

Abstract
Clostridium perfringens enterotoxin (CPE) has significant medical importance due to its involvement in several common human gastrointestinal diseases. This 35 kDa single polypeptide toxin consists of two domains: a C-terminal domain involved in receptor binding and an N-terminal domain involved in oligomerization, membrane insertion and pore formation. The action of CPE starts with its binding to receptors, which include certain members of the claudin tight junction protein family; bound CPE then forms a series of complexes, one of which is a pore that causes the calcium influx responsible for host cell death. Recent studies have revealed that CPE binding to claudin receptors involves interactions between the C-terminal CPE domain and both the 1st and 2nd extracellular loops (ECL-1 and ECL-2) of claudin receptors. Of particular importance for this binding is the docking of ECL-2 into a pocket present in the C-terminal domain of the toxin. This increased understanding of CPE interactions with claudin receptors is now fostering the development of receptor decoy therapeutics for CPE-mediated gastrointestinal disease, reagents for cancer therapy/diagnoses and enhancers of drug delivery.
Keywords: Clostridium perfringens, Enterotoxin, Claudins, Receptors, Pore
1. Introduction: The medical importance of Clostridium perfringens enterotoxin (CPE)
The Gram-positive, spore-forming anaerobe Clostridium perfringens produces at least 18 different toxins. However, considerable diversity exists between the toxin arsenals of different Clostridium perfringens strains. Those variations in toxin production are exploited by a historical classification scheme [1] that assigns C. perfringens isolates to one of five types (A–E), based upon their production of four (alpha, beta, epsilon and iota toxins) typing toxins [1]. Although not used for toxinotyping classification, C. perfringens enterotoxin (CPE) is arguably the most medically important toxin made by this bacterium. CPE is known to be produced by all C. perfringens types except type B.
CPE-producing type A strains are responsible for the diarrhea and abdominal cramps of C. perfringens type A food poisoning, which is currently the 2nd most common bacterial foodborne disease in developed countries [2]. For example, nearly a million cases of C. perfringens type A food poisoning occur annually in the USA, with economic losses approaching half a billion dollars per year [3,4]. Substantial evidence supports the involvement of CPE in C. perfringens type A food poisoining. For example, lysates from sporulating, but not vegetative cultures, of CPE-producing food poisoning strains cause gastrointestinal (GI) pathology (intestinal histologic damage and fluid loss) in animal models [5], which is informative because CPE is only produced during sporulation [2]. Furthermore, sporulating lysates from a cpe null mutant of a food poisoning strain failed to cause any GI pathology in animal models and complementation reversed this attenuation [5]. In addition, physiologically-relevant concentrations, of highly purified CPE are sufficient to fully recapitulate, in animal models, the GI effects caused by sporulating culture lysates of CPE-producing type A food poisoning strains [5,6].
During the past decade, two exceptionally severe C. perfringens type A food poisoning outbreaks occurred in the USA, resulting in the deaths of several relatively young and healthy patients [7,8]. Both of those atypical outbreaks occurred in psychiatric hospitals, where (prior to developing food poisoning) the affected patients had been treated with psychotropic drugs that can induce fecal impaction or constipation side-effects. It is believed that these food poisoning cases became so severe because this preexisting fecal impaction/constipation had interfered with development of the diarrhea typically associated with C. perfringens type A food poisoning. Impairment of diarrhea would limit the natural flushing of CPE (and perhaps other C. perfringens toxins) from the GI tract, thereby prolonging the presence of the toxin(s) in the intestines, which could promote absorption of toxin(s) from the intestines into the circulation and thereby lead to a deadly enterotoxemia. This hypothesis has received direct experimental support from a study [9] using mouse small intestinal ligated loops as an animal model of static GI tract conditions. That mouse study demonstrated lethal absorption of purified CPE from the small intestines into the circulation. The absorbed enterotoxin then bound to internal organs, including the liver and kidneys. Death of the mice correlated with the appearance of high potassium levels in their blood, an effect probably due to CPE effects on internal organs. This CPE-induced hyperpotassemia then likely induced death of the mice by cardiac arrest.
Recent studies [10] suggested that CPE may also contribute to some cases of enteritis necroticans, a food-borne human disease caused by C. perfringens type C strains. Historically, enteritis necroticans is most-often associated with the Papua New Guinea highlands, where it is known locally as pigbel [10]. Beta toxin is clearly important for enteritis necroticans [10,11]. However, many type C strains associated with enteritis necroticans produce CPE, as well as beta toxin [10]. Recent studies [10] using toxin knock-out mutants and purified toxins showed that, when produced by type C strains, CPE can act synergistically with beta toxin [10]. The mechanism of this toxin synergism remains to be determined.
In addition to its involvement in food-borne human disease, CPE–producing C. perfringens type A strains cause ~5–10% of all cases of human nonfoodborne GI infections, including antibiotic-associated diarrhea (AAD) [1]. CPE appears to be an important contributor to these infections since, i) sporulating, but not vegetative, culture lystates of a nonfoodborne GI disease strain caused histologic damage and fluid accumulation in animal models and ii) inactivating the cpe gene in that strain produced sporulating culture lysates that are unable to cause GI pathology in animal models and this attenuation was reversible by complementation [5]. CPE-associated nonfoodborne GI disease cases are usually longer in duration and more serious than a typical case of C. perfringens type A food poisoning [1,12]. Recent in vitro adherence studies [13] suggest that these disease differences may reflect variations in host cell adherence ability amongst CPE-producing type A strains, i.e., nonfoodborne GI disease isolates may be better intestinal colonizers than most food poisoning strains.
All CPE-associated human diseases are infections, not intoxications [1,2]. During these illnesses, CPE is produced when C. perfringens cells sporulate in the intestine. After synthesis of CPE during sporulation, the enterotoxin accumulates in the cytoplasm of the mother cell. At the completion of sporulation the mother cell lyses, causing concomitant release of both the mature spore and CPE into the intestinal lumen. The toxin then binds and exerts its action, as described below.
2. Overview of CPE action
While the interactions between CPE and its receptors are described in detail later, this topic will now be briefly introduced in the context of overviewing CPE-induced cytotoxicity (Fig. 1). CPE action starts when this toxin binds to certain members of the claudin family of host proteins [14–18]. Claudins can form fibrils that modulate the structure and function of mammalian tight junctions [19–21]. However, some claudins are located on the cell surface outside of tight junctions [22] and it is those non-junctional claudins that are believed to be primarily important in CPE binding, at least initially [22].
Fig. 1. The action of C. perfringens enterotoxin.

CPE first binds to claudin receptors on the exposed surface of host cells to form a small complex. Approximately 6 small complexes (which also contain nonreceptor claudins, not shown) oligomerize into a large prepore complex on the membrane surface, followed by formation of an active pore named CH-1. High CPE doses cause a massive calcium influx that strongly activates cytoplasmic calpain to induce cell death via oncosis. At lower CPE doses, there is less calcium influx and a milder cytoplasmic calpain activation, which causes a classical caspase-3 mediated apoptosis. Cell death produces morphologic damage that exposes the basolateral surface of the cell, allowing CPE access to additional receptors to form more CH-1. This also results in formation of a second large CPE complex named CH-1 that contains occludin; this can trigger internalization of occludin and claudin into the cytoplasm. Modified with permission from Ref. [75].
Once bound to claudin receptors, CPE becomes localized in a series of complexes, starting with a small (~90 kDa) complex that contains, in addition to the enterotoxin, CPE receptor claudins and (possibly) claudins that are not CPE receptors [23,24]. On the surface of the host cell cytoplasmic membrane, approximately six of these small CPE complexes oligomerize to form a prepore [25,26]. β hairpin loops from the CPE molecules in this prepore then rapidly assemble into a β-barrel that inserts into the membrane, creating an active pore complex named CH-1. CH-1 is ~450 kDa in size and contains (at minimum) six CPE molecules, along with CPE receptor claudins and (possibly) claudins that are not receptors for the enterotoxin [25].
The CH-1 CPE pore is permeable to small molecules (<200 Da), including calcium [27]. Influx of calcium through this CPE pore activates calmodulin and calpain [27]. Activation of those host proteins then triggers cell death by oncosis (when host cells are exposed to high CPE treatment doses, where a massive calcium influx occurs) or classical caspase-3-mediated apoptosis (when host cells are exposed to lower CPE treatment doses, where the calcium influx is more modest).
Dying CPE-treated cells also develop morphologic damage, which exposes claudins located on the basolateral surface of either the dying cell or adjacent cells [28]. This exposure of basolateral surfaces permits more CPE binding, additional formation of CH-1 pores, and interaction of some receptor-bound CPE with occludin, another tight junction protein, to form an ~600 kDa CPE complex named CH-2 [25,29]. The contribution of CH-2 to CPE action is unclear, but it may help to explain why CPE induces occludin (and claudin) internalization into the cytoplasm of Caco-2 cells [25].
CPE-induced host cell death induces substantial damage in the intestines, including epithelial desquamation and severe villus blunting (Fig. 2). Several lines of evidence suggest that this damage triggers fluid and electrolyte loss (which manifest as diarrhea) in the small intestine and, perhaps, the colon. For example, in the CPE-treated rabbit small intestine, there is a close temporal correlation between the onset of fluid/electrolyte damage and the development of intestinal damage [30]. In addition, only those CPE doses capable of causing intestinal damage can produce fluid and electrolyte losses in the rabbit small intestine [31]. Last, an attenuated CPE variant that binds to intestinal receptors failed to cause either damage or fluid transport changes in rabbit small intestinal loops (Fig. 2).
Fig. 2. Small intestinal effects of CPE or C-CPE.

Left panel shows control rabbit small intestinal loops treated with Ringers solution. L is lumen; LP is lamina propria. Middle panel shows the effects of CPE on rabbit small intestinal loops. Note the epithelial desquamation and villus destruction. Right panel shows the effects of CPE168-319 (C-CPE) on rabbit small intestinal loops. Note the absence of damage. Final magnification of each photomicrograph is 400×. Used with permission from Ref. [76].
3. The CPE protein
3.1. Introduction
CPE, a 319 amino acid (35.3 kDa) single polypeptide, has a unique primary amino acid sequence, except for some limited homology (of still unknown significance) with the Antp70/C1 protein of Clostridium botulinum [2,32,33]. The primary amino acid sequence of CPE is highly conserved amongst CPE-producing type A, C and D strains [34]. However, a few type E strains produce a CPE variant that has (relative to classical CPE) ten amino acid differences of unknown phenotypic consequence [35].
3.2. Mapping CPE functional regions
Early biochemical and recombinant DNA studies [36,37] demonstrated that the C-terminal half of CPE is non-cytotoxic, and does not damage the intestine (Fig. 2), yet possesses strong receptor binding activity. Follow-up recombinant DNA and synthetic peptide studies then determined that the extreme C-terminal region (amino acids 290–319) of the toxin is required for receptor binding [38]. Later site-directed mutagenesis work identified several residues (Y306, Y310, Y312 and L315) within this extreme CPE C-terminal receptor-binding region that are key for receptor binding by the toxin [39]. Site-directed mutagenesis [40] studies also demonstrated the involvement of additional C-terminal CPE residues (e.g. L223, R227, and L254) in receptor binding [39].
Full-length CPE has cytotoxic activity for cultured cells. However, deletion mutagenesis studies showed that the first 45 amino acids of CPE are not important for this cytotoxicity, with removal of those N-terminal sequences actually increasing toxin activity by 2–3 fold [41,42]. This enhancement may have pathophysiologic relevance since purified trypsin and chymotrypsin can induce a similar 2–3 fold-increase in CPE activity by removing the first 25 or 37 N-terminal amino acids, respectively, from the enterotoxin [43,44]. Trypsin- and/or chymotrypsin-induced proteolytic activation of CPE, which could occur in the intestine during natural disease, is attributable to increased CH-1 pore formation by the N-terminal truncated CPE [45]. Specifically, residues 1–45 of CPE are thought to reduce cytotoxicity by sterically interfering with the CPE amino acid 45–53 region (see below) involved in oligomerization, a prerequisite for CH-1 pore formation [26,45].
Deleting amino acids beyond residue 45 from the N-terminus completely abrogates the cytotoxic activity of CPE [41]. Site-directed mutagenesis studies [42] explained this phenotype by identifying several amino acids located between amino acids D45 and G53 of the toxin as being critical for CPE oligomerization. Specifically, introduction of a D48A mutation into CPE completely blocked oligomerization and cytotoxicity, while a I53A CPE variant was highly attenuated for CH-1 formation.
An N-terminal region named TM1 spans from amino acids 80 to 106 of CPE and possesses the alternating hydrophilic and hydrophobic amino acid residues typical of a β-hairpin loop (Fig. 3). This characteristic of the TM1 region is informative for understanding CPE structure vs. function relationships because β-hairpin loops are known to assemble into a β-barrel structure that inserts into host plasma membranes during pore formation by several other pore-forming toxins. When site-directed mutagenesis of the TM1 region was performed [46], several CPE variants were identified that showed wild-type toxin binding and oligomerization activity, yet were highly attenuated for cytotoxicity. Directly supporting the hypothesis that the TM1 region of CPE is a β-hairpin loop involved in beta barrel formation for insertion into membranes during CH-1 pore formation (Fig. 3), several of those TM1 site-directed variants were impaired for membrane insertion while other TM1 variants inserted yet did not form a functional pore [46].
Fig. 3. Model for CPE insertion into membranes.

A) Model for the TM1 region sequence (CPE amino acids 90–106), depicting the TM1 region as an unfolded β-hairpin with alternating hydrophobic and hydrophilic residues. The circled amino acids were shown to be important for CPE membrane insertion or pore formation by site-directed mutagenesis [46]. Used with permission from Ref. [46]. B) Model for CPE insertion and pore formation. β-hairpins extend from each of ~6 CPE proteins in the prepore to assemble a β-barrel that inserts into membranes to create an active pore.
3.3. Structure of the CPE protein
Initial insights into CPE protein structure were provided when the C-terminal half of CPE (C-CPE, i.e., residues 194–319) was crystalized and its structure solved by x-ray diffraction [47]. This work revealed that the C-CPE domain is a nine-stranded, mostly antiparallel, β-sandwich. Residues implicated, by site-directed mutagenesis [39], as being important for binding of CPE to its receptor were shown to line a pocket/groove located on the surface of C-CPE. This work also determined that the C-CPE domain shares some structural similarity with the receptor binding domains of several other toxins made by spore-forming bacteria, e.g., some members of the large Cry toxin family of Bacillus thuringiensis.
More recently, two research groups [48,49] independently solved the structure of the entire CPE protein by x-ray diffraction (Fig. 4). Those studies demonstrated that the enterotoxin consists of the C-terminal C-CPE receptor-binding domain and an N-terminal domain comprised of two halves. The first 34 residues of native CPE have no interpretable electron density, indicating they are likely disordered, which may explain why these extreme N-terminal sequences have the ability to partially interfere with CPE oligomerization (see the preceding section). The D48-G53 sequences critical for CPE oligomerization mapped to the junction of the two halves of the N-terminal domain of the toxin. The TM1 region mediating membrane insertion is largely contained within an alpha helix located in the N-terminal domain of the toxin. During CH-1 pore formation, this alpha helix presumably unfolds into a β-hairpin loop that assembles with similar loops from other CPE molecules present in the prepore to form a β-barrel for penetration into the membrane and pore formation.
Fig. 4. Structure of the CPE protein.
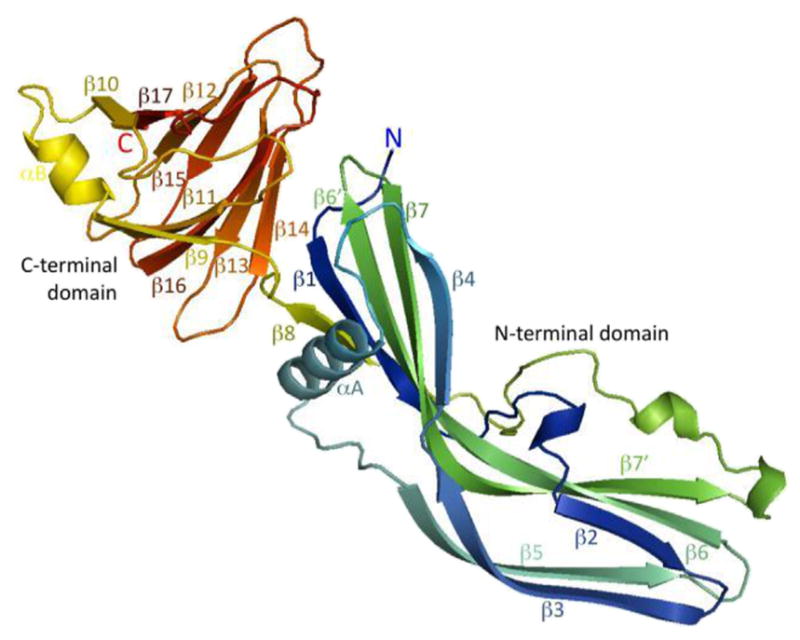
The N-terminal domain of CPE is responsible for oligomerization and contains the TM1 region (localized around the central alpha helical region of the N-terminal domain) that unwinds into a β-hairpin to form a β-barrel for membrane insertion and pore formation. The C-terminal domain of CPE is responsible for receptor binding activity. Reproduced with permission from Ref. [49].
Furthermore, solving the complete CPE structure revealed that this enterotoxin belongs structurally to the aerolysin pore-forming toxin family, which also includes (for example) C. perfringens epsilon toxin [48,49]. Specifically, the β-pore forming toxin members of this family share structurally similar domain(s) involved in oligomerization and membrane insertion/pore formation. However, CPE is a relatively unusual member of this family since, in the soluble monomer form, its eventual β-hairpin loop is apparently located in an alpha helix rather than the β-strand found in soluble toxin monomers of other members of this toxin family. The receptor binding domain(s) of the aerolysin toxin family members vary considerably, consistent with these toxins binding to different receptors [48,49].
4. Claudins: an introduction
The first claudins were discovered in 1998 by Tsukita’s research group [50]. Currently, humans are thought to produce 27 different members of the claudin family, which are single polypeptides that typically range in size from ~21 to 27 kDa [51]. Recent structural biology studies [52,53] confirmed computer-modeling predictions [54] that claudins are comprised of a four transmembrane domain bundle and two extracellular loops (referred to as ECL-1 and ECL-2). Both ECL-1 and ECL-2 are thought to mediate intercellular interactions [55,56], while paracellular permeability gating (see next paragraph) is mainly attributed to ECL-1 [55,56]. Claudins also have a short (~7 amino acid) N-terminal sequence and a longer (20–60 amino acid) C-terminal tail, both located in the cytoplasm of host cells [56]. The C-terminal tail of most claudins fosters interactions with scaffolding proteins, like zonula occludens-1 (ZO-1), via its PDZ motif (Post-synaptic density 95, Discs large ZO-1) [55,56]. Considerable amino acid sequence diversity exists amongst the cytoplasmic tails of various claudins, so antibodies specific for a particular claudin are often raised against synthetic peptides containing the tail sequence of that claudin.
In epithelial and endothelial cells, claudins can form fibrils that are key components of tight junctions [55,57]. Claudins play a key role in the “fence” function of tight junctions by helping to form the barrier that modulates paracellular permeability and separates the apical and basolateral surfaces of polarized cells. However, some claudins are also important contributors to the “gate” properties of tight junctions as they regulate paracellular ion fluxes by forming ion-selective pores [55,56]. Expression of claudins varies considerably amongst different tissues and gradients of claudin expression even exist within tissues; for example, claudin expression patterns vary along the crypt to villus axis of the small intestine [58]. Last, individual epithelial or endothelial cells often produce multiple members of the claudin family.
5. CPE interactions with claudins
5.1. Mapping CPE binding regions of claudins
Extensive early work demonstrated that CPE binds to protein receptors on sensitive host cells [59]. Key insights into the identity of the CPE receptor(s) were provided by the expression cloning studies of Katahira et al. [14,60]. Using transfected mouse L929 fibroblasts, which are naturally insensitive to CPE because they cannot bind the toxin, Katahira et al. showed that expression of either of two ~20 kDa Vero cell proteins created transfectant cells that were highly sensitive to this toxin due to strong CPE binding [14,60].
This early work on CPE receptor identification had been performed concurrently with Tsukita’s discovery of claudins [50], so it soon became apparent that the two Vero cell CPE receptors identified by Katahira et al. were claudins-3 and -4. Additional studies, also employing fibroblast transfectant cells, then established that not all claudins are CPE receptors, e.g., transfectant cells expressing claudins-1 or -2 are CPE-insensitive because they are unable to bind the toxin [14,15,24,60]. To date, known CPE receptor claudins include claudins-3, -4, -6, -7, -8, -9 and -14 [61]. However, claudins-1, -2, -5, -10,-13, and 20–24 are not CPE receptors, at least at physiologically-relevant toxin concentrations [61]. Amongst the claudin receptors, not all bind CPE with equal affinity. For example, claudins-3 and -4 are high affinity CPE receptors, while claudins-8 and -14 bind CPE more weakly [18,54].
Studies then evaluated which portion(s) of a claudin receptor is essential for CPE binding. Using chimeric claudins expressed in fibroblast transfectant cells, it was shown that the C-terminal half of claudins plays an essential role in CPE binding and action [15]. Specifically, fibroblast transfectant cells expressing a chimeric claudin consisting of the N-terminal half of claudin-1 (not a CPE receptor) and the C-terminal half of claudin-3 (a CPE receptor) were killed by CPE; however, this toxin had no effect on fibroblasts expressing the converse chimeric claudin consisting of the N-terminal half of claudin-3 and the C-terminal half of claudin-1. Additional studies using chimeric claudins then specifically mapped this CPE-binding region in the C-terminal half of receptor claudins to the ECL-2 region [24].
Several synthetic peptide mapping and site-directed mutagenesis studies have now dissected which residues in the ECL-2 region of receptor claudins are important for CPE binding [18,24,54]. Peptide mapping approaches identified an ECL-2 motif (NPLVP) that plays a role in CPE binding to claudin-3 [54]. Bioinformatic analyses noted that the N residue in this ECL-2 CPE-binding motif is highly conserved amongst receptor claudins, while the equivalent residue is never N (it is usually a D or S) in claudins that are not CPE receptors [24]. Site-directed mutagenesis then demonstrated that this N residue is a critical yes/no determinant for CPE binding to claudins by showing that changing the natural D residue in claudin-1 to an N converts claudin-1 to a strong CPE binding receptor when expressed by fibroblast transfectant cells. Conversely, changing the natural N residue in claudin-4 to D destroyed the CPE binding ability of claudin-4 when expressed by fibroblast transfectant cells [24].
There are also now insights into why some claudins (e.g., claudin-8 and -14) bind CPE but at relatively low affinity despite their possessing the ECL-2 N residue that is the yes/no determinant for CPE binding. Site-directed mutagenesis studies [18] showed that neighboring residues located near the critical ECL-2 N residue can modulate CPE binding affinity (Fig. 5). For example, a P residue is located next to the critical N in the claudin-4 ECL-2 CPE binding motif, but the equivalent residue is an S in claudin-8. Changing that natural S residue in claudin-8 to P produced a claudin-8 variant that, when expressed by fibroblast transfectant cells, binds CPE at similar high affinity as wild-type claudin-4. The converse was also true, i.e., changing the natural P residue in the claudin-4 ECL-2 to S produced a claudin-4 variant that bound CPE much less strongly than natural claudin-4 when expressed by fibroblast transfectant cells. Interestingly, claudin-14 also binds CPE relatively weakly even though it has both the critical N and the adjacent P residues in the ECL-2 CPE binding motif [18]. Site-directed mutagenesis studies [18] showed that the weak CPE binding of claudin-14 is attributable to the presence of an N residue, rather than the equivalent D residue of claudin-4, located near the CPE-binding motif. This finding establishes that some residues outside the classical ECL-2 binding motif can also impact CPE binding affinity.
Fig. 5. Claudin ECL-2 sequences that modulate CPE binding affinity and cytotoxic activity.

Top, Comparison of claudin ECL-2 region sequences for claudins that bind CPE strongly, weakly or not at all (as indicated). The critical Asn residue that is a yes/no determinant for claudin binding to CPE is denoted by a single asterisk. Bottom, an illustrative example of how changing a single amino acid residue near this critical Asn in the ECL-2 region significantly affects CPE binding properties and cytotoxicity. In this example, changing the natural proline of claudin-4 (double asterisk in top panel) to the serine of claudin-8 greatly reduced CPE sensitivity of transfectants; this phenotype was due to decreased CPE binding (not shown). Conversely, changing the natural serine of claudin-8 to the proline of claudin-4 greatly increased CPE sensitivity of transfectants and this phenotype was due to increased CPE binding (not shown). Modified with permission from Ref. [18].
5.2. Structural biology of CPE: claudin receptor interactions
Structural biology studies have now provided important direct insights into the binding of CPE to receptor claudins. The first such study [62] used a synthetic peptide corresponding to the ECL-2 sequences shown to be important for the binding of this enterotoxin (see preceding paragraph). When the structure of this peptide bound to CPE was solved by x-ray crystallography, it confirmed earlier computer predictions [55] by demonstrating that the CPE binding motif sequence present in the ECL-2 of a receptor claudin docks into the putative claudin-binding groove on the surface of the C-terminal domain of CPE (C-CPE).
Understanding CPE: receptor interaction was then further advanced by a recent x-ray diffraction study [52] that solved the structure of C-CPE bound to a claudin receptor. This structure (Fig. 6) supported the physiologic relevance of ECL-2 synthetic peptide/CPE findings described above by showing that, in the context of an intact claudin receptor, the ECL-2 binding motif also docks in a groove located on the surface of C-CPE. Interactions between C-CPE and ECL-2 were determined to involve a number of hydrophobic interactions. Solving the structure of C-CPE bound to a claudin receptor also provided an explanation for why the N residue of the ECL-2 binding motif is critical for CPE binding, i.e., this residue apparently hydrogen bonds with several CPE residues present in the binding pocket of receptor claudins.
Fig. 6. Structure of the C-CPE binding domain bound to a claudin receptor.

Shown is a claudin oriented in a host cell plasma membrane. The claudin consists of four alpha helixes and two extracellular loops (locations of the ECL-1 and ECL-2 loops are indicated). Not shown is the C-terminal cytoplasmic tail of claudins. Note that the C-CPE binding domain (beta sandwich at top of structure) interacts with both of the ECLs present in a receptor claudin. However, it is the interaction of a binding cavity on C-CPE with the ECL-2 loop that dictates whether a claudin is a CPE receptor. Reproduced with permission from Ref. [52].
Interestingly, the structural biology analysis [52] of C-CPE bound to a claudin receptor also detected interactions between C-CPE and the ECL-1 of claudins. Site-directed mutagenesis then showed that ECL-1 residues A39-I41 are important for C-CPE binding to claudin receptors. Therefore, it is now apparent that C-CPE (and presumably CPE) interacts with both ECL-1 and ECL-2 during receptor binding. However, the ECL-1 amino acid residues important for CPE binding are relatively conserved amongst most claudins, including claudins that are not CPE receptors, so ECL-2 is of predominant importance for determining the ability of a claudin to bind CPE, with ECL-1 contributing to this binding by enlarging the hydrophobic contact area.
6. Applications from understanding CPE: claudin receptor interactions
Why study CPE interactions with claudin receptors? The rapid increase in understanding of CPE: receptor interactions is already suggesting several translational medical applications. For example, this information could lead to the development of potential therapeutics against CPE-mediated intestinal disease. Towards that goal, a synthetic peptide corresponding to the ECL-2 region of the CPE receptor claudin-4 was shown to inhibit CPE action, both on cultured cells and in rabbit small intestinal loops (6). This inhibition was specific since a synthetic peptide corresponding to the ECL-2 region of claudin-1 (not a CPE receptor) did not reduce CPE action in vitro or in vivo. These results suggest that claudin receptor-based ECL-2 peptides should be further investigated for their potential application as receptor decoy therapeutics against CPE-mediated disease, particularly for treating chronic and severe AAD cases.
Antigen delivery to mucosal-associated lymphoid tissue (MALT) is the requisite first step for induction of mucosal immunity by vaccines. Located in the mucosa, MALT contains several cell types, including M cells, which take up antigen and present it to underlying antigen presenting cells for processing. Given the critical role of M cells in mucosal immunity, targeting ligands to M cells is an attractive strategy for improving mucosal immunizations. Interestingly, M cells show significant expression of claudin-4, a strong CPE receptor [63]. While fusion proteins containing active CPE cannot be used for immunizations due to toxicity issues, the presence of claudin-4 on M cells suggested that a targeting strategy using nontoxic, but claudin binding-capable, CPE derivatives might be useful for preparing fusion proteins for use in improved mucosal vaccines. Several published studies now support this approach. For example, C-CPE has been fused with pneumococcal surface protein A (PspA), an important antigen for inducing protective immunity against Streptococcus pneumoniae infection [63]. When administered intranasally, this vaccine induced immune responses that were protective against infection by this important bacterial pathogen. To cite a second example using this approach, a peptide corresponding to the nontoxic C-terminal 30 amino acids of CPE was used as part of a conjugate vaccine to protect against coxsackie virus B3 infection in mice [64].
Although nontoxic, C-CPE can break apart tight junctions [65,66]. This ability has been exploited to increase intestinal drug absorption via increased paracellular transport. It has been reported [67] that C-CPE has 400-fold more drug absorption-enhancing potency than clinically-used enhancers. Efforts are currently underway [68] to modify C-CPE for stronger binding or more specificity, e.g., to target a C-CPE derivative to a single claudin. On a cautionary note, some hepatic toxicity has been noted in mice receiving an i.v. injection of C-CPE [69].
A final translational field where knowledge of CPE binding interactions is being exploited concerns cancer therapy and diagnosis [16,70]. Many fatal cancers are derived from epithelial tissues and claudins are often overexpressed in those cancers. For example, overexpression of the CPE receptor claudin-4 is common in breast, prostate, pancreatic and ovarian cancers [70]. Tumor cells over-expressing claudin-4 are highly sensitive to CPE in vitro. Direct injection of CPE into tumors can reduce tumor volume significantly [16,71]. As an alternative approach, C-CPE has been coupled with other cytotoxic proteins to obtain potent killing properties against tumors expressing claudin receptors for CPE [16,70–72].
There are obvious safety concerns regarding the systemic use of CPE for treating non-solid tumors and metastatic cancers since many normal cells also produce claudin CPE receptors. One approach to overcome this limitation has involved use of C-CPE in combination with traditional anti-cancer agents [16,70]. For example, at relatively low doses, C-CPE sensitizes ovarian cancer cells to the killing effects of carboplatin, a traditional anticancer agent [73].
Besides employing claudin-directed CPE-based agents as cancer therapeutics, there is now considerable interest in utilizing labeled C-CPE as a diagnostic tool for tumor imaging [16,70]. These C-CPE-based imaging agents bind strongly to tumors consisting of cancer cells that overexpress claudin CPE receptors, such as claudin-4. For example, fluorescently-labeled C-CPE has been used to visualize micrometastatic human ovarian cancer tumor implants in mice [74].
7. Concluding remarks
The binding of CPE to claudin receptors has become a paradigm for understanding interactions between a toxin and its receptors. Furthermore, knowledge of CPE: receptor interactions holds promise for developing improved therapeutics against CPE-mediated intestinal disorders. Last, illustrating the serendipity of science, improved understanding CPE: receptor interactions is leading to translational medical applications for cancer therapy/diagnostics and drug delivery that could mature in the near future.
Acknowledgments
Preparation of this review was supported, in part, by grant AI019844-33 from the National Institute of Allergy and Infectious Diseases.
References
- 1.McClane BA, Uzal FA, Miyakawa MF, Lyerly D, Wilkins TD. The enterotoxic clostridia. In: Dworkin M, Falkow S, Rosenburg E, Schleifer H, Stackebrandt E, editors. The Prokaryotes. 3. Springer NY press; New York: 2006. pp. 688–752. [Google Scholar]
- 2.McClane BA, Robertson SL, Li J. Clostridium perfringens . In: Doyle MP, Buchanan RL, editors. Food Microbiology: Fundamentals and Frontiers. 4. ASM press; Washington D.C: 2013. pp. 465–489. [Google Scholar]
- 3.Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson M, Roy S, Jones JL, Griffin PM. Foodborne illness acquired in the United States-major pathogens. Emerg Infect Dis. 2011;17:7–15. doi: 10.3201/eid1701.P11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoffmann S, Batz MB, Morris JG., Jr Annual cost of illness and quality-adjusted life year losses in the United States due to 14 foodborne pathogens. J Food Prot. 2012;75:1292–1302. doi: 10.4315/0362-028X.JFP-11-417. [DOI] [PubMed] [Google Scholar]
- 5.Sarker MR, Carman RJ, McClane BA. Inactivation of the gene (cpe) encoding Clostridium perfringens enterotoxin eliminates the ability of two cpe-positive C. perfringens type A human gastrointestinal disease isolates to affect rabbit ileal loops. Mol Microbiol. 1999;33:946–958. doi: 10.1046/j.1365-2958.1999.01534.x. [DOI] [PubMed] [Google Scholar]
- 6.Shrestha A, Robertson SL, Garcia J, Beingasser J, McClane BA, Uzal FA. A synthetic peptide corresponding to the extracellular loop 2 region of claudin-4 protects against Clostridium perfringens enterotoxin in vitro and in vivo. Infect Immun. 2014;82 doi: 10.1128/IAI.02453-14. http://dx.doi.org/10.1128/IAI.02453-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bos J, Smithee L, McClane BA, Distefano RF, Uzal F, Songer JG, Mallonee S, Crutcher JM. Fatal necrotizing colitis following a foodborne outbreak of enterotoxigenic Clostridium perfringens type A infection. Clin Infect Dis. 2005;40:E78–E83. doi: 10.1086/429829. [DOI] [PubMed] [Google Scholar]
- 8.Centers for Disease Control and Prevention. Fatal foodborne Clostridium perfringens illness at a state psychiatric hospital–Louisiana 2010. Morb Mortal Wkly Rep. 2012;61:605–608. [PubMed] [Google Scholar]
- 9.Caserta JA, Robertson SL, Saputo J, Shrestha A, McClane BA, Uzal FA. Development and application of a mouse interstinal loop model to study the in vivo action of Clostridium perfringens enterotoxin. Infect Immun. 2011;79:3020–3027. doi: 10.1128/IAI.01342-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma M, Gurjar A, Theoret JR, Garcia JP, Beingesser J, Freedman JC, Fisher DJ, McClane BA, Uzal FA. Synergistic effects of Clostridium perfringens enterotoxin and beta toxin in rabbit small intestinal loops. Infect Immun. 2014;82:2958–2970. doi: 10.1128/IAI.01848-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sayeed S, Uzal FA, Fisher DJ, Saputo J, Vidal JE, Chen Y, Gupta P, Rood JI, McClane BA. Beta toxin is essential for the intestinal virulence of Clostridium perfringens type C disease isolate CN3685 in a rabbit ileal loop model. Mol Microbiol. 2008;67:15–30. doi: 10.1111/j.1365-2958.2007.06007.x. [DOI] [PubMed] [Google Scholar]
- 12.Carman RJ. Clostridium perfringens in spontaneous and antibiotic-associated diarrhoea of man and other animals. Rev Med Microbiol. 1997;8(Suppl 1):S43–S5. [Google Scholar]
- 13.Li J, McClane BA. Contributions of NanI sialidase to Caco-2 cell adherence by Clostridium perfringens type A and C strains causing human intestinal disease. Infect Immun. 2014;82:4620–4630. doi: 10.1128/IAI.02322-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katahira J, Sugiyama H, Inoue N, Horiguchi Y, Matsuda M, Sugimoto N. Clostridium perfringens enterotoxin utilizes two structurally related membrane proteins as functional receptors in vivo. J Biol Chem. 1997;272:26652–26658. doi: 10.1074/jbc.272.42.26652. [DOI] [PubMed] [Google Scholar]
- 15.Fujita K, Katahira J, Horiguchi Y, Sonoda N, Furuse M, Tskuita S. Clostridium perfringens enterotoxin binds to the second extracellular loop of claudin-3, a tight junction membrane protein. FEBS Lett. 2000;476:258–261. doi: 10.1016/s0014-5793(00)01744-0. [DOI] [PubMed] [Google Scholar]
- 16.English DPSA. Claudin overexpression in ovarian cancer: potential targets for Clostridium perfringens enterotoxin (CPE) based diagnosis and therapy. Int J Mol Sci. 2013;14:10412–10437. doi: 10.3390/ijms140510412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lal-Nag M, Battis M, Santin AD, Morin PJ. Claudin-6: a novel receptor for CPE-mediated cytotoxicity in ovarian cancer. Oncogenesis. 2012;1:e33. doi: 10.1038/oncsis.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shrestha A, McClane BA. Human claudin-8 and -14 are receptors capable of conveying the cytotoxic effects of Clostridium perfringens enterotoxin. mBio. 2013;4(1):e00594–12. doi: 10.1128/mBio.00594-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gunzel D, Yu AS. Claudins and the modulation of the tight junction permeability. Physiol Rev. 2013;93:525–569. doi: 10.1152/physrev.00019.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krause G, Winkler L, Mueller SL, Haseloff RF, JP, Blasig IE. Structure and function of claudins. Biochim Biophys Acta. 2008;1778:631–645. doi: 10.1016/j.bbamem.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 21.Furuse M. Molecular basis of the core structure of tight junctions. Cold Spring Har Perspect Biol. 2010;2:a002907. doi: 10.1101/cshperspect.a002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winkler L, Gehring C, Wenzel A, Muller SL, Piehl C, Krause G, Blasig IE, Piontek J. Molecular determinants of the interaction between Clostridium perfringens enterotoxin fragments and claudin-3. J Biol Chem. 2009;284:18863–18872. doi: 10.1074/jbc.M109.008623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wieckowski EU, Wnek AP, McClane BA. Evidence that an ~50kDa mammalian plasma membrane protein with receptor-like properties mediates the amphiphilicity of specifically-bound Clostridium perfringens enterotoxin. J Biol Chem. 1994;269:10838–10848. [PubMed] [Google Scholar]
- 24.Robertson SL, Smedley JG, 3rd, McClane BA. Identification of a claudin-4 residue important for mediating the host cell binding and action of Clostridium perfringens enterotoxin. Infect Immun. 2010;78:505–517. doi: 10.1128/IAI.00778-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robertson SL, Smedley JG, 3rd, Singh U, Chakrabarti G, Van Itallie CM, Anderson JM, McClane BA. Compositional and stoichiometric analysis of Clostridium perfringens enterotoxin complexes in Caco-2 cells and claudin 4 fibroblast transfectants. Cell Microbiol. 2007;9:2734–2755. doi: 10.1111/j.1462-5822.2007.00994.x. [DOI] [PubMed] [Google Scholar]
- 26.Smedley JG, 3rd, Uzal FA, McClane BA. Identification of a prepore large-complex stage in the mechanism of action of Clostridium perfringens enterotoxin. Infect Immun. 2007;75:2381–2390. doi: 10.1128/IAI.01737-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chakrabarti G, McClane BA. The importance of calcium influx, calpain and calmodulin for the activation of CaCo-2 cell death pathways by Clostridium perfringens enterotoxin. Cell Microbiol. 2005;7:129–146. doi: 10.1111/j.1462-5822.2004.00442.x. [DOI] [PubMed] [Google Scholar]
- 28.Singh U, Mitic LL, Wieckowski E, Anderson JM, McClane BA. Comparative biochemical and immunochemical studies reveal differences in the effects of Clostridium perfringens enterotoxin on polarized CaCo-2 cells versus vero cells. J Biol Chem. 2001;276:33402–33412. doi: 10.1074/jbc.M104200200. [DOI] [PubMed] [Google Scholar]
- 29.Singh U, Van Itallie CM, Mitic LL, Anderson JM, McClane BA. CaCo-2 cells treated with Clostridium perfringens enterotoxin form multiple large complex species, one of which contains the tight junction protein occludin. J Biol Chem. 2000;275:18407–18417. doi: 10.1074/jbc.M001530200. [DOI] [PubMed] [Google Scholar]
- 30.Sherman S, Klein E, McClane BA. Clostridium perfringens type A enterotoxin induces concurrent development of tissue damage and fluid accumulation in the rabbit ileum. J Diarrheal Dis Res. 1994;12:200–207. [PubMed] [Google Scholar]
- 31.McDonel JL. In vivo effects of Clostridium perfringens enterotoxin on the rat ileum. Infect Immun. 1974;10:1156–1162. doi: 10.1128/iai.10.5.1156-1162.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Czeczulin JR, Hanna PC, McClane BA. Cloning, nucleotide sequencing, and expression of the Clostridium perfringens enterotoxin gene in Escherichia coli. Infect Immun. 1993;61:3429–3439. doi: 10.1128/iai.61.8.3429-3439.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Melville SB, Collie RE, McClane BA. Regulation of enterotoxin production in Clostridium perfringens. In: Rood JI, McClane BA, Songer JG, Titball R, editors. The Molecular Genetics and Pathogenesis of the Clostridia. London: 1997. pp. 471–487. [Google Scholar]
- 34.Li J, Miyamoto K, Sayeed S, McClane BA. Organization of the cpe locus in CPE-positive Clostridium perfringens type C and D isolates. PLoS One. 2010;5:e10932. doi: 10.1371/journal.pone.0010932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyamoto K, Yumine N, Mimura K, Nagahama M, Li J, McClane BA, Akimoto S. Identification of novel Clostridium perfringens type E strains that carry an iota toxin plasmid with a functional enterotoxin gene. PLoS One. 2011;6:e20376. doi: 10.1371/journal.pone.0020376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanna PC, Wnek AP, McClane BA. Molecular cloning of the 3′ half of the Clostridium perfringens enterotoxin gene and demonstration that this region encodes receptor-binding activity. J Bacteriol. 1989;171:6815–6820. doi: 10.1128/jb.171.12.6815-6820.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Horiguchi Y, Akai T, Sakaguchi G. Isolation and function of a Clostridium perfringens enterotoxin fragment. Infect Immun. 1987;55:2912–2915. doi: 10.1128/iai.55.12.2912-2915.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanna PC, Mietzner TA, Schoolnik GK, McClane BA. Localization of the receptor-binding region of Clostridium perfringens enterotoxin utilizing cloned toxin fragments and synthetic peptides. The 30 C-terminal amino acids define a functional binding region. J Biol Chem. 1991;266:11037–11043. [PubMed] [Google Scholar]
- 39.Harada M, Kondoh M, Ebihara C, Takahashi A, Komiya E, Fujii M, Mizuguchi H, Tsunoda S, Horiguchi Y, Yagi K, Watanabe Y. Role of tyrosine residues in modulation of claudin-4 by the C-terminal fragment of Clostridium perfringens enterotoxin. Biochem Pharmacol. 2007;73:206–214. doi: 10.1016/j.bcp.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 40.Veshnyakova A, Piontek J, Protze J, Waziri N, Heise I, Krause G. Mechanism of Clostridium perfringens enterotoxin interaction with claudin-3/-4 protein suggests structural modifications of the toxin to target specific claudins. J Biol Chem. 2012;287:1698–1708. doi: 10.1074/jbc.M111.312165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kokai-Kun JF, McClane BA. Deletion analysis of the Clostridium perfringens enterotoxin. Infect Immun. 1997;65:1014–1022. doi: 10.1128/iai.65.3.1014-1022.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smedley JG, 3rd, McClane BA. Fine-mapping of the N-terminal cytotoxicity region of Clostridium perfringens enterotoxin by site-directed mutagenesis. Infect Immun. 2004;72:6914–6923. doi: 10.1128/IAI.72.12.6914-6923.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hanna PC, Wieckowski EU, Mietzner TA, McClane BA. Mapping functional regions of Clostridium perfringens type A enterotoxin. Infect Immun. 1992;60:2110–2114. doi: 10.1128/iai.60.5.2110-2114.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Granum PE, Richardson M. Chymotrypsin treatment increases the activity of Clostridium perfringens enterotoxin. Toxicon. 1991;29:445–453. doi: 10.1016/0041-0101(91)90227-i. [DOI] [PubMed] [Google Scholar]
- 45.Kokai-Kun JF, Benton K, Wieckowski EU, McClane BA. Identification of a Clostridium perfringens enterotoxin region required for large complex formation and cytotoxicity by random mutagenesis. Infect Immun. 1999;67:6534–6541. doi: 10.1128/iai.67.11.5634-5641.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen J, Theoret JR, Shrestha A, Smedley JG, 3rd, McClane BA. Cysteine scanning mutagenesis supports the importance of Clostridium perfringens enterotoxin amino acids 80-106 for membrane insertion and pore formation. Infect Immun. 2012;80:4078–4088. doi: 10.1128/IAI.00069-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Itallie CM, Betts L, Smedley JG, 3rd, McClane BA, Anderson JM. Structure of claudin binding domain of Clostridium perfringens enterotoxin. J Biol Chem. 2008;283:268–274. doi: 10.1074/jbc.M708066200. [DOI] [PubMed] [Google Scholar]
- 48.Kitadokoro K, Nishimura K, Kamitani S, Fukui-Miyazaki A, Toshima H, Abe H, Kamata Y, Sugita-Konishi Y, Yamamoto S, Karatani H, Horiguchi Y. Crystal structure of Clostridium perfringens enterotoxin displays features of beta-pore-forming toxins. J Biol Chem. 2011;286:19549–19555. doi: 10.1074/jbc.M111.228478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Briggs DC, Naylor CE, Smedley JG, 3rd, Lukoyanova N, Robertson S, Moss DS, McClane BA, Basak AK. Structure of the food-poisoning Clostridium perfringens enterotoxin reveals similarity to the aerolysin-like pore-forming toxins. J Mol Biol. 2011;413:138–149. doi: 10.1016/j.jmb.2011.07.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsukita S, Furuse M, Itoh M. Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol. 2001;2:285–293. doi: 10.1038/35067088. [DOI] [PubMed] [Google Scholar]
- 51.Mineta K, Yamamoto Y, Yamazaki Y, Tanaka H, Tada Y, Saito K, Tamura A, Michihiro I, Toshinori E, Takeuchi K, Tsukita S. Predicted expansion of the claudin multigene family. FEBS Lett. 2011;585:606–612. doi: 10.1016/j.febslet.2011.01.028. [DOI] [PubMed] [Google Scholar]
- 52.Saitoh Y, Suzuki H, Tani K, Nishikawa K, Irei K, Ogura Y, Tamura A, Tsukita S, Fujiyoshi Y. Structural insight into tight junction disassembly by Clostridium perfringens enterotoxin. Science. 2015;347:775–778. doi: 10.1126/science.1261833. [DOI] [PubMed] [Google Scholar]
- 53.Suzuki H, Nishizawa T, Tani K, Yamazaki Y, Tamura A, Ishitani R, Dohmae N, Tsukita S, Nureki O, Fujiyoshi Y. Crystal structure of a claudin provides insight into the architecture of tight junctions. Science. 2014;18:304–307. doi: 10.1126/science.1248571. [DOI] [PubMed] [Google Scholar]
- 54.Veshnyakova A, Protze J, Rossa J, Blasig IE, Krause G, Piontek J. On the interactin of Clostridium perfringens enterotoxin with claudins. Toxins. 2010;2:1336–1356. doi: 10.3390/toxins2061336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krause G, Protze J, Piontek J. Assembly and function of claudins: structure-function relationships based on homology models and crystal structures. Semin Cell Dev Biol. 2015;42:3–12. doi: 10.1016/j.semcdb.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 56.Koval M. Differential pathways of claudin oligomerization and integraton into tight junction. Tissue Barriers. 2013;13:e24518. doi: 10.4161/tisb.24518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Van Itallie CM, Anderson JM. Architecture of tight junctions and principles of molecular composition. Semin Cell Dev Biol. 2014;0:157–165. doi: 10.1016/j.semcdb.2014.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu Z, Ding L, Lu Q, YHC Claudins in intestines: distribution and functional significance in health and diseases. Tissue Barriers. 2013;1:e24978. doi: 10.4161/tisb.24978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McDonel JL. Toxins of Clostridium perfringens types A, B, C, D, and E. In: Dorner F, Drews H, editors. Pharmacology of Bacterial Toxins. Pergamon Press; Oxford: 1986. pp. 477–517. [Google Scholar]
- 60.Katahira J, Inoue N, Horiguchi Y, Matsuda M, Sugimoto N. Molecular cloning and functional characterization of the receptor for Clostridium perfringens enterotoxin. J Cell Biol. 1997;136:1239–1247. doi: 10.1083/jcb.136.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mitchell LA, Koval M. Specificity of interaction between Clostridium perfringens enterotoxin and claudin-family tight junction proteins. Toxins. 2010;2:1595–1611. doi: 10.3390/toxins2071595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yelland TS, Naylor C, Bagoban T, Savva Cg, Moss DS, McClane BA, Blasig IE, Popoff M, Basak AK. Structure of a C. perfringens enterotoxin mutant in complex with a modified claudin-2 extracellular loop2. J Mol Biol. 2014;426:3134–3147. doi: 10.1016/j.jmb.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Suzuki H, Watari A, Hashimoto E, Yonemitsu M, Kiyono H, Yagi K, Kondoh M, Kunisawa J. C-Terminal Clostridium perfringens enterotoxin-mediated antigen delivery for nasal pneumococcal vaccine. PLoS One. 2015;10:e0126352. doi: 10.1371/journal.pone.0126352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ye T, Yue Y, Fan X, Dong C, Xu W, Xiong S. M cell-targeting strategy facilitates mucosal immune response and enhances protection against CVB3-induced viral myocarditis elicited by chitosan-DNA vaccine. Vaccine. 2014;32:4457–4465. doi: 10.1016/j.vaccine.2014.06.050. [DOI] [PubMed] [Google Scholar]
- 65.Kojima T, Kyuno D, Sawada N. Targeting claudin-4 in human pancreatic cancer. Expert Opin Ther Targets. 2012;16:881–887. doi: 10.1517/14728222.2012.708340. [DOI] [PubMed] [Google Scholar]
- 66.Suzuki H, Kondoh M, Takahashi A, Yagi K. Proof of concept for claudin-targeted drug development. Ann N Y Acad Sci. 2012;1258:65–70. doi: 10.1111/j.1749-6632.2012.06503.x. [DOI] [PubMed] [Google Scholar]
- 67.Kondoh M, Takahashi A, Fujii M, Yagi K, Watanabe Y. A novel strategy for a drug delivery system using a claudin modulator. Biol Pharm Bull. 2006;29:1783–1789. doi: 10.1248/bpb.29.1783. [DOI] [PubMed] [Google Scholar]
- 68.Takahashi A, Komiya E, Kakutani H, Yoshida T, Fujii M, Horiguchi Y, Mizuguchi H, Tsutsumi Y, Tsunoda S, Koizumi N, Isoda K, Yagi K, Watanabe Y, Kondoh M. Domain mapping of a claudin-4 modulator, the C-terminal region of C-terminal fragment of Clostridium perfringens enterotoxin, by site-directed mutagenesis. Biochem Pharmacol. 2008;75:1639–1648. doi: 10.1016/j.bcp.2007.12.016. [DOI] [PubMed] [Google Scholar]
- 69.Li X, Saeki R, Watari A, Yagi K, Kondoh M. Tissue distribution and safety evaluation of a claudin-targeting molecule, the C-terminal fragment of Clostridium perfringens enterotoxin. Euro J Pharm Sci. 2014;52:132–137. doi: 10.1016/j.ejps.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 70.Gao Z, McClane BA. Use of Clostridium perfringens enterotoxin and the enterotoxin receptor-binding domain (C-CPE) for cancer treatment: opportunities and challenges. J Toxicol. 2012;2012:981626. doi: 10.1155/2012/981626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Michl P, Buchholz M, Rolke M, Kunsch S, Lohr M, McClane B, Tsukita S, Leder G, Adler G, Gress TM. Claudin-4: a new target for pancreatic cancer treatment using Clostridium perfringens enterotoxin. Gastroenterology. 2001;121:678–684. doi: 10.1053/gast.2001.27124. [DOI] [PubMed] [Google Scholar]
- 72.Saeki R, Kondoh M, Kakutani H, Tsunoda S, Mochizuki Y, Hamakubo T, Horiguchi Y, Yagi K. A novel tumor targeted therapy using a claudin-4-targeting molecule. Mol Pharmacol. 2009;76:918–926. doi: 10.1124/mol.109.058412. [DOI] [PubMed] [Google Scholar]
- 73.Gao Z, Xu X, McClane BA, Zeng Q, Litkouhi B, Welch WR, Berkowitz RS, Mok SC, Garner EI. C terminus of Clostridium perfringens enterotoxin downregulates CLDN4 and sensitizes ovarian cancer cells to Taxol and Carboplatin. Clin Cancer Res. 2011;17:1065–1074. doi: 10.1158/1078-0432.CCR-10-1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cocco ESE, Gasparrini S, Lopez S, Schwab CL, Bellone S, Bortolomai I, Sumi NJ, Bonazzoli E, Nicoletti R, Deng Y, Saltzman WM, Zeiss CJ, Centritto F, Black JD, Silasi DA, Ratner E, Azodi M, Rutherford TJ, Schwartz PE, Pecorelli S, Santin AD. Clostridium perfringens enterotoxin C-terminal domain labeled to fluorescent dyes for in vivo visualization of micrometastatic chemotherapy-resistant ovarian cancer. Int J Cancer. 2015;137:2618–2629. doi: 10.1002/ijc.29632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McClane BA, Lyerly DM, Wilkins TD. Enterotoxic clostridia: Clostridium perfringens type A and Clostridium difficile. In: Fischetti VA, Novick RP, Ferretti JJ, Portnoy DA, Rood J, editors. Gram-positive Pathogens. 3. ASM press; Washington, DC: 2006. pp. 703–714. [Google Scholar]
- 76.Smedley JG, 3rd, Saputo J, Parker JC, Fernandez-Miyakawa ME, Robertson SL, McClane BA, Uzal FA. Noncytotoxic Clostridium perfringens enterotoxin (CPE) variants localize CPE intestinal binding and demonstrate a relationship between CPE-induced cytotoxicity and enterotoxicity. Infect Immun. 2008;76:3793–3800. doi: 10.1128/IAI.00460-08. [DOI] [PMC free article] [PubMed] [Google Scholar]