

Abstract
Introduction Gorham–Stout Disease (GSD) is a rare disorder of bony destruction due to lymphangiomatosis, and is often triggered by hormones. One complication of GSD is the development of chylothorax, which carries a high mortality rate. Very little experience has been published to guide management in GSD during pregnancy to optimize both fetal and maternal health.
Case Study A 20-year-old woman with known GSD presented with shortness of breath at 18 weeks of pregnancy, due to bilateral chylothoraces which required daily drainage. To minimize chylous fluid formation, she was placed on bowel rest with total parenteral nutrition (limiting lipid intake) and received octreotide to decrease splanchnic blood flow and chylous fluid drainage. Treatment options were limited due to her pregnancy. Twice daily home chest tube drainage of a single lung cavity, total parenteral nutrition, octreotide, and albumin infusions allowed successful delivery of a healthy 37 weeks' gestation infant by cesarean delivery.
Discussion This case illustrates the management of a rare clinical disease of bone resorption and lymphangiomatosis complicated by bilateral, refractory chylothoraces, triggered by pregnancy, in whom treatment options are limited, and the need for a multidisciplinary health care team to ensure successful maternal and fetal outcomes.
Keywords: pregnancy, chylothorax, Gorham–Stout disease, total parenteral nutrition, octreotide
Gorham–Stout disease (GSD) is a rare and sporadic bone disorder characterized by destruction of the osseous matrix by the overgrowth of lymphatic vessels. The exact etiology of this disease is unknown. The first reported case of this condition was in 1838 by Jackson, who describes a case of a young man with a gradually vanishing humerus.1 It was not until 1955 that Gorham and Stout presented the first case of “vanishing bone disease” in an article where they described the disease and its association with angiomatosis.2 Since the initial description, over 200 cases have been reported worldwide in the medical literature. One of the more significant complications of GSD is the formation of chylothorax. The incidence of chylothorax occurs in roughly 20% of patients with GSD and is associated with a high mortality rate.3 Here, we present a rare and complicated case of a 20-year-old pregnant woman with GSD.
Case Study
Our patient was a 20-year-old woman with a known history of GSD, diagnosed at the age of 14 years shortly after the onset of puberty, after she developed left arm pain and weakness; on imaging, she was found to have an absence of her left clavicle, scapula, and superior aspect of her humerus. She never received treatment and was lost to follow-up until she presented to an outside hospital at 18 weeks' gestation of her first pregnancy with shortness of breath, paroxysmal nocturnal dyspnea, and four-pillow orthopnea. On evaluation, she had absent breath sounds halfway up her lungs bilaterally. Chest X-ray revealed an absence of the majority of her left clavicle, scapula, and humeral head as well as bilateral pleural effusions (Fig. 1). Therapeutic thoracentesis revealed a milky white fluid with laboratory analysis consistent with chyle. She had a rapid accumulation of chylous fluid despite daily drainage. During this hospital course, in an attempt to decrease the formation of chylous fluid, she was treated with alendronate injections (to prevent bone resorption), however, it had minimal effect. Due to persistent symptoms, she received 15 thoracenteses over the course of 2 weeks. At this time, she was transferred to our institution for further management.
Fig. 1.
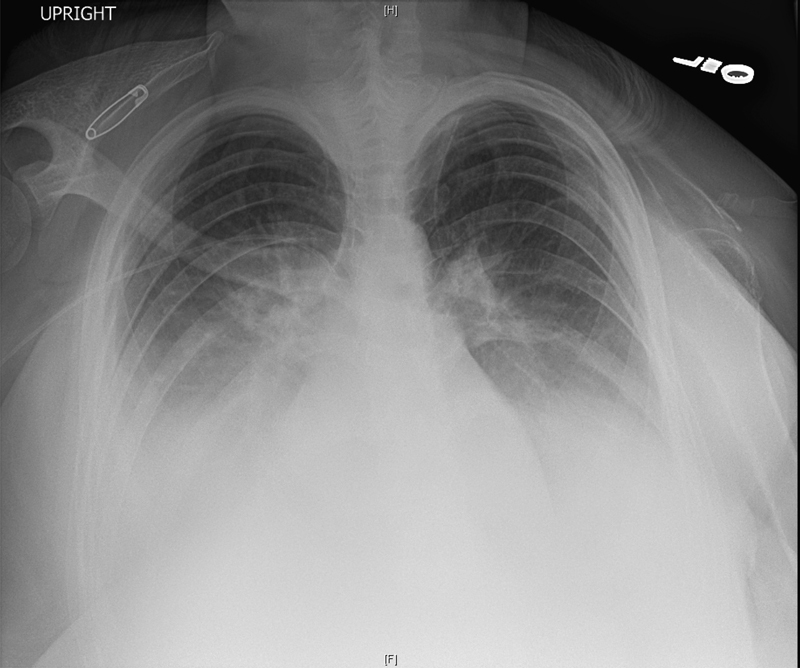
Chest X-ray demonstrating bilateral pleural effusions, absence of left clavicle, and portion of the scapula, degeneration of head of humerus.
On arrival, she was tachypneic, tachycardic, and uncomfortable appearing. She was using accessory muscles and was unable to lie flat. Laboratory studies were notable for albumin of 1.5 and analysis of her pleural fluid was consistent with chylothorax (triglycerides 985 mg/dL, total protein 1,266 g/dL, lactate dehydrogenase 94 IU/L, glucose 129 mg/dL, and cholesterol 60 mg/dL). She had a left-sided chest tube placed to allow for daily drainage of chylous fluid, which provided her symptomatic relief. There appeared to be a communication between both pleural spaces such that draining the left also lowered the pleural effusion on the right and did not require separate drainage. A multidisciplinary team was convened to determine the best care plan. Surgical options such as thoracic duct ligation or pleurodesis were discussed, but felt to be too risky during the second trimester. Medical therapies such as interferon, sirolimus, and thalidomide, which have been shown to have some success,4 were deferred given her pregnancy. She was placed on octreotide injections and started on total parental nutrition (TPN) with a < 5 g oral fat diet. The TPN helped decrease the rate of fluid accumulation as well as decrease the wasting of fat and protein into the pleural fluid. With these two interventions, her symptoms improved and average oxygen saturation increased from the low to high 90s. She was discharged with TPN and chest tube management with instructions to drain 1,000 to 1,500 mL twice a day. Due to the massive protein loss, she sustained from her chest tubes, her albumin remained low despite TPN protein replacement. This likely contributed to the development of a polymicrobial groin abscess as well as an upper extremity deep vein thrombosis. She had several ultrasounds to assess fetal growth, which initially did not show any evidence of intrauterine growth restriction, despite her severe hypoalbuminemia and malnutrition. However, she was admitted at 37 weeks for scheduled cesarean delivery; fetal ultrasound at that time showed intrauterine growth restriction (9th percentile) and oligohydramnios with an amniotic fluid index of 5 cm. She underwent a cesarean delivery at 37+4 and delivered a 2,270 g girl, Apgar scores were 8 and 9 at 1 and 5 minutes, respectively. Postpartum she continued to drain 3 to 4 L/d from her chest tube. Her baby girl has done well and is developing normally.
Discussion
GSD is a rare disease caused by the uncontrolled destructive proliferation of vascular or lymphatic tissue resulting in osteolysis of the skeletal system. The disease shows no clear inheritance pattern and can occur in patients of any age, with most cases occurring in children or adults less than 40, and the disease shows no clear sex predilection.5 6 However, some authors report that it is more common in males.7
Clinical features of GSD include bone pain, skeletal deformities, and muscle pain.8 GSD has been described throughout the body but most commonly affects the upper arm, shoulder girdle, and mandible. When the disease burden affects the ribs, scapular, or thoracic vertebrae this may result in the development of a chylothorax. Chylothorax has been described in roughly 17 to 25% of the patients2 9 and is thought to be a direct result of lymphatic invasion into the pleural cavity or thoracic duct.3 Based on small sample size, estimates suggest that chylothorax treated with medical management alone carries with it a 69% mortality, which improves to 36% mortality if surgical intervention is undertaken.3 Death generally occurs as a result of massive loss of protein, which leads to malnutrition and infection.3 10 While our patient had the scapular and clavicular disease for many years, it is not clear why she did not develop chylothorax until she became pregnant. To our knowledge, there have been no documented cases of pregnancy triggering chylothorax in GSD. There are case reports of idiopathic chylothorax in pregnancy, this has been theorized to be from changes in intrathoracic pressure or elevated estrogen.11
Diagnosis of GSD is often challenging as presentations vary depending on the location of the disease. General laboratory studies tend to be normal with alkaline phosphatase often slightly elevated.12 Diagnosis is usually based on radiologic evidence and histopathology. Initially on plain films, the bone lesions resemble osteoporotic lesions with intramedullary or subcortical radiolucent foci. These foci subsequently increase to involve the cortex, joints, and soft tissue leading to further loss of bone.2 12 13 Pathological fractures can occur at any time and regeneration of bone is rarely seen. Histologically, Gorham and Stout found that bone was replaced by fibrous tissue containing numerous dilated endothelial line vessels.14 With the advent of immunohistochemistry, these endothelial vessels were further characterized as lymphatic vessels.15
Theories as to the underlying mechanism of GSD are varied and controversial. Growth factors such as vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFDF), and platelet-derived growth factor (PDGF) drive lymphatic endothelial cell proliferation via the mechanistic target of the rapamycin (mTOR) pathway.4 The rampant growth of lymphatic tissue leads to bone absorption, potentially through compression2 of bone or local hypoxia.16 Some data suggest that osteoblasts are absent in affected bone and that they do not appear to respond to the presence of bone lesions in GSD by rebuilding.2 17 The role of osteoclasts is less clear; while several studies have cited increased levels of osteoclast activity18 19; others have noted a scarcity of osteoclasts around bone lesions.2 17 Many of the targets for therapy are informed by the understanding the lymphatic endothelial cell (LEC) proliferation pathway. Interferon inhibits PDGF and bFDF; thalidomide interferes with PDGF and sirolimus inhibits LEC proliferation through the mTOR pathway.
There are currently multiple strategies to treat GSD, depending on the severity and extent of organ involvement, and include surgery, radiotherapy, and medical therapy. Surgery is most commonly used in cases of chylothorax to help reduce pleural fluid buildup as well as for large symptomatic lesions. Interventions include pleurectomy, pleurodesis, or thoracic duct ligation.12 Radiation has been shown to help reduce disease burden and is commonly used in addition to surgery.20 21 Common medications used to treat GSD target the lymphatic endothelial cell proliferation pathway and include interferon α2b, anti-VEGF antibody (bevacizumab), sirolimus, and thalidomide.4 8 12 22 23 24 Interferon α2b has antiangiogenic characteristics and inhibit the formation of lymphatic vessels, bevacizumab is a recombinant human monoclonal antibody that blocks angiogenesis by inhibiting VEGF. Sirolimus (rapamycin) often used for immunosuppression during transplantation and for cancer therapy specifically blocks the function of the mTOR pathway and has been shown to have antiangiogenic properties. Thalidomide has both immunomodulatory and antiangiogenic characteristics.25 In patients with chylothorax, strategies are also used to decrease the production of chyle such as placing patients on a low (< 5 g) oral fat diet as triglycerides absorbed by the gastrointestinal tract go on to form chylous fluid. Patients on a minimal fat diet are then supplemented with TPN. Octreotide injections can also be used to decrease the flow of lymph and its return to the thoracic duct, thus decreasing chylothorax.26
Our patient did well on a combination of octreotide and TPN, both of which are acceptable in pregnancy, and was able to carry her fetus to 37 weeks. This case illustrates a rare clinical disease of bone resorption and lymphangiomatosis complicated by bilateral, refractory chylothoraces. In addition, it presents the difficulty of treating and managing a poorly understood complication in a pregnant woman in whom treatment options are limited.
References
- 1.Jackson J B. A boneless arm. Boston Med Surg J. 1838;18:368–369. [Google Scholar]
- 2.Dellinger M T, Garg N, Olsen B R. Viewpoints on vessels and vanishing bones in Gorham-Stout disease. Bone. 2014;63:47–52. doi: 10.1016/j.bone.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 3.Tie M L, Poland G A, Rosenow E C III. Chylothorax in Gorham's syndrome. A common complication of a rare disease. Chest. 1994;105(1):208–213. doi: 10.1378/chest.105.1.208. [DOI] [PubMed] [Google Scholar]
- 4.Hagendoorn J, Yock T I, Borel Rinkes I H, Padera T P, Ebb D H. Novel molecular pathways in Gorham disease: implications for treatment. Pediatr Blood Cancer. 2014;61(3):401–406. doi: 10.1002/pbc.24832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Möller G, Priemel M, Amling M, Werner M, Kuhlmey A S, Delling G. The Gorham-Stout syndrome (Gorham's massive osteolysis). A report of six cases with histopathological findings. J Bone Joint Surg Br. 1999;81(3):501–506. doi: 10.1302/0301-620x.81b3.9468. [DOI] [PubMed] [Google Scholar]
- 6.Kiran D N, Anupama A. Vanishing bone disease: a review. J Oral Maxillofac Surg. 2011;69(1):199–203. doi: 10.1016/j.joms.2010.05.088. [DOI] [PubMed] [Google Scholar]
- 7.Hu P, Yuan X G, Hu X Y, Shen F R, Wang J A. Gorham-Stout syndrome in mainland China: a case series of 67 patients and review of the literature. J Zhejiang Univ Sci B. 2013;14(8):729–735. doi: 10.1631/jzus.B1200308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y, Zhong D R, Zhou P R. et al. Gorham-Stout disease: radiological, histological, and clinical features of 12 cases and review of literature. Clin Rheumatol. 2016;35(3):813–823. doi: 10.1007/s10067-014-2780-2. [DOI] [PubMed] [Google Scholar]
- 9.Kose M, Pekcan S, Dogru D. et al. Gorham-Stout Syndrome with chylothorax: successful remission by interferon alpha-2b. Pediatr Pulmonol. 2009;44(6):613–615. doi: 10.1002/ppul.20849. [DOI] [PubMed] [Google Scholar]
- 10.Riantawan P, Tansupasawasdikul S, Subhannachart P. Bilateral chylothorax complicating massive osteolysis (Gorham's syndrome) Thorax. 1996;51(12):1277–1278. doi: 10.1136/thx.51.12.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brunelli A, Catalini G, Fianchini A. Pregnancy exacerbating unsuspected mediastinal lymphangioleiomyomatosis and chylothorax. Int J Gynaecol Obstet. 1996;52(3):289–290. doi: 10.1016/0020-7292(95)02619-3. [DOI] [PubMed] [Google Scholar]
- 12.Patel D V. Gorham's disease or massive osteolysis. Clin Med Res. 2005;3(2):65–74. doi: 10.3121/cmr.3.2.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nikolaou V S, Chytas D, Korres D, Efstathopoulos N. Vanishing bone disease (Gorham-Stout syndrome): A review of a rare entity. World J Orthod. 2014;5(5):694–698. doi: 10.5312/wjo.v5.i5.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gorham L W, Stout A P. Massive osteolysis (acute spontaneous absorption of bone, phantom bone, disappearing bone); its relation to hemangiomatosis. J Bone Joint Surg Am. 1955;37-A(5):985–1004. [PubMed] [Google Scholar]
- 15.Lala S, Mulliken J B, Alomari A I, Fishman S J, Kozakewich H P, Chaudry G. Gorham-Stout disease and generalized lymphatic anomaly—clinical, radiologic, and histologic differentiation. Skeletal Radiol. 2013;42(7):917–924. doi: 10.1007/s00256-012-1565-4. [DOI] [PubMed] [Google Scholar]
- 16.Heyden G, Kindblom L G, Nielsen J M. Disappearing bone disease. A clinical and histological study. J Bone Joint Surg Am. 1977;59(1):57–61. [PubMed] [Google Scholar]
- 17.Dickson G R, Hamilton A, Hayes D, Carr K E, Davis R, Mollan R A. An investigation of vanishing bone disease. Bone. 1990;11(3):205–210. doi: 10.1016/8756-3282(90)90215-k. [DOI] [PubMed] [Google Scholar]
- 18.Hirayama T, Sabokbar A, Itonaga I, Watt-Smith S, Athanasou N A. Cellular and humoral mechanisms of osteoclast formation and bone resorption in Gorham-Stout disease. J Pathol. 2001;195(5):624–630. doi: 10.1002/path.989. [DOI] [PubMed] [Google Scholar]
- 19.Choma N D, Biscotti C V, Bauer T W, Mehta A C, Licata A A. Gorham's syndrome: a case report and review of the literature. Am J Med. 1987;83(6):1151–1156. doi: 10.1016/0002-9343(87)90959-4. [DOI] [PubMed] [Google Scholar]
- 20.Heyd R, Micke O, Surholt C. et al. Radiation therapy for Gorham-Stout syndrome: results of a national patterns-of-care study and literature review. Int J Radiat Oncol Biol Phys. 2011;81(3):e179–e185. doi: 10.1016/j.ijrobp.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 21.Dunbar S F, Rosenberg A, Mankin H, Rosenthal D, Suit H D. Gorham's massive osteolysis: the role of radiation therapy and a review of the literature. Int J Radiat Oncol Biol Phys. 1993;26(3):491–497. doi: 10.1016/0360-3016(93)90968-2. [DOI] [PubMed] [Google Scholar]
- 22.Grunewald T G, Damke L, Maschan M. et al. First report of effective and feasible treatment of multifocal lymphangiomatosis (Gorham-Stout) with bevacizumab in a child. Ann Oncol. 2010;21(8):1733–1734. doi: 10.1093/annonc/mdq331. [DOI] [PubMed] [Google Scholar]
- 23.Nir V, Guralnik L, Livnat G. et al. Propranolol as a treatment option in Gorham-Stout syndrome: a case report. Pediatr Pulmonol. 2014;49(4):417–419. doi: 10.1002/ppul.22869. [DOI] [PubMed] [Google Scholar]
- 24.Brodszki N, Länsberg J K, Dictor M. et al. A novel treatment approach for paediatric Gorham-Stout syndrome with chylothorax. Acta Paediatr. 2011;100(11):1448–1453. doi: 10.1111/j.1651-2227.2011.02361.x. [DOI] [PubMed] [Google Scholar]
- 25.Guba M, von Breitenbuch P, Steinbauer M. et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med. 2002;8(2):128–135. doi: 10.1038/nm0202-128. [DOI] [PubMed] [Google Scholar]
- 26.Bender B, Murthy V, Chamberlain R S. The changing management of chylothorax in the modern era. Eur J Cardiothorac Surg. 2016;49(1):18–24. doi: 10.1093/ejcts/ezv041. [DOI] [PubMed] [Google Scholar]