

Abstract
We herein report a new catalytic method for intermolecular olefin aminofluorination using earth-abundant iron catalysts and nucleophilic fluoride ion. This method tolerates a broad range of unfunctionalized olefins, especially nonstyrenyl olefins that are incompatible with existing olefin aminofluorination methods. This new iron-catalyzed process directly converts readily available olefins to internal vicinal fluoro carbamates with high regioselectivity (N vs F), many of which are difficult to prepare using known methods. Preliminary mechanistic studies demonstrate that it is possible to exert asymmetric induction using chiral iron catalysts and that both an iron-nitrenoid and carbocation species may be reactive intermediates.
Graphical Abstract

INTRODUCTION
Selective incorporation of fluorine into organic molecules is an emerging area with increasing importance in organic synthesis because fluorine atoms are present in numerous pharmaceuticals and biological probes.1 In particular, intermolecular olefin functionalization via fluorine atom transfer represents a convenient approach to synthesize high-value building blocks with fluorine-containing stereogenic centers.2 Among a variety of olefin fluorination reactions, the intermolecular amino-fluorination of unfunctionalized olefins is of great value since it can directly convert simple olefins to vicinal fluoro primary amines which are important structural motifs in medicinal chemistry.3 However, development of this catalytic reaction has been challenging; only a few methods were reported, and most of them are exclusively compatible with styrenyl olefins.4
Amid these discoveries, Stavber reported Ritter-type electrophilic olefin vicinal fluoro acetamidation in acetonitrile using electrophilic fluorination reagent Accufluor NFTh, through β-fluorocarbenium intermediates, to afford terminal fluorides; however, the substrate scope was predominantly limited to styrenyl olefins.4a Liu reported palladium-catalyzed styrene aminofluorination using electrophilic NFSI, which affords terminal sulfonamido fluorides;4b the same group later documented a related styrene aminofluorination using AgF that provides internal sulfonamido fluorides.4e
The existing intermolecular olefin aminofluorination methods are mechanistically illuminating; however, there still exist significant gaps for this important transformation. First, there has yet to be developed a method for catalytic amino-fluorination of a broad range of unf unctionalized olef ins (especially for nonstyrenyl olefins). Next, the feasibility of asymmetric catalysis for intermolecular aminofluorination of unfunctionalized olefins has not yet been demonstrated.5 Additionally, the use of earth-abundant iron catalysts to promote olefin aminofluorination with simple nucleophilic fluoride ion is desirable,6 especially considering potential applications in 18F PET imaging; however, iron-catalyzed intermolecular olefin fluorination using nucleophilic fluoride remains unreported,7 presumably because of the high BDE of the Fe–F bond,8 the low solubility of iron fluoride species, and low nucleophilicity of solvated fluoride ion in organic solvents.9
Herein, we report an iron-catalyzed intermolecular amino-fluorination method for a broad range of unfunctionalized olefins, including nonstyrenyl olefins, using fluoride ion (Scheme 1). This new reaction readily affords a variety of synthetically valuable vicinal fluoro carbamates with high regioselectivity (N vs F), many of which are difficult to prepare using existing methods.10 We further demonstrate that it is possible to modulate the enantioselectivity for aminofluorination of cyclic olefins. Additionally, preliminary mechanistic studies suggest a possible new olefin aminofluorination pathway in which both an iron-nitrenoid and carbocation species may be involved in a stepwise nitrogen and fluorine atom transfer.
Scheme 1.

Iron-Catalyzed Intermolecular Olefin Aminofluorination Using Fluoride Ion
RESULTS AND DISCUSSION
We previously reported an iron-catalyzed method for intra-molecular olefin aminofluorination using fluoride ion in which iron(II)–bidentate ligand complexes catalyze the reaction in the presence of Et3N·3HF (Scheme 2A);11 however, this type of catalyst was ineffective to catalyze the intermolecular olefin aminofluorination because of lack of reactivity (Scheme 2B). We also reported an iron-catalyzed method for intermolecular olefin aminohydroxylation using iron(II)–tridentate ligand complexes;12 however, these catalysts were unable to promote the intermolecular olefin aminofluorination in the presence of Et3N·3HF, possibly due to catalyst deactivation (Scheme 2B). We therefore explored a range of catalysts, oxidants, and fluorination reagents to search for a stable yet reactive catalyst in the presence of the nucleophilic fluoride ion.
Scheme 2.

Iron-Catalyzed Intramolecular Olefin Aminofluorination and Early Attempts for Intermolecular Olefin Aminofluorination
To determine the possible cause of the aforementioned catalyst deactivation (Scheme 2B), we exposed an in situ generated Fe(BF4)2–L1 catalyst (1:1 ratio), previously discovered for styrene aminohydroxylation, to an intra-molecular aminofluorination condition in the absence of any olefin or oxidant (Scheme 3). Insoluble FeF2 and a soluble iron complex were obtained, and X-ray crystallographic analysis revealed its structure as Fe(L1)2(BF4)2, which, surprisingly, is inactive for styrene aminohydroxylation.13 This result suggests that fluoride species may disrupt the Fe(BF4)2–L1 complex through iron sequestering and therefore lead to the inactive Fe(L1)2(BF4)2 catalyst formation.
Scheme 3.

Experiments To Explore the Putative Fluoride-Induced Catalyst Deactivation
During the discovery of the aforementioned iron-catalyzed intramolecular olefin aminofluorination, we identified that electrophilic XtalFluor-E14 effectively suppresses the competing olefin aminohydroxylation, presumably by sequestering the benzoate generated during the N–O bond cleavage (eq 1).11,14
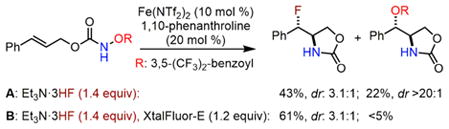 |
(1) |
We also noted, through NMR titration experiments, that XtalFluor-E and Et3N·3HF15 may form dynamic complexes (Scheme 4),16 which could attenuate the concentration of Et3N·3HF and therefore significantly slow down the putative fluoride-induced catalyst deactivation.
Scheme 4.

Proposed Complex Formation from Et3N·3HF and XtalFluor-E
To test this hypothesis, we selected styrene 1 as a model substrate for catalyst discovery (Table 1). We first observed that the Fe(NTf2)2–L1 complex, a catalyst for styrene amino-oxygenation, led to inefficient aminofluorination when a 1:1 mixture of XtalFluor-E and Et3N·3HF was used: vicinal amino fluoride 3 was obtained in a low yield (16%) together with the amino-oxygenation product 4 (entry 1). Notably, there was no reaction in the absence of an iron catalyst; both 1 and 2a were stable toward XtalFluor-E and Et3N·3HF, and they were fully recovered (entry 2).
Table 1.
Catalyst Discovery for the Intermolecular Olefin Aminofluorination

| |||||||
|---|---|---|---|---|---|---|---|
| entrya | Fe(X)2 | ligand | 2 | Et3N·3HF (equiv) | XtalFluor-E (equiv) | yield (3)b | yield (4)b |
| 1 | Fe(NTf2)2 | L1 | 2a | 1.5 | 1.5 | 16% | 30% |
| 2c | none | none | 2a | 1.5 | 1.5 | NA | NA |
| 3 | Fe(NTf2)2 | L1 | 2a | 1.5 | 2.5 | 42% | 17% |
| 4 | Fe(NTf2)2 | L1 | 2b | 1.5 | 2.5 | 45% | 23% |
| 5d | Fe(NTf2)2 | L1 | 2b | 1.5 | 3.0 | 23% | 52% |
| 6e | Fe(NTf2)2 | L1 | 2b | none | 1.5 | NA | NA |
| 7d | Fe(NTf2)2 | L1 | 2c | 1.5 | 2.5 | 46% | 44% |
| 8d | Fe(NTf2)2 | L1 | 2d | 1.5 | 2.5 | 73% | <5% |
| 9d | Fe(NTf2)2 | L1 | 2d | 2.4 | 4.0 | 81% | <5% |
| 10d | Fe(NTf2)2 | L2 | 2d | 2.4 | 4.0 | 78% | <5% |
| 11f | 6: Fe(L1)(BF4)2(MeCN)(H2O)2 | none | 2d | 1.5 | 2.5 | 88% | <5% |
Reactions were carried out under N2 at −15 °C in 2 h, and 4 Å activated molecular sieves (powder) were used to remove deleterious moisture, unless stated otherwise. The reaction was quenched with saturated NaHCO3 solution.
Isolated yield.
No reaction; 1 and 2a were recovered.
The yield of 5 is 8–12%.
No desired product observed, and 5 (42%) was isolated.
Preformed complex 6 was used as the catalyst in CH2Cl2 in the absence of MeCN; −15 °C, 2 h; the yield of 5 is less than 5%.
We also observed a higher yield of 3 when the XtalFluor-E/Et3N·3HF ratio was increased to 1.6:1 (entry 3, XtalFluor-E (2.5 equiv), Et3N·3HF (1.5 equiv), 42% yield). Suspecting that an extra equivalent of XtalFluor-E may sequester the carboxylate generated during the N–O bond cleavage, we replaced 2a with 2b, from which we expected a more nucleophilic benzoate to be generated. A similar yield of 3 and a better mass balance were observed (entry 4). Interestingly, a 2:1 mixture of XtalFluor-E/Et3N·3HF resulted in a decreased yield of 3 and isolation of imidazoline 5, presumably through MeCN (cosolvent) incorporation (entry 5). Notably, no desired product was observed in the absence of Et3N·3HF, and only 5 was isolated (entry 6).
In order to attenuate the neighboring carbamate group’s participation, we switched 2b to more electrophilic 2c and 2d and observed further improved yield of 3 (entries 7–8, 46–73% yield). Furthermore, an excess amount of both XtalFluor-E and Et3N·3HF is beneficial to the yield of 3 (entry 9, 81% yield), and the Fe(NTf2)–L2 complex provides essentially the same yield (entry 10, 78% yield). To our surprise, Fe(L1)-(MeCN)(H2O)2(BF4)2 (6), a crystalline solid that was easily prepared and handled outside of a glovebox (Figure 1), was found to effectively catalyze styrene aminofluorination in the absence of the MeCN cosolvent; 3 was isolated in an excellent yield without using an excess amount of fluorination reagent (entry 11, 88%).13
Figure 1.

Crystal structure of Fe(L1)(MeCN)(H2O)2(BF4)2 (6).
To better understand the structure–reactivity relationship of fluoride donors, we explored a range of variations using other nucleophilic fluorination reagents under the optimized conditions (Table 2). First, replacements of Et3N·3HF with Et3N·HF, Et3N·2HF, TBAF·(tBuOH)4, KF, or AgF all lead to unproductive decomposition of 2d (entry 1).17 Next, replacement of XtalFluor-E with XtalFluor-M also affords 3d, albeit in a lower yield (entry 2, 65% yield). Furthermore, using benzoyl fluoride instead of Et3N·3HF/XtalFluor-E does not afford the desired product 3, but rather leads to benzoylation of 2d (entry 3). Furthermore, using DFI,18 PhenoFluor,19 or DAST,20 reagents that can presumably sequester benzoates, leads to unproductive decomposition of 2d (entries 4–5). Surprisingly, the DAST/XtalFluor-E (1:1) combination can lead to full conversion of 1 and generation of amino fluoride 3d (entry 6, 51% yield). This result corroborates the proposal that Et3N·3HF and XtalFluor-E may form dynamic complexes, which are compatible with the iron catalyst (Scheme 4).
Table 2.
Structure–Reactivity Relationship Studies of Nucleophilic Fluorination Reagents
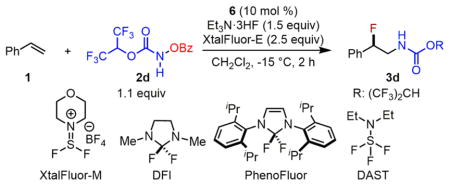
| ||
|---|---|---|
| entrya | variation from the std conditions | observations |
| 1 | replace Et3N·3HF with Et3N·HF, Et3N·2HF, TBAF(tBuOH)4, or AgF | decomposition of 2d, no desired product |
| 2 | replace XtalFluor-E with XtalFluor-M | 3d (65%) with full conversion |
| 3 | replace Et3N·3HF/XtalFluor-E with benzoyl fluoride | 3d (<5%) with 66% conversionb |
| 4 | replace Et3N·3HF/XtalFluor-E with DFI or PhenoFluor | decomposition of 2d, no desired product |
| 5 | replace Et3N·3HF/XtalFluor-E with DAST | decomposition of 2d, no desired product |
| 6 | replace Et3N·3HF/XtalFluor-E (1.5 equiv/2.5 equiv) with DAST/XtalFluor-E (1.5 equiv/ 1.0 equiv) | 3d (51%) with full conversion |
Reactions were carried out under N2 at −15 °C in 2 h, and 4 Å activated molecular sieves (powder) were used to remove deleterious moisture, unless stated otherwise.
Amino-oxygenation product 4d (42%) and benzoylated 2d (24%) were isolated.
Upon discovering the optimal catalytic conditions, we explored the substrate scope of this new reaction (Table 3). First, a range of styrenes with various electronic properties are compatible with this method, and they were converted to internal amino fluorides with excellent yields (entries 1a–1d). Additionally, ene-yne and α-methylstyrene are both excellent substrates (entries 2–3).
Table 3.
Substrate Scope for the Iron-Catalyzed Olefin Aminofluorination
| ||||
|---|---|---|---|---|
| entrya | olefins | reaction time | vincinal fluoro carbamates | yield |
| 1 |
|
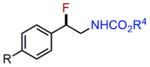
|
||
| 2h | a:R=H | 88% | ||
| 4h | b: R=Meb | 82% | ||
| 3h | c: R=Br | 87% | ||
| 4h | d: R=CO2Meb,c | 81% | ||
| 2 |
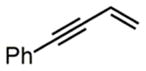
|
4h |
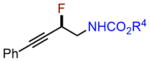
|
75% |
| 3b |
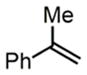
|
3h |
|
83% |
| 4 |
|
5h |
|
82% |
| 5c,d |
|
3h |
|
65% |
| 6e,f |
|
4h |
|
65% |
| 7d |
|
6h |

|
84% |
| 8b |
|
6h |

|
91% |
| 9c,g |
|
6h |
|
72% |
| 10b,c |

|
3h |
|
71% |
| 11f,h |
|
2h |
|
62% |
Reactions were carried out under N2 with preformed iron(II)–L1 catalysts (10 mol %), unless stated otherwise.
XtalFluor-E (3.0 equiv)/Et3N·3HF (1.8 equiv).21
Catalyst loading: 15 mol %.
XtalFluor-E (4.0 equiv)/Et3N·3HF (2.4 equiv).21
Catalyst loading: 20 mol %.
Additional XtalFluor-E (2.5 equiv)/Et3N·3HF (2.5 equiv) were added after 1 h.22
XtalFluor-E (5.0 equiv)/Et3N·3HF (3.0 equiv).21
Catalyst loading: 30 mol %, XtalFluor-E (1.5 equiv)/Et3N·3HF (0.9 equiv).
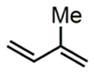
Next, we evaluated olefins that are incompatible with existing aminofluorination methods. An allyl silane with a labile C–Si bond in the presence of fluoride ion was converted to an internal amino fluoride, which has a nitrogen, fluorine, and silicon-based triad with excellent yield and regioselectivity (entry 4, 82% yield). A silyl dienol and an ethyl dienoate, both with labile functional groups, were also tolerated by this method to produce amino allylic fluorides in reasonable yields (entries 5–6). Isoprene aminofluorination provided a pair of vicinal amino allylic fluorides with an excellent yield and good regioselectivity (entry 7, 84% yield). Furthermore, a linear aliphatic diene was converted to both 1,2- and 1,4-amino fluorides with excellent combined yield (entry 8, 91% yield).
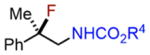
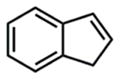
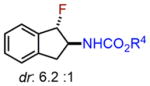
We subsequently explored indene aminofluorination using the Fe(NTf2)2–L1 complex: an anti-2-amino fluoride was isolated with excellent regioselectivity and significant diastereoselectivity (entry 9, 72% yield, dr: 6.2:1).23 Moreover, we evaluated isolated olefins, including 1,1-disubstituted and monosubstituted olefins, both of which are significantly less reactive. With increased catalyst loading and the amount of fluorination reagents, both of them were converted to internal amino fluorides with reasonable yields and high regioselectivity (entries 10–11, 62–71% yield). Notably, this new method enables the synthesis of a range of structurally diverse vicinal amino fluorides for the first time (described in entries 4, 5, 7, 10, and 11).
The furnished vicinal fluoro carbamates, both from styrenyl and aliphatic olefins, such as 7 and 9, can be easily derivatized to N-Boc protected vicinal fluoro amines 8 and 10 in excellent yields under mild conditions (Scheme 5).13
Scheme 5.

Derivatization of Olefin Aminofluorination Products to N-Boc-Protected Vicinal Fluoro Amines
With the success of this aminofluorination method for simple unfunctionalized olefins, we further explored this new reaction with complex olefins, including myrcene which contains both a trisubstituted olefin and a conjugated diene, and (−)-β-pinene (Scheme 6). The aminofluorination of myrcene readily affords both a standard 1,2-addition product 11b and a 1,6-addition product 11a presumably through cyclization. We also note that the nitrogen atom was selectively transferred to the same terminal positions in both 11a and 11b. To our surprise, the aminofluorination of (−)-β-pinene does not afford any 1,2-addition product, but rather the same 1,6-addition product 11a presumably through rearrangement. Notably, complete stereochemistry retention is observed in this aminofluorination–rearrangement cascade.24
Scheme 6.

Iron-Catalyzed Aminofluorination of Myrence and (−)-β-Pinene
Fluorinated amphetamines are of great interest to biological studies of the central nervous system; therefore, we evaluated the aminofluorination of trans-β-methylstyrene (Scheme 7).25 Interestingly, although the Fe(BF4)2–L1 complex 6 promotes undesirable C–H abstraction, a less sterically hindered Fe(NTf2)2–L2 complex catalyzes efficient aminofluorination to afford 12 (Scheme 7, 72% yield, dr: 2.0:1).13
Scheme 7. Iron-Catalyzed Aminofluorination of trans-β-Methyl-styrene.

aReaction condition: Fe(NTf2)2 (15 mol %), L2 (15 mol %), XtalFluor-E (2.5 equiv)/Et3N·3HF (1.5 equiv), 4 Å MS, CH2Cl2/MeCN, −15 °C, 1 h; then, another portion of XtalFluor-E (1.5 equiv)/Et3N·3HF (1.5 equiv), 0 °C, 3 h.
In order to demonstrate the potential practicality of this reaction, we explored the aminofluorination of indene and isoprene on a large scale (Scheme 8). To our pleasure, both catalytic reactions can be scaled up to gram scale with consistent yield and regio- and stereoselectivity.13
Scheme 8.

Iron-Catalyzed Gram-Scale Aminofluorination of Indene and Isoprene
The catalytic enantioselective aminofluorination of unfunctionalized olefins has been an unsolved synthetic challenge;26 therefore, we explored the feasibility of achieving asymmetric induction using chiral iron catalysts. As a first step, we studied asymmetric indene aminofluorination as a proof of concept, which may provide valuable mechanistic insights (Table 4). The Fe(NTf2)2–chiral ligand L3 complex catalyzes indene aminofluorination with 2d and affords anti-2-amino fluoride 7d with significant ee (entry 1, 60% ee). Although less sterically hindered acyloxyl carbamates (2b and 2c) lead to further increased ee for indene aminofluorination, both reactions evidently suffer from lower reactivity (entries 2–3, 78–83% ee). In order to achieve enhanced reactivity, the benzoyl activating group in 2c was switched to 3,5-bis(trifluoromethyl)-benzoyl group in 2e. To our surprise, a dramatically decreased ee was observed (entry 4, 55% ee). Fortunately, indene aminofluorination with acyloxyl carbamate 2f, which has a 2,4-dichloro-benzoyl activating group, provides 7 with a synthetically useful yield and high ee (entry 5, 59% yield, 84% ee). Other chiral ligands, including tridentate L4 and L5, were also evaluated, and they all provided inferior results.
Table 4.
Catalytic Asymmetric Indene Aminofluorination
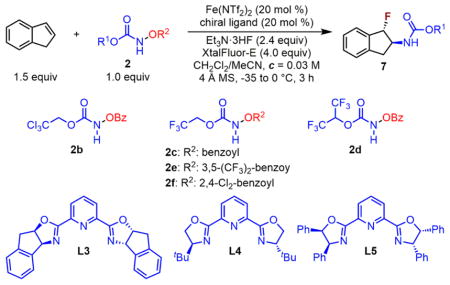
| |||||
|---|---|---|---|---|---|
| entrya | 2 | ligand | yieldb | drc | ee (major/minor)d |
| 1 | 2d | L3 | 48% | 5.2:1 | 60%/60% |
| 2 | 2b | L3 | 38% | 2.5:1 | 78%/77% |
| 3 | 2c | L3 | 35% | 2.4:1 | 83%/82% |
| 4 | 2e | L3 | 55% | 2.2:1 | 55%/55% |
| 5 | 2f | L3 | 59% | 3.0:1 | 84%/84% |
| 6 | 2f | L4 | 32% | 1.8:1 | 36%/37% |
| 7 | 2f | L5 | 39% | 1.9:1 | 32%/30% |
Unless stated otherwise, the reactions were carried out under a nitrogen atmosphere with 4 Å molecular sieves.
Isolated yield.
dr was determined by 1H NMR.
Enantiomeric excess (ee) was measured by HPLC with chiral columns; the absolute stereochemistry was determined by X-ray crystallographic analysis of 7d (R: (CF3)2CH).
These results also provide valuable mechanistic insight. The acyloxyl moiety on 2 proves crucial for stereochemical communication in the iron-catalyzed indene aminofluorination (Table 4, entries 3–5). This observation suggests that the benzoate generated during N–O bond cleavage may be involved in the ee-determining step. Additionally, since the same ee was observed for both diastereomers of 7, it is likely that the C–N bond forming step is ee-determining.
In order to formulate a mechanistic model, we carried out several control experiments to gain more insight into both the C–N and C–F bond forming steps (Scheme 9). First, two analogues of 2 in which the N–H moiety was replaced with either the N–Me or N–Ac groups were subjected to standard reaction conditions (Scheme 9, eq 2). Neither was reactive toward the aminofluorination, which suggests that the N–H moiety in 2 is critical for its activation.
Scheme 9. Control Experiments To Probe for Mechanistic Insight.

a6 (15 mol %), XtalFluor-E (2.5 equiv)/Et3N·3HF (1.5 equiv), −15°C, CH2Cl2. b6 (15 mol %), XtalFluor-E (4.0 equiv)/Et3N·3HF (2.4 equiv), −40 °C, CH2Cl2. c6 (15 mol %), XtalFluor-E (3.0 equiv)/Et3N·3HF (1.8 equiv), −35 to 0 °C, CH2Cl2. R1: 2,4-Cl2-benzoyl, R2: CF3CH2, R3: (CF3)2CH.
Next, the aminofluorination of a cyclopropyl-substituted olefin 15 afforded only the ring-opening products 16 and 17 (Scheme 9, eq 3). This result, together with the observed 1,6-myrcene aminofluorination, suggests that the reaction may proceed through a stepwise process and that a carbo-radical species may be an intermediate. To obtain more mechanistic details beyond the C–N bond forming step, we further evaluated a 1,1-disubstituted olefin 18 and observed both 1,2-amino fluoride 19 and 1,3-aminofluoride 20 which is presumably obtained through 1,2-hydride shift from a carbocation (Scheme 9, eq 4). This result suggests that a carbocation may also be involved in the reaction pathway.
Furthermore, benzoylated morpholine 21 was identified as a side product in styrene aminofluorination using Et3N·3HF/ XtalFluor-M (Scheme 9, eq 5). We suspected that 21 might be derived from the interaction between XtalFluor-M and the benzoic acid that was generated during the iron-catalyzed N–O bond cleavage; therefore, a control experiment between benzoic acid and XtalFluor-M was conducted, from which 21 was indeed isolated (Scheme 9, eq 6). These results suggest that XtalFluor-E or XtalFluor-M sequesters benzoic acids to promote efficient fluorine atom transfer.
On the basis of a range of mechanistic evidence presented in Scheme 9, a mechanistic working hypothesis for the iron-catalyzed olefin aminofluorination is proposed in Scheme 10. First, the iron catalyst may reductively cleave the N–O bond in 2, which can possibly be converted to an iron-nitrenoid A. The iron-nitrenoid may initiate a radical addition with olefin 1 to afford a carbo-radical species B. Subsequently, the high-valent iron intermediate may further oxidize the radical, presumably through single electron transfer (SET),27 to generate a carbocation C1. When the concentration of a suitable fluoride donor is low, C1 may be efficiently intercepted by the neighboring carbamate group to afford amino-oxygenation product 4.
Scheme 10.

Mechanistic Working Hypothesis of the Iron-Catalyzed Intermolecular Olefin Aminofluorination Using Fluoride Ion
However, in the presence of a suitable nucleophilic fluoride and benzoate sequestering reagent (e.g., Et3N·3HF/XtalFluor-E), carbocation C1 may be rapidly converted to C2, which can be captured and converted to amino fluoride 3; therefore, the aminofluorination pathway will override the competing amino-oxygenation pathway. Since oxazoline 4 may also be converted to carbocation C1 under reaction conditions which are strongly Lewis acidic, it is possible that this additional pathway may also operate, for a certain type of substrates, to afford the olefin aminofluorination product 3.
CONCLUSION
In conclusion, we reported a catalytic method for intermolecular olefin aminofluorination using earth-abundant iron catalysts and nucleophilic fluoride ion. This method tolerates a broad range of unfunctionalized olefins, including nonstyrenyl olefins that are incompatible with existing aminofluorination methods. It also readily affords a variety of synthetically valuable vicinal fluoro carbamates with high regioselectivity. Notably, many of them are difficult to prepare using existing methods. We systematically described the catalytic discovery, structure–reactivity relationship studies of fluoride donors, substrate scope and limitation of this method, derivatization of vicinal fluoro carbamates under mild conditions, practical procedures for gram-scale olefin aminofluorination, chiral catalyst discovery for asymmetric olefin aminofluorination, and preliminary mechanistic studies. These studies demonstrate that it is feasible for asymmetric induction using chiral iron catalysts for aminofluorination of unfunctionalized olefins, in which both an iron nitrenoid and a carbocation species may be possible reactive intermediates. Our current efforts focus on both mechanistic understanding and synthetic applications of this new reaction.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (GM110382) and Georgia State University. H.X. is an Alfred P. Sloan Research Fellow. We also thank a referee for insightful comments and suggestions.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b07221.
Experimental procedure, characterization data for all new compounds, selected NMR spectra and HPLC traces (PDF)
si_002.cif for compound 6 (CIF)
si_003.cif for Fe(L1)2(BF4)2 (CIF)
si_004.cif for compound 7d (CIF)
si_005.cif for compound 11a (CIF)
References
- 1.For a selected reference of fluorine in medicinal chemistry, see: Müller K, Faeh C, Diederich F. Science. 2007;317:1881. doi: 10.1126/science.1131943.
- 2.For iron-mediated intermolecular olefin hydrofluorination, see: Barker TJ, Boger DL. J Am Chem Soc. 2012;134:13588. doi: 10.1021/ja3063716.For recent references of catalytic olefin difluorination, see: Banik SM, Medley JW, Jacobsen EN. J Am Chem Soc. 2016;138:5000. doi: 10.1021/jacs.6b02391.Banik SM, Medley JW, Jacobsen EN. Science. 2016;353:51. doi: 10.1126/science.aaf8078.Molnár IG, Gilmour R. J Am Chem Soc. 2016;138:5004. doi: 10.1021/jacs.6b01183.For selected references of styrene hydrofluorination, see: Emer E, Pfeifer L, Brown JM, Gouverneur V. Angew Chem, Int Ed. 2014;53:4181. doi: 10.1002/anie.201310056.Wilger DJ, Grandjean JMM, Lammert TR, Nicewicz DA. Nat Chem. 2014;6:720. doi: 10.1038/nchem.2000.For olefin azido-fluorination, see: Li Z, Zhang C, Zhu L, Liu C, Li C. Org Chem Front. 2014;1:100.For styrene fluorosulfonylation, see: Yuan Z, Wang HY, Mu X, Chen P, Guo YL, Liu G. J Am Chem Soc. 2015;137:2468. doi: 10.1021/ja5131676.
- 3.For a selected reference of vicinal fluoro amines in medicinal chemistry, see: Hagmann WK. J Med Chem. 2008;51:4359. doi: 10.1021/jm800219f.
- 4.Stavber S, Pecăn TS, Papež M, Zupan M. Chem Commun. 1996:2247.In this report, the substrate scope was predominantly limited to styrenyl olefins with two exceptions. First, tetramethylethylene. Second, 1-octene was converted to an inseparable mixture of 1,2- and 1,3-terminal fluorides. For recent references related to intermolecular styrene aminofluorination, see: Qiu S, Xu T, Zhou J, Guo Y, Liu G. J Am Chem Soc. 2010;132:2856. doi: 10.1021/ja909716k.Zhang H, Song Y, Zhao J, Zhang J, Zhang Q. Angew Chem, Int Ed. 2014;53:11079. doi: 10.1002/anie.201406797.Saavedra-Olavarria J, Arteaga GC, López JJ, Pérez EG. Chem Commun. 2015;51:3379. doi: 10.1039/c4cc10162f.Chen P, Liu G. Eur J Org Chem. 2015;2015:4295.For a recent review of intramolecular olefin aminofluorination, see: Serguchev YA, Ponomarenko MV, Ignat’ev NV. J Fluorine Chem. 2016;185:1.
- 5.For a reference of asymmetric fluoro aminoxylation of enals using chiral diphenyl prolinol-type of organocatalysts, see: Appayee C, Brenner-Moyer SE. Org Lett. 2010;12:3356. doi: 10.1021/ol101167z.
- 6.The aminofluorination of terminal olefins using nucleophilic fluorination reagents affords internal amino fluorides with the inverse regioselectivity obtained using electrophilic fluorination reagents.
- 7.For iron-mediated olefin hydrofluorination with electrophilic Selectfluor, see ref 2a. For iron-catalyzed benzylic C–H fluorination with Selectfluor, see: Bloom S, Pitts CR, Woltornist R, Griswold A, Holl MG, Lectka T. Org Lett. 2013;15:1722. doi: 10.1021/ol400424s.For other selected examples of radical fluorination that are not promoted by iron catalysts, see: Rueda-Becerril M, Chatalova Sazepin C, Leung JCT, Okbinoglu T, Kennepohl P, Paquin J-F, Sammis GM. J Am Chem Soc. 2012;134:4026. doi: 10.1021/ja211679v.Liu W, Huang X, Cheng MJ, Nielsen RJ, Goddard WA, Groves JT. Science. 2012;337:1322. doi: 10.1126/science.1222327.Xia JB, Zhu C, Chen C. J Am Chem Soc. 2013;135:17494. doi: 10.1021/ja410815u.Zhang C, Li Z, Zhu L, Yu L, Wang Z, Li C. J Am Chem Soc. 2013;135:14082. doi: 10.1021/ja408031s.Ventre S, Petronijevic FR, MacMillan DWC. J Am Chem Soc. 2015;137:5654. doi: 10.1021/jacs.5b02244.
- 8.BDE for Fe–F is 447 kJ/mol, while BDEs for Fe–Cl and Fe–Br are 336 and 243 kJ/mol, respectively. See: Luo Y-R. Comprehensive Handbook of Chemical Bond Energies. CRC Press; Boca Raton, FL: 2007. p. 803.
- 9.The nonsolvated fluoride ion is exceptionally nucleophilic; for a reference, see: Sun H, DiMagno SG. J Am Chem Soc. 2005;127:2050. doi: 10.1021/ja0440497.
- 10.For existing vicinal fluoro acetamide and sulfonamide synthesis from olefins, see ref 4. For selected examples of fluoro amine synthesis from other precursors, see: Duthion BR, Pardo DG, Cossy J. Org Lett. 2010;12:4620. doi: 10.1021/ol1019579.Schulte ML, Lindsley CW. Org Lett. 2011;13:5684. doi: 10.1021/ol202415j.Wade TN. J Org Chem. 1980;45:5328.Kalow JA, Schmitt DE, Doyle AG. J Org Chem. 2012;77:4177. doi: 10.1021/jo300433a.Sladojevich F, Arlow SI, Tang P, Ritter T. J Am Chem Soc. 2013;135:2470. doi: 10.1021/ja3125405.Davies SG, Fletcher AM, Frost AB, Roberts PM, Thomson JE. Org Lett. 2015;17:2254. doi: 10.1021/acs.orglett.5b00880.Zhang Q, Stockdale DP, Mixdorf JC, Topczewski JJ, Nguyen HM. J Am Chem Soc. 2015;137:11912. doi: 10.1021/jacs.5b07492.
- 11.Lu DF, Liu GS, Zhu CL, Yuan B, Xu H. Org Lett. 2014;16:2912. doi: 10.1021/ol501051p.For related iron-catalyzed enantioselective olefin aminochlorination and diastereoslective aminobromination, see: Zhu CL, Tian JS, Gu ZY, Xing GW, Xu H. Chem Sci. 2015;6:3044. doi: 10.1039/c5sc00221d.Tian JS, Zhu CL, Chen YR, Xu H. Synthesis. 2015;47:1709. doi: 10.1055/s-0034-1378719.
- 12.Lu DF, Zhu CL, Jia ZX, Xu H. J Am Chem Soc. 2014;136:13186. doi: 10.1021/ja508057u.For a related iron-catalyzed intramolecular olefin aminohydroxylation, see: Liu GS, Zhang YQ, Yuan YA, Xu H. J Am Chem Soc. 2013;135:3343. doi: 10.1021/ja311923z.
- 13.For experimental details, see the Supporting Information.
- 14.For synthesis of XtalFluor-E, see: L’Heureux A, Beaulieu F, Bennett C, Bill DR, Clayton S, LaFlamme F, Mirmehrabi M, Tadayon S, Tovell D, Couturier M. J Org Chem. 2010;75:3401. doi: 10.1021/jo100504x. XtalFluor-E is hygroscopic; see Supporting Information for correct handling and storage. XtalFluor-E has no effect on the catalytic activity of the iron–bi-dentate ligand complexes in the intramolecular olefin aminofluorination. Full conversion of the substrate was observed in the absence of XtalFluor-E (eq 1).
- 15.For a reference describing the structure of Et3N·3HF, see: Wiechert D, Mootz D, Franz R, Siegemund G. Chem - Eur J. 1998;4:1043.
- 16.XtalFluor-E is prepared using DAST and BF3·Et2O. For experimental details, see the Supporting Information. For reactivity of these complexes, see entry 11 of Table 1 and entry 6 of Table 2.
- 17.Acyloxyl carbamate 2d decomposes under weakly basic conditions. We suspect that the reaction medium may be slightly basic due to solvation of fluoride ion with deleterious moisture in these reagents.
- 18.Hayashi H, Sonoda H, Fukumura K, Nagata T. Chem Commun. 2002:1618. doi: 10.1039/b204471d. [DOI] [PubMed] [Google Scholar]
- 19.Tang P, Wang W, Ritter T. J Am Chem Soc. 2011;133:11482. doi: 10.1021/ja2048072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Middleton WJ. J Org Chem. 1975;40:574. [Google Scholar]
- 21.An excess amount of XtalFluor-E/Et3N·3HF was applied to suppress the rapid competing olefin aminohydroxylation.
- 22.An additional amount of XtalFluor-E/Et3N·3HF was used to convert oxazolines to corresponding amino fluorides.
- 23.For a diastereoselective indene aminofluorination to afford 1-amino fluoride, which is the regioisomer of 7, see ref 4e.
- 24.The ee for (−)-β-pinene is 92%, and the ee for 11a is 91%. For experimental details, see the Supporting Information.
- 25.For a selected example of fluorinated amphetamine synthesis, see: Cresswell AJ, Davies SG, Lee JA, Roberts PM, Russell AJ, Thomson JE, Tyte MJ. Org Lett. 2010;12:2936. doi: 10.1021/ol100862s.
- 26.For iron-catalyzed asymmetric intramolecular olefin amino-fluorination, see ref 11. For a recent review of asymmetric fluorocyclization, see: Wolstenhulme JR, Gouverneur V. Acc Chem Res. 2014;47:3560. doi: 10.1021/ar500282z.For other selected examples of asymmetric fluorination, see: Beeson TD, MacMillan DWC. J Am Chem Soc. 2005;127:8826. doi: 10.1021/ja051805f.Kalow JA, Doyle AG. J Am Chem Soc. 2010;132:3268. doi: 10.1021/ja100161d.Rauniyar V, Lackner AD, Hamilton GL, Toste FD. Science. 2011;334:1681. doi: 10.1126/science.1213918.For asymmetric olefin difluroination, see ref 2b–d. For asymmetric fluoro aminoxylation of enals using organocatalysis, see ref 5. For a recent review of asymmetric olefin aminohalogenation, see: Chemler SR, Bovino MT. ACS Catal. 2013;3:1076. doi: 10.1021/cs4001249.
- 27.(a) Kharasch MS, Sosnovsky G. J Am Chem Soc. 1958;80:756. [Google Scholar]; (b) Kochi JK. Science. 1967;155:415. doi: 10.1126/science.155.3761.415. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.