

Abstract
Graft-versus-host disease (GVHD) is a significant cause of non-relapse mortality after allogeneic hematopoietic cell transplantation (allo-HCT). Existing strategies to prevent and treat GVHD are incomplete, where a significant portion of allo-HCT recipients developed this complication. Despite this, one such therapy has emerged involving the use of regulatory T cells (Tregs) to control GVHD. The use of natural Tregs (nTregs) yielded positive pre-clinical results and are actively under investigation to reduce GVHD. However, broad application of this approach may require standardization of Treg expansion methods and dosing. Inducible Tregs (iTregs) can be seamlessly generated, but controversial pre-clinical findings and phenotype instability have hampered their translation into the clinic. Here, we review the current biological differences between nTregs and iTregs, as well as their effects on GVHD and graft-versus-leukemia (GVL) responses. We conclude by exploring the idea of combinational cellular therapies for the prevention of GVHD and preservation of GVL.
Keywords: Graft-versus-host disease (GVHD), nTregs, iTregs, Cellular therapy
INTRODUCTION
Allogeneic hematopoietic cell transplantation (allo-HCT) provides a reconstituted, healthy immune system for patients suffering from bone marrow failure syndromes and hematological malignancies such as leukemias, lymphomas, and myelomas. Donors are identified by high-resolution typing of class I and II human leukocyte antigen (HLA), and typically selected by recipient matching at HLA-A, -B, -C, -DRB1, DQB1, and –DPB1 [1]. Disparity within the major HLA, or even minor histocompatibility antigens [2], may stimulate donor T cells to induce GVHD. However, this is offset by the anti-cancer graft-versus-leukemia (GVL) effect of the allograft. The pathophysiology of GVHD is complex, involving many different T-helper cell types which contribute to disease manifestation; we refer the readers to our extensive review discussing the characteristics of these cells [3].
In brief, following conditioning, damage to host tissues causes the release of pro-inflammatory cytokines and danger-associated molecular pattern molecules (DAMPs), which in turn activate recipient antigen-presenting cells (APCs).
These host APCs then present host antigens to the donor T cells, which rapidly expand and differentiate into effector T cells (Teffs). Following differentiation, Teffs migrate to the GVHD target organs (skin, liver, lung, and gut) and cause end organ damage [3]. Despite extensive advancements in HLA matching, immunosuppressive drugs, and conditioning therapies, many patients that receive allo-HCT still succumb to primary disease (37%), GVHD (20%), or infection (17%), respectively [4]. Clearly, there is room for improving the success of allo-HCT. Many clinicians and scientists have begun to embrace the concept of harnessing our own suppressive immune cells, T regulatory cells (Tregs), to improve recipient survival and quality of life [5–7]. A delicate balance exists between GVL and GVHD responses, with too much suppression leading to tumor relapse and too little suppression leading to alloreactivity and end organ damage (Figure 1). Alas, balancing these fine cellular mechanisms has yet to be realized. Nonetheless, Tregs, with their ability to acquire antigen specificity, may be the answer clinicians and scientists have been looking for.
Figure 1. Delicate Balance between GVH and GVL responses.
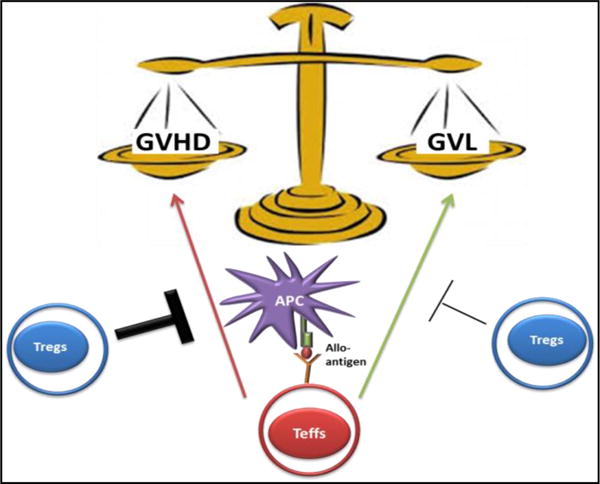
Following allogeneic HSCT, effector T cells within the graft inoculum recognize non-hematopoietic and hematopoietic allo-antigens presented by host and/or donor APCs resulting in both graft-versus-host (GVH) and graft-versus-leukemia (GVL) responses. Treg therapy could improve outcomes in allo-HSCT by greatly inhibiting Teffs cells causing GVHD with little or partial inhibition of the GVL effect.
Tregs are relatively young, first being described as “suppressor T cells” in the 1970’s by Gershon and Kondo, who conducted elegant experiments illustrating that induction of tolerance was dependent on thymus-derived lymphocytes, and not B cells [8,9]. However, due to the inability to clearly characterize this suppressive lymphocyte population, controversial findings within the I-J region [10], and limitations in scientific techniques, the “suppressor T cells” fell off the scientific map for 12 years. In 1982, Sakaguchi and colleagues, while studying the effects of neonatal thymectomy on normal immune homeostasis, stumbled upon a very important discovery: within the CD4 T lymphocyte compartment were cells capable of causing autoimmune disease and those capable of preventing it [11]. Thirteen years later, Sakaguchi was able to distinguish a reliable cell surface marker (CD25) which could differentiate between the protective CD4 T cells (CD25hi) fraction from the pathologic CD4 cells (CD25low) [12]. However, activated T cells can also express CD25, therefore negating the exclusivity of CD25 as marker for Tregs [13]. Luckily, advances in intracellular staining techniques allowed for the discovery of Foxp3 (a member of the forkhead winged helix family), the master transcription factor for determining Treg fate and suppressive function [14]. The specificity of Foxp3 to the Treg lineage was solidified by the finding that patients suffering from the autoimmune disease immunedysregulation polyendocrinopathy enteropathy X-linked syndrome (IPEX) had inherited germline mutations within the FOXP3 gene, which resulted in non-functional Tregs [15]. Scrufy mice, harboring a deletion of the Foxp3 gene, also display a lymphoproliferative disease characterized by multiorgan damage. The ability to definitively isolate and study Tregs (CD4+CD25+Foxp3+) in autoimmune diseases clearly shows that the major function of these cells is to maintain immune homeostasis.
Characteristics of T regulatory cells
Development and Generation
With the identification of Foxp3, studies on Tregs increased exponentially and soon after we would find that regulatory cells of the immune system were not just confined to expression of Foxp3 or even the T cell compartment. Over the years, multiple different flavors of regulatory cells have been discovered: Tr1 cells [16], CD8+-Tregs [17,18] myeloid derived suppressor cells (MDSC) [19], and B cells (B10 cells) [20].
In this review, we will focus on CD4+CD25+Foxp3+ regulatory T cells. As stated in the introduction, early neonatal thymectomy on day 3 versus day 7 of life pointed to the thymus as a major tissue associated with generation of Treg [21]. Experiments transferring the CD25+CD4+ Tregs from the periphery and the resulting abolition of autoimmune disease in Scurfy mice [14] hinted that the Treg pool was actually comprised of two distinct subsets. Indeed, it is now widely accepted that Tregs can be either naturally derived from the thymus (nTregs) or converted from naïve CD4+CD25− T cells in the periphery termed as inducible Tregs (iTregs).
Both nTregs and iTregs have differential requirements for their generation, which helps characterize these two distinct subsets. nTregs are derived exclusively from the thymus. Upon recognition of self-antigen/self-MHC (major histocompatibility complex) with high affinity [22,23], co-stimulation from CD28/B7 interactions [24] and IL-2 (although not required) [25], nTregs begin to increase expression of Foxp3 and acquire suppressive function [26,27]. iTregs, on the other hand, arise in the periphery from a population of naïve T cells, and therefore do not recognize self-antigens with high affinity [28]. Instead, during chronic antigen exposure, including microbes in the gut and with suboptimal co-stimulation through CD28/B7, iTregs initiate the expression of Foxp3. In contrast to nTregs, iTregs require the presence of exogenous cytokines, IL-2 [25] and TGFβ [28], to fully differentiate into the commonly known suppressor T cells. Retinoic acid, (RA) produced by CD103+ dendritic cells (DC) in the gut, has also been shown to further drive conventional T cells to express Foxp3 [29,30] (Figure 2).
Figure 2. Comparison of nTregs and iTregs: Generation, Suppressive Mechanism, and Stability.
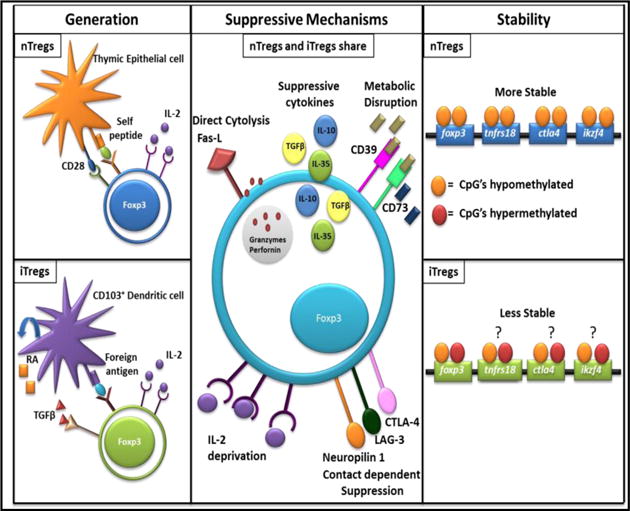
For generation nTregs and iTregs are distinct, with nTregs requiring recognition of self-antigen, costimulation, and IL-2; whereas iTregs recognize foreign antigen and require IL-2, TGFβ, and RA. nTregs and iTregs share suppressive mechanisms, broadly defined as direct cytolysis, suppressive cytokines, metabolic disruption, IL-2 deprivation, and contact dependent suppression. nTregs are more stable than iTregs with a fully demethylated CNS2 region with the foxp3 gene whereas iTregs sometimes display a partially methylated CNS2.
Suppressive Mechanisms
While nTregs and iTregs may differ in their requirements for generation, they utilize a multitude of similar mechanisms in order to maintain immune homeostasis [31,32] (Figure 2).
Tregs are activated via TCR engagement, which is absolutely necessary to mediate their suppressive function in vivo. In an elegant study using inducible genetic ablation of cell surface TCR complexes, Levine and colleagues found that TCR stimulation was surprisingly not required for Foxp3 expression, stability, or the ability of Tregs to consume IL-2 [33]. Instead, TCR activation is necessary for the expression of a limited number of genes, like IRF4, that are required for activated Tregs to maintain suppressive function [33]. The suppressive mechanisms of Tregs can be broadly classified into contact-dependent or contact-independent suppression. Contact-dependent suppression involves the expression of inhibitory molecules: CTLA-4 (cytotoxic T-lymphocyte-associated protein 4), LAG-3 (lymphocyte activation gene 3), and Neuropilin-1. CTLA-4 inhibits expression of the costimulatory markers CD80/CD86 on the surface of APCs through trans-endocytosis [34], and thus results in decreased proliferation of T cells. Specific deletion of CTLA4 in Tregs resulted in decreased suppressive function [35]. LAG-3 binds to MHC-Class II with a high affinity [31] on immature DCs and inhibits their maturation and co-stimulatory capacity [36]. Neuropilin-1, a recently discovered component of the Treg suppressive arsenal, was found to potentiate long-lasting interactions between Tregs and DCs. Neuropilin-1 ablation resulted in attenuated Treg suppressive function [37,38].
In conjunction with contact-dependent suppression, Tregs utilize contact-independent mechanisms that create an immunosuppressive milieu which can counteract the inflammatory milieu. A brief list of such mechanisms include the secretion on anti-inflammatory cytokines (IL-10, TGFβ, and IL-35), IL-2 consumption, release of granzymes, and generation of adenosine through ectoenzymes CD39/CD73 on the Treg surface (Figure 2). IL-10, an immunoregulatory cytokine, seems to act as a tissue- specific suppressive mechanism utilized by Tregs at intestinal interfaces. In an induced colitis model, IL-10 deficient Tregs could not protect mice during transfer of CD45RBhighCD4+ T cells [39]. Likewise, Rubtsov and colleagues generated a specific IL-10 ablation within Foxp3 expressing cells and found 40% of IL-10 deficient mice developed spontaneous colitis by 6 months of age. However, these same mice did not develop systemic autoimmunity [40]. The major function of TGFβ-mediated Treg suppression is surprisingly through contact-dependent, but APC-independent, induction of infectious tolerance via a process of converting naïve or Teffs into suppressive CD4+Foxp3+ suppressor T cells [41]. IL-35, much like TGFβ, has been implicated in conferring infectious tolerance by inducing iTr35 regulatory cells, which mediate suppression via IL-35 [42].
Interestingly, high expression of CD25 (IL-2 receptor alpha chain) not only aids in the identification of Tregs but also allows Tregs to non-specifically sequester IL-2 from the inflammatory microenvironment. This effect was illustrated when addition of common-γ chain cytokines reversed Treg-mediated T-cell apoptosis in vitro and in vivo [43]. Since Tregs require activation through TCR signaling, it is no surprise that they also express the ectoenzymes CD39/CD73, which convert extracellular adenosine triphosphate (ATP) into adenosine [44,45]. Tregs utilize adenosine by increasing its concentration within the inflammatory microenvironment, which increases adenosine binding to A2A adenosine receptors expressed on DCs and T cells. This leads to a subsequent increase of cyclic AMP, which results in inhibition of DCs and T cells [46]. Finally, Tregs can cause direct apoptosis of Teffs through the release of granzymes [47].
With regards to GVL/GVHD responses, the role of granzymes generated by Tregs is complex. Ley and colleagues found that granzyme B-expressing Tregs specifically accumulated in the tumor microenvironment and directly caused granzyme-mediated apoptosis of NK and CD8 Teffs, inhibiting tumor clearance. How [48] ever, some years later, Ley also noted that Tregs do not use granzyme B to mediate apoptosis in controlling Teffs during GVHD [49]. More recently, granzyme A was shown to be critical for Tregs in controlling intestinal GVHD. In this study, mice treated with Tregs deficient for granzyme A failed to rescue hosts from gastrointestinal GVHD [50]. IL-10 was also found to be a key factor utilized by nTregs to suppress GVHD, as CD4+CD25+ Tregs from IL-10−/− mice were ineffective in alleviating acute GVHD [51]. Homing to lymph nodes and target organs via CCR5 expression is also indispensable for the ability of Tregs to suppress GVHD. Genetic ablation of CCR5 negates the Treg’s ability to attenuate GVHD [52]. Another important molecule that is required for Tregs to suppress GVHD is CD62L, when CD62L is expressed at low levels on Tregs they cannot effectively home to the lymph nodes and suppress early activation of Teffs [53,54]. Hence, it seems Tregs use a vast repertoire of suppressive mechanisms to regulate immune reactions in a context and tissue-specific manner. Further research is needed to exploit these aspects of Treg suppression for maximal therapeutic efficacy.
Stability
In order to effectively incorporate nTregs or iTregs as a cellular therapy, whether for GVHD or autoimmune disorders, strict precautions must be taken to ensure patient safety. The advantage of cellular therapy is that these Tregs arise naturally to promote immune homeostasis. Therefore, off-target side effects, like those seen with pharmacological therapy, should be reduced significantly. However, two different lineage-tracing studies revealed that Foxp3 expression could be lost in a subset of Tregs, referred to as “ex-Tregs”.
The degree of stability varied based on the tracking system deployed by each lab, in one study when Foxp3 was tagged using NOD BAC transgenic mice expressing GFP-Cre within the Foxp3 promoter crossed with ROSA-LSL-YFP mice, allowing Tregs to be labeled with YFP before loss of Foxp3-GFP, the investigators reported 10–15% of Tregs to be GFP−YFP+ “ex-Tregs” [55].
When another group used a tamoxifen-inducible GFP-Cre fusion with the estrogen receptor mutant (GFP-creERt2) crossed with ROSA-LSL-YFP, allowing for transient tagging of Tregs, they reported 96% of Tregs to be stable GFP+YFP+ even under inflammatory conditions [56]. The discrepancy between these two lineage-tracing studies is still under active investigation. Hesitation among clinicians and scientists began after these initial lineage-tracing studies and was amplified with the finding that nTregs can lose expression of Foxp3 after repeated rounds of ex vivo stimulation [57,58]. Taking these finding into account, a major concern becomes apparent: how can we ensure the Treg cellular therapy remains suppressive and safe if the master transcription factor and regulator of suppressive function, Foxp3, is lost?
The questions surrounding the environmental factors, external stimuli, and intrinsic mechanisms that maintain or negate the expression and stability of Foxp3 have become extraordinarily prevalent in the field of Treg research, and still remain a hot topic of debate. Recently, numerous extensive reviews have explored the notion of Treg stability versus Treg plasticity, with the general consensus being that Tregs possess the ability to display both of these characteristics depending on the microenvironmental signals they receive [59,60]. Treg stability can be generally separated into two subsets: the epigenetic control of Foxp3 (gene regulation) and the stability of Foxp3 (transcription factor maintenance). Classically, a stable Treg’s genetic signature consisted of highly demethylated CpG islands within the conserved non-coding sequence 2 (CNS2) in the Treg-specific demethylation region (TSDR), with nTregs displaying fully demethylated CNS2 and iTregs displaying partially demethylated CNS2 regions [61]. However, the field of Treg genetic stability has moved from a Foxp3 centric view to a multiple Treg-signature gene view, termed “nTreg-Me” by Ohkura et al. [62]. In these experiments, it was demonstrated that CpG hypomethylation of four Treg signature genes: Foxp3, Tnfrs18 (GITR), Ctla4, and Ikzf4 (Eos) was independent of Foxp3 expression and occurred following strong and/or chronic TCR signaling. Importantly, it was found that cells expressing Foxp3, but without a full nTreg-Me signature, can lose stability and become plastic, secreting proinflammatory cytokines (62) (Figure 2). In line with this study was the establishment of the Treg-quintet: a complex of five redundant transcription factors that act in conjunction with Foxp3 to fully establish the Treg-signature [63]. Any one of these factors, Eos, IRF4, GATA-1, Lef-1, and Stab1 can help stabilize Foxp3 after it binds its target site, resulting in either repression of IL-2 or enhancement of CTLA-4 expression, thus fully committing the cell to the Treg phenotype.
Given that expression of the Foxp3 protein itself ensures inheritable maintenance of the Treg phenotype through direct binding to the CNS2 in a Cbfb-Runx1 demethylation dependent manner [61], any investigators have shifted their focus to identifying what factors contribute to the stability of Foxp3 expression. Recently, some key negative (CDK2 and Stub1) and positive (PTEN and Ezh2) regulators have emerged. Cyclin-dependent kinase 2 (CDK2) was found to phosphorylate Foxp3, which then recruits the E3 ubiquitin ligase Scf/Fwb7. Furthermore, when CDK2 was genetically deleted, the half-life of Foxp3 was dramatically increased, resulting in a more potently suppressive Treg [64]. Likewise, the E3 ubiquitin ligase, Stub1, was found to polyubiquinate Foxp3 in a heat shock protein 70-dependent fashion during inflammatory responses [65]. Silencing of Stub1 decreased the degradation of Foxp3 and enhanced protection from T cell mediated colitis in mice [65]. Conversely, phosphatase and tensin homolog (PTEN) deficiency lead to a loss of CD25 expression, and eventual loss of Foxp3 expression and suppressive function. This effect can likely be attributed to overt signaling through PI3(K), a direct target of PTEN [66,67]. Finally, the chromatin-modifying enzyme (Ezh2) was found to aid Foxp3 in binding to repression target genes (IL-2 and IFNγ) in order to silence them. Genetic ablation of Ezh2 lead to a decrease in Foxp3+ cells in non-lymphoid tissues and expression of genes resembling Teffs at those sites [68]. Hence, Ezh2 deficiency in this context failed to protect mice from autoimmune colitis [69]. More specifically, Ezh2 may impacts Tregs in tissue specific manner as Ezh2 deficient Tregs displayed reduced expansion on the spleen and lymph nodes, but not in the thymus and lamina propria [69]. Furthermore, He et al. demonstrated that Ezh2 plays an important role in Treg survival and expansion post BMT [70]. Extensive research is needed to understand exactly what can make, and more importantly, maintain a stable Treg phenotype if we hope to one day apply Treg therapy in a clinical setting.
Harnessing Tregs for Cellular Therapy in GVHD
nTregs
Given their natural presence, high stability, and important function in maintaining homeostasis, nTregs were the first subset of Tregs to be explored as an option for cellular therapy. The uncontrolled immune activation, high likelihood of disease (GVHD), limited therapeutic options, and steroid refraction that surround allo-HCT made nTregs an ideal candidate for a potential therapeutic. Initial experiments in pre-clinical models found that donor-type CD25+CD4+ Tregs could suppress lethal acute GVHD in BALB/c recipients, but only if a high ratio of 1:1 (Tregs: Teffs) was maintained [51]. The knowledge that nTregs only account for 5–10% of the total CD4 T-cell population and that a high number was needed to achieve GVHD attenuation made it clear that nTregs would need to be expanded ex vivo in order to achieve a more effective therapy. A seminal study from Blazar’s group in 2002 tested ex vivo polyclonal activated and expanded nTregs in three different models of lethal acute GVHD [71]. Importantly, this study established that nTregs can be expanded (67-fold) to sufficient numbers that can attenuate GVHD, thus offering a solution to the problem of low circulating nTregs. To further assess clinical applicability, investigators strove to see if nTregs would suppress the beneficial GVL effect. Using two different tumor models, A20 and BCL1, it was demonstrated that freshly isolated CD4+CD25+ Tregs did not impair the ability of Teffs to clear tumor at a 1:1 ratio. However, if the Teffs dose was below a certain threshold, the tumor relapsed [72].
With the strong preclinical findings indicating that nTregs could functionally attenuate GVHD while maintaining GVL, the field moved quickly to translate murine findings to human nTregs. Levings isolated CD4+CD25+ human nTregs from peripheral blood and expanded them with IL-2 and allogeneic feeder cells. These expanded nTregs remained unresponsive to allogeneic DCs and anti-CD3 activation, while maintaining the ability to suppress autologous CD25− T cells in vitro [73]. nTreg expansion of 100-fold was reached by Godfrey in 2004, using cell-sized dynabeads with anti-CD3 and anti-CD28 attached, CD4 feeder cells, and IL-2 [74]. It was found that these activated and expanded nTregs could potently suppress DC-driven allogeneic mixed lymphocyte reactions by 90%, and completely prevent the secretion of pro- inflammatory cytokines [74]. Since cord blood transplants are often used in the clinic, researchers also tested whether nTreg isolation and expansion from this source could also be effective. Cord blood was found to contain a larger CD25bright population compared to adult peripheral blood, in which the population was CD25dim indicating a non-suppressive function. These nTregs displayed a comparable growth rate to peripheral nTregs, and were also potently suppressive against allogeneic CD4+CD25− Teffs [75]. Lastly, based on the finding that human nTregs could be expanded more robustly using anti-CD3 loaded artificial APCs and could potently suppress xenogeneic GVHD [76], the first clinical trials were initiated for nTreg therapy for the treatment of GVHD.
Recently, a new concept has emerged regarding the expansion of nTreg cells for cellular therapy: selective expansion of the alloreactive nTregs within an apheresis product. This more personalized approach, using nTregs specific for both HLA-mismatched [7] and HLA-matched but minor antigen mismatched (miHAgs) [77], yielded a high number of potently suppressive nTregs. These results have initiated the first clinical trial using personalized nTregs to prevent acute GVHD [6].
In 2009, the first patients were treated with ex vivo expanded CD4+CD25+CD127− nTregs from donor peripheral blood [78]. In this initial trial, only two patients were enrolled, as nTreg therapy could only be initiated once standard immunosuppression failed. One patient developed acute GVHD and displayed transient alleviation of disease; however, the Treg source became exhausted and the patient later succumbed to multiorgan failure [78]. The other patient developed chronic GVHD. Yet, once nTreg therapy was initiated, a significant reduction in symptoms was observed [78]. Even though the sample size was very small, this study lead to the first dose escalation study for ex vivo expanded nTregs isolated from umbilical cord blood [79]. A dosing of 1, 3, 10, or 30 × 106 Tregs/kg was tested. Of the 23 patients enrolled, 17 patients received their target dose and no dose-limited toxicities were observed. A modest reduction in acute GVHD was observed in the 23 patients, compared with historical controls (43% vs 61%, respectively) [79]. In a very bold clinical trial, freshly isolated nTregs from donor peripheral blood were administrated four days prior to transplant, followed by no post-transplant immunosuppression. Of the 26 patients enrolled, only 2 developed GVHD. Given that no immunosuppression was used, this trial proved that nTregs could be used as a prophylactic for GVHD [80]. However, 13 of the 26 patients died within 3 months post-transplant from other co-morbidities. These three clinical trials have opened the door to a realm of possibilities for Treg therapy. However, there are still improvements that need to be made. For instance, the expansion potential of nTregs remains a major obstacle, as 5 patients did not receive sufficient cell doses [79]. Also, despite the success of using freshly isolated nTregs, a high ratio of 2:1 (Treg: Teff) was still needed to prevent GVHD [80].
iTregs
The study of iTregs in pre-clinical models of GVHD has been restricted to in vitro generation of iTregs due to the fact that an adequate marker to fully distinguish nTregs from iTregs has not been established. Given that conventional T cells comprise a larger percentage of peripheral blood or cord blood products and have an increased activation capacity compared to nTreg cells, protocols to polarize these cells into iTregs are currently being investigated. It is now well established that conventional CD4 T cells isolated from peripheral lymphoid organs can begin to express Foxp3 upon polyclonal stimulation with anti-CD3/anti-CD28 in the presence of TGFβ and IL-2 [28,81,82]; and the addition of retinoic acid (RA) can further enhance the expression of Foxp3 [29].
Unlike nTreg preclinical findings, which displayed similar results even across different expansion and GVHD models, there is still considerable controversy in the literature regarding iTreg therapy for the prevention or treatment of GVHD. This controversy seems to encompass differences in activation reagents, polarizing cytokines, and infusion schedule (Table 1). iTregs generated using polyclonal antigen-specific [86,87]/allo-antigen specific iTregs [88,89]. Beres et al illustrated that a high percentage of conversion can be achieved using polyclonal activation; however, even at a 1:1 (Treg: Teff) ratio, these iTregs could not effectively attenuate acute GVHD [83]. They claim that the ineffectiveness of iTreg therapy directly stems from the loss of Foxp3 expression. This finding agrees with the subsequent study by Zhang et al, which showed that polyclonal activated iTregs failed to protect recipient mice and could even be pathogenic if systemic rapamycin and IL-2 complexes were not co-administrated [84]. Despite these two pre-clinical findings, Hippen et al was able to induce naïve T cells from human peripheral blood products, and generated 240 × 109 iTregs after stimulation with KT64/86 cells (a K562 cell-based artificial APC with expression of CD86 and high affinity Fc receptor loaded with anti-CD3); these iTregs potently suppressed xenogeneic GVHD [85]. Alternatively, we have shown that using OT-II and HY-transgenic naïve T cells stimulated with either OVA [86] and HY peptide [87], induced a large amount of antigen-specific iTregs that potently suppress acute GVHD, even at low Treg: Teff ratios in both cases. This higher potency is attributed to the ability of antigen-specific iTregs to recognize antigen, as these antigen-specific iTregs failed to protect recipient mice when the cognate antigen was not expressed. This further emphasizes that continuous activation of Tregs through TCR engagement is essential for their suppressive function. In a non-irradiation BMT model, when naïve B6 T cells were used to generate induced alloreactive iTregs with BALB/c BM-derived mature DCs [88], the generated iTregs proved ineffective in protecting mice from GVHD. This was due primarily to loss of Foxp3 expression. In contrast, when CD11c+ splenic DCs [89] were used to generate induced alloreactive iTregs in the same manner, mice had significantly attenuated GVHD, and these iTregs were able to persist for 6 months in recipient mice. We have adapted the method established by Sela et al. and generated alloreactive CD4 iTregs, and have found these iTregs to be potently suppressive, and effectively attenuate GVHD in a major MHC-mismatched irradiated BMT model (unpublished observations). It is no surprise that antigen-specific iTregs are more potent and suppressive than polyclonal iTregs. According to a recent study, the two different activation signals impart different phenotypic profiles to each iTreg [90]. Physiologically activated iTregs displayed better control of Th1 responses as well as a broader range of chemokine and chemokine receptor expression than anti-CD3/CD28 activated iTregs [90]. This is a potential explanation for the differences seen between investigators with regards to the iTregs ability to attenuate GVHD.
Table 1. Summation of Differences in iTreg Pre-Clinical Results.
Differences in activation cue, polarizing cytokines, and infusion schedule have results in dramatically different results for iTreg therapy. Many more tests on GVL function are highly warranted.
| Investigator | Activation Reagent | Polarizing Cytokines | Infusion Schedule | GVHD Outcome | GVL Outcome |
|---|---|---|---|---|---|
| Koenecke et al 2009 | BM-derived mature DCs | IL-2 TGFβ RA | Tregs + Teff Day 0 | No attenuation | Not tested |
| Beres et al 2011 | Plate bound anti-CD3 Soluble anti-CD28 |
IL-2 TGFβ RA | Tregs + Teff Day 0 | No attenuation | Not tested |
| Semple et al 2011 | OVA peptide | IL-2 TGFβ RA | Tregs + Teff Day 0 | Significant attenuation | Not tested |
| Sela et al 2011 | CD11c+ DCs |
IL-2 TGFβ RA | Tregs (multiple infusions) Teff Day 0 | Significant attenuation | Not tested |
| Hippen et al 2011 | KT64/86 With anti-CD3 |
IL-2 TGFβ Rapamycin | Tregs + Teff Day 0 | Significant attenuation xenogeneic | Not tested |
| Zhang et al 2013 | Plate bound anti-CD3 Soluble anti-CD28 |
IL-2 TGFβ Rapamycin | Tregs + Teff Day 0 | Attenuation when stabilized by rapamycin/IL-2 complexes | Tregs impair CML clearance |
| Li et al 2015 | HY peptide | IL-2 TGFβ RA | Tregs Day 0 Teff Day 3 | Significant attenuation | Preserved GVL function |
| Heinrichs et al 2015 | CD11c+ DCs |
IL-2 IGFβ RA | Tregs Day 0 Teff Day 3 | Significant attenuation | Tregs impair P815 clearence |
Differences in the polarizing conditions would also account for the discrepancy seen in iTreg therapy in controlling GVHD. IL-2 and TGFβ are present throughout all experiments performed, however, some investigators use activation (anti-CD3/anti-CD28) [83,85] are inferior. rapamycin [84,85], while others use RA [83,86–89]. Since rapamycin has been shown to preferentially suppress Teffs while allowing for the growth/conversion of iTregs, the addition of this compound to generation conditions should yield a more pure population of iTregs [91]. Yet, our lab and others have proven that RA greatly increases the amount of naïve T cells converted into iTregs, which exhibit potent suppressive function. An important reason for this is that RA has been shown to increase the histone acetylation and methylation within the CNS elements of the Foxp3 promoter region, thus increasing accessibility of binding partners to the Foxp3 promoter [92].
Finally, the infusion schedule seems to play a major part in determining the degree of GVHD attenuation using iTreg therapy. Almost all studies use iTregs as a prophylactic therapy, as iTregs have yet to be shown to be beneficial as a treatment modality.
Most investigators infuse iTregs with T-cell depleted bone marrow and CD25-depleted Teffs within 24 hours of irradiation [83,84,88,89]. Noting the observation that initial infusion of nTregs two days prior to Teffs infusion resulted in a robust expansion of nTregs and a 10-fold decrease in the amount of Tregs needed to attenuate GVHD [93], we strove to apply this infusion schedule to iTreg therapy.
Indeed, we found infusion of iTregs prior to Teffs greatly increased the potency of iTregs in attenuating GVHD [87]. Despite these conflicting results, the first dose escalation in a clinical trial using iTregs [85] will be tested in adults receiving non-myeloablative HLA-identical sibling donor transplantation [94]. We are eagerly awaiting the outcome of this trial, as it will further contribute to our understanding of iTreg cellular therapy.
Preserved or Compromised GVL
Although attenuation of GVHD is the main focus of investigators in assessing the potential for Treg therapy, suppression of Teffs can only reach a certain threshold before these cells are unable to clear recipients of residual tumor cells (the GVL effect). In fact, the increase in Treg numbers in the peripheral blood and/or tumor microenvironment positively correlates with tumor relapse or growth in mice and humans [95,96]. With regards to nTreg therapy, pre-clinical models show contrasting results depending on the type of tumor tested. In models using A20 [72,97] and BCL1 [72], Tregs did not inhibit the GVL effect. However, the GVL effect was only slightly inhibited in a model using P815 mastocytoma [97]. Be that as it may, initial nTreg clinical trials indicated no increased incidence of tumor relapse compared to historical controls [79]. iTregs, on the other hand, seem to be more complex. Zhang et al. found that polyclonal activated CD4 iTregs, despite being unable to attenuate GVHD without the addition of rapamycin, also impaired the capacity of Teffs to clear primary myeloid blast crisis CML. This impairment was not due to rapamycin administration, as mice treated only with rapamycin did not succumb to tumor mortality [84]. In our lab, we found HY-specific iTregs could attenuate GVHD and still maintain the GVL effect, even against pre-established P815 mastocytoma tumors [87]. However, our recent data shows that alloreactive CD4 iTregs, when infused three days prior to Teffs, significantly impairs the GVL effect (unpublished findings). We aim to further elucidate the mechanism that underlies CD4 iTregs impairment of GVL function.
Improving iTreg Therapy
CD8 iTregs
A less understood population of suppressor T cells is derived from the CD8 T-cell lineage [17,18]. Surprisingly, after allogeneic BMT in murine models, significant populations of CD8+CD25+Foxp3+ iTregs have been shown to emerge early after transplantation [98,99], but not after syngeneic transplant. These CD8 iTregs were found to express similar suppressive molecules as CD4 iTregs (GITR, CD44, CTLA-4, and CD25), and could potentially be substitutes for CD4 iTregs to attenuate GVHD [98].
Though, CD8 iTregs did express increased levels of α4β7 when compared to CD4 iTregs [99]. Importantly, when these CD8 iTregs were isolated from recipient mice and used as a prophylactic in secondary recipients, they were able to significantly attenuate GVHD [99]. To compare these findings to human samples, patients’ peripheral blood was analyzed 6 months post-transplant and, surprisingly, no CD8+CD25+Foxp3+ iTregs were found. Authors later found that all patients had received cyclosporine as a prophylactic and thus concluded CD8 iTregs were acutely sensitive to cyclosporine treatment [99]. Future experiments are needed to see if this population arises in patients receiving various prophylactic therapies, such as rapamycin.
Currently, only two groups have published pre-clinical experimental data using in vitro generated CD8 iTregs to attenuate GVHD, with one result contradicting the other. While testing polyclonal CD4 iTregs, Zhang et al. simultaneously generated polyclonal CD8 iTregs, and found them to be equally pathogenic due to loss of Foxp3 expression 3 weeks post-transplant [84]. They also found CD8 iTregs to be less responsive to Foxp3 stabilization, using rapamycin and IL-2 complex treatments, as compared to CD4 iTregs [84]. Due to the inability to attenuate GVHD, the GVL function was not assessed in that study. In contrast, CD8 iTreg therapy by Zheng and colleagues involved isolating naïve human CD8+CD25−CD45RA+CD45RO− and generating alloreactive CD8 iTregs (termed CD8hi) by stimulating solely with hCD40-B cells (100). These CD8hi iTregs potently suppressed GVHD induced by hPBMC injected into Rag2−/−γc−/− mice (humanized mouse model of GVHD). Authors used lymphoblastoid cell line (LCL) to assess CD8hi ability to maintain the GVL effect. Infusion of CD8hi iTregs did not impair the GVL effect, as LCL tumor was cleared within the blood of recipient mice versus PBS treated controls [100]. Interestingly, it was found CD8hi iTregs had direct a cytotoxic effect against LCL tumors through Fas- FasL, perforin, and granzyme B pathways; inhibition of any of the three negated the lysis of LCL tumors in vitro. The cytotoxic effect of CD8 iTregs directly correlates with our own findings using murine alloreactive CD8 iTregs in GVHD. We found that CD8 iTregs possess some direct toxicity against P815 mastocytoma, yet not enough to fully eradicate tumor without Teff cell infusion (unpublished findings). Lastly, These CD8 iTregs were moderately effective at GVHD attenuation.
Two heads are better than one: Combinational Therapy
The dichotomy we have seen between CD4 and CD8 iTregs, especially with regards to GVL and GVHD responses, raises the question as to whether these cells can work together to optimize the outcome of allo-HCT? As our knowledge about cellular and biological processes continues to expand, clinicians and scientists have moved from a singular approach in order to incorporate a combinational therapeutic approach in a vast majority of disease models. Alloreactive CD4 iTregs are able to potently attenuate GVHD, yet severely compromise GVL function. Alloreactive CD8 iTregs only modestly attenuate GVHD, but possess GVL capability (unpublished findings). We hypothesized that combining these two cellular therapies would result in attenuation of GVHD while preserving the GVL effect. Indeed, we found that in allogeneic BMTs, a combination of CD4 iTregs and CD8 iTregs was effectively able to decrease GVHD while maintaining GVL (unpublished findings). The precise mechanisms underlying the ability of this combination therapy to mediate this effect are still under investigation. With regards to combinational therapy, two investigators have found that the addition of Rapamycin and IL-2 complexes in conjunction with iTreg infusion creates optimal attenuation of GVHD [84,99]. We believe that even more beneficial combinational therapies will emerge for GVHD in years to come.
Modifying Tregs
Tregs are the master regulators of balance in our immune systems. Given their natural function, we have tried to exploit them to control immune disorders characterized by unbridled inflammation (namely autoimmunity/GVHD). However, isolation, expansion, and reinfusion of Tregs did not result in an adequate therapy. Thus, investigators are eagerly testing new strategies to increase the specificity, stability, and activity of Tregs. Chimeric antigen receptor (CAR) modified T cells have shown great promise for increasing the antitumor effects in acute and chronic B cell malignancies [101,102] as well as some solid tumors [103]. Since Tregs are themselves derived from the same lymphocyte progenitors, it is tempting to envision the use of CARs to increase Treg specificity and stability. In this regard, specificity was easily achieved, illustrated by two studies using hapten-specific CAR Tregs that were more potent in alleviating experimental colitis than unmodified Tregs [104,105]. Given the increased potency of antigen-specific iTregs compared to polyclonal iTregs, it would be ideal to find a way to engineer iTregs that could specifically suppress responses against tissue damage (GVHD), while ignoring responses to tumor antigens (GVL). To increase stability, silencing of Stub1, a molecule that ubquinates Foxp3, was tested by infecting Tregs with lentivirus containing sh-Stub1 (silencing RNA). It was found that sh-Stub1 Tregs were more stable during experimental colitis induction [65]. Likewise, Restifo and colleagues established BACH2 as a key partner for Foxp3 stability, as genetic deletion resulted in Tregs inability to suppress lethal inflammation in RAG KO mice [106]. Therefore, Retrovirally induced expression of BACH2 in iTregs could potentially increase their stability and should be further investigated.
Concluding Remarks
The field of regulatory T cell therapy has come a long way since their discovery in 1970. However, there is still a long way to go. Although nTregs are an effective source for therapy, their low proliferative potential remains a major issue. Additionally, the expansion of GMP-grade nTreg for use in allo-HCT recipients requires significant clinical infrastructure and coordination. iTregs can be generated rapidly, but a consensus on their stability and ability to suppress GVHD has not been reached. With the field trending towards investigating the differential abilities between CD4 vs. CD8 iTregs with regards to GVHD and GVL responses, the convergence of these two therapies seems inevitable. The coming clinical trials involving both alloreactive nTregs and polyclonal iTregs will give us detailed insight into the next steps for improving iTreg cellular therapy for the treatment of GVHD. Furthermore, genetically engineering Tregs opens a new avenue to optimize or tailor Treg therapy in the near future.
Acknowledgments
We would like to thank all past and current members of the Yu Lab for their intellectual support. The research in the Yu Lab was supported in part by the US National Institutes of Health (R01 CA11816, CA143812, AI 082685, and CA169116).
Abbreviations
- allo-HCT
Allogeneic Hematopoietic Cell Transplantation
- GVHD
Graft-Versus-Host Disease
- GVL
Graft-Versus-Leukemia
- Tregs
Regulatory T Cells
- Ntregs
Natural T Regulatory Cells
- Itregs
Inducible T Regulatory Cells
- Teffs
Effector T Cells
- HLA
Human Leukocyte Antigen
- Apcs
Antigen Presenting Cells
- IPEX
Immunedysregulation Polyendocrinopathy Enteropathy X-Linked Syndrome
- MHC
Major Histocompatibility Complex
- MDSC
Myeloid Derived Suppressor Cells
- RA
Retinoic Acid
- TCR
T Cell Receptor
- CTLA-4
Cytotoxic T-Lymphocyte-Associated Protein 4
- LAG-3
Lymphocyte Activation Gene 3
- ATP
Adenosine Triphosphate
- DC
Dendritic Cells
- CNS2
Conserved Non-Coding Sequence 2
- TSDR
Treg Specific Demethylation Region
- CDK2
Cyclin-Dependent Kinase 2
- Ezh2
Chromatin-Modifying Enzyme
- PTEN
Phosphatase and Tensin Homolog, LCL: Lymphoblastoid Cell Line
- Mihags
Minor Mismatch Antigens
- Damps
Danger-Associated Molecular Pattern
References
- 1.Pidala J, Lee SJ, Ahn KW, Spellman S, Wang HL, et al. Nonpermissive HLA-DPB1 mismatch increases mortality after myeloablative unrelated allogeneic hematopoietic cell transplantation. Blood. 2014;124:2596–606. doi: 10.1182/blood-2014-05-576041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goulmy E, Schipper R, Pool J, Blokland E, Falkenburg JH, et al. Mismatches of minor histocompatibility antigens between HLA-identical donors and recipients and the development of graft-versus-host disease after bone marrow transplantation. N Engl J Med. 1996;334:281–285. doi: 10.1056/NEJM199602013340501. [DOI] [PubMed] [Google Scholar]
- 3.Fu J, Heinrichs J, Yu XZ. Helper T-cell differentiation in graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Arch Immunol Ther Exp (Warsz) 2014;62:277–301. doi: 10.1007/s00005-014-0284-z. [DOI] [PubMed] [Google Scholar]
- 4.Pasquini MC, Z X. Current uses and outcomes of hematopoietic stem cell transplantation: 2014 CIBMTR Summary Slides 2014 [Google Scholar]
- 5.Brunstein CG, Blazar BR, Miller JS, Cao Q, Hippen KL, et al. Adoptive transfer of umbilical cord blood-derived regulatory T cells and early viral reactivation. Biol Blood Marrow Transplant. 2013;19:1271–1273. doi: 10.1016/j.bbmt.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anasetti C. Ex-vivo Expanded Donor Regulatory T Cells for Prevention of Acute Graft-Versus-Host Disease. 2015 clinicaltrails.gov.
- 7.Veerapathran A, Pidala J, Beato F, Yu XZ, Anasetti C. Ex vivo expansion of human Tregs specific for alloantigens presented directly or indirectly. Blood. 2011;118:5671–5680. doi: 10.1182/blood-2011-02-337097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gershon RK, Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology. 1970;18:723–737. [PMC free article] [PubMed] [Google Scholar]
- 9.Gershon RK, Cohen P, Hencin R, Liebhaber SA. Suppressor T cells. J Immunol. 1972;108:586–590. [PubMed] [Google Scholar]
- 10.Kronenberg M, Steinmetz M, Kobori J, Kraig E, Kapp JA, et al. RNA transcripts for I-J polypeptides are apparently not encoded between the I-A and I-E subregions of the murine major histocompatibility complex. Proc Natl Acad Sci USA. 1983;80:5704–5708. doi: 10.1073/pnas.80.18.5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakaguchi S, Takahashi T, Nishizuka Y. Study on cellular events in post-thymectomy autoimmune oophoritis in mice. II. Requirement of Lyt-1 cells in normal female mice for the prevention of oophoritis. J Exp Med. 1982;156:1577–1586. doi: 10.1084/jem.156.6.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 13.Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10:490–500. doi: 10.1038/nri2785. [DOI] [PubMed] [Google Scholar]
- 14.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 15.Gambineri E, Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX), a syndrome of systemic autoimmunity caused by mutations of FOXP3, a critical regulator of T-cell homeostasis. Curr Opin Rheumatol. 2003;15:430–435. doi: 10.1097/00002281-200307000-00010. [DOI] [PubMed] [Google Scholar]
- 16.Roncarolo MG, Bacchetta R, Bordignon C, Narula S, Levings MK. Type 1 T regulatory cells. Immunol Rev. 2001;182:68–79. doi: 10.1034/j.1600-065x.2001.1820105.x. [DOI] [PubMed] [Google Scholar]
- 17.Guillonneau C, Picarda E, Anegon I. CD8+ regulatory T cells in solid organ transplantation. Curr Opin Organ Transplant. 2010;15:751–756. doi: 10.1097/MOT.0b013e32834016d1. [DOI] [PubMed] [Google Scholar]
- 18.Cortesini R, LeMaoult J, Ciubotariu R, Cortesini NS. CD8+CD28− T suppressor cells and the induction of antigen-specific, antigen-presenting cell-mediated suppression of Th reactivity. Immunol Rev. 2001;182:201–206. doi: 10.1034/j.1600-065x.2001.1820116.x. [DOI] [PubMed] [Google Scholar]
- 19.Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. 2013;13:739–752. doi: 10.1038/nrc3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Candando KM, Lykken JM, Tedder TF. B10 cell regulation of health and disease. Immunol Rev. 2014;259:259–272. doi: 10.1111/imr.12176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asano M, Toda M, Sakaguchi N, Sakaguchi S. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J Exp Med. 1996;184:387–396. doi: 10.1084/jem.184.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, et al. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol. 2001;2:301–306. doi: 10.1038/86302. [DOI] [PubMed] [Google Scholar]
- 23.Lio CW, Hsieh CS. Becoming self-aware: the thymic education of regulatory T cells. Curr Opin Immunol. 2011;23:213–219. doi: 10.1016/j.coi.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 24.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 25.D’Cruz LM, Klein L. Development and function of agonist-induced CD25+FOXP3+ regulatory T cells in the absence of interleukin 2 signaling. Nat Immunol. 2005;6:1152–1159. doi: 10.1038/ni1264. [DOI] [PubMed] [Google Scholar]
- 26.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 27.Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med. 2005;201:723–735. doi: 10.1084/jem.20041982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen W, Jin W, Hardegen N, Lei KJ, Li L, et al. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med. 2007;204:1765–1774. doi: 10.1084/jem.20070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, et al. A functionally specialized population of mucosal CD103+ DCs induces FOXP3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of treg-mediated T cell suppression. Front Immunol. 2012;3:51. doi: 10.3389/fimmu.2012.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shevach EM, McHugh RS, Piccirillo CA, Thornton AM. Control of T-cell activation by CD4+ CD25+ suppressor T cells. Immunol Rev. 2001;182:58–67. doi: 10.1034/j.1600-065x.2001.1820104.x. [DOI] [PubMed] [Google Scholar]
- 33.Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol. 2014;15:1070–1078. doi: 10.1038/ni.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332:600–603. doi: 10.1126/science.1202947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, et al. CTLA-4 control over FOXP3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 36.Liang B, Workman C, Lee J, Chew C, Dale BM, et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J Immunol. 2008;180:5916–5926. doi: 10.4049/jimmunol.180.9.5916. [DOI] [PubMed] [Google Scholar]
- 37.Sarris M, Andersen KG, Randow F, Mayr L, Betz AG. Neuropilin-1 expression on regulatory T cells enhances their interactions with dendritic cells during antigen recognition. Immunity. 2008;28:402–413. doi: 10.1016/j.immuni.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Solomon BD, Mueller C, Chae WJ, Alabanza LM, Bynoe MS. Neuropilin-1 attenuates autoreactivity in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2011;108:2040–2045. doi: 10.1073/pnas.1008721108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J Exp Med. 1999;190:995–1004. doi: 10.1084/jem.190.7.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 41.Andersson J, Tran DQ, Pesu M, Davidson TS, Ramsey H, et al. CD4+ FOXP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J Exp Med. 2008;205:1975–1981. doi: 10.1084/jem.20080308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chaturvedi V, Collison LW, Guy CS, Workman CJ, Vignali DA. Cutting edge: Human regulatory T cells require IL-35 to mediate suppression and infectious tolerance. J Immunol. 2011;186:6661–6666. doi: 10.4049/jimmunol.1100315. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+FOXP3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. 2007;8:1353–1362. doi: 10.1038/ni1536. [DOI] [PubMed] [Google Scholar]
- 44.Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, et al. Expression of ectonucleotidase CD39 by FOXP3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225–1232. doi: 10.1182/blood-2006-12-064527. [DOI] [PubMed] [Google Scholar]
- 45.Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, et al. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5′-adenosine monophosphate to adenosine. J Immunol. 2006;177:6780–6786. doi: 10.4049/jimmunol.177.10.6780. [DOI] [PubMed] [Google Scholar]
- 46.Ernst PB, Garrison JC, Thompson LF. Much ado about adenosine: adenosine synthesis and function in regulatory T cell biology. J Immunol. 2010;185:1993–1998. doi: 10.4049/jimmunol.1000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol. 2005;174:1783–1786. doi: 10.4049/jimmunol.174.4.1783. [DOI] [PubMed] [Google Scholar]
- 48.Cao X, Cai SF, Fehniger TA, Song J, Collins LI, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. 2007;27:635–646. doi: 10.1016/j.immuni.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 49.Cai SF, Cao X, Hassan A, Fehniger TA, Ley TJ. Granzyme B is not required for regulatory T cell-mediated suppression of graft-versus-host disease. Blood. 2010;115:1669–1677. doi: 10.1182/blood-2009-07-233676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Velaga S, Ukena SN, Dringenberg U, Alter C, Pardo J, et al. Granzyme A Is Required for Regulatory T-Cell Mediated Prevention of Gastrointestinal Graft-versus-Host Disease. PLoS One. 2015;10:e0124927. doi: 10.1371/journal.pone.0124927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoffmann P, Ermann J, Edinger M, Fathman CG, Strober S. Donor-type CD4(+)CD25(+) regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J Exp Med. 2002;196:389–399. doi: 10.1084/jem.20020399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wysocki CA, Jiang Q, Panoskaltsis-Mortari A, Taylor PA, McKinnon KP, et al. Critical role for CCR5 in the function of donor CD4+CD25+ regulatory T cells during acute graft-versus-host disease. Blood. 2005;106:3300–3307. doi: 10.1182/blood-2005-04-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taylor PA, Panoskaltsis-Mortari A, Swedin JM, Lucas PJ, Gress RE, et al. 0 L-Selectin(hi) but not the L-selectin(lo) CD4+25+ T-regulatory cells are potent inhibitors of GVHD and BM graft rejection. Blood. 2004;104:3804–3812. doi: 10.1182/blood-2004-05-1850. [DOI] [PubMed] [Google Scholar]
- 54.Ermann J, Hoffmann P, Edinger M, Dutt S, Blankenberg FG, et al. Only the CD62L+ subpopulation of CD4+CD25+ regulatory T cells protects from lethal acute GVHD. Blood. 2005;105:2220–2226. doi: 10.1182/blood-2004-05-2044. [DOI] [PubMed] [Google Scholar]
- 55.Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 2009;10:1000–1007. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, et al. Stability of the regulatory T cell lineage in vivo. Science. 2010;329:1667–1671. doi: 10.1126/science.1191996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoffmann P, Boeld TJ, Eder R, Huehn J, Floess S, et al. Loss of FOXP3 expression in natural human CD4+CD25+ regulatory T cells upon repetitive in vitro stimulation. Eur J Immunol. 2009;39:1088–1097. doi: 10.1002/eji.200838904. [DOI] [PubMed] [Google Scholar]
- 58.Duarte JH, Zelenay S, Bergman ML, Martins AC, Demengeot J. Natural Treg cells spontaneously differentiate into pathogenic helper cells in lymphopenic conditions. Eur J Immunol. 2009;39:948–955. doi: 10.1002/eji.200839196. [DOI] [PubMed] [Google Scholar]
- 59.Sawant DV, Vignali DA. Once a Treg, always a Treg? Immunol Rev. 2014;259:173–191. doi: 10.1111/imr.12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hori S. Lineage stability and phenotypic plasticity of Foxp3(+) regulatory T cells. Immunol Rev. 2014;259:159–172. doi: 10.1111/imr.12175. [DOI] [PubMed] [Google Scholar]
- 61.Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, et al. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010;463:808–812. doi: 10.1038/nature08750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity. 2012;37:785–799. doi: 10.1016/j.immuni.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 63.Fu W, Ergun A, Lu T, Hill JA, Haxhinasto S, et al. A multiply redundant genetic switch ‘locks in’ the transcriptional signature of regulatory T cells. Nat Immunol. 2012;13:972–980. doi: 10.1038/ni.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morawski PA, Mehra P, Chen C, Bhatti T, Wells AD. Foxp3 protein stability is regulated by cyclin-dependent kinase 2. J Biol Chem. 2013;288:24494–24502. doi: 10.1074/jbc.M113.467704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen Z, Barbi J, Bu S, Yang HY, Li Z, et al. The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity. 2013;39:272–285. doi: 10.1016/j.immuni.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huynh A, DuPage M, Priyadharshini B, Sage PT, Quiros J, et al. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol. 2015;16:188–196. doi: 10.1038/ni.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shrestha S, Yang K, Guy C, Vogel P, Neale G, et al. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat Immunol. 2015;16:178–187. doi: 10.1038/ni.3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.DuPage M, Chopra G, Quiros J, Rosenthal WL, Morar MM, et al. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity. 2015;42:227–238. doi: 10.1016/j.immuni.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang XP, Jiang K, Hirahara K, Vahedi G, Afzali B, et al. EZH2 is crucial for both differentiation of regulatory T cells and T effector cell expansion. Sci Rep. 2015;5:10643. doi: 10.1038/srep10643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.He S, Xie F, Liu Y, Tong Q, Mochizuki K, et al. The histone methyltransferase Ezh2 is a crucial epigenetic regulator of allogeneic T-cell responses mediating graft-versus-host disease. Blood. 2013;122:4119–4128. doi: 10.1182/blood-2013-05-505180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Taylor PA, Lees CJ, Blazar BR. The infusion of ex vivo activated and expanded CD4(+)CD25(+) immune regulatory cells inhibits graft-versus-host disease lethality. Blood. 2002;99:3493–3499. doi: 10.1182/blood.v99.10.3493. [DOI] [PubMed] [Google Scholar]
- 72.Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, et al. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat Med. 2003;9:1144–1150. doi: 10.1038/nm915. [DOI] [PubMed] [Google Scholar]
- 73.Levings MK, Sangregorio R, Roncarolo MG. Human cd25(+)cd4(+) t regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J Exp Med. 2001;193:1295–1302. doi: 10.1084/jem.193.11.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Godfrey WR, Ge YG, Spoden DJ, Levine BL, June CH, et al. In vitro-expanded human CD4(+)CD25(+) T-regulatory cells can markedly inhibit allogeneic dendritic cell-stimulated MLR cultures. Blood. 2004;104:453–461. doi: 10.1182/blood-2004-01-0151. [DOI] [PubMed] [Google Scholar]
- 75.Godfrey WR, Spoden DJ, Ge YG, Baker SR, Liu B, et al. Cord blood CD4(+)CD25(+)-derived T regulatory cell lines express FoxP3 protein and manifest potent suppressor function. Blood. 2005;105:750–758. doi: 10.1182/blood-2004-06-2467. [DOI] [PubMed] [Google Scholar]
- 76.Hippen KL, Merkel SC, Schirm DK, Sieben CM, Sumstad D, et al. Massive ex vivo expansion of human natural regulatory T cells (T(regs)) with minimal loss of in vivo functional activity. Sci Transl Med. 2011;3:83ra41. doi: 10.1126/scitranslmed.3001809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Veerapathran A, Pidala J, Beato F, Betts B, Kim J, et al. Human regulatory T cells against minor histocompatibility antigens: ex vivo expansion for prevention of graft-versus-host disease. Blood. 2013;122:2251–2261. doi: 10.1182/blood-2013-03-492397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Trzonkowski P, Bieniaszewska M, Juscinska J, Dobyszuk A, Krzystyniak A, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin Immunol. 2009;133:22–26. doi: 10.1016/j.clim.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 79.Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. 2011;117:1061–1070. doi: 10.1182/blood-2010-07-293795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. 2011;117:3921–3928. doi: 10.1182/blood-2010-10-311894. [DOI] [PubMed] [Google Scholar]
- 81.Davidson TS, DiPaolo RJ, Andersson J, Shevach EM. Cutting Edge: IL-2 is essential for TGF-beta-mediated induction of FOXP3+ T regulatory cells. J Immunol. 2007;178:4022–4026. doi: 10.4049/jimmunol.178.7.4022. [DOI] [PubMed] [Google Scholar]
- 82.Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, et al. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25− T cells through Foxp3 induction and down-regulation of Smad7. J Immunol. 2004;172:5149–5153. doi: 10.4049/jimmunol.172.9.5149. [DOI] [PubMed] [Google Scholar]
- 83.Beres A, Komorowski R, Mihara M, Drobyski WR. Instability of Foxp3 expression limits the ability of induced regulatory T cells to mitigate graft versus host disease. Clin Cancer Res. 2011;17:3969–3983. doi: 10.1158/1078-0432.CCR-10-3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang P, Tey SK, Koyama M, Kuns RD, Olver SD, et al. Induced regulatory T cells promote tolerance when stabilized by rapamycin and IL-2 in vivo. J Immunol. 2013;191:5291–5303. doi: 10.4049/jimmunol.1301181. [DOI] [PubMed] [Google Scholar]
- 85.Hippen KL, Merkel SC, Schirm DK, Nelson C, Tennis NC, et al. Generation and large-scale expansion of human inducible regulatory T cells that suppress graft-versus-host disease. Am J Transplant. 2011;11:1148–1157. doi: 10.1111/j.1600-6143.2011.03558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Semple K, Yu Y, Wang D, Anasetti C, Yu XZ. Efficient and selective prevention of GVHD by antigen-specific induced Tregs via linked-suppression in mice. Biol Blood Marrow Transplant. 2011;17:309–318. doi: 10.1016/j.bbmt.2010.12.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li J, Heinrichs J, Haarberg K, Semple K, Veerapathran A, et al. HY-Specific Induced Regulatory T Cells Display High Specificity and Efficacy in the Prevention of Acute Graft-Versus-Host Disease. J Immunol. 2015 doi: 10.4049/jimmunol.1401250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Koenecke C, Czeloth N, Bubke A, Schmitz S, Kissenpfennig A, et al. Alloantigen-specific de novo-induced FOXP3+ Treg revert in vivo and do not protect from experimental GVHD. Eur J Immunol. 2009;39:3091–3096. doi: 10.1002/eji.200939432. [DOI] [PubMed] [Google Scholar]
- 89.Sela U, Olds P, Park A, Schlesinger SJ, Steinman RM. Dendritic cells induce antigen-specific regulatory T cells that prevent graft versus host disease and persist in mice. J Exp Med. 2011;208:2489–2496. doi: 10.1084/jem.20110466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhao C, Shi G, Vistica BP, Hinshaw SJ, Wandu WS, et al. Induced regulatory T-cells (iTregs) generated by activation with anti-CD3/CD28 antibodies differ from those generated by the physiological-like activation with antigen/APC. Cell Immunol. 2014;290:179–184. doi: 10.1016/j.cellimm.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 91.Gao W, Lu Y, El Essawy B, Oukka M, Kuchroo VK, et al. Contrasting effects of cyclosporine and rapamycin in de novo generation of alloantigen-specific regulatory T cells. Am J Transplant. 2007;7:1722–1732. doi: 10.1111/j.1600-6143.2007.01842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lu L, Ma J, Li Z, Lan Q, Chen M, et al. All-trans retinoic acid promotes TGF-beta-induced Tregs via histone modification but not DNA demethylation on Foxp3 gene locus. PLoS One. 2011;6:e24590. doi: 10.1371/journal.pone.0024590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nguyen VH, Zeiser R, Dasilva DL, Chang DS, Beilhack A, et al. In vivo dynamics of regulatory T-cell trafficking and survival predict effective strategies to control graft-versus-host disease following allogeneic transplantation. Blood. 2007;109:2649–2656. doi: 10.1182/blood-2006-08-044529. [DOI] [PubMed] [Google Scholar]
- 94.MacMillan M. Inducible Regulatory T Cells (iTregs) in Non-Myeloablative Sibling Donor Peripheral Blood Stem Cell Transplantation 2015 [Google Scholar]
- 95.Nishikawa H, Jager E, Ritter G, Old LJ, Gnjatic S. CD4+ CD25+ regulatory T cells control the induction of antigen-specific CD4+ helper T cell responses in cancer patients. Blood. 2005;106:1008–1011. doi: 10.1182/blood-2005-02-0607. [DOI] [PubMed] [Google Scholar]
- 96.Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999;163:5211–5218. [PubMed] [Google Scholar]
- 97.Trenado A, Charlotte F, Fisson S, Yagello M, Klatzmann D, et al. Recipient-type specific CD4+CD25+ regulatory T cells favor immune reconstitution and control graft-versus-host disease while maintaining graft-versus-leukemia. J Clin Invest. 2003;112:1688–1696. doi: 10.1172/JCI17702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Beres AJ, Haribhai D, Chadwick AC, Gonyo PJ, Williams CB, et al. CD8+ FOXP3+ regulatory T cells are induced during graft-versus-host disease and mitigate disease severity. J Immunol. 2012;189:464–474. doi: 10.4049/jimmunol.1200886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Robb RJ, Lineburg KE, Kuns RD, Wilson YA, Raffelt NC, et al. Identification and expansion of highly suppressive CD8(+)FoxP3(+) regulatory T cells after experimental allogeneic bone marrow transplantation. Blood. 2012;119:5898–5908. doi: 10.1182/blood-2011-12-396119. [DOI] [PubMed] [Google Scholar]
- 100.Zheng J, Liu Y, Liu M, Xiang Z, Lam KT, et al. Human CD8+ regulatory T cells inhibit GVHD and preserve general immunity in humanized mice. Sci Transl Med. 2013;5:168ra9. doi: 10.1126/scitranslmed.3004943. [DOI] [PubMed] [Google Scholar]
- 101.Porter DL, Kalos M, Zheng Z, Levine B, June C. Chimeric Antigen Receptor Therapy for B-cell Malignancies. J Cancer. 2011;2:331–332. doi: 10.7150/jca.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Elinav E, Adam N, Waks T, Eshhar Z. Amelioration of colitis by genetically engineered murine regulatory T cells redirected by antigen-specific chimeric receptor. Gastroenterology. 2009;136:1721–1731. doi: 10.1053/j.gastro.2009.01.049. [DOI] [PubMed] [Google Scholar]
- 105.Elinav E, Waks T, Eshhar Z. Redirection of regulatory T cells with predetermined specificity for the treatment of experimental colitis in mice. Gastroenterology. 2008;134:2014–2024. doi: 10.1053/j.gastro.2008.02.060. [DOI] [PubMed] [Google Scholar]
- 106.Roychoudhuri R, Hirahara K, Mousavi K, Clever D, Klebanoff CA, et al. BACH2 represses effector programs to stabilize T(reg)-mediated immune homeostasis. Nature. 2013;498:506–510. doi: 10.1038/nature12199. [DOI] [PMC free article] [PubMed] [Google Scholar]