

Abstract
We have previously found that thalamostriatal axodendritic terminals are reduced as early as 1 month of age in heterozygous Q140 HD mice (Deng et al., 2013). As cholinergic interneurons are a major target of thalamic axodendritic terminals, we examined the VGLUT2 immunolabeled thalamic input to striatal cholinergic interneurons in heterozygous Q140 males at 1 and 4 months of age, using choline acetyltransferase (ChAT) immunolabeling to identify cholinergic interneurons. Although blinded neuron counts showed that ChAT+ perikarya were normal in abundance in Q140 mice, size measurements indicated they were significantly smaller. Sholl analysis further revealed the dendrites of Q140 ChAT+ interneurons were significantly fewer and shorter. Consistent with the light microscopic data, ultrastructural analysis showed that the number of ChAT+ dendritic profiles per unit area of striatum was significantly decreased in Q140 striata, as was the abundance of VGLUT2+ axodendritic terminals making synaptic contact with ChAT+ dendrites per unit area of striatum. The density of thalamic terminals along individual cholinergic dendrites was, however, largely unaltered, indicating that the reduction in the areal striatal density of axodendritic thalamic terminals on cholinergic neurons was due to their dendritic territory loss. These results show that the abundance of thalamic input to individual striatal cholinergic interneurons is reduced early in the lifespan of Q140 mice, raising the possibility that this may occur in human HD as well. Since cholinergic interneurons differentially affect striatal direct versus indirect pathway spiny projection neurons, their reduced thalamic excitatory drive may contribute to early abnormalities in movement in HD.
Keywords: Huntington’s disease, Corticostriatal, Thalamostriatal, Premanifest, Pathology, Cholinergic Interneurons, Striatum, AB_2301731, AB_90650
Graphical abstract
Our results show that the dendrites of striatal cholinergic interneurons are fewer and shorter, and the abundance of thalamic input to individual striatal cholinergic interneurons as a result less, in young Q140 Huntington’s disease mice. Since cholinergic interneurons play a key role in basal ganglia motor function and plasticity, our results raise the possibility that these early abnormalities in them contribute to early abnormalities in movement in Huntington’s disease.

Introduction
Premanifest HD individuals are impaired in the initiation and/or execution of motor tasks (Siemers et al., 1996; de Boo et al., 1997; Kirkwood et al., 1999, 2000; Blekher et al., 2004; Rao et al., 2008, 2011; Biglan et al., 2009; Bechtel et al., 2010; Delval et al., 2011; Tabrizi et al., 2011; Turner et al., 2011). Motor symptoms in premanifest HD develop in parallel with gradual loss of cerebral and striatal white matter (Kipps et al., 2005; Reading et al., 2005; Ciarmiello et al., 2006; Paulsen et al., 2006; Rosas et al., 2006; Hobbs et al., 2010a; Aylward et al., 2011; Dumas et al., 2012), reductions in striatal volume (Aylward, 1987; Aylward et al., 2012, 2013; Tabrizi et al., 2013), increasing striatal hypometabolism (Grafton et al., 1992; Ciarmiello et al., 2006), and reduced striatal activation during behavioral tasks (Paulsen et al., 2004; Wolf et al., 2012). Although imaging studies show premanifest striatal volume loss, suggesting possible striatal neuron loss, the few neuropathological studies of premanifest striatum have reported only some variable neuronal loss in the head of the dorsal caudate, and little or no neuron loss has been described for the motor striatum (Vonsattel et al., 1985; Albin et al., 1991; Vonsattel and DiFiglia, 1998).
To evaluate the possibility that loss of cortical and thalamic input occurs early in the lifespan of HD gene carriers and contributes to premanifest symptoms, we have used ultrastructural methods to study thalamic and cortical input to striatum in a precise genetic mimic of human HD, the heterozygous Q140 knock-in mouse. In a prior electron microscopic (EM) immunohistochemical single-label study, we found significant deficiencies in thalamic input to the spines and dendrites of striatal neurons by 1 month of age (Deng et al., 2013). These findings suggest that loss of thalamostriatal terminals may contribute to impairments in premanifest HD. Among the neuronal targets of thalamic input to striatum are striatal projection neurons (SPNs), which receive synaptic input from thalamus on their spines and dendrites. In a subsequent EM immunohistochemical double-label study we specifically assessed the thalamic input to SPNs in heterozygous Q140 mice, and confirmed it is deficient at 4 months of age (Deng et al., 2014). Thalamic input to striatum, however, also prominently ends on the dendrites of cholinergic interneurons (Lapper and Bolam, 1992). In the present study, we thus used EM double-label methods to determine if axodendritic thalamic input to the dendrites of cholinergic interneurons was deficient in heterozygous Q140 mice. Our results indicate that a deficiency in thalamic input to individual cholinergic interneurons occurs early in the lifespan of these mice, in large part due to their reduced dendritic branching. Given the important role of cholinergic interneurons for striatal function, this has implications for the pathophysiology of premanifest HD motor deficits.
Materials and Methods
Animals
Results from 13 wild-type male (WT) and 13 heterozygous male Q140 mice (obtained from JAX, Bar Harbor, Maine) are presented here, and all animal use was carried out in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals, Society for Neuroscience Guidelines, and University of Tennessee Health Science Center Guidelines. Eight 1-month old and 5 4-month old mice were studied for both genotypes. Because deficiencies in the thalamostriatal axodendritic projections were evident already at 1 month, the Q140 mice and WT mice analyzed included both 1 and 4 month-old mice. Heterozygous HD mutants were studied because the human disease most commonly occurs due to a single allelic defect. Note that the repeat length in the Q140 mice used had undergone a spontaneous reduction during breeding at JAX, and the average CAG repeat length in our eight 1-month old Q140 mice was 127.8, and our five 4-month old Q140 mice was 128.6. We refer to our mutant mice as Q140, despite the slightly shorter CAG repeat length, to relate our findings to other work on the same mutant strain (in which the first exon of mouse huntingtin was replaced with a human equivalent with ~140 CAG repeats) (Menalled et al., 2003), as others have done as well (Hickey et al., 2008, 2012; Lerner et al. 2012). For histological analysis, mice were deeply anesthetized with 0.2ml of 35% chloral hydrate in saline, and then exsanguinated by transcardial perfusion with 30 ml of 6% dextran in sodium phosphate buffer (PB), followed by 200 ml of 3.5% paraformaldehyde - 0.6% glutaraldehyde - 15% saturated picric acid in PB (pH 7.4). The brain of each mouse was removed, postfixed overnight in 3.5% paraformaldehyde - 15% saturated picric acid in PB. The right side of the brain was used for light microscopic (LM), and the left for electron microscopic (EM) double-label analysis. These animals had previously also been used in our prior LM and EM studies focusing on inputs to SPNs (Deng et al., 2013, 2014). For LM studies, forebrain was sectioned frozen at 35 μm with a sliding microtome, and for EM studies forebrain was sectioned at 50μm on a vibratome, as described previously.
Immunolabeling for VGLUT2 and ChAT
Sections were pretreated with 1% sodium borohydride in 0.1 M PB for 30 minutes followed by incubation in 0.5% H2O2 solution in 0.1 M PB for 30 minutes. To carry out double-label immunohistochemistry for EM viewing, sections were incubated overnight at room temperature in primary antisera containing guinea pig anti-VGLUT2 (diluted 1:2000) and goat anti-ChAT (1:400), with 0.1M PB containing 10% normal horse serum, 4% normal goat serum, 1.5% bovine serum albumin, and 0.02% Triton X-100. Sections were then rinsed and incubated in a mixture of biotinylated donkey anti-guinea pig IgG diluted 1:100 (to detect guinea pig anti-VGLUT2) and donkey anti-goat IgG diluted 1:100 (to detect goat anti-ChAT), at room temperature for one hour. This was followed by incubation in a mixture containing avidin-biotin complex (ABC) (to detect guinea pig anti-VGLUT2) and goat peroxidase-antiperoxidase (PAP) at a 1:500 dilution (to detect goat anti-ChAT), in 0.1M PB (pH7.4) at room temperature for two hours. The sections were rinsed between secondary and ABC and/or PAP incubations in three five-minute washes of PB. Subsequent to the ABC and/or PAP incubation, the sections were rinsed with three to six 10-minute washes in 0.1M PB, and a peroxidase reaction using diaminobenzidine (DAB) carried out. After the PB rinses, the sections were immersed for 10 minutes in 0.05% DAB (Sigma, St. Louis, MO) in 0.1M PB (pH7.2). Hydrogen peroxide was then added to a final concentration of 0.01%, and the sections were incubated in this solution for an additional 10 minutes, and washed six times in PB. The double-label tissue was prepared for EM analysis as described below. Additional series of sections were immunolabeled for ChAT alone and prepared for LM viewing, using procedures as described previously (Deng et al., 2013).
Preparation of Tissue for EM
Following immunolabeling as described above, sections processed for EM viewing were rinsed in 0.1M sodium cacodylate buffer (pH 7.2), postfixed for 1 hour in 2% osmium tetroxide (OsO4) in 0.1 M sodium cacodylate buffer, dehydrated in a graded series of ethyl alcohols, impregnated with 1% uranyl acetate in 100% alcohol, and flat-embedded in Spurr’s resin (Electron Microscopy Sciences, Fort Washington, PA). For the flat-embedding, the sections were mounted on microslides pretreated with liquid releasing factor (Electron Microscopy Sciences, Fort Washington, PA). Pieces of embedded tissue were cut from the dorsolateral (motor) striatum and glued to carrier blocks, and ultrathin sections were cut from these specimens with a Reichert ultramicrotome. The sections were mounted on mesh grids, stained with 0.4% lead citrate and 4.0% uranyl acetate using an LKB Ultrastainer, and finally viewed and images captured with a JEOL 2000EX electron microscope.
Antibodies
The immunogen for guinea pig VGLUT2 antibody (Chemicon AB5907) was aa565-582 of the C-terminus of rat VGLUT2, and the antibody is highly specific for VGLUT2 (Table 1), as described in detail previously (Lei et al., 2013; Deng et al., 2014). The goat anti-ChAT antibody (Chemicon AB144) is directed against human ChAT and is highly selective for ChAT (Table 1), as described in detail previously (Shiromani et al., 1987; Coulon et al., 2011).
Table 1.
Detail on the animal host, commercial source, catalogue number, target antigen, Journal of Comparative Neurology Registry Identification, and working concentration for the antibodies used in the present study. Information on antibody specificity testing is provided in the text, and in the cited prior papers using these antibodies, and in the papers cited in the Journal of Comparative Neurology Antibody Database.
Primary Antibodies Used
| Antigen | Description of Immunogen |
Source, Host Species, Cat. #, Clone or Lot#, RRID |
Concentration Used |
|---|---|---|---|
| Vesicular glutamate transporter 2 (VGluT2) |
rat VGLUT2 C-terminus (aa565-582): VQESAQDAYSYKDRDDYS |
Millipore/Chemicon, guinea pig polyclonal, Cat# AB5907, RRID: AB_2301731 |
1:1000 (LM) 1:5000 (EM) |
| Choline acetyltransferase (ChAT) |
Human placental ChAT | Chemicon, goat polyclonal, Cat# AB144, RRID: AB_90650 |
1:400 (LM) 1:400 (EM) |
Counts and Size Measurement of ChAT+ perikarya in striatum
Sections immunolabeled for ChAT from seven WT and 8 Q140 1-month old mice were used for LM counts to compare the abundance of ChAT+ perikarya in WT versus mutant striatum. The levels chosen for analysis ranged from the rostral tip of the striatum to the anterior of commissure, and thus represent the anterior two-thirds of striatum, approximately. Low-power images (4x) containing striatum, in which cholinergic neurons were clear and visible, were taken using a digital camera, and each striatum reconstructed digitally. ChAT+ neurons in each striatum were then counted and the area of striatum measured. Counts were corrected for double-counting of structures at the section surface according to the Abercrombie method (Guillery and Herrup, 1997). The relative number of ChAT neurons in striatum for each case was then calculated based on the thickness of the sections, the number of sections cut, and the number of striatal sections counted. For perikarya size, the area of 11-19 ChAT+ perikarya was measured in striata from 7 1-month old WT mice and 8 1-month old Q140 mice using ImageJ, and the diameter for each perikaryon calculated from these using the formula, diameter = 2 × the square root of area/π.
Sholl analysis of ChAT+ neurons in striatum
A total 7 WT and 8 Q140 5-week old mice were used for Sholl analysis (Sholl, 1953; Ferreira et al., 2014) of ChAT+ neurons in striatum, ranging from its rostral tip to the anterior commissure. Ten to 20 immunolabeled ChAT interneurons from dorsolateral striatum from each case were randomly selected, and their neuronal cell body and all processes were drawn separately with a 40x objective using camera lucida. Each drawing was scanned using an Epson Scanner (Epson Perfection 4490 Photo) and used for Sholl analysis (number of dendritic branch intersections with concentric circles of increasing radii from the soma). The Sholl analysis was performed using the Simple Neurite Tracer (SNT) or the Sholl analysis plugin from Fiji (http://fiji.sc/Sholl_Analysis). In image J, the 2D drawing of each ChAT+ neuron was analyzed automatically using the Sholl analysis function. To study the number of branches as a function of the distance from the cell body, a series of concentric circles (10μm distance), which were centered on the neuronal body, were placed on each drawing and the intersections of dendrites with these circles counted.
EM Analysis
Blinded analysis and quantification was carried out on digital EM images of random fields from dorsolateral somatomotor striatum (Fig. 1). Ten WT and 10 mutant mice were studied, equally divided between 1-month and 4-month old animals. Typically, 25-30 EM images that in total encompassed 400-450 μm2 of dorsolateral striatum were analyzed per animal. This typically yielded 45-55 thalamic terminals per animal in WT mice, but fewer in mutant mice, as described in the Results section. We focused on dorsolateral striatum because matrix compartment neurons of this region are important for motor function, because it is poor in striosomes, and because it is the major target of intralaminar thalamus (Berendse and Groenewegen, 1990; Gerfen, 1992; Desban et al., 1993; Wang et al., 2007). We performed the analysis on the upper 5 microns of the sections, in which labeling was optimal, but avoided the very surface, where histology was poor. Fields were examined at the EM level and images of all cholinergic dendrites encountered were captured. These images were then used to determine the abundance of VGLUT2 synaptic contacts on cholinergic dendrites. Only terminals with an overt synaptic contact possessing a PSD (post-synaptic density) were counted and measured, since all VGLUT2 terminals are excitatory and their synaptic contacts evidenced by the presence of vesicles in the terminal and a PSD in the dendrite (Deng et al., 2013; Lei et al., 2013). Note that immunolabeled VGLUT2 synaptic terminals in striatum could be readily distinguished from immunolabeled cholinergic terminals since the latter form symmetric contacts and only rarely contact cholinergic neurons in any case (Phelps et al., 1985). The size of VGLUT2+ terminals was determined by measuring them at their widest diameter parallel to and 0.1μm before the postsynaptic density, and spines were identifiable by their small size, continuity with dendrites, prominent postsynaptic density, and/or the presence of spine apparatus (Wilson et al., 1983). Dendrites were identifiable by their size, oval or elongate shape, and the presence of microtubules and mitochondria.
Figure 1.

Images of ChAT+ cholinergic interneurons in the right striatum of WT (A) and Q140 (B) mice, and box-and-whicker plots comparing their mean abundance per striatum at the levels assessed (C) and comparing their perikaryal diameter (D) between WT and mutant mice. No significant difference in the abundance of striatal ChAT+ perikarya was found between Q140 and WT mice (C), but the perikaryal diameter was slightly but significantly less (asterisk) in the mutants.
Statistical Analysis
The data are presented as group means (±SEM), unless otherwise stated. The Kolmogorov-Smirnov test was used to assess the normality for each type of measurement for WT and Q140 mice. All data were normally distributed, except for the results for the WT group in the Sholl analysis, for which there was one outlier mouse. A Mann-Whitney U test was thus performed on the Sholl data. All other statistical comparisons between Q140 and WT mice were made using t-tests.
Results
Light microscopic analysis showed that striatal cholinergic interneuron abundance in the Q140 mice was 98.2% of that in the age-matched WT mice, and not significantly different (p=0.3724) than in WT mice (Fig. 1). The soma diameter of ChAT+ interneurons was, however, significantly decreased (p=0.0081) in Q140 heterozygous mice to 91.0% of WT (Fig. 1). Examination of immunolabeled sections suggested that the dendritic branching of cholinergic interneurons in Q140 striatal was somewhat impoverished (Fig. 2). Sholl analysis of 104 WT neurons and 120 Q140 neurons confirmed that the dendritic branches of ChAT+ striatal neurons were significantly fewer and shorter (p=0.021) in Q140 mice (Fig. 3). For example, the abundance of total intersections for Q140 ChAT+ dendrites was only 70.2% of that in WT mice. Moreover, cholinergic neurons tended to have fewer intersections at all distances from the cell body, indicating that they were less branched overall, as well as shorter.
Figure 2.

Images of ChAT+ cholinergic interneurons in the striatum of WT (A) and Q140 (B) mice, showing the slightly smaller size and lessened dendritic branching in the Q140 mice.
Figure 3.
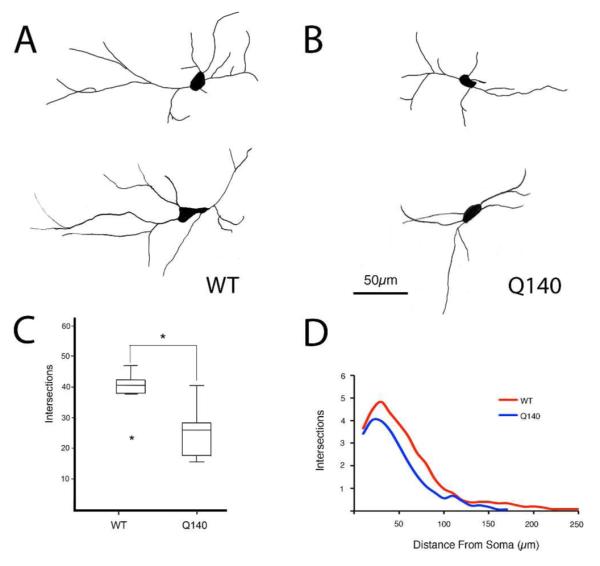
Representative camera lucida drawings of ChAT+ cholinergic interneurons at high power in the striatum of WT (A) and Q140 (B) mice, a box-and-whicker plot showing total dendrite intersections (C), and a graph showing dendrite intersections as a function of distance from the soma (D) in WT and mutant mice based on Sholl analysis. The results show that dendritic arborizations of ChAT+ interneurons were significantly decreased in Q140 heterozygous mice. One outlier is evident for the WT mice in C.
Consistent with the LM evidence of dendritic arbor reduction, EM analysis revealed that the number of individual ChAT+ dendritic profiles per unit area was decreased significantly in Q140 striata (Table 2, Fig. 4). Similarly, the average area occupied by individual ChAT+ dendritic profiles in striatum at the EM level was 1.874 μm2 in WT mice, but was significantly reduced by 22.6% in Q140 mice (Table 2). Similarly, the total overall area occupied by ChAT+ dendritic profiles was 7.67% of the total striatal area sampled in WT mice, but was significantly reduced by 32.4% in Q140 mice (Table 2). Thus, EM analysis confirmed the approximately 30% deficiency in ChAT+ dendrites seen in Q140 striatum by LM analysis.
Table 2.
Table comparing Q140 mice and WT mice for the EM results on the total overall area occupied by ChAT+ dendritic profiles in dorsolateral striatum, the average area of individual ChAT+ dendritic profiles in striatum, and the number of ChAT+ dendritic profiles in striatum. Note that all three parameters show a reduction for cholinergic dendrites in Q140 mice. The Q140 and WT mice are also compared for VGLUT2+ synaptic terminals on ChAT+ dendritic profiles per unit area of striatum, and the abundance of VGLUT2+ terminals per unit area of ChAT+ dendrite. These results show that the frequency of VGLUT2+ terminals ending on ChAT+ dendrites per unit area of striatum is significantly reduced, but their abundance per unit area of ChAT+ dendrites is not significantly reduced. These results show that cholinergic neurons possess fewer and shorter dendritic branches in Q140 mice, and as a result individual cholinergic striatal neurons receive fewer synaptic terminals from thalamus.
EM Data on Cholinergic Neuron Dendrites and VGLUT2+ Terminals on Cholinergic Dendrites
| Morphological Parameter – Cholinergic Dendrites |
WT Mean (±SEM) |
Q140 Mean (±SEM) |
Q140 as Percent of WT |
P Value* |
|---|---|---|---|---|
| Number of ChAT+ Dendritic Profiles (per μm2) |
0.0417 ±0.002 |
0.0357 ±0.001 |
85.7%* | 0.019 |
| Average Size of Individual ChAT+ Dendrites (μm2) |
1.874 ±0.14 |
1.451 ±0.09 |
77.4%* | 0.022 |
| % Image Area Occupied by ChAT+ Dendrites |
7.673 ±0.61% |
5.186 ±0.37% |
67.6%* | 0.003 |
|
Morphological Parameter – VGLUT2+ Terminals on ChAT+ Dendrites |
WT Mean
(±SEM) |
Q140 Mean
(±SEM) |
Q140 as Percent
of WT |
P Value * |
| Abundance of VGLUT2+ Synaptic Terminals Contacting ChAT+ Dendrites (per μm2) |
0.00273 ±0.0003 |
0.00176 ±0.0002 |
64.5%* | 0.013 |
| Abundance of VGLUT2+ Synaptic Terminals per ChAT+ Dendrite Unit Area (μm2) |
0.0385 ±0.006 |
0.0363 ±0.005 |
94.3% | 0.781 |
Significant differences between WT and Q140 mice are shown in bold in the last column.
Figure 4.

EM images with cholinergic processes highlighted, showing that fewer ChAT+ profiles are typically present per unit area in Q140 striatum than in WT striatum.
To determine if the thalamic connectivity of striatal cholinergic interneurons was changed along with their dendrite loss, striatum was processed for VGLUT2 and ChAT double-labeling immunohistochemistry and examined at the EM level. Numerous VGLUT2+ terminals were found in WT and Q140 mice, most of which made axospinous contact with striatal projection neurons (Fig. 5). Nonetheless, a small percent of VGLUT2+ terminals were observed to make asymmetric synaptic contact with ChAT+ dendrites in WT mice at 1 and 4 months of age (Figs. 5, 6). In general, each ChAT+ dendrite fragment seen at the EM level only possessed one VGLUT2+ synaptic contact. The cholinergic dendrite fragments were typically 4-5 μm long and 1-2 μm wide. Examination of ChAT+ dendrites in Q140 mice also revealed many VGLUT2+ synaptic contacts, but their abundance appeared to be less than in WT mice. Blinded analysis revealed that there was an overall decrease in the number of VGLUT2+ synaptic terminals on ChAT+ dendrites in Q140 striatum per unit area of striatum (Table 2). EM double-label studies showed that the abundance of VGLUT2+ axodendritic terminals making synaptic contact with ChAT+ dendrites per unit area of striatum was decreased by 35.5% at 1 to 4 months of age in the Q140 mice overall (Table 2). The difference was statistically significant. We noted that the loss of VGLUT2+ terminals on ChAT+ dendrites per unit area of striatum was proportional to the loss in ChAT+ dendrite coverage of striatum, which suggested that the loss of axodendritic VGLUT2+ terminals ending on cholinergic dendrites per unit area of striatum might simply reflect loss of dendritic target rather than loss of terminals per se. Consistent with this, the abundance of VGLUT2+ terminals per unit area of ChAT+ dendrite was very similar between Q140 and WT mice, and not significantly different. Thus, the density of thalamic terminals along individual cholinergic dendrites was largely unaltered, but a loss of axodendritic thalamic terminals on individual cholinergic neurons occurred due to the dendrite territory loss.
Figure 5.

Representative EM images showing VGLUT2+/ChAT+ double-immunolabeling in striatum in WT mice at 1 month of age. Image A shows VGLUT2+ immunolabeled terminals (arrow) and ChAT+ immunolabeled dendrites (den). Image B presents a high power view of the framed selection in image A, showing a VGLUT+ synaptic terminal on a ChAT+ dendrite.
Figure 6.

Representative EM images of VGLUT2+/ChAT double-immunolabeling in striatum in WT (A) and Q140 (B) mice at 1 month of age. VGLUT2+ immunolabeled synaptic terminals are indicated by arrows. ChAT+ dendrites are indicated by the abbreviation “den”. Both images are at the same magnification. The areal abundance of VGLUT2+ synaptic terminals on ChAT+ dendrites was determined for Q140 and WT mice at 1 and 4 months of age, and found to be less.
Discussion
Cholinergic interneurons represent 1-2% of all striatal neurons and correspond to the tonically active striatal neurons (TANs) detected in electrophysiological recording studies (Pisani et al., 2007; Smith et al., 2011). They receive their major excitatory input from the thalamus, as well as an input from midbrain dopaminergic neurons that exerts an inhibitory effect via D2 receptors on cholinergic interneurons (Pisani et al., 2007; Lapper and Bolam, 1992). Cholinergic interneurons innervate both direct and indirect pathway striatal projection neurons (SPNs), and modulate projection neuron responsivity and plasticity at corticostriatal synapses (Pisani et al., 2007; Ding et al., 2010; Smith et al., 2011). Thalamic activation of cholinergic interneurons, in particular, is thought to play a role in attentional shifts to salient or novel environmental stimuli mediated by striatum. Because of their important role in regulating striatal projection neuron activity and plasticity, abnormalities in cholinergic striatal interneurons would be expected to adversely affect striatal function.
We found that cholinergic interneurons are not significantly reduced in numbers in Q140 heterozygous mice compared to WT mice at 1-4 months of age; however, their perikarya are 9.0% smaller in diameter. Moreover, our LM Sholl analysis showed that cholinergic neuron dendritic arbors are 30% fewer in Q140 heterozygous mice than in WT mice at 1-4 months of age. Our EM analysis confirmed that the dendrites of cholinergic interneurons are thinner and fewer at this age in Q140 heterozygous mice. Our EM analysis also showed there are 35% fewer VGLUT2+ terminals on cholinergic interneuron dendrites in heterozygous Q140 striatum than in WT striatum at this age, largely due to the ChAT+ dendrite loss. Such early abnormalities in thalamic input to striatal cholinergic interneurons are of significance because they may contribute to premanifest HD symptoms, if they also occur in humans.
Our findings of abnormalities in cholinergic striatal interneuron dendritic branching and thalamic input in HD, but preservation of neuronal numbers, is consistent with prior studies in humans and mouse models of HD. For example, Smith and coworkers (2006) reported that in spite of a normal number of cholinergic interneurons in the striatum in R6/1 HD transgenic mice and in post-mortem striatal tissue from HD patients, the levels of both the vesicular acetylcholine transporter (VAChT) and ChAT are markedly decreased. In the case of the R6/1 mice, the defect was already evident early in the life span. Similarly, other studies of HD models (Vetter et al., 2003; Holley et al., 2015) and HD patients (Spokes, 1980; Ferrante et al., 1987; Suzuki et al., 2001) have reported preserved neuron numbers but depressed ChAT activity and VAChT binding. Farrar et al. (2011) have noted that activation of excitatory inputs to striatal cholinergic interneurons is dysfunctional in R6/2 HD mice, and suggested that the reduced levels of extracellular striatal ACh in HD striatum may reflect abnormalities in the excitatory innervation of cholinergic interneurons. Although the functional implication of reduced total thalamic input per cholinergic neuron but generally conserved density of thalamic input that we have observed in Q140 mice needs to be assessed experimentally, it may be that the diminished thalamostriatal input in Q140 mice occurs in R6/2 mice as well (and HD in general) and contributes to a reduced activation and acetylcholine release from these neurons. Holley et al. (2015) have also reported enhanced inhibitory responses in striatal cholinergic interneurons in R6/2 mice, as well as a trend toward reduced thalamic responsiveness, that could contribute to reduced acetylcholine release.
Numerous imaging studies have reported cortical thinning in premanifest HD, coupled with cortical white matter loss (DiProspero et al., 2004; Kipps et al., 2005; Reading et al., 2005; Rosas et al., 2005, 2006; Ciarmiello et al., 2006; Paulsen et al., 2006; Hobbs et al., 2010a; Aylward et al., 2011; Dumas et al., 2012), reductions in striatal volume (Aylward, 1987; Aylward et al., 2012, 2013; Tabrizi et al., 2013), and striatal hypoactivation (Grafton et al., 1992; Paulsen et al., 2004; Ciarmiello et al., 2006; Wolf et al., 2012). Although premanifest cortical and striatal neuron loss have not been quantified, they are generally thought to be minimal (Vonsattel et al., 1985; Augood et al., 1997; Vonsattel and DiFiglia, 1998; Glass et al., 2000; Deng et al., 2004; Nopoulos, et al., 2010). We have previously shown that loss of cortical and thalamic input to SPNs occurs in heterozygous Q140 knock-in males, suggesting that some of the white matter loss in premanifest HD may also reflect loss of cortical and thalamic input to SPNs, particularly to direct pathway SPNs in the case of the cortical input (Deng et al., 2013, 2014). The present results show that cholinergic striatal interneurons as well show a substantial deficiency in thalamic input in heterozygous Q140 knock-in males, in this case early in the lifespan.
Although the loss of cortical input to dSPNs may help explain the hypokinesia seen very early in the course of HD before striatal neuron loss, as in our 12-month old Q140 heterozygous mice (Deng et al., 2014), hyperactivity in a rearing test has been reported in homozygous Q140 mice at 1 month of age (Menalled et al., 2003). Hyperactivity in a rearing test has not, however, been observed at 2.5 months of age in Q140 heterozygous mice, in whom the phenotype is slowed compared to Q140 homozygous mice (Rising et al., 2011). Thus, it is uncertain that Q140 heterozygous mice show an early hyperactivity similar to that reported in Q140 homozygous mice. In any event, the basis of the rearing hyperactivity in homozygous Q140 mice at 1 months of age is uncertain, and not likely to be attributable to cortical input loss since the loss we observed in heterozygous Q140 mice does not occur until much later (Deng et al., 2014). In our prior EM single-label study (Deng et al., 2013), we found a deficiency in thalamic input to striatal dendrites already at 1 month of age, which persists beyond this age. The present study shows that loss of input to cholinergic neurons accounts for this at least in part, since we found that cholinergic striatal interneurons in heterozygous Q140 mice show an approximately 35% deficiency in axodendritic thalamic input at 1-4 month of age. If a similar phenomenon occurs in homozygous Q140 mice (which seems likely), and there are no compensatory changes, it may explain the early hyperkinesia reported for Q140 mice, as the loss of thalamic input to cholinergic neurons would be predicted to more greatly lessen the responses of iSPNs than dSPNs to cortical drive (Smith et al., 2011), which models of basal ganglia function predict should cause hyperkinesia (Albin et al., 1989). The early rearing hyperkinesia at 2 months in homozygous Q94 knock-in mice (Menalled et al., 2002) and the open field hyperactivity at 3 months in heterozygous YAC128 mice (Slow et al., 2003), both before striatal projection neuron loss, might be explainable by this mechanism as well. An early deficiency in large axodendritic thalamic terminals on iSPNs, as suggested by our findings in Deng et al. (2014), may also contribute to the early hyperkinesia. Subsequent loss of cortical input to dSPNs may lead to hypokinesia eventually becoming the major motor manifestation during manifest stages in Q140 mice (Deng et al., 2014; Fowler and Muma, 2015).
Given the thalamic atrophy and hypometabolism reported in premanifest HD, which eventually progresses to intralaminar thalamic neuron loss (Heinsen et al., 1996), an early deficiency in thalamic input to striatum may also occur in human HD victims (Paulsen et al., 2004; Feigin et al., 2007; Aylward et al., 2011). How this defect might affect behavior is uncertain, but the present results suggest that it might contribute to a very early defect in cholinergic neuron function, which would then affect SPN function, given the prominent input of cholinergic neurons to SPNs. Of note in this regard is the finding that m1 and m4 type muscarinic receptors, which are found on SPNs, are significantly reduced in abundance in human HD striatum (Cha et al., 1998; Kuhn et al., 2007; Strand et al., 2007). Of relevance to the basis of the thalamostriatal shortfall in Q140 mice, our finding that thalamostriatal input to cholinergic interneurons is already deficient at 1-4 months of age in Q140 mice raises the possibility that the deficiency involves an early developmental defect rather than a later pathological event. The development of imaging approaches for visualizing human cholinergic nerve terminals in vivo using (–)-5-18F-Fluoroethoxybenzovesamicol (Petrou et al., 2014) makes it possible to determine if, in fact, cholinergic interneuron abnormalities occur early in the course of HD. Whether such a putative abnormality might be caused by a direct action of the mutation on cholinergic interneurons or by an action mediated via other neuron types is uncertain, but it is clear that striatal cholinergic interneurons express high levels of huntingtin (Fusco et al., 1999) and that the mutation can drive abnormal development of striatal projection neurons (Molero et al., 2009). In any event, given the apparent role of the thalamic input to striatum in attentional mechanisms and the demonstrated role of cholinergic neurons in striatal plasticity and learning (Lim et al., 2014; Pisani et al., 2007), early deficits in thalamic input to striatal cholinergic neurons are likely to have consequences for motor performance and learning.
Acknowledgments
We thank Marion Joni, Kathy Troughton, and Yunming Hu for technical assistance. Supported by the Cure Huntington’s Disease Initiative Foundation (AR), NIH NS28721 (AR), NIH NS57722 (AR), and The Methodist Hospitals Endowed Professorship in Neuroscience (AR).
Footnotes
The authors have no financial interest in the research reported here.
References
- Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- Albin RL, Qin Y, Young AB, Penney JB, Chesselet MF. Preproenkephalin messenger RNA-containing neurons in striatum of patients with symptomatic and presymptomatic Huntington's disease: an in situ hybridization study. Ann Neurol. 1991;30:542–549. doi: 10.1002/ana.410300406. [DOI] [PubMed] [Google Scholar]
- Augood SJ, Faull RL, Emson PC. Dopamine D1 and D2 receptor gene expression in the striatum in Huntington's disease. Ann Neurol. 1997;42:215–221. doi: 10.1002/ana.410420213. [DOI] [PubMed] [Google Scholar]
- Aylward EH. Change in MRI striatal volumes as a biomarker in preclinical Huntington's disease. Brain Res Bull. 2007;72:152–158. doi: 10.1016/j.brainresbull.2006.10.028. [DOI] [PubMed] [Google Scholar]
- Aylward EH, Liu D, Nopoulos PC, Ross CA, Pierson RK, Mills JA, Long JD, Paulsen JS, PREDICT-HD Investigators and Coordinators of the Huntington Study Group Striatal volume contributes to the prediction of onset of Huntington disease in incident cases. Biol Psychiat. 2012;71:822–828. doi: 10.1016/j.biopsych.2011.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylward EH, Harrington DL, Mills JA, Nopoulos PC, Ross CA, Long JD, Liu D, Westervelt HK, Paulsen JS, PREDICT-HD Investigators and Coordinators of the Huntington Study Group Regional atrophy associated with cognitive and motor function in prodromal Huntington disease. J Huntington Dis Res. 2013;2:477–489. doi: 10.3233/JHD-130076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylward EH, Nopoulos PC, Ross CA, Langbehn DR, Pierson RK, Mills JA, Johnson HJ, Magnotta VA, Juhl AR, Paulsen JS. PREDICT-HD Investigators and Coordinators of Huntington Study Group (HSG). Longitudinal change in regional brain volumes in prodromal Huntington disease. J Neurol Neurosurg Psychiatry. 2011;82:405–410. doi: 10.1136/jnnp.2010.208264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechtel N, Scahill RI, Rosas HD, Acharya T, van den Bogaard SJ, Jauffret C, Say MJ, Sturrock A, Johnson H, Onorato CE, Salat DH, Durr A, Leavitt BR, Roos RA, Landwehrmeyer GB, Langbehn DR, Stout JC, Tabrizi SJ, Reilmann R. Tapping linked to function and structure in premanifest and symptomatic Huntington disease. Neurology. 2010;75:2150–2160. doi: 10.1212/WNL.0b013e3182020123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendse HW, Groenewegen HJ. Organization of the thalamostriatal projections in the rat, with special emphasis on the ventral striatum. J Comp Neurol. 1990;299:187–228. doi: 10.1002/cne.902990206. [DOI] [PubMed] [Google Scholar]
- Biglan KM, Ross CA, Langbehn DR, Aylward EH, Stout JC, Queller S, Carlozzi NE, Duff K, Beglinger LJ, Paulsen JS, PREDICT-HD Investigators of the Huntington Study Group (HSG) Motor abnormalities in premanifest persons with Huntington's disease: the PREDICT-HD study. Mov Disord. 2009;24:1763–1772. doi: 10.1002/mds.22601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blekher TM, Yee RD, Kirkwood SC, Hake AM, Stout JC, Weaver MR, Foroud TM. Oculomotor control in asymptomatic and recently diagnosed individuals with the genetic marker for Huntington's disease. Vision Res. 2004;44:2729–2736. doi: 10.1016/j.visres.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Cha JH, Kosinski CM, Kerner JA, Alsdorf SA, Mangiarini L, Davies SW, Penney JB, Bates GP, Young AB. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human Huntington disease gene. Proc Natl Acad Sci USA. 1998;95:6480–6485. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciarmiello A, Cannella M, Lastoria S, Simonelli M, Frati L, Rubinsztein DC, Squitieri F. Brain white-matter volume loss and glucose hypometabolism precede the clinical symptoms of Huntington's disease. J Nucl Med. 2006;47:215–222. [PubMed] [Google Scholar]
- Coulon P, Bras H, Vinay L. Characterization of last-order premotor interneurons by transneuronal tracing with rabies virus in the neonatal mouse spinal cord. J Comp Neurol. 2011;519:3470–3487. doi: 10.1002/cne.22717. [DOI] [PubMed] [Google Scholar]
- de Boo GM, Tibben A, Lanser JB, Jennekens-Schinkel A, Hermans J, Maat-Kievit A, Roos RA. Early cognitive and motor symptoms in identified carriers of the gene for Huntington disease. Arch Neurol. 1997;54:1353–1357. doi: 10.1001/archneur.1997.00550230030012. [DOI] [PubMed] [Google Scholar]
- Delval A, Bleuse S, Simonin C, Delliaux M, Rolland B, Destee A, Defebvre L, Krystkowiak P, Dujardin K. Are gait initiation parameters early markers of Huntington's disease in pre-manifest mutation carriers? Gait & Posture. 2011;34:202–207. doi: 10.1016/j.gaitpost.2011.04.011. [DOI] [PubMed] [Google Scholar]
- Deng YP, Albin RL, Penney JB, Young AB, Anderson KD, Reiner A. Differential loss of striatal projection systems in Huntington's disease: a quantitative immunohistochemical study. J Chem Neuroanat. 2004;27:143–164. doi: 10.1016/j.jchemneu.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Deng YP, Wong T, Bricker-Anthony C, Deng B, Reiner A. Loss of corticostriatal and thalamostriatal synaptic terminals precedes striatal projection neuron pathology in heterozygous Q140 Huntington’s disease mice. Neurobiol Dis. 2013;60:89–107. doi: 10.1016/j.nbd.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng YP, Wong T, Wan JY, Reiner A. Differential loss of thalamostriatal and corticostriatal input to striatal projection neuron types prior to overt motor symptoms in the Q140 knock-in mouse model of Huntington's disease. Front Syst Neurosci. 2014;8:198. doi: 10.3389/fnsys.2014.00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desban M, Kemel ML, Glowinski J, Gauchy C. Spatial organization of patch and matrix compartments in the rat striatum. Neuroscience. 1993;57:661–667. doi: 10.1016/0306-4522(93)90013-6. [DOI] [PubMed] [Google Scholar]
- Ding JB, Guzman JN, Peterson JD, Goldberg JA, Surmeier DJ. Thalamic gating of corticostriatal signaling by cholinergic interneurons. Neuron. 2010;67:294–307. doi: 10.1016/j.neuron.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiProspero NA, Chen EY, Charles V, Plomann M, Kordower JH, Tagle DA. Early changes in Huntington's disease patient brains involve alterations in cytoskeletal and synaptic elements. J Neurocytol. 2004;33:517–533. doi: 10.1007/s11068-004-0514-8. [DOI] [PubMed] [Google Scholar]
- Dumas EM, van den Bogaard SJ, Ruber ME, Reilman RR, Stout JC, Craufurd D, Hicks SL, Kennard C, Tabrizi SJ, van Buchem MA, van der Grond J, Roos RA. Early changes in white matter pathways of the sensorimotor cortex in premanifest Huntington's disease. Human Brain Mapping. 2012;33:203–212. doi: 10.1002/hbm.21205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar A, Callahan JW, Abercrombie ED. Reduced striatal acetylcholine efflux in the R6/2 mouse model of Huntington's disease: an examination of the role of altered inhibitory and excitatory mechanisms. Exp Neurol. 2011;232:119–125. doi: 10.1016/j.expneurol.2011.08.010. [DOI] [PubMed] [Google Scholar]
- Feigin A, Tang C, Ma Y, Mattis P, Zgaljardic D, Guttman M, Paulsen JS, Dhawan V, Eidelberg D. Thalamic metabolism and symptom onset in preclinical Huntington's disease. Brain. 2007;130:2858–2867. doi: 10.1093/brain/awm217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Beal MF, Kowall NW, Richardson EP, Jr, Martin JB. Sparing of acetylcholinesterase-containing striatal neurons in Huntington's disease. Brain Res. 1987;411:162–166. doi: 10.1016/0006-8993(87)90694-9. [DOI] [PubMed] [Google Scholar]
- Ferreira T, Blackman A, Oyrer J, Jayabal A, Chung A, Watt A, Sjöström J, van Meyel D. Neuronal morphometry directly from bitmap images. Nature Methods. 2014;11:982–984. doi: 10.1038/nmeth.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler SC, Muma NA. Use of a force-sensing automated open field apparatus in a longitudinal study of multiple behavioral deficits in CAG140 Huntington's disease model mice. Behav Brain Res. 2015;294:7–16. doi: 10.1016/j.bbr.2015.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco FR, Chen Q, Lamoreaux WJ, Figueredo-Cardenas G, Jiao Y, Coffman JA, Surmeier DJ, Honig MG, Carlock LR, Reiner A. Cellular localization of huntingtin in striatal and cortical neurons in rats: lack of correlation with neuronal vulnerability in Huntington's disease. J Neurosci. 1999;19:1189–1202. doi: 10.1523/JNEUROSCI.19-04-01189.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Ann Rev Neurosci. 1992;15:285–320. doi: 10.1146/annurev.ne.15.030192.001441. [DOI] [PubMed] [Google Scholar]
- Glass M, Dragunow M, Faull RLM. The pattern of neurodegeneration in Huntington’s disease: a comparative study of cannabinoid, dopamine, adenosine and GABAa receptor alterations in the human basal ganglia in Huntington’s disease. Neuroscience. 2000;97:505–519. doi: 10.1016/s0306-4522(00)00008-7. [DOI] [PubMed] [Google Scholar]
- Grafton ST, Mazziotta JC, Pahl JJ, St George-Hyslop P, Haines JL, Gusella J, Hoffman JM, Baxter LR, Phelps ME. Serial changes of cerebral glucose metabolism and caudate size in persons at risk for Huntington's disease. Arch Neurol. 1992;49:1161–1167. doi: 10.1001/archneur.1992.00530350075022. [DOI] [PubMed] [Google Scholar]
- Guillery RW, Herrup K. Quantification without pontification: choosing a method for counting objects in sectioned tissues. J Comp Neurol. 1997;386:2–7. doi: 10.1002/(sici)1096-9861(19970915)386:1<2::aid-cne2>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Hedreen JC, Folstein SE. Early loss of neostriatal striosome neurons in Huntington’s disease. J Neuropathol Exp Neurol. 1995;54:105–120. doi: 10.1097/00005072-199501000-00013. [DOI] [PubMed] [Google Scholar]
- Heinsen H, Rüb U, Bauer M, Ulmar G, Bethke B, Schüler M, Böcker F, Eisenmenger W, Götz M, Korr H, Schmitz C. Nerve cell loss in the thalamic mediodorsal nucleus in Huntington's disease. Acta Neuropathol. 1999;97:613–622. doi: 10.1007/s004010051037. [DOI] [PubMed] [Google Scholar]
- Hickey MA, Kosmalska A, Enayati J, Cohen R, Zeitlin S, Levine MS, Chesselet MF. Extensive early motor and non-motor behavioral deficits are followed by striatal neuronal loss in knock-in Huntington's disease mice. Neuroscience. 2008;157:280–295. doi: 10.1016/j.neuroscience.2008.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey MA, Zhu C, Medvedeva V, Lerner RP, Patassini S, Franich NR, Maiti P, Frautschy SA, Zeitlin S, Levine MS, Chesselet MF. Improvement of neuropathology and transcriptional deficits in CAG 140 knock-in mice supports a beneficial effect of dietary curcumin in Huntington's disease. Mol Neurodegener. 2012;7:12. doi: 10.1186/1750-1326-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs NZ, Barnes J, Frost C, Henley SM, Wild EJ, Macdonald K, Barker RA, Scahill RI, Fox NC, Tabrizi SJ. Onset and progression of pathologic atrophy in Huntington disease: a longitudinal MR imaging study. Am J Neuroradiol. 2010a;31:1036–1041. doi: 10.3174/ajnr.A2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holley SM, Joshi PR, Parievsky A, Galvan L, Chen JY, Fisher YE, Huynh MN, Cepeda C, Levine MS. Enhanced GABAergic Inputs Contribute to Functional Alterations of Cholinergic Interneurons in the R6/2 Mouse Model of Huntington’s Disease. eNeuro. 2015;2:e0008. doi: 10.1523/ENEURO.0008-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipps CM, Duggins AJ, Mahant N, Gomes L, Ashburner J, McCusker EA. Progression of structural neuropathology in preclinical Huntington's disease: a tensor based morphometry study. J Neurol Neurosurg Psychiatry. 2005;76:650–655. doi: 10.1136/jnnp.2004.047993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood SC, Siemers E, Bond C, Conneally PM, Christian JC, Foroud T. Confirmation of subtle motor changes among presymptomatic carriers of the Huntington disease gene. Arch Neurol. 2000;57:1040–1044. doi: 10.1001/archneur.57.7.1040. [DOI] [PubMed] [Google Scholar]
- Kirkwood SC, Siemers E, Stout JC, Hodes ME, Conneally PM, Christian JC, Foroud T. Longitudinal cognitive and motor changes among presymptomatic Huntington disease gene carriers. Arch Neurol. 1999;56:563–568. doi: 10.1001/archneur.56.5.563. [DOI] [PubMed] [Google Scholar]
- Kuhn A, Goldstein DR, Hodges A, Strand AD, Sengstag T, Kooperberg C, Becanovic K, Pouladi MA, Sathasivam K, Cha JH, Hannan AJ, Hayden MR, Leavitt BR, Dunnett SB, Ferrante RJ, Albin R, Shelbourne P, Delorenzi M, Augood SJ, Faull RL, Olson JM, Bates GP, Jones L, Luthi-Carter R. Mutant huntingtin's effects on striatal gene expression in mice recapitulate changes observed in human Huntington's disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum Mol Gen. 2007;16:1845–1861. doi: 10.1093/hmg/ddm133. [DOI] [PubMed] [Google Scholar]
- Lapper SR, Bolam JP. Input from the frontal cortex and the parafascicular nucleus to cholinergic interneurons in the dorsal striatum of the rat. Neuroscience. 1992;51:533–545. doi: 10.1016/0306-4522(92)90293-b. [DOI] [PubMed] [Google Scholar]
- Lei W, Deng YP, Liu BB, Mu SH, Guley NM, Wong T, Reiner A. A confocal laser scanning microscopy and ultrastructural study of VGLUT2 thalamic input to striatal projection neurons in rats. J Comp Neurol. 2013;521:1354–1377. doi: 10.1002/cne.23235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner RP, Trejo Martinez Ldel C, Zhu C, Chesselet MF, Hickey MA. Striatal atrophy and dendritic alterations in a knock-in mouse model of Huntington's disease. Brain Res Bull. 2012;87:571–578. doi: 10.1016/j.brainresbull.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SA, Kang UJ, McGehee DS. Striatal cholinergic interneuron regulation and circuit effects. Front Synaptic Neurosci. 2014;6:22. doi: 10.3389/fnsyn.2014.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled LB, Sison JD, Wu Y, Olivieri M, Li XJ, Li H, Zeitlin S, Chesselet MF. Early motor dysfunction and striosomal distribution of huntingtin microaggregates in Huntington's disease knock-in mice. J Neurosci. 2002;22:8266–8276. doi: 10.1523/JNEUROSCI.22-18-08266.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington's disease with 140 CAG repeats. J Comp Neurol. 2003;465:11–26. doi: 10.1002/cne.10776. [DOI] [PubMed] [Google Scholar]
- Molero AE, Gokhan S, Gonzalez S, Feig JL, Alexandre LC, Mehler MF. Impairment of developmental stem cell-mediated striatal neurogenesis and pluripotency genes in a knock-in model of Huntington’s disease. Proc Natl Acad Sci USA. 2009;106:21900–21905. doi: 10.1073/pnas.0912171106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nopoulos PC, Aylward EH, Ross CA, Johnson HJ, Magnotta VA, Juhl AR, Pierson RK, Mills J, Langbehn DR, Paulsen JS, PREDICT-HD Investigators Coordinators of Huntington Study Group (HSG) Cerebral cortex structure in prodromal Huntington disease. Neurobiol Dis. 2010;40:544–554. doi: 10.1016/j.nbd.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen JS, Magnotta VA, Mikos AE, Paulson HL, Penziner E, Andreasen NC, Nopoulos PC. Brain structure in preclinical Huntington's disease. Biol Psychiatry. 2006;59:57–63. doi: 10.1016/j.biopsych.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Paulsen JS, Zimbelman JL, Hinton SC, Langbehn DR, Leveroni CL, Benjamin ML, Reynolds NC, Rao SM. fMRI biomarker of early neuronal dysfunction in presymptomatic Huntington's Disease. Am J Neuroradiol. 2004;25:1715–1721. [PMC free article] [PubMed] [Google Scholar]
- Petrou M, Frey KA, Kilbourn MR, Scott PJ, Raffel DM, Bohnen NI, Müller ML, Albin RL, Koeppe RA. In vivo imaging of human cholinergic nerve terminals with (–)-5-18F-Fluoroethoxybenzovesamicol: Biodistribution, dosimetry, and tracer kinetic analyses. J Nucl Med. 2014;55:396–404. doi: 10.2967/jnumed.113.124792. [DOI] [PubMed] [Google Scholar]
- Phelps PE, Houser CR, Vaughn JE. Immunocytochemical localization of choline acetyltransferase within the rat neostriatum: a correlated light and electron microscopic study of cholinergic neurons and synapses. J Comp Neurol. 1985;238:286–307. doi: 10.1002/cne.902380305. [DOI] [PubMed] [Google Scholar]
- Pisani A, Bernardi G, Ding J, Surmeier DJ. Re-emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci. 2007;30:545–553. doi: 10.1016/j.tins.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Rao AK, Gordon AM, Marder KS. Coordination of fingertip forces during precision grip in premanifest Huntington's disease. Mov Disord. 2011;26:862–869. doi: 10.1002/mds.23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao AK, Muratori L, Louis ED, Moskowitz CB, Marder KS. Spectrum of gait impairments in presymptomatic and symptomatic Huntington's disease. Mov Disord. 2008;23:1100–1107. doi: 10.1002/mds.21987. [DOI] [PubMed] [Google Scholar]
- Reading SA, Yassa MA, Bakker A, Dziorny AC, Gourley LM, Yallapragada V, Rosenblatt A, Margolis RL, Aylward EH, Brandt J, Mori S, van Zijl P, Bassett SS, Ross CA. Regional white matter change in pre-symptomatic Huntington's disease: a diffusion tensor imaging study. Psychiatry Res. 2005;140:55–62. doi: 10.1016/j.pscychresns.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Rising AC, Xu J, Carlson A, Napoli VV, Denovan-Wright EM, Mandel RJ. Longitudinal behavioral, cross-sectional transcriptional and histopathological characterization of a knock-in mouse model of Huntington's disease with 140 CAG repeats. Exp Neurol. 2011;228:173–182. doi: 10.1016/j.expneurol.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas HD, Hevelone ND, Zaleta AK, Greve DN, Salat DH, Fischl B. Regional cortical thinning in preclinical Huntington disease and its relationship to cognition. Neurology. 2005;65:745–747. doi: 10.1212/01.wnl.0000174432.87383.87. [DOI] [PubMed] [Google Scholar]
- Rosas HD, Koroshetz WJ, Chen YI, Skeuse C, Vangel M, Cudkowicz ME, Caplan K, Marek K, Seidman LJ, Makris N, Jenkins BG, Goldstein JM. Evidence for more widespread cerebral pathology in early HD: an MRI-based morphometric analysis. Neurology. 2003;60:1615–1620. doi: 10.1212/01.wnl.0000065888.88988.6e. [DOI] [PubMed] [Google Scholar]
- Rosas HD, Tuch DS, Hevelone ND, Zaleta AK, Vangel M, Hersch SM, Salat DH. Diffusion tensor imaging in presymptomatic and early Huntington's disease: Selective white matter pathology and its relationship to clinical measures. Mov Disord. 2006;21:1317–1325. doi: 10.1002/mds.20979. [DOI] [PubMed] [Google Scholar]
- Shiromani PJ, Armstrong DM, Bruce G, Hersh LB, Groves PM, Gillin JC. Relation of pontine choline acetyltransferase immunoreactive neurons with cells which increase discharge during REM sleep. Brain Research Bulletin. 1987;18:447–455. doi: 10.1016/0361-9230(87)90019-0. [DOI] [PubMed] [Google Scholar]
- Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- Siemers E, Foroud T, Bill DJ, Sorbel J, Norton JA, Jr, Hodes ME, Niebler G, Conneally PM, Christian JC. Motor changes in presymptomatic Huntington disease gene carriers. Arch. Neurol. 1996;53:487–492. doi: 10.1001/archneur.1996.00550060029011. [DOI] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- Smith R, Chung H, Rundquist S, Maat-Schieman ML, Colgan L, Englund E, Liu YJ, Roos RA, Faull RL, Brundin P, Li JY. Cholinergic neuronal defect without cell loss in Huntington’s disease. Hum Mol Genet. 2006;15:3119–3131. doi: 10.1093/hmg/ddl252. [DOI] [PubMed] [Google Scholar]
- Smith Y, Surmeier DJ, Redgrave P, Kimura M. Thalamic contributions to basal ganglia-related behavioral switching and reinforcement. J Neurosci. 2011;31:16102–16106. doi: 10.1523/JNEUROSCI.4634-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spokes EG. Neurochemical alterations in Huntington’s chorea: a study of post-mortem brain tissue. Brain. 1980;103:179–210. doi: 10.1093/brain/103.1.179. [DOI] [PubMed] [Google Scholar]
- Strand AD, Baquet ZC, Aragaki AK, Holmans P, Yang L, Cleren C, Beal MF, Jones L, Kooperberg C, Olson JM, Jones KR. Expression profiling of Huntington’s disease models suggests that brain-derived neurotrophic factor depletion plays a major role in striatal degeneration. J Neurosci. 2007;27:11758–11768. doi: 10.1523/JNEUROSCI.2461-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Desmond TJ, Albin RL, Frey KA. Vesicular neurotransmitter transporter in Huntington’s disease: initial observations and comparison with traditional synaptic markers. Synapse. 2001;41:329–336. doi: 10.1002/syn.1089. [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Scahill RI, Durr A, Roos RA, Leavitt BR, Jones R, Landwehrmeyer GB, Fox NC, Johnson H, Hicks SL, Kennard C, Craufurd D, Frost C, Langbehn DR, Reilmann R, Stout JC, TRACK-HD Investigators Biological and clinical changes in premanifest and early stage Huntington's disease in the TRACK-HD study: the 12-month longitudinal analysis. Lancet Neurol. 2011;10:31–42. doi: 10.1016/S1474-4422(10)70276-3. [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Scahill RI, Owen G, Durr A, Leavitt BR, Roos RA, Borowsky B, Landwehrmeyer B, Frost C, Johnson H, Craufurd D, Reilmann R, Stout JC, Langbehn DR, TRACK-HD Investigators Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington's disease in the TRACK-HD study: analysis of 36 month observational data. Lancet Neurol. 2013;12:637–647. doi: 10.1016/S1474-4422(13)70088-7. [DOI] [PubMed] [Google Scholar]
- Turner TH, Goldstein J, Hamilton JM, Jacobson M, Pirogovsky E, Peavy G, Corey-Bloom J. Motor abnormalities in premanifest persons with Huntington's disease: the PREDICT-HD study. J Mot Behav. 2011;43:295–302. doi: 10.1080/00222895.2011.580390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter JM, Jehle T, Heinemeyer J, Franz P, Behrens PF, Jackisch R, Landwehrmeyer GB, Feuerstein TJ. Mice transgenic for exon 1 of Huntington’s disease: properties of cholinergic and dopaminergic presynaptic function in the striatum. J Neurochem. 2003;85:1054–1063. doi: 10.1046/j.1471-4159.2003.01704.x. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M. Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr. Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- Wang HB, Deng YP, Reiner A. In situ hybridization histochemical and immunohistochemical evidence that striatal projection neurons co-containing substance P and enkephalin are overrepresented in the striosomal compartment of striatum in rats. Neurosci Lett. 2007;425:195–199. doi: 10.1016/j.neulet.2007.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CJ, Groves PM, Kitai ST, Linder JC. Three-dimensional structure of dendritic spines in the rat neostriatum. J Neurosci. 1983;3:383–388. doi: 10.1523/JNEUROSCI.03-02-00383.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf RC, Grön G, Sambataro F, Vasic N, Wolf ND, Thomann PA, Saft C, Landwehrmeyer GB, Orth M. Brain activation and functional connectivity in premanifest Huntington's disease during states of intrinsic and phasic alertness. Human Brain Mapping. 2012;33:2161–2173. doi: 10.1002/hbm.21348. [DOI] [PMC free article] [PubMed] [Google Scholar]