 and dependent on CD4+ and CD8+ T cells.16,41 Our studies however, investigate the use of membrane associated IL-12 (GPI-IL-12) instead of recombinant, soluble IL-12 which demonstrates comparable in vivo efficacy without the observed toxicity of IL-12 administration.13,16
Despite the ∼85% homology between human and mouse HER-242 and the conservation of a HER-2-specific CTL epitope between species,43 we recognize that expression of this xenoantigen in our experimental model is likely to mediate the robust HER-2 specific immunity that is induced endogenously as well as following vaccination. While our studies were conducted in BALB/c mice which are non-tolerant to HER2/neu, evaluation of vaccine efficacy in non-tolerant mice can be helpful in determining the most potent vaccines from a collection of potential vaccine candidates.44 However, in order to more accurately recapitulate the tolerogenic immune responses elicited by cancer patients against the self-antigen HER-2, further investigation of the efficacy of our combination therapy is needed in HER-2/neu transgenic (tolerant) mice.45,46
Our pre-clinical findings demonstrate significant prophylactic as well as therapeutic efficacy following a single dose of cellular vaccines adjuvanted with membrane associated proteins (B7-1 and GPI-IL-12) and their combination effect with a clinically investigated immune checkpoint blockade therapy, anti-PD-L1, which led to complete regression in 50% of treated mice and sustained protection against secondary challenge. Our combination approach effectively modulated endogenous immune responses toward a more protective phenotype, which appeared primarily cellular-mediated. Taken together our studies provide evidence for future investigation of the use of the combination therapy PD-L1 blockade and cellular vaccination expressing glycolipid proteins as a potential treatment regime for breast cancer.
Materials and Methods
Tumor model and animals
D2F2, a mouse mammary tumor line derived from a single tumor from the transplantable hyperplastic alveolar nodule D2 line, was previously transfected with the wild-type human HER-2 gene to establish the D2F2/E2 cell line.9 This tumor cell line expresses a low, but detectable level of the endogenous mouse ErbB-2 receptor. D2F2/E2 tumor cells were a kind gift of Dr. Wei-Zen Wei (Wayne State University, Detroit, MI) and was cultured in DMEM media (Cellgro) supplemented with 10% cosmic calf serum, 1% penicillin streptomycin and 400 µg/ml G418. Stable transfectants expressing immune stimulatory molecules (ISMs), transmembrane B7-1 (B7-1), glycolipid (GPI) IL-12 and GPI-GM-CSF, were established as previously described10 and maintained under 5 µg/ml blasticidin selection. HER-2 negative breast cancer cell line, 4TO7, derived from a mammary adenocarcinoma in BALB/c mice, were maintained in DMEM media with 10% cosmic calf serum and 1% penicillin streptomycin. All cell culture reagents were purchased from Invitrogen. Female BALB/c mice, 6–8 weeks of age, were purchased from Jackson Laboratories and were maintained in accordance with IACUC approved institutional guidelines and protocols.
Direct challenge and prophylactic vaccination studies
For direct challenge studies, mice were injected subcutaneously (s.c.) on the hind flank with 2 × 105 live D2F2/E2 cells expressing the GPI-ISMs. Tumor-free mice were re-challenged on the contralateral flank with an additional 2 × 105 live D2F2/E2 wild-type (WT) cells 5 weeks after initial tumor challenge. For prophylactic vaccination studies, mice were vaccinated (s.c.) with 2 × 105 irradiated (80 Gy) D2F2/E2 cellular vaccines (Gammacell40 Caesium 137 irradiation unit) 2 weeks prior to tumor challenge with 2 × 105 D2F2/E2 WT tumor cells on the opposite hind flank. Irradiated cellular vaccines expressing single ISMs (1 × 105 each) were co-injected to create dual ISM expressing vaccines. Tumor-free mice were subjected to a secondary challenge 77 d after the primary challenge. Mice were monitored for tumor growth and were euthanized when tumors became ulcerated or the tumor size reached >2 cm2. Tumor size (mm2) was measured using Vernier calipers with 2 × 2 perpendicular measurements.
Therapeutic vaccination and program death ligand-1(PD-L1) blockade
Therapeutic vaccination with 2×105 irradiated ISM-expressing cellular vaccines was administered (s.c.) on the contralateral flank 1 week after D2F2/E2 tumor challenge either alone or in combination with 100µg anti-PD-L1 antibody (BioXcell) which was administered intraperitoneally (i.p.) on days 7, 10 and 13 post challenge. Rat IgG antibody (Jackson ImmunoResearch) was administered as a control. Mice were monitored as mentioned previously. Tumor-free mice were subjected to a secondary challenge with D2F2/E2 WT cells 57 d after the initial tumor challenge.
In vivo cellular depletion
CD4+ and CD8+ cells were depleted by i.p. injection of 200µg of anti-CD4 (clone GK1.5) and anti-CD8 (clone 2.43) antibodies in PBS at days 6, 10 and 13 post D2F2/E2 tumor cell challenge. Cellular depletion was confirmed by flow cytometry analysis of peripheral blood (data not shown). Depletion antibodies were purchased from BioXcell.
Serum and plasma analysis
Serum and plasma were collected from mice 24hours following direct live cell challenge. Cytokines present in circulation were detected in diluted serum and plasma (1:10) using a standard cytokine sandwich ELISA (eBioscience) as per the manufacturer's instructions. Antigen-specific antibodies were measured by a cell ELISA using serum collected at the following time points: 5 weeks post primary tumor challenge (direct challenge studies), 2 weeks post irradiated cellular vaccination (prophylactic studies), 6 weeks post primary tumor challenge (therapeutic studies) or 2 weeks post tumor challenge (in vivo cell depletion studies). Diluted serum (as indicated) was incubated with 5×104 D2F2/E2 cells for 1hr at 4°C in triplicate. After which, goat anti-mouse HRP-conjugated antibodies against total mouse IgG or subclass-specific mouse IgG1 and IgG2a (Southern Biotech) was incubated with the tumor cells for 1hr at 4°C at a dilution of 1:2000. TMB substrate (Biolegend) was used for detection and sulfuric acid was used as the stop solution. Absorbance (OD) was measured at 415 or 450nm in a microplate reader (Molecular Devices).
Cytotoxic T lymphocyte (CTL) assay
Spleens were harvested from mice 2 weeks following vaccination, subjected to mechanical dissociation and passed through a nylon mesh cell strainer to obtain a single cell suspension. Red blood cells (RBCs) were removed following osmotic water lysis for 20 seconds. Cytotoxic T lymphocytes (CTLs) were generated following a 5 day co-culture of isolated splenocytes with mitomycin C-treated (50µg/mL) D2F2/E2 cells as stimulator cells at a ratio of 5:1. Recombinant IL-2 (10U/mL) was added on day 2 of re-stimulation. After 5 days, dead cells were removed by centrifugation using Histopaque-1077 separation (Sigma). CTL activity was quantified using the CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega) according to the manufacturer's instructions with breast cancer lines D2F2/E2, D2F2 and 4TO7 cells as target cells at indicated effector: target ratios. In brief, CTLs were cultured with tumor cells for 4–5 hrs at 37°C. After which, the supernatant was removed and incubated for 30 mins at room temperature with the substrate to assay for LDH release. Total cytotoxicity was calculated as follows:
Isolation and cellular phenotyping of spleen cells and tumor-infiltrating lymphocytes (TILs)
Spleens and tumors were harvested from D2F2/E2 tumor-bearing mice >50 d post challenge. Splenocytes were isolated as previously described, Fc blocked (clone 2.4G2) and stained with directly conjugated antibodies (eBioscience) for 25 min at 4°C. Tumors were harvested, mechanically dissociated, enzymatically digested using collagenase type III (Sigma) for 2 h at 37°C and passed through a nylon cell strainer to obtain a single cell suspension. Enrichment of TILs was achieved by centrifugation with a discontinuous Percoll (Sigma-Aldrich) gradient (44% and 67%). Lymphocytes were collected from the gradient interface and subsequently dual-stained with directly conjugated antibodies for multi-colored analysis using a FACSCalibur flow cytometer and FlowJo software. The directly conjugated antibodies (eBioscience) used included:
Characterization of in vitro cultured and freshly isolated tumor cells
Cells from freshly isolated D2F2/E2 tumors were harvested and single cell suspensions were prepared as previously mentioned. Surface expression of PD-L1 on in vitro cultured cells and freshly isolated tumors was determined by direct immunofluorescence staining (clone MIH5) and analyzed by flow cytometry analysis.
Statistical analysis
Differences between tumor growth curves and serum antibody responses were analyzed using ANOVA or the Student's t test, respectively. Values of p < 0.05 were considered significant. For tumor-free survival studies, Kaplan-Meier survival curves were plotted and analyzed. All graphs and statistical calculations were analyzed using Prism software (GraphPad).
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
The authors would like to thank Dr. Rafi Ahmed and Jaina Patel for critical analysis of the manuscript.
Funding
The study was supported by National Institutes of Health (NIH) funding: R01 CA138993–01A1 (PS) and F31 CA165897–01 (EB).
References
- 1.Labarthe MC, Halanek N, Birchall L, Russell N, Desel C, Todryk S, Peters MJ, Lucas A, Falkenberg FW, Dalgleish AG, et al.. The biological effects of syngeneic and allogeneic cytokine-expressing prophylactic whole cell vaccines and the influence of irradiation in a murine melanoma model. Cancer Immunol Immunother 2006; 55:277-88; PMID:16158275; http://dx.doi.org/ 10.1007/s00262-005-0061-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Srivatsan S, Patel JM, Bozeman EN, Imasuen IE, He S, Daniels D, Selvaraj P. Allogeneic tumor cell vaccines: the promise and limitations in clinical trials. Hum Vaccin Immunother 2014; 10:52-63; PMID:24064957; http://dx.doi.org/ 10.4161/hv.26568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al.. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711-23; PMID:20525992; http://dx.doi.org/ 10.1056/NEJMoa1003466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, Weber JS, Joshua AM, Hwu WJ, Gangadhar TC, et al.. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 2014; 384:1109-17; PMID:25034862; http://dx.doi.org/ 10.1016/S0140-6736(14)60958-2 [DOI] [PubMed] [Google Scholar]
- 5.Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, Brahmer JR, Lawrence DP, Atkins MB, Powderly JD, et al.. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014; 32:1020-30; PMID:24590637; http://dx.doi.org/ 10.1200/JCO.2013.53.0105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al.. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012; 366:2455-65; PMID:22658128; http://dx.doi.org/ 10.1056/NEJMoa1200694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nanda R, Chow L, Dees EC, Berger R, Gupta S, Geva R, Pusztai L, Dolled-Filhart M, Emancipator K, Gonzalez EJ, et al.. A phase Ib study of pembrolizumab (MK-3475) in patients with advanced triple-negative breast cancer. In: Proceedings of the Thirty-Seventh Annual CTRC-AACR San Antonio Breast Cancer Symposium: 2014 Dec 9-13; San Antonio, TX. Philadelphia, PA: AACR; Cancer Res 2015;75(9 Suppl):Abstract nr S1-09. [Google Scholar]
- 8.Sabatier R, Finetti P, Mamessier E, Adelaide J, Chaffanet M, Ali HR, Viens P, Caldas C, Birnbaum D, Bertucci F. Prognostic and predictive value of PDL1 expression in breast cancer. Oncotarget 2014; 2015 Mar 10;6(7):5449-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei WZ, Shi WP, Galy A, Lichlyter D, Hernandez S, Groner B, Heilbrun L, Jones RF. Protection against mammary tumor growth by vaccination with full-length, modified human ErbB-2 DNA. Int J Cancer 1999; 81:748-54; PMID:10328228; http://dx.doi.org/ 10.1002/(SICI)1097-0215(19990531)81:5%3c748::AID-IJC14%3e3.0.CO;2-6 [DOI] [PubMed] [Google Scholar]
- 10.Bozeman EN, Cimino-Mathews A, Machiah DK, Patel JM, Krishnamoorthy A, Tien L, Shashidharamurthy R, Selvaraj P. Expression of membrane anchored cytokines and B7-1 alters tumor microenvironment and induces protective antitumor immunity in a murine breast cancer model. Vaccine 2013; 31:2449-56; PMID:23541884; http://dx.doi.org/ 10.1016/j.vaccine.2013.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veenstra JJ, Gibson HM, Littrup PJ, Reyes JD, Cher ML, Takashima A, Wei WZ. Cryotherapy with concurrent CpG oligonucleotide treatment controls local tumor recurrence and modulates HER2/neu immunity. Cancer Res 2014; 74:5409-20; PMID:25092895; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-0501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol 2006; 3:269-80; PMID:16683005; http://dx.doi.org/ 10.1038/ncponc0509 [DOI] [PubMed] [Google Scholar]
- 13.Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, Sosman JA, Dutcher JP, Vogelzang NJ, Ryan JL. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood 1997; 90:2541-8; PMID:9326219 [PubMed] [Google Scholar]
- 14.Kusumoto M, Umeda S, Ikubo A, Aoki Y, Tawfik O, Oben R, Williamson S, Jewell W, Suzuki T. Phase 1 clinical trial of irradiated autologous melanoma cells adenovirally transduced with human GM-CSF gene. Cancer Immunol Immunother 2001; 50:373-81; PMID:11676397; http://dx.doi.org/ 10.1007/s002620100213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi KJ, Kim JH, Lee YS, Kim J, Suh BS, Kim H, Cho S, Sohn JH, Kim GE, Yun CO. Concurrent delivery of GM-CSF and B7-1 using an oncolytic adenovirus elicits potent antitumor effect. Gene Ther 2006; 13:1010-20; PMID:16525479; http://dx.doi.org/ 10.1038/sj.gt.3302759 [DOI] [PubMed] [Google Scholar]
- 16.Coughlin CM, Wysocka M, Kurzawa HL, Lee WM, Trinchieri G, Eck SL. B7-1 and interleukin 12 synergistically induce effective antitumor immunity. Cancer Res 1995; 55:4980-7; PMID:7585539 [PubMed] [Google Scholar]
- 17.McHugh RS, Nagarajan S, Wang YC, Sell KW, Selvaraj P. Protein transfer of glycosyl-phosphatidylinositol-B7-1 into tumor cell membranes: a novel approach to tumor immunotherapy. Cancer Res 1999; 59:2433-7; PMID:10344754 [PubMed] [Google Scholar]
- 18.Cayeux S, Beck C, Aicher A, Dorken B, Blankenstein T. Tumor cells cotransfected with interleukin-7 and B7.1 genes induce CD25 and CD28 on tumor-infiltrating T lymphocytes and are strong vaccines. Eur J Immunol 1995; 25:2325-31; PMID:7545119; http://dx.doi.org/ 10.1002/eji.1830250831 [DOI] [PubMed] [Google Scholar]
- 19.Cayeux S, Beck C, Dorken B, Blankenstein T. Coexpression of interleukin-4 and B7.1 in murine tumor cells leads to improved tumor rejection and vaccine effect compared to single gene transfectants and a classical adjuvant. Hum Gene Ther 1996; 7:525-9; PMID:8800747; http://dx.doi.org/ 10.1089/hum.1996.7.4-525 [DOI] [PubMed] [Google Scholar]
- 20.Muenst S, Schaerli AR, Gao F, Daster S, Trella E, Droeser RA, Muraro MG, Zajac P, Zanetti R, Gillanders WE, et al.. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res Treat 2014; 146:15-24; PMID:24842267; http://dx.doi.org/ 10.1007/s10549-014-2988-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muenst S, Soysal SD, Gao F, Obermann EC, Oertli D, Gillanders WE. The presence of programmed death 1 (PD-1)-positive tumor-infiltrating lymphocytes is associated with poor prognosis in human breast cancer. Breast Cancer Res Treat 2013; 139:667-76; PMID:23756627; http://dx.doi.org/ 10.1007/s10549-013-2581-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pilon-Thomas S, Mackay A, Vohra N, Mule JJ. Blockade of programmed death ligand 1 enhances the therapeutic efficacy of combination immunotherapy against melanoma. J Immunol 2010; 184:3442-9; http://dx.doi.org/ 10.4049/jimmunol.0904114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ha SJ, Mueller SN, Wherry EJ, Barber DL, Aubert RD, Sharpe AH, Freeman GJ, Ahmed R. Enhancing therapeutic vaccination by blocking PD-1-mediated inhibitory signals during chronic infection. J Exp Med 2008; 205:543-55; PMID:18332181; http://dx.doi.org/ 10.1084/jem.20071949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li F, Zhao C, Wang L. Molecular-targeted agents combination therapy for cancer: developments and potentials. Int J Cancer 2014; 134:1257-69; PMID:23649791; http://dx.doi.org/ 10.1002/ijc.28261 [DOI] [PubMed] [Google Scholar]
- 25.Luu T, Chung C, Somlo G. Combining emerging agents in advanced breast cancer. Oncologist 2011; 16:760-71; PMID:21543509; http://dx.doi.org/ 10.1634/theoncologist.2010-0345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, Thatcher N, Wagstaff J, Zielinski C, Faulkner I, et al.. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol 2014; 11:509-24; PMID:25001465; http://dx.doi.org/ 10.1038/nrclinonc.2014.111 [DOI] [PubMed] [Google Scholar]
- 27.Hanna MG Jr., Hoover HC Jr., Vermorken JB, Harris JE, Pinedo HM. Adjuvant active specific immunotherapy of stage II and stage III colon cancer with an autologous tumor cell vaccine: first randomized phase III trials show promise. Vaccine 2001; 19:2576-82; PMID:11257395; http://dx.doi.org/ 10.1016/S0264-410X(00)00485-0 [DOI] [PubMed] [Google Scholar]
- 28.Simons JW, Mikhak B, Chang JF, DeMarzo AM, Carducci MA, Lim M, Weber CE, Baccala AA, Goemann MA, Clift SM, et al.. Induction of immunity to prostate cancer antigens: results of a clinical trial of vaccination with irradiated autologous prostate tumor cells engineered to secrete granulocyte-macrophage colony-stimulating factor using ex vivo gene transfer. Cancer Res 1999; 59:5160-8; PMID:10537292 [PubMed] [Google Scholar]
- 29.Rao JB, Chamberlain RS, Bronte V, Carroll MW, Irvine KR, Moss B, Rosenberg SA, Restifo NP. IL-12 is an effective adjuvant to recombinant vaccinia virus-based tumor vaccines: enhancement by simultaneous B7-1 expression. J Immunol 1996; 156:3357-65 [PMC free article] [PubMed] [Google Scholar]
- 30.Armstrong CA, Botella R, Galloway TH, Murray N, Kramp JM, Song IS, Ansel JC. Antitumor effects of granulocyte-macrophage colony-stimulating factor production by melanoma cells. Cancer Res 1996; 56:2191-8; PMID:8616871 [PubMed] [Google Scholar]
- 31.Postow MA, Callahan MK, Wolchok JD. Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol 2015; 33:1974-82; PMID:25605845; http://dx.doi.org/ 10.1200/JCO.2014.59.4358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, Rietz C, Flies DB, Lau JS, Zhu G, et al.. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res 2005; 65:1089-96; PMID:15705911 [PubMed] [Google Scholar]
- 33.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al.. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014; 515:568-71; PMID:25428505; http://dx.doi.org/ 10.1038/nature13954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al.. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366:2443-54; PMID:22658127; http://dx.doi.org/ 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fu J, Malm IJ, Kadayakkara DK, Levitsky H, Pardoll D, Kim YJ. Preclinical evidence that PD-1 blockade cooperates with cancer vaccine TEGVAX to elicit regression of established tumors. Cancer Res 2014; 74(15):4042-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li B, VanRoey M, Wang C, Chen TH, Korman A, Jooss K. Anti-programmed death-1 synergizes with granulocyte macrophage colony-stimulating factor–secreting tumor cell immunotherapy providing therapeutic benefit to mice with established tumors. Clin Cancer Res 2009; 15:1623-34; PMID:19208793; http://dx.doi.org/ 10.1158/1078-0432.CCR-08-1825 [DOI] [PubMed] [Google Scholar]
- 37.Chakraborty M, Abrams SI, Camphausen K, Liu K, Scott T, Coleman CN, Hodge JW. Irradiation of tumor cells up-regulates Fas and enhances CTL lytic activity and CTL adoptive immunotherapy. J Immunol 2003; 170:6338-47; http://dx.doi.org/ 10.4049/jimmunol.170.12.6338 [DOI] [PubMed] [Google Scholar]
- 38.Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K, Jackson V, Hamada H, Pardoll D, Mulligan RC. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci U S A 1993; 90:3539-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soiffer R, Hodi FS, Haluska F, Jung K, Gillessen S, Singer S, Tanabe K, Duda R, Mentzer S, Jaklitsch M, et al.. Vaccination with irradiated, autologous melanoma cells engineered to secrete granulocyte-macrophage colony-stimulating factor by adenoviral-mediated gene transfer augments antitumor immunity in patients with metastatic melanoma. J Clin Oncol 2003; 21:3343-50; PMID:12947071; http://dx.doi.org/ 10.1200/JCO.2003.07.005 [DOI] [PubMed] [Google Scholar]
- 40.Eager R, Nemunaitis J. GM-CSF gene-transduced tumor vaccines. Mol Ther 2005; 12:18-27; PMID:15963916; http://dx.doi.org/ 10.1016/j.ymthe.2005.02.012 [DOI] [PubMed] [Google Scholar]
- 41.Pizzoferrato E, Chu NR, Hawley TS, Lieu FH, Barber BH, Hawley RG, Watts TH, Berinstein NL. Enhanced immunogenicity of B cell lymphoma genetically engineered to express both B7-1 and interleukin-12. Hum Gene Ther 1997; 8:2217-28; PMID:9449375; http://dx.doi.org/ 10.1089/hum.1997.8.18-2217 [DOI] [PubMed] [Google Scholar]
- 42.Nagata Y, Furugen R, Hiasa A, Ikeda H, Ohta N, Furukawa K, Nakamura H, Furukawa K, Kanematsu T, Shiku H. Peptides derived from a wild-type murine proto-oncogene c-erbB-2/HER2/neu can induce CTL and tumor suppression in syngeneic hosts. J Immunol 1997; 159:1336-43 [PubMed] [Google Scholar]
- 43.Ikuta Y, Okugawa T, Furugen R, Nagata Y, Takahashi Y, Wang L, Ikeda H, Watanabe M, Imai S, Shiku H. A HER2/NEU-derived peptide, a K(d)-restricted murine tumor rejection antigen, induces HER2-specific HLA-A2402-restricted CD8(+) cytotoxic T lymphocytes. Int J Cancer 2000; 87:553-8; PMID:10918197; http://dx.doi.org/ 10.1002/1097-0215(20000815)87:4%3c553::AID-IJC15%3e3.0.CO;2-8 [DOI] [PubMed] [Google Scholar]
- 44.Landuzzi L, Antognoli A, Nicoletti G, Croci S, Palladini A, Ianzano ML, Murgo A, Stivani V, Grosso V, Nanni P, et al.. HER-2/neu tolerant and non-tolerant mice for fine assessment of antimetastatic potency of dendritic cell-tumor cell hybrid vaccines. Vaccine 2011; 29:4690-7; PMID:21569812; http://dx.doi.org/ 10.1016/j.vaccine.2011.04.096 [DOI] [PubMed] [Google Scholar]
- 45.Nanni P, Landuzzi L, Nicoletti G, De Giovanni C, Rossi I, Croci S, Astolfi A, Iezzi M, Di Carlo E, Musiani P, et al.. Immunoprevention of mammary carcinoma in HER-2/neu transgenic mice is IFN-gamma and B cell dependent. J Immunol 2004; 173:2288-96; http://dx.doi.org/ 10.4049/jimmunol.173.4.2288 [DOI] [PubMed] [Google Scholar]
- 46.De Giovanni C, Nicoletti G, Landuzzi L, Astolfi A, Croci S, Comes A, Ferrini S, Meazza R, Iezzi M, Di Carlo E, et al.. Immunoprevention of HER-2/neu transgenic mammary carcinoma through an interleukin 12-engineered allogeneic cell vaccine. Cancer Res 2004; 64:4001-9; PMID:15173014; http://dx.doi.org/ 10.1158/0008-5472.CAN-03-2984 [DOI] [PubMed] [Google Scholar]