

Abstract
Cancer initiating cells (CIC) undergo asymmetric growth patterns that increase phenotypic diversity and drive selection for chemotherapeutic resistance and tumor relapse. WNT signaling is a hallmark of colon CIC, often caused by APC mutations, which enable activation of β-catenin and MYC. Accumulating evidence indicates that long non-coding RNAs (lncRNA) contribute to the stem-like character of colon cancer cells. In this study, we report enrichment of the lncRNA RBM5-AS1/LUST during sphere formation of colon CIC. Its silencing impaired WNT signaling, whereas its overexpression enforced WNT signaling, cell growth and survival in serum-free media. RBM5-AS1 has been little characterized previously and we determined it to be a nuclear-retained transcript that selectively interacted with β-catenin. Mechanistic investigations showed that silencing or overexpression of RBM5-AS1 caused a respective loss or retention of β-catenin from TCF4 complexes bound to the WNT target genes SGK1, YAP1 and MYC. Our work suggests that RBM5-AS1 activity is critical for the functional enablement of colon cancer stem-like cells. Further, it defines the mechanism of action of RBM5-AS1 in the WNT pathway via physical interactions with β-catenin, helping organize transcriptional complexes that sustain colon CIC function.
Keywords: LUST, Colon cancer initiating cells, Long non-coding RNA, Wingless, β Catenin, C-MYC, RNA interactions
INTRODUCTION
Colorectal cancer (CRC) represents one of the most common tumor types and the third leading cause of cancer mortalities in the United States (1). The bulk of a tumor consists of rapidly proliferating, post-mitotic, differentiated cells and a very small population of Cancer Initiating Cells (CICs) or Cancer Stem Cells (CSCs). CICs/CSCs show a remarkable capacity for self-renewal and asymmetrical tumor cell growth and have been identified in several cancers, including human colon malignancies (2). Representative colon CICs (CCICs) surface markers and combined phenotypes have been used to isolate presumed CCIC populations, including the enrichment of CD133, CD44, CD24 and CD29 as cell surface antigens, and CD24+CD44+, EphB2high, EpCAMhigh/CD44+/CD166+, ALDH+, LGR5+, and CD44v6+ as phenotypic marks of stem cells (3). From a molecular perspective, a hallmark of CCICs is represented by a constitutively active Wingless (WNT)/β-catenin signaling pathway (4). Active WNT signaling is critical for intestinal stem cells, crypt cell proliferation and turnover; however, “stem-like” characteristics result from the convergence of cell-intrinsic features, extracellular signals, and stochastic events that continuously shape the self-renewing compartment (3). In particular, the epigenetic programming of transcription involves lncRNAs, in addition to chromatin proteins and DNA. LncRNAs have various roles (for review (5)) with previous reports demonstrating their direct involvement in regulating, as well as maintaining, pluripotent states at the chromatin level (6). Interestingly, the WNT-pathway can be modulated by lncRNAs in several tumor types (7). Within the context of a population of CCICs, we hypothesize that lncRNAs coordinate the chromatin architecture to include specific lncRNAs that facilitate re-programming of the epigenome, thereby, enabling the emergence of stem–like progenitors in colon cancer.
We identify the RBM5 anti-sense (RBM5-AS1) transcript or LUST (Luca-15 Specific Transcript), herein, referenced as LUST as a specific lncRNA elevated in CCICs population. The loss of LUST lncRNA results in the progressive differentiation of CCICs, whereas, ectopic expression corresponds with resistance to cellular differentiation and the stable maintenance of CICCs population. Moreover, LUST appears to selectively direct β-catenin transactivation via a TCF4 reporter system facilitating its capacity to target gene expression. Moreover, we find that LUST reinforces the chromatin association between β-catenin and TCF4 on specific targets CMYC, CCND1, SGK1 and YAP1 to provide a cell growth advantage, reflecting CCICs in vivo. These findings demonstrate that the LUST transcript is a novel lncRNA involving the recruitment and function of β-catenin in CCICs, and regulates WNT pathway by promoting “stemness” maintenance.
MATERIALS AND METHODS
Cells and reagents
Human LS174T (ATCC #CL-188), SW480 (ATCC #CCL-228), HT-29 (ATCC #HTB-38), CaCo-2 (ATCC #HTB-37), DLD-1 (ATCC #CCL-221) and HCT 116 (ATCC #CCL-247) colon cancer cell lines were purchased from the American Type Culture Collection (ATCC) between the years 2013–2014 and propagated and passaged as adherent cell cultures according to instructions provided by ATCC. For all cell lines the cells were received from ATCC as early passages and guidelines for authentication were followed as described previously (8). However, no additional steps to authenticate were taken. All documentation related to the cell lines obtained can be acquired through ATCC. Cells were maintained in adherent conditions, at 37°C in humidified atmosphere containing 5% CO2. The medium was changed twice a week, cells were passaged using 0.05% trypsin/EDTA (Corning) and preserved at early passages. Mycoplasma detection was routinely tested by qPCR methods (9).
Flow-cytometry analysis and cell sorting
HT-29, LS174T and SW480 colon cancer cells were stained using FITC-conjugated CD24, PE-coniugated CD166, APC-coniugated CD133 (BD Biosciences), and PE-Cy7-conjugated CD44 (BioLegend). Samples were analyzed on a BD LSRII flow-cytometer (Bekton Dikinson, Franklin Lakes, NJ, USA). Fluorescence-activated cell sorting of HT-29 cells was performed using BD FACSAria II (Bekton Dikinson). Analysis of cytometric data was performed using FACSDiva software (Bekton Dikinson) (see Supplemental Information).
In vitro Colonospheres formation assay
Spheres formed with colon carcinoma cells (HT-29, LS174T, SW480, DLD-1, HCT116) were obtained as previously described (4) with minor modifications provided in Supplemental Information.
RNA extraction, qRT-PCR and RNA-Seq
Total RNA was extracted from HT-29, CaCo-2, LS174T, SW480 and HT-29 derived colonospheres using Trizol and the RNeasy MiniKit (Qiagen) according to the manufacturer’s protocol. Reverse transcription was performed using PrimeScript RT Reagent kit (Takara #6130). Quantitative PCR (qRT-PCT) was performed using the GoTaq® qPCR Master Mix (Promega). Hypoxanthine-guanine phosphoribosyltransferase (HPRT) gene was used as housekeeping gene for normalization. Sequences of all the primers used for qRT-PCR are listed in (Table S2). RNA-Seq data sets from Illumina HiSeq2500 sequencing approaches have been deposited in the NCBI GEO under the accession number GSE69236, NCBI website: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=sbibkwgutdgzxyn&acc=GSE69236. Gene ontology analysis was performed as previously described (10)
LncRNA LUST knockdown and overexpression
LncRNA LUST knockdown was performed using LNA longRNA GapmeR (Exiqon, #300600). Four different probes directed against lncRNA LUST transcript and one unspecific Negative control probe were used. The construct pcDNA3-LUST was used and generated as previously described (11). The sequences of the oligonucleotides and their LNATM spiking patterns were designed using Exiqon’s GapmeR Design Algorithm: (http://www.exiqon.com/ls/Pages/GDTSequenceInput.aspx?SkipCheck=true).
TOPFlash dual luciferase assay
HT-29 cells were transiently transfected using Lipofectamine (Invitrogen) according to the manufacturer’s instructions, using 250ng of the TOPFlash reporter gene construct (M50 Super 8× TOPFlash, Plasmid #12456, Addgene) and 500ng of pcDNA3-LUST and/or 500ng of pcDNA-β-catenin construct. Luciferase reporter gene expression was measured according to the manufacturer’s protocol (Dual-luciferase Reporter assay System, Promega). The luciferase activity was normalized to Renilla luciferase activity from co-transfected internal control plasmid pRL-CMV.
Immunoblotting for proteins
For western blot analysis, 30µg of protein lysate were analyzed by SDS-PAGE, transferred to PVDF membranes (Bio-Rad) and blotted with indicated antibodies followed by ECL detection (Thermo Scientific).. Western blot assays were performed using the fallowing commercially available antibodies, at the indicated concentrations: anti–β-actin (Sigma, A5441, 1:1,000), anti-α-tubulin (Sigma, T5168, 1:1,000), anti-β-catenin (Bethyl Laboratories, A302-012A, 1:1,000), anti-active-β-catenin (Millipore, 05-665, 1:1,000), anti-cyclin D1 (CCND1, Abcam, ab16663, 1:1,000), anti-c-myc (Cell Signaling, 5605, 1:1,000).
Cell tansfections
For lncRNA LUST knockdown, cells were transfected using 300pmole of LNA GapmeRs and Lipofectamine (Invitrogen) according to the manufacturer’s protocol. For lncRNA LUST overexpression, pcDNA3 and pcDNA3-LUST construct were transiently transfected into HT-29 cells using 4µg of DNA. For colonospheres formation assay, HT-29, LS 174T, SW-480, DLD-1 and HCT116 cells were transiently transfected with pcDNA3 control or pcDNA-LUST and then seeded in a low-attachment plate
Nuclear fractionation and fluorescence in situ hybridization
Nuclear fractionation was performed as previously described (12). For lncRNA LUST localization, RNA-FISH was performed using customized probes purchased from Exiqon, following the manufacturer’s instructions for use.
Cross-linked RNA immunoprecipitation (RIP), Chromatin Immunoprecipitation (ChIP), Photoactivatable-Ribonucleoside-Enhanced Crosslinking and Immunoprecipitation (PAR-CLIP) and Sequential (-ChIP)
Essentially all experiments performed in this section have been previously described (10,13) and performed using 5µg of anti-β-catenin (Bethyl Laboratories, A302-012A) and 5µg of Rabbit Control IgG-ChIP Grade (Abcam, ab46540). ChIP was performed as previously described (14) using 6µg of anti-β-catenin (Bethyl Laboratories, A302-012A) and equal amount of Rabbit Control IgG-ChIP Grade (Abcam, ab46540). The Sequential-ChIP (or Re-ChIP) studies were performed as described (15). PAR-CLIP was performed as described in (10,16).
Mouse xenograft studies
2 × 106 HT-29 transfected cells were recovered, placed in 1 × PBS and inoculated by subcutaneous engraftment in the hind rump of male immunodeficient CB17-SCID mice (Charles River Laboratories) between 8–10 weeks of age. Tumor cell growth was established directly from HT-29 cells manipulated either by transfection with the expression vector for the LUST RNA transcript or by depleting LUST using locked nucleic RNAs (purchased through Exiqon). The growth of the tumors was followed by means of caliper measurements. Monitoring of tumor growth was performed at least twice per week using calipers. Details on analyses of tumor volume and survival are described within Supplemental Information.
Chromatin Isolation by RNA Purification (ChIRP)
ChIRP experiments were performed as previously described (17,18). 25 probes were generated against the RBM5-AS1 (LUST), RBM5-S1 (sense), and HOTAIR transcripts. Bound chromatin was detected by qPCR from either HOXD3, MYC 5′, MYC 3′, CCND1, SGK1 promoter-specific primers. Sequences of RBM5-AS1 and RBM5, and HOTAIR RNA probes are listed in Table S2.
Statistical Analysis
All experiments were performed in triplicate at least 3 times. All values were expressed as mean ± SEM. Statistical analysis was performed by the unpaired Student’s t test. A probability value of p ≤ 0.05 was considered statistically significant.
RESULTS
β-Catenin target genes are stimulated within HT-29 CCICs compared to their more differentiated counterparts
Here, we confirmed the expression of surface markers (2,19) on 3 different colon cancer cell lines chosen for their differentiated status: SW480 (poorly differentiated), HT-29 (moderately differentiated) and LS174T (well differentiated). Within the HT-29 cell line we identified two subpopulations co-expressing all four surface markers with signal intensity corresponding respectively as “bright” and “dim” (CD24, CD44, CD133 and CD166, Figure 1A). For LS174T and SW480 colon cancer cell lines, two different subpopulations could be discriminated as well, based on the signal intensity of CD24 and CD44 co-expression (bright and dim). However, the intensity of CD133 and CD166 antigens was lower compared to that of the HT-29 subpopulation (Figure 1A). To assess the ability of self-renewal of undifferentiated (CD24bright/CD44bright) versus differentiated (CD24dim/CD44dim) HT-29 cell subpopulations, we performed in vitro colon carcinoma spheres assay as previously described (2,19,20). Colon cancer progenitor cells can grow as spherical aggregates that, in the presence of serum or extracellular matrix, differentiate upon growth factor removal (21–23). Toward this aim, we first isolated HT-29 CD24bright/CD44bright (CCICs) and CD24dim/CD44dim (the differentiated counterpart) by Fluorescence-activated cell sorting (FACS) (Figure 1B) and then growth the sorted cells in suspension, in serum-free media, to induce spheres formation (19). After 3 weeks of culture, we obtained spheres formed as aggregates of exponentially growing undifferentiated cells CD24bright/CD44bright (CCICs) that were larger in terms of size and number compared to the ones obtained from the CD24dim/CD44dim colon cancer cells, as expected and previously reported (24) (Figure 1C). Based on WNT/β-catenin signaling transcripts as a prominent signature characteristic of some cancer stem cells (25), we performed RNA-Seq on CD24bright/CD44bright (CCICs) and CD24dim/CD44dim (a differentiated comparison) sorted subpopulations from HT-29 colon cancer cell line, and spheres derived from the CD24bright/CD44bright subpopulation, to explore pathways that are dys-regulated. Approximately, 3000 genes were elevated in CCICs, when compared to the differentiated population, indicated with a fold change >1.5 (Figure 1D, upper left panel). Analysis of the elevated genes identifies them among regulation of cell proliferation, cell migration and other biological processes characteristic of cancer cell phenotype (Figure 1D, lower left panel). Approximately 2000 genes resulted to be repressed between CD24bright/CD44bright and CD24dim/CD44dim subpopulations (Figure 1D, upper right panel), involved as well in regulation of cell proliferation or cellular metabolism (Figure 1D, lower right panel). We then analyzed the expression of genes defining colonic mucosa cells differentiation, such as MUCIN 2 (MUC2) or Keratin B20 (KB20/KRT80) that resulted down-regulated, or colon cancer stem cells markers, in particular CD24, CD44, CD166 and ALDH1A1 that were up-regulated (Figure 1E, shown as the green and red bars, respectively). Members of the canonical WNT-mediated signaling such as ASCL2, IGFBP2, LGR4, DKK1, MYCL1, FGFR2, SP5, MMP7 and the receptor EPHB3 were elevated in the CCICs compared to the more differentiated counterpart, with a fold increase >1.5 (Figure 1E–F, shown as the blue bars), as shown form the IGV tracks (Figure S1). RNA-Seq performed on spheres derived from CCICs was used to compare the expression of WNT-target genes and stem-like markers with the subpopulation (CD24bright/CD44bright) they were originated from. The expression of ASCL2, MYCL1, LGR4, EPHB3, CLDN1, CD44, CD24, ALDH1A1 and AKR1B10 was strongly increased during spheres formation. Genes involved in differentiation, i.e. Kruppel-like factor 9 (KLF9), KB20 and the β-catenin-regulated gene Peptidyl Arginine Deiminase, Type 1 (PADI1), were the most suppressed (Fold change >1.5) transcripts in both, spheres and the parental subpopulation, as well as the retinoic acid receptor responder protein 1 (RARRES1), known to have a tumor suppressor role (26). Intriguingly, two lncRNAs: KCNQ1OT1 and LUST were increased in this process, and the overexpression of LUST seemed to be more sphere’s specific (Figure 1G). Furthermore, when cells were induced to differentiate, the mRNA levels of CCDN1, LGR4 and CMYC were reduced (Figure 1H), showing that WNT-signaling is highly and selectively active during HT-29 cells spheres formation, and that the activity is reduced upon FBS-induced differentiation, confirming its role in maintenance of colon cancer cell spheres (27).
Figure 1. RNA-Seq analysis of CCICs and more differentiated counterpart in HT-29 cell line reveals deregulation of WNT-signaling and non-coding RNAs.
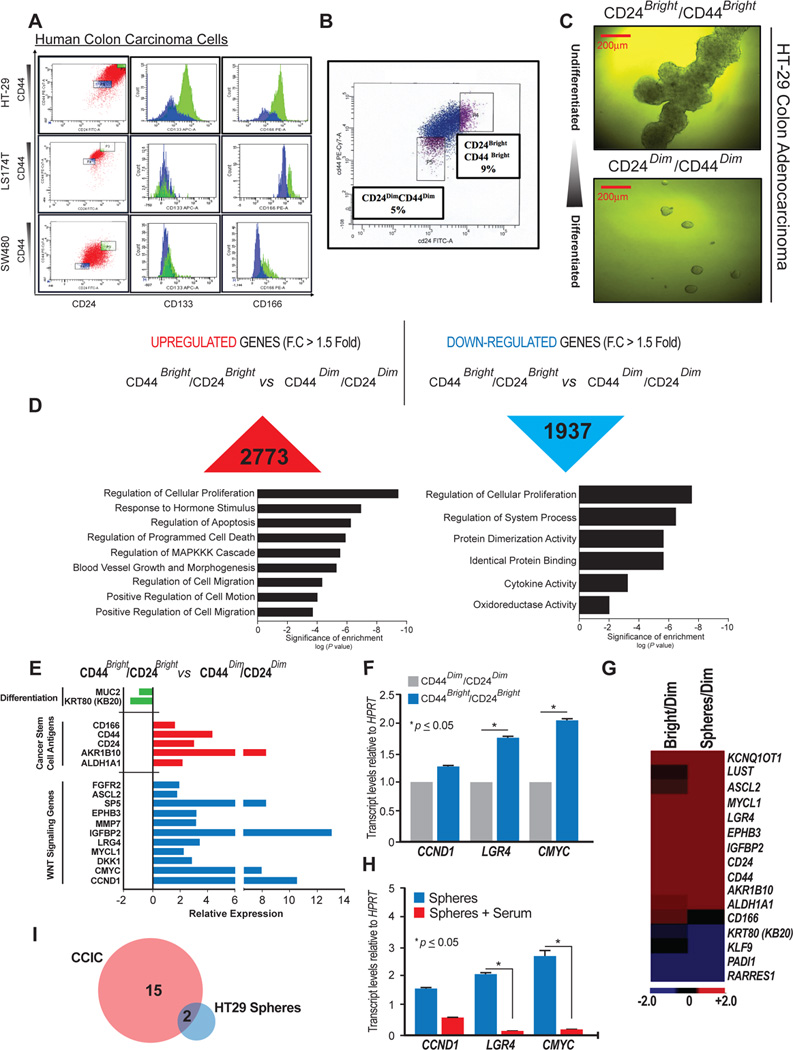
A) FACS analysis of HT-29, LS174T and SW480 colon cancer cells. Plots are gated on CD24bright CD44bright (CCICs) and CD24dim CD44dim cells (more differentiated cells). Histograms show CD133 and CD166 surface antigens expression in CCICs (green) and more differentiated cell (blue) subpopulations for the above cells lines. B) Sorting of CCICs and more differentiated cells from HT-29 colon cell line, plots are gated on CD24bright CD44bright and CD24dim CD44dim cells, percentage of sorted populations are shown. C) Spheres formed with colon carcinoma cells obtained from CD24bright CD44bright and CD24dim CD44dim sorted cells; D) (Upper panels): number of up-regulated (left) and down-regulated (right) genes with a >1.5 fold change between CD24Bright/CD44Bright and CD24Dim/CD44Dim subpopulations; (Lower panels): gene ontology analysis of up-regulated (left) and down-regulated genes (right); E) differential expression of genes between CD24Bright/CD44Bright and CD24Dim/CD44Dim subpopulations. Fold change >1.5 p<0.01; F) qRT-PCR validation of WNT-target genes G) heat map showing elevated (red) and repressed genes (blue) in CD24Bright/CD44Bright compared to CD24Dim/CD44Dim subpopulations, and in spheres formed with colon carcinoma cells compared to the CD24Dim/CD44Dim subpopulation. Fold change >1.5 p<0.01; H) qRT-PCR validation of WNT-target genes differentially expressed in spheres formed with colon carcinoma cells and FBS-induced differentiation cells. For qRT-PCR, HPRT was used as housekeeping control gene for normalization. Error bars indicate the standard error of the mean (SEM). *p<0.05; I) Venn Diagram showing overlapping elevated lncRNAs between CCICs and HT-29 derived spheres.
The lncRNA LUST transcript expression corresponds strongly with the stem-like capacity of CCICs
Recent data have shown that dys-regulation of coding as well as non-coding RNAs contribute to CCIC generation, reviewed in (28), and recently has been demonstrated that in cancer the WNT-signaling can be additionally regulated by lncRNAs through cell-autonomous mechanisms (29). In order to investigate the involvement of lncRNAs in colon cancer initiating cells maintenance and their role in the WNT-signaling regulation, we performed a lncRNAs profiler array analysis on CCICs and the more differentiated counterpart isolated from HT-29 colon cancer cells. Several lncRNAs were dramatically elevated in CCICs compared to their differentiated counterpart (Table S1), with an increase higher than 2.5 fold.
Among these lncRNAs, LUST, and KCNQ1OT1 were overexpressed, as confirmed by RNA-Seq also in spheres derived from HT29 cells (Figure 1G and I). To address the functional role of LUST in cancer stem cell maintenance, we analyzed by qRT-PCR the lncRNA and stem-like markers expression in spheres derived from CCICs and in FBS-induced differentiation cells. During the spheres formation process, the lncRNA LUST expression resulted strongly increased (Figure 2A, left). Of note, the increase appears to be significant already after 14 days in culture, when the stem-like markers CD24 and CD44 mRNA levels (Figure 2A, middle and right) are not elevated yet, and reaches a 10 fold change increase when cells are grown for 5 weeks in the same conditions (Figure 2A, left). This result suggests that LUST up-regulation could be an early event in the sphere formation process. As expected, when differentiation of the spheres is induced by adding FBS for additional 48h (20), the mRNA expression levels of conventional cancer stem cells markers, CD24 and CD44, is abrogated (Figure 2A, middle and right panel), confirming the loss of the stem-like potential. Interestingly, the expression level of the lncRNA LUST drastically decreased (Figure 2A, left panel) and the loss of markers CD24 and CD44, shown at the protein level (Figure 2B, blue population), strongly correlates with the switch from spheroid toward more adherent cell morphology (Figure 2C, upper and lower respectively).
Figure 2. The lncRNA LUST overexpression is an early event in the spheres formation process.
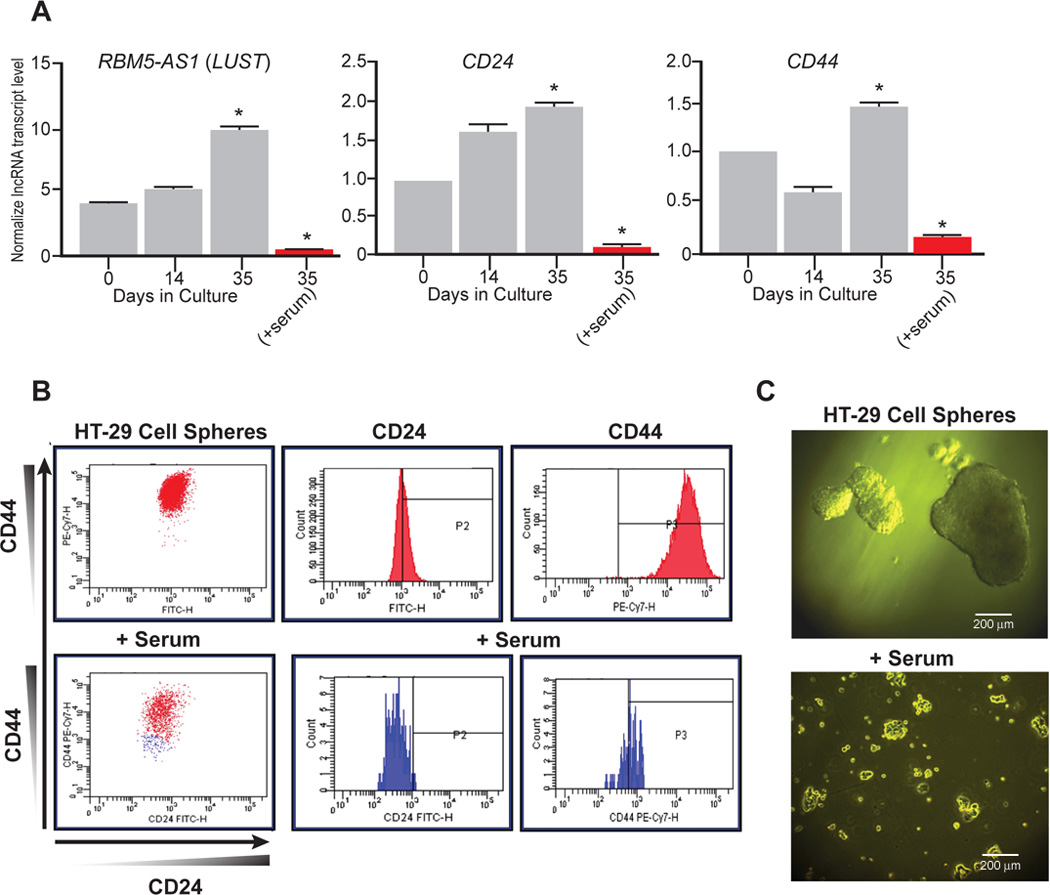
A) LUST, CD24 and CD44 mRNA levels measured by qRT-PCR in HT-29 derived spheres formed with colon carcinoma cells, respectively after 14 and 35 days cultured in ultra-low attachment conditions and in FBS-induced differentiation cells. HPRT transcript was used as housekeeping control gene for normalization. Error bars indicate the standard error of the mean (SEM). *p≤0.05; B) FACS analysis of spheres formed with colon adenocarcinoma cell lines (upper panels) and FBS-induced differentiated cells (lower panels) showing corresponding loss of expression of stem-like markers (blue population). Histograms report the mean fluorescence intensity (MFI, range 0–10,000) of CD24 (FITC) and CD44 (PE-Cy7); C) Representative images of HT-29-derived spheres formed with colon adenocarcinoma cells with characteristic spheroid morphology (top) and differentiated spheres showing adherent morphology upon FBS-induced differentiation (bottom).
WNT-signaling activation is impaired upon LUST knock-down
We performed loss-of-function assays in HT-29 cells by using two distinct locked nucleic acid (LNA) RNA GapmeRs that knocked down the expression of LUST to ~60 and ~50% respectively, compared to the control unspecific probe (Figure 3A), and analyzed the expression of WNT-signaling target genes by RT-qPCR. While control cells retained the expression of WNT-target genes, a strong reduction of mRNA transcripts such as AXIN2, CCND1, CD44, and TCF4 as well as CD24 mRNA levels occurs upon LUST knockdown (Figure 3B). A reduction at the protein level was seen for CCND1 and C-MYC (Figure 3C) and, of note, LUST knockdown caused also a reduction of active β-catenin (Figure 3C).
Figure 3. The lncRNA LUST is required for WNT-signaling target genes activation.
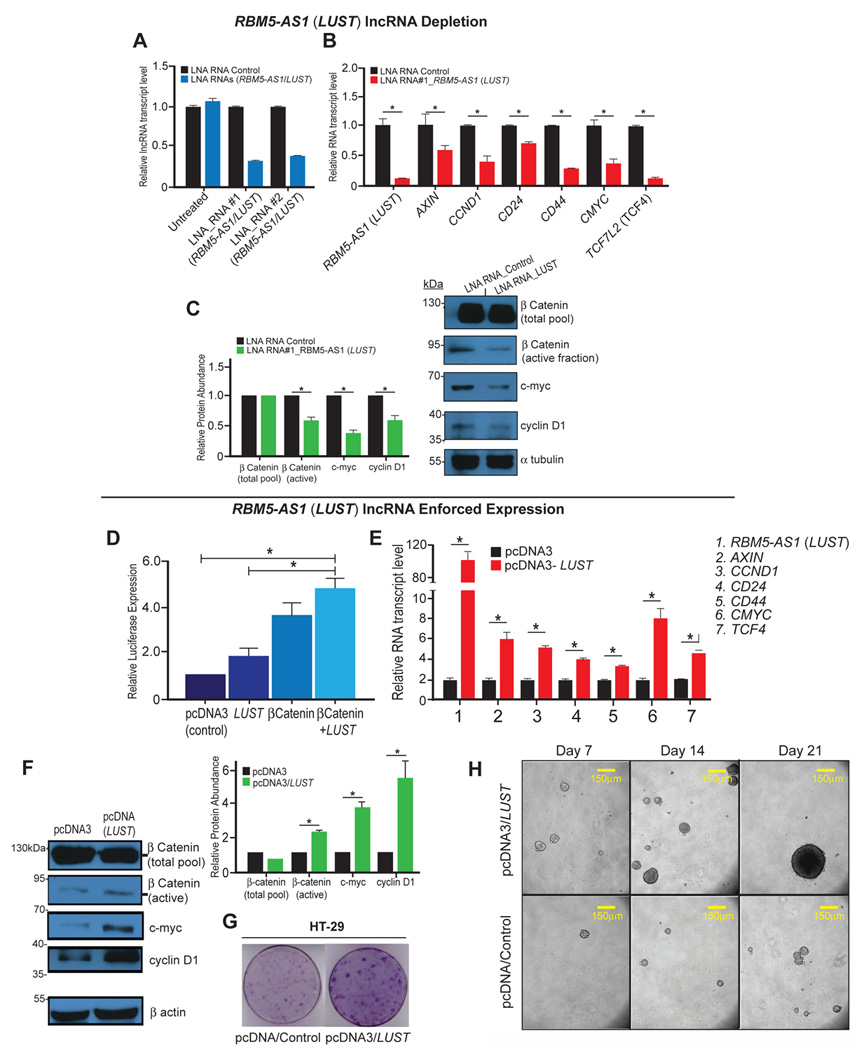
A) Knockdown of the lncRNA LUST using two different Anti-sense oligo probes (LNA RNAs). An unspecific probe was used as negative control (LNA RNA Control); B) Relative mRNA levels of WNT-target genes upon LUST knockdown, analyzed by qRT-PCR. C) WB analysis (right) and Relative Protein Abundance (left) of total β-catenin, active β-catenin, c-Myc and Cyclin D1 in control cells and in LUST knockdown cells, α-tubulin was used as a loading control. D) Relative luciferase activity measured in TOPFlash-HT-29 cells transiently transfected with pcDNA3-LUST, pcDNA3-β-catenin, or pcDNA3-LUST and pcDNA3-β-catenin; E) relative mRNA levels of WNT-target genes upon LUST overexpression analyzed by qRT-PCR; F) WB analysis (left) and Relative Protein Abundance (right) of total β-catenin, active β-catenin, c-Myc and Cyclin D1 in pcDNA3 control cells and in LUST overexpressing cells; β-actin was used as a loading control For qRT-PCR experiments HPRT was used as housekeeping control gene for normalization. Error bars indicate the standard error of the mean (SEM). *p≤0.05. G) Soft agar colony formation assay for HT-29 pcDNA3 control and LUST overexpressing cells H), Representative images of spheres formed with colon adenocarcinoma cells derived from pcDNA3, and pcDNA3-LUST transfected cells. Cells were cultured in ultra-low attachment conditions, in serum free media for 7, 14 and 21 days.
LUST overexpression potentiates WNT/β-catenin signaling cascade and accelerates spheroids formation across multiple colon carcinoma cell lines
We then performed dual luciferase assay using TOPFlash promoter (a reporter plasmid containing multiple copies of wild-type Tcf4-binding sites), transiently integrated in HT-29 cells (TOPFlash-HT-29 cells). Relative luciferase activity was measured in TOPFlash-HT-29 cells transfected with pcDNA3-LUST, pcDNA3-β-catenin, or pcDNA3-LUST and pcDNA3-β-catenin. The lncRNA LUST alone as well as β-catenin, were able to activate TCF reporter and induce luciferase activity. Interestingly, the co-transfection of pcDNA3-LUST and pcDNA3-β-catenin induced a significant strong increase in luciferase activity compared to pcDNA3-LUST and pcDNA3-β-catenin alone. These results demonstrated that the lncRNA LUST and β-catenin synergistically activate WNT/β-catenin signaling (Figure 3D). To further evaluate the LUST-mediated regulation of WNT-signaling in colon cancer, we analyzed the mRNA levels of WNT-signaling target genes upon LUST overexpression. pcDNA3 and pcDNA3-LUST constructs were transiently transfected into HT-29 cells and RNA samples were analyzed through qRT-PCR. The expression of AXIN2, CCND1, MYC, TCF4 and of the stemness markers CD24 and CD44 was significantly elevated under these conditions (Figure 3E). Although total β-catenin protein level remains stable, we verified a strong increase of active β-catenin, MYC and CCND1 expression, (Figure 3F), showing that the increase is induced at both, mRNA and protein levels and therefore demonstrating that LUST enhances WNT-signaling activation. Moreover, transient overexpression of LUST in HT29 cells corresponds with more profound colony formation when evaluated by crystal violet on soft agar (Figure 3G). To investigate a key role of LUST in tumor initiation, we performed in vitro spheres formation assay from HT-29 cells, and showed that LUST overexpression promotes an earlier onset of spheroids compared to the control. In fact, HT-29 LUST overexpressing cells are able to form spheres within 7 days in serum-free media culture conditions and the full spherical morphology is reached within 14 days. Instead, HT-29 cells transfected with empty vector required a longer timeframe corresponding to 21 days (Figure 3H).
We extended our study using four additional human colon cancer cell lines, to further validate the role of LUST as a colon cancer stem cells regulator. Exogenous LUST overexpression in LS174T and SW-480 (both APCwild-type/CTNNB1mutant) and in DLD-1 and HT116 (both APCmutant/CTNNB1wild-type), confirmed the mRNA levels induction of CCND1, CD44, C-MYC and TCF4, as well as the protein level increases of c-MYC, CCND1 and active β-catenin (Figure 4A–B). Even in this case, we could notice an earlier onset of spheroids formation after only 14 days in culture (Figure 4C–E–F–G). These results collectively demonstrate that LUST is a lncRNA transcript that we consider as a key component in regulating CCICs spheroid formation.
Figure 4. LUST expression promotes spheroid formation across multiple human colon adenocarcinoma cell lines.
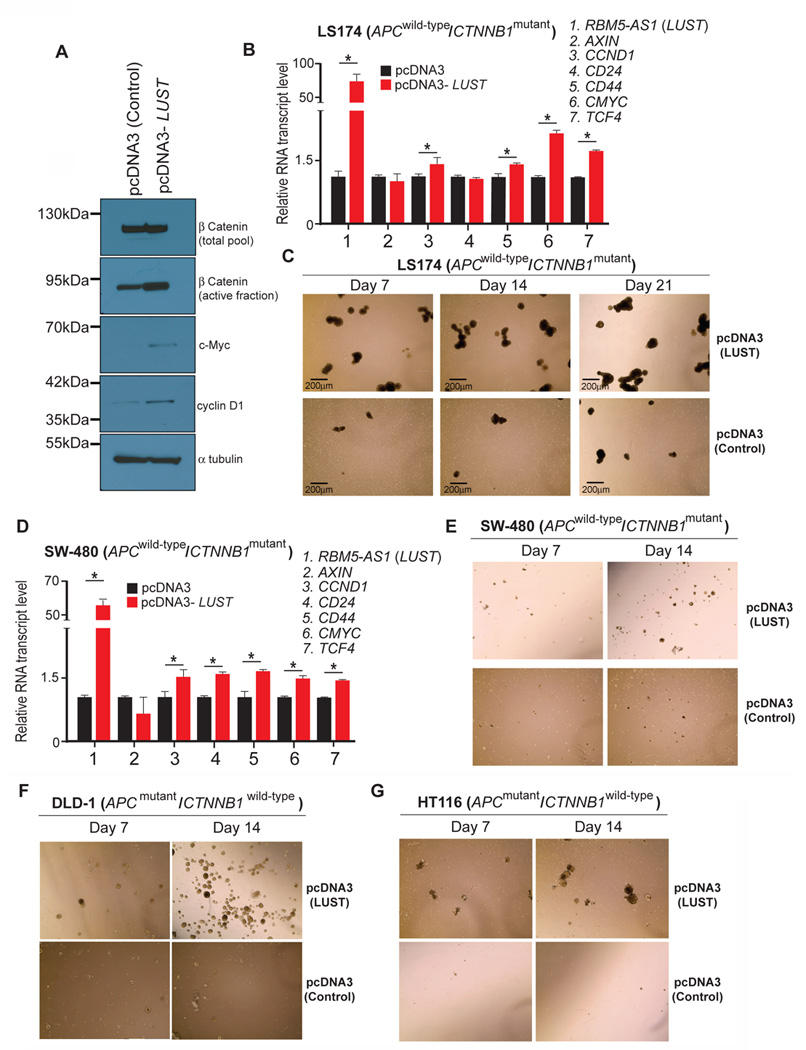
A) WB analysis of total cellularβ-catenin, active β-catenin, c-Myc and Cyclin D1 in LS174T control cells and LUST overexpressing cells, α-tubulin was used as a loading control. B) qRT-PCR validation of WNT-target genes expression upon LUST overexpression, C) Representative images of colonospheres formed with colon cancer cells derived from pcDNA3, and pcDNA3-LUST transfected cells. D) Relative mRNA levels of WNT-target genes upon LUST overexpression in SW-480 colon cancer cells, analyzed by qRT-PCR. E–G), Representative images of spheres formed respectively with SW-480, DLD-1 and HT116 colon cancer cells, derived from pcDNA3 and pcDNA3-LUST transfected cells. Cells were cultured in “ultra-low” attachment conditions, in serum free media for 7, 14 and 21 days. For qRT-PCR experiments HPRT was used as housekeeping control gene for normalization. Error bars indicate the standard error of the mean (SEM). *p≤0.05.
LUST is a nuclear-retained lncRNA that directly interacts with β-catenin and enhances occupation of β-catenin at promoters associated with WNT-signaling target genes
LUST lncRNA localization resulted to be mostly nuclear (Figure 5A), and RNA-FISH performed in HT-29 cells transiently transfected with pcDNA3 or pcDNA3-LUST confirmed the nuclear expression of the transcript (magenta color) (Figure 5B). A role for lncRNAs with WNT/β-catenin signaling in cancer has been indicated (29,30). Therefore, we analyzed the role played by LUST in the regulation of β-catenin activity in cancer initiating cells, by performing RNA Immunoprecipitation (RIP). Results demonstrated that the lncRNA LUST strongly binds to β-catenin (Figure 5C and Figure S2). To determine the activity of the lncRNA as a transcriptional regulator of WNT-signaling, we performed β-catenin ChIP experiments in HT-29 LUST overexpressing and control cells. Analysis of ChIP-qPCR indicates that LUST induces the enrichment of β-catenin at the promoters of WNT-signaling target genes as MYC, CXXC, YAP1, HDAC4 and SGK1, previously shown to be β-catenin targets (31) (Figure 5D). For additional evidence where the binding of LUST with β-catenin could be detected, we performed RIP assay (32) in CaCo-2 cells that harbor both, APC and β-catenin mutations (33). PAR-CLIP assays, performed in CaCo-2 cells, validated that the binding of β-catenin to the lncRNA is selective when the enrichment is compared with other known RNA-protein interactions of EZH2 and CBX7 (Figure S2). To assess RNA binding affinities we examined the binding of nuclear proteins isolated from CaCo-2 cells to biotinylated RNA probe representing the LUST transcript, and show from the shifted band, that the RNA-nuclear protein complex involves β-catenin and TCF4 (Figure S3A). Moreover, we show that affinity purified β-catenin protein selectively binds the LUST transcript and as a control fails to bind the sense RBM5 transcript (Figure S3B). These results suggest that the lncRNA LUST play a role in CICC maintenance by regulating the WNT signaling pathway, hence we sought to investigate an additional role as a prognostic marker in colorectal cancer. We compared survival from 187 patients with a graded stage III or greater of colorectal cancer progression following resection, obtained from The Cancer Genome Atlas (TCGA) database (34). We indicate that LUST and AKR1B10 transcript abundance corresponds with a poorer survival outcome (35) in patients following surgical resection, since the expression of AKR1B10, corresponds with mortality in patients with colon adenocarcinoma. Weaker outcomes in survival were shown when the expression of either LUST or AKR1B10 transcripts were above the median from all patients from TCGA datasets, recorded following surgical resection (Figure S4) using algorithms previously described (34,36). Therefore, to validate the role of LUST in directing tumor cell fate we generated a xenograft mouse model using HT-29 cells that were depleted of LUST (LNA_RMB5-AS1), HT-29 cells that were transiently overexpressing (xEx_RBM5-AS1), and relative control (LNA_Control). We find a strong link of RBM5-AS1 expression with HT-29 cell tumor growth from xenograft implants into immunocompromised mice, in both volume and survival outcomes. This suggest that, in vivo, LUST expression in HT-29 cells leads to increased tumor cell growth, whereas, depletion corresponds with tumor growth loss and better survival rate (Figure 5G).
Figure 5. The nuclear lncRNA LUST recruits β-catenin promoting WNT-signaling activation.
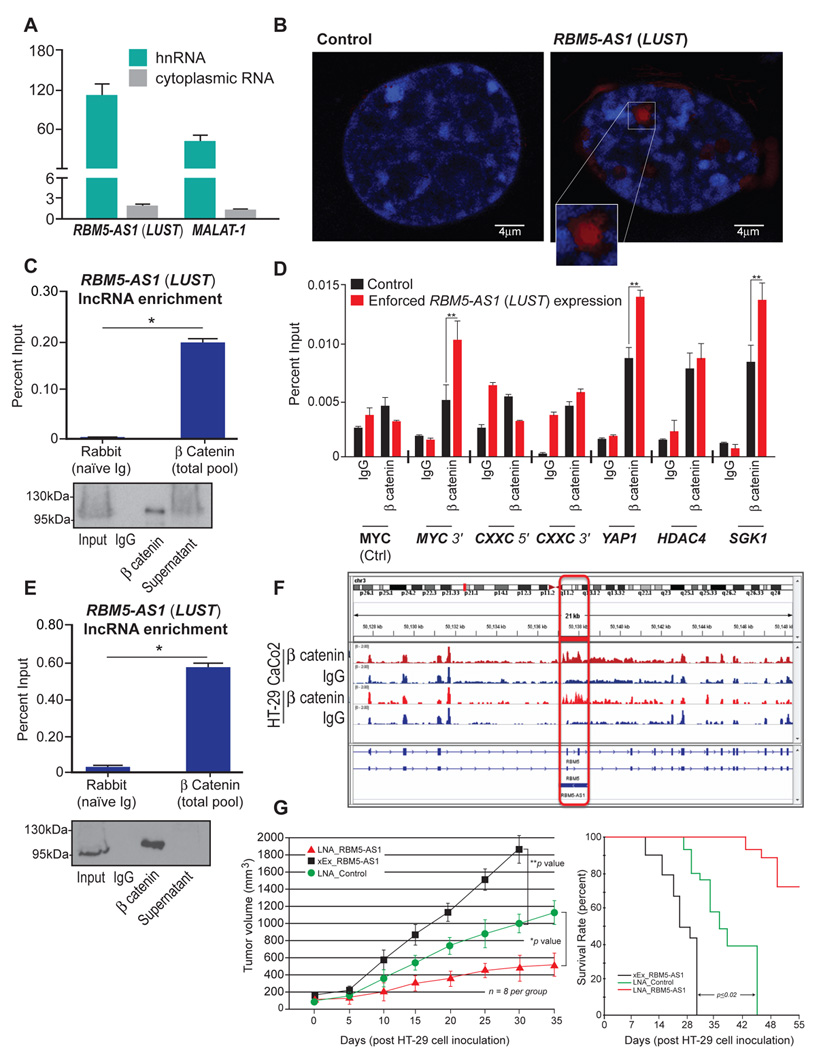
A) Relative subcellular abundance of LUST transcript determined by qRT-PCR; MALAT-1 was used as positive control for nuclear localization, HPRT was used as housekeeping control gene for normalization; B) Single molecule localization of LUST in HT-29 pcDNA3 (left) or pcDNA3-LUST (right) cells. Nuclei are stained with DAPI (blue), lncRNA LUST was detected using LNA double-DIG mRNA probe (red). C) RNA-Immunoprecipitation (RIP) assay showing the physical association between LUST and β-catenin in HT-29 cells. β-catenin was immuno-precipitated from nuclear extracts of formaldehyde-crosslinked HT-29 cells and associated RNA was detected by qRT-PCR. Enrichment of LUST binding to β-catenin is shown as % input; RIP-WB showing β-catenin immunoprecipitation; D) ChIP-RT-qPCR analysis of β-catenin binding at the MYC, CXXC, YAP1, HDAC4 and SGK1 loci. Enrichment of β-catenin is shown as % input; E) RIP assay showing the physical association between LUST and β-catenin in CaCo-2 cells. Enrichment of LUST binding to β-catenin is shown as % input; RIP-WB showing β-catenin immunoprecipitation. (C–E) IgG was used as negative control. Error bars indicate the standard error of the mean (SEM)*p<0.05; F) IGV tracks of LUST lncRNA bound to β-catenin recovered after immunoprecipitation. Peaks respectively correspond to β-catenin and Input in CaCo-2 and HT-29 colon cancer cell lines. G) Tumor progression and sizes (left) were evaluated following the orthotopically-introduced HT-29 cells, upon either LUST overexpression or by LNA depletion, into male CB17-SCID mice. Mice were followed over a period of 35 days (n=12 for each group) for tumor volume and for 55 days for overall survival. Tumor volume was monitored by using calipers during tumor progression and measured from the mice three times weekly.
LUST targets CMYC, CCND1 and SGK1 by reinforcing TCF4:β–catenin interactions
Finally, to assess the mechanism by which LUST transcript can target chromatin, we generated biotin-tagged oligonucleotide probes that were tiled across the RBM5-AS1 transcript and performed chromatin isolation by RNA purification (ChIRP) as previously described (17,18). Subsequent quantification using qPCR determined that the LUST transcript is capable of selectively target the MYC promoter at the 3′ end, as well as CCND1 and SGK1 promoters. As expected, RBM5, as a sense transcript, showed no enrichment at the mentioned regions. HOXD3-4 genomic site, known to be HOTAIR target site (37) was used as negative control (Figure 6A). Furthermore, we verified the selectivity of our ChRIP assay by testing the enrichment of RNA-bound chromatin for β–catenin/TCF4 target loci through qPCR. Specifically, we examined CCND1, SGK1, and YAP1 loci, based on the ChIP evidence we provided from the CCIC population previously characterized in Figures 1 and 2. Results now demonstrate how the LUST transcript targets and modulates the chromatin context surrounding specific β-catenin targets such as CMYC, CCND1, YAP1 and SGK1 (Figure 6B).
Figure 6. The RBM5-AS1 (LUST) transcript reinforces chromatin-associated β-catenin to TCF4 in HT-29 colon carcinoma cells.
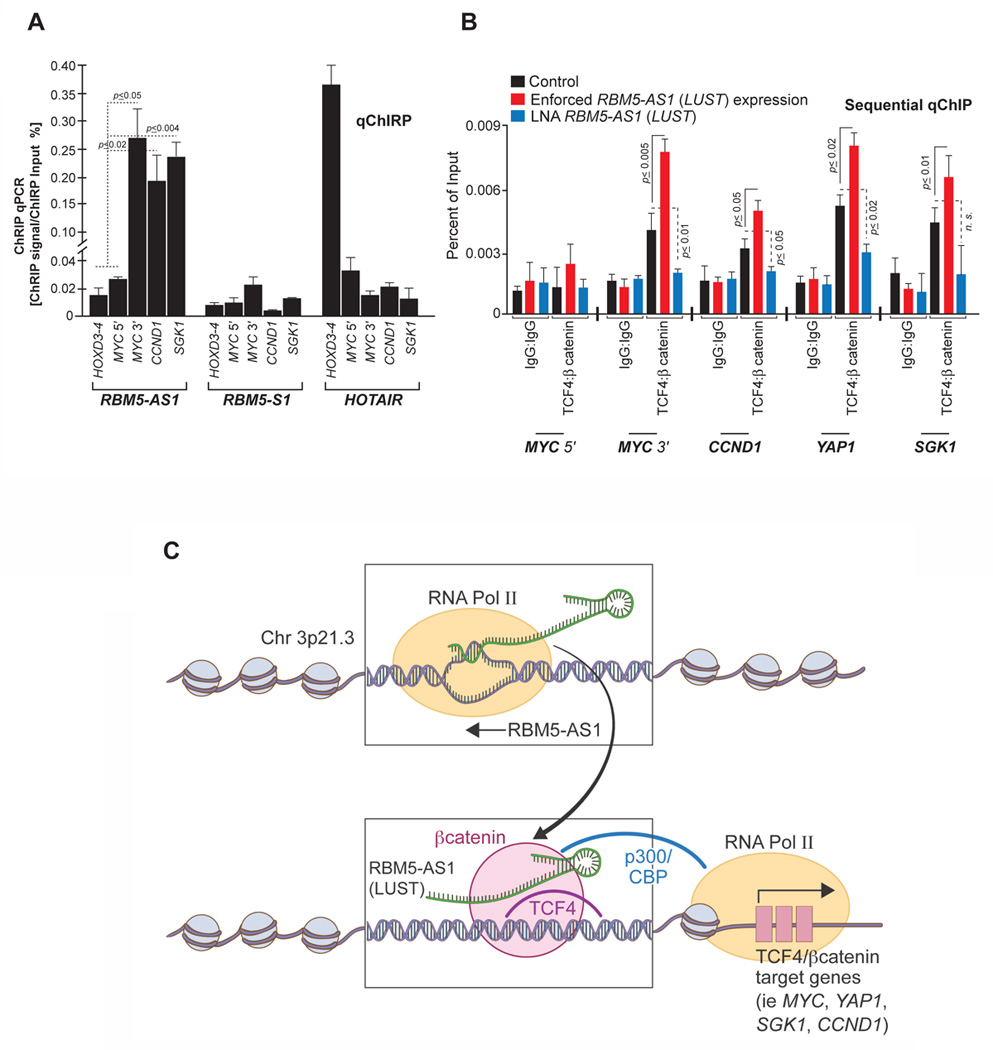
A) RBM5-AS1, RBM5-S1 and HOTAIR Chromatin isolation by RNA purification (ChIRP)-qPCR in HT-29 cells. HOXD3-4 and MYC 5’ are used as negative controls. B) Sequential ChIP (Re-ChIP) was performed to independently and sequentially select for interactions between TCF4 and β-catenin on the chromatin targets indicated. Comparisons were made between HT-29 cells transfected with pcDNA3 control, pcDNA3-LUST or LNA RNAs against RBM5-AS1 (LUST). Naïve mouse and rabbit IgG were used as negative controls. C) Model schematic depicting role of the LUST lncRNA to facilitate expression of WNT signal-induced transcription.
DISCUSSION
In this study, we isolated by FACS the CD24brightCD44bright and the CD24dimCD44dim subpopulations from HT-29 colon cancer cell line and identified them respectively as CCICs and more differentiated counterpart. Through RNA sequencing analysis we identified several coding and long non-coding RNA (lncRNA) transcripts differentially expressed and confirmed a dys-regulation of WNT-signaling in CCICs (4). LncRNAs can regulate gene expression by diverse mechanisms (38) and their involvement in colorectal cancer has been demonstrated (39). We defined LUST, a lncRNA, among the most highly express genes in CCICs, as responsible of promoting CCICs self-renewal. Here, we show a function of LUST as a co-transcriptional activator of the WNT signaling by facilitating β-catenin binding to the TCF4 transcription factor. The interaction between β-catenin and LUST amplifies the signaling to maintain self-renewal of CCICs. Constitutive activation of WNT signaling is a hallmark of CCICs as indicated previously (4). Therefore, we now define LUST as a transcriptional regulator of WNT-signaling during the process of spheroids formation. During this process, in fact, the mRNA expression levels of CD24 and CD44 increase, as evidence of cancer stem cell-like potential and enrichment of CCICs (27), while the increased expression of LUST, also confirmed by RNA-seq analysis, is further induced within only 14 days in culture, when the so-called “stemness” markers levels are not increased yet. This expression reaches a 10-fold change increase when cells are grown for five weeks in the same conditions, suggesting that LUST expression is an early event in the sphere formation process. The differentiation process induced in spheres derived from colon carcinoma cells (19,20) show that both mRNA and protein expression levels of the “stem-like” markers CD24 and CD44 is abrogated, confirming reduced CCIC-like potential. Interestingly, LUST expression strongly decreases upon induced differentiation, showing as evidence of its involvement with stem-like characteristics. Our loss-of-function assays demonstrate that LUST inhibition impairs the transcriptional activation of WNT-signaling target genes AXIN2, CCND1, MYC and of stemness markers CD24 and CD44, suggesting the loss of stem-like character; moreover the reduction of CCND1 and MYC gene product levels may indicate a reduction of cell proliferation (40,41). LUST knockdown reduces active β-catenin, confirming the decrease of WNT-signaling activation. The dual luciferase assay performed on LUST overexpressing cells, showed that the lncRNA alone as well as β-catenin, is able to activate the TCF-4 reporter mini-gene. The increased luciferase activity in cells co-transfected with pcDNA3-LUST and pcDNA3-β-catenin, instead, demonstrates that the LUST transcript and β-catenin coordinately regulate WNT/β-catenin signaling. Ectopic expression of the transcript in HT-29 cells induces WNT-signaling target genes and stem-like markers transcriptional activation, and the increase is also reflected at the protein level. Moreover, LUST overexpression induces enrichment of β-catenin at the promoters of WNT-signaling target genes and facilitates an earlier spheres formation process in several colorectal cancer cell lines, further demonstrating the important role of LUST in CICCs maintenance by modulating the pathway activation. Nuclear/cytoplasmic RNA fractionation and RNA-FISH experiments revealed that LUST is a nuclear lncRNA, to conceptualize a transcriptional or chromatin-based role.
HT-29 colon cancer cells harbor an APC inactivating mutation (as a deletion of the carboxyl terminus at residue 1555) (42), inducing the cells to be inert to WNT ligands, but carry constitutively active β-catenin/TCF4 transcription (43). We anticipated that lncRNAs have a direct role during the spheres formation process in CCICs maintenance by regulating the WNT-mediated signaling. Previous studies reported that β-catenin could selectively bind RNA (44) (45). The strong enrichment of LUST binding to β-catenin suggests that LUST could bind to a mutated isoform of the same protein. CaCo-2 cells harbor both APC and β-catenin mutations (33), and we show that even in this case LUST binds to β-catenin. We demonstrated that LUST enforces the binding of β-catenin and TCF4 facilitating oncogenic transcription. Furthermore, xenograft experiments and survival data from human patients suggest a reliable role of this transcript in tumor initiation and growth. This is the first detailed characterization of LUST localization and role in CCICs self-renewal, and established a co-activating regulatory model whereby LUST function is critical for the transcriptional activation of the WNT-signaling targets (Figure 6C), essential to insure CCICs maintenance.
Supplementary Material
Acknowledgments
We thank Dr. A. Alonso of the Weill-Cornell College of Medicine’s Epigenomic Sequencing Core for support for RNA-Seq and RIP-Seq studies. We acknowledge expertise from Stephen Hearn from the CSHL for help conducting the RNA-FISH studies. We thank Jill Gregory for providing the model illustration.
Grant Support: Senior Scholar Award in Aging (AG-SS-2482-10) to M.J.W. from the Ellison Medical Foundation, awards 5RO1 CA154903 and 5RO1 HL103967 from the NIH to M.J.W.
Footnotes
*Conflict of Interest Statement: All the authors of this manuscript declare that they have no conflict of interest.
REFERENCES
- 1.Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA: a cancer journal for clinicians. 2014;64(2):104–117. doi: 10.3322/caac.21220. [DOI] [PubMed] [Google Scholar]
- 2.Yeung TM, Gandhi SC, Wilding JL, Muschel R, Bodmer WF. Cancer stem cells from colorectal cancer-derived cell lines. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(8):3722–3727. doi: 10.1073/pnas.0915135107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeuner A, Todaro M, Stassi G, De Maria R. Colorectal cancer stem cells: from the crypt to the clinic. Cell stem cell. 2014;15(6):692–705. doi: 10.1016/j.stem.2014.11.012. [DOI] [PubMed] [Google Scholar]
- 4.Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nature cell biology. 2010;12(5):468–476. doi: 10.1038/ncb2048. [DOI] [PubMed] [Google Scholar]
- 5.Shen XH, Qi P, Du X. Long non-coding RNAs in cancer invasion and metastasis. Mod Pathol. 2015;28(1):4–13. doi: 10.1038/modpathol.2014.75. [DOI] [PubMed] [Google Scholar]
- 6.Ghosal S, Das S, Chakrabarti J. Long noncoding RNAs: new players in the molecular mechanism for maintenance and differentiation of pluripotent stem cells. Stem cells and development. 2013;22(16):2240–2253. doi: 10.1089/scd.2013.0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herriges MJ, Swarr DT, Morley MP, Rathi KS, Peng T, Stewart KM, et al. Long noncoding RNAs are spatially correlated with transcription factors and regulate lung development. Genes & development. 2014;28(12):1363–1379. doi: 10.1101/gad.238782.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freedman LP, Gibson MC, Ethier SP, Soule HR, Neve RM, Reid YA. Reproducibility: changing the policies and culture of cell line authentication. Nat Methods. 2015;12(6):493–497. doi: 10.1038/nmeth.3403. [DOI] [PubMed] [Google Scholar]
- 9.Dobrovolny PL, Bess D. Optimized PCR-based detection of mycoplasma. J Vis Exp. 2011;(52) doi: 10.3791/3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aguilo F, Zhang F, Sancho A, Fidalgo M, Di Cecilia S, Vashisht A, et al. Coordination of m(6)A mRNA Methylation and Gene Transcription by ZFP217 Regulates Pluripotency and Reprogramming. Cell stem cell. 2015;17(6):689–704. doi: 10.1016/j.stem.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rintala-Maki ND, Sutherland LC. Identification and characterisation of a novel antisense non-coding RNA from the RBM5 gene locus. Gene. 2009;445(1–2):7–16. doi: 10.1016/j.gene.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 12.Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142(3):409–419. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moran VA, Niland CN, Khalil AM. Co-Immunoprecipitation of long noncoding RNAs. Methods in molecular biology (Clifton, NJ) 2012;925:219–228. doi: 10.1007/978-1-62703-011-3_15. [DOI] [PubMed] [Google Scholar]
- 14.Gomes AQ, Nolasco S, Soares H. Non-coding RNAs: multi-tasking molecules in the cell. Int J Mol Sci. 2013;14(8):16010–16039. doi: 10.3390/ijms140816010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeng L, Zhang Q, Li S, Plotnikov AN, Walsh MJ, Zhou MM. Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature. 2010;466(7303):258–262. doi: 10.1038/nature09139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, et al. PAR-CliP--a method to identify transcriptome-wide the binding sites of RNA binding proteins. J Vis Exp. 2010;(41) doi: 10.3791/2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell. 2011;44(4):667–678. doi: 10.1016/j.molcel.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chu C, Quinn J, Chang HY. Chromatin isolation by RNA purification (ChIRP) J Vis Exp. 2012;(61) doi: 10.3791/3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445(7123):111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 20.Ricci-Vitiani L, Fabrizi E, Palio E, De Maria R. Colon cancer stem cells. Journal of molecular medicine (Berlin, Germany) 2009;87(11):1097–1104. doi: 10.1007/s00109-009-0518-4. [DOI] [PubMed] [Google Scholar]
- 21.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 22.Vescovi AL, Parati EA, Gritti A, Poulin P, Ferrario M, Wanke E, et al. Isolation and cloning of multipotential stem cells from the embryonic human CNS and establishment of transplantable human neural stem cell lines by epigenetic stimulation. Experimental neurology. 1999;156(1):71–83. doi: 10.1006/exnr.1998.6998. [DOI] [PubMed] [Google Scholar]
- 23.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes & development. 2003;17(10):1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nature medicine. 2006;12(10):1167–1174. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- 25.Vermeulen L, Todaro M, de Sousa Mello F, Sprick MR, Kemper K, Perez Alea M, et al. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(36):13427–13432. doi: 10.1073/pnas.0805706105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jing C, El-Ghany MA, Beesley C, Foster CS, Rudland PS, Smith P, et al. Tazarotene-induced gene 1 (TIG1) expression in prostate carcinomas and its relationship to tumorigenicity. Journal of the National Cancer Institute. 2002;94(7):482–490. doi: 10.1093/jnci/94.7.482. [DOI] [PubMed] [Google Scholar]
- 27.Kanwar SS, Yu Y, Nautiyal J, Patel BB, Majumdar AP. The Wnt/beta-catenin pathway regulates growth and maintenance of colonospheres. Molecular cancer. 2010;9:212. doi: 10.1186/1476-4598-9-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eades G, Zhang YS, Li QL, Xia JX, Yao Y, Zhou Q. Long non-coding RNAs in stem cells and cancer. World journal of clinical oncology. 2014;5(2):134–141. doi: 10.5306/wjco.v5.i2.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Q, Jiang H, Ping C, Shen R, Liu T, Li J, et al. Exploring the Wnt Pathway-Associated LncRNAs and Genes Involved in Pancreatic Carcinogenesis Driven by Tp53 Mutation. Pharmaceutical research. 2015;32(3):793–805. doi: 10.1007/s11095-013-1269-z. [DOI] [PubMed] [Google Scholar]
- 30.Ji Q, Liu X, Fu X, Zhang L, Sui H, Zhou L, et al. Resveratrol inhibits invasion and metastasis of colorectal cancer cells via MALAT1 mediated Wnt/beta-catenin signal pathway. PloS one. 2013;8(11):e78700. doi: 10.1371/journal.pone.0078700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bottomly D, Kyler SL, McWeeney SK, Yochum GS. Identification of {beta}-catenin binding regions in colon cancer cells using ChIP-Seq. Nucleic Acids Res. 2010;38(17):5735–5745. doi: 10.1093/nar/gkq363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ule J, Jensen K, Mele A, Darnell RB. CLIP: a method for identifying protein-RNA interaction sites in living cells. Methods (San Diego, Calif) 2005;37(4):376–386. doi: 10.1016/j.ymeth.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 33.Ilyas M, Tomlinson IP, Rowan A, Pignatelli M, Bodmer WF. Beta-catenin mutations in cell lines established from human colorectal cancers. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(19):10330–10334. doi: 10.1073/pnas.94.19.10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cline MS, Craft B, Swatloski T, Goldman M, Ma S, Haussler D, et al. Exploring TCGA Pan-Cancer data at the UCSC Cancer Genomics Browser. Sci Rep. 2013;3:2652. doi: 10.1038/srep02652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsunaga T, Wada Y, Endo S, Soda M, El-Kabbani O, Hara A. Aldo-Keto Reductase 1B10 and Its Role in Proliferation Capacity of Drug-Resistant Cancers. Front Pharmacol. 2012;3:5. doi: 10.3389/fphar.2012.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang L, Feng Z, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26(1):136–138. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- 37.Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129(7):1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang G, Lu X, Yuan L. LncRNA: a link between RNA and cancer. Biochimica et biophysica acta. 2014;1839(11):1097–1109. doi: 10.1016/j.bbagrm.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 39.Nissan A, Stojadinovic A, Mitrani-Rosenbaum S, Halle D, Grinbaum R, Roistacher M, et al. Colon cancer associated transcript-1: a novel RNA expressed in malignant and pre-malignant human tissues. International journal of cancer Journal international du cancer. 2012;130(7):1598–1606. doi: 10.1002/ijc.26170. [DOI] [PubMed] [Google Scholar]
- 40.Brocardo MG, Borowiec JA, Henderson BR. Adenomatous polyposis coli protein regulates the cellular response to DNA replication stress. Int J Biochem Cell Biol. 2011;43(9):1354–1364. doi: 10.1016/j.biocel.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 41.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398(6726):422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 42.Hinoi T, Yamamoto H, Kishida M, Takada S, Kishida S, Kikuchi A. Complex formation of adenomatous polyposis coli gene product and axin facilitates glycogen synthase kinase-3 beta-dependent phosphorylation of beta-catenin and down-regulates beta-catenin. J Biol Chem. 2000;275(44):34399–34406. doi: 10.1074/jbc.M003997200. [DOI] [PubMed] [Google Scholar]
- 43.Scholer-Dahirel A, Schlabach MR, Loo A, Bagdasarian L, Meyer R, Guo R, et al. Maintenance of adenomatous polyposis coli (APC)-mutant colorectal cancer is dependent on Wnt/beta-catenin signaling. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(41):17135–17140. doi: 10.1073/pnas.1104182108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edwards TA, Pyle SE, Wharton RP, Aggarwal AK. Structure of Pumilio reveals similarity between RNA and peptide binding motifs. Cell. 2001;105(2):281–289. doi: 10.1016/s0092-8674(01)00318-x. [DOI] [PubMed] [Google Scholar]
- 45.Kim I, Kwak H, Lee HK, Hyun S, Jeong S. beta-Catenin recognizes a specific RNA motif in the cyclooxygenase-2 mRNA 3'-UTR and interacts with HuR in colon cancer cells. Nucleic Acids Res. 2012;40(14):6863–6872. doi: 10.1093/nar/gks331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.