

Abstract
The endocannabinoid (eCB) system has emerged as one of the most important mediators of physiological and pathological reward-related synaptic plasticity. eCBs are retrograde messengers that provide feedback inhibition, resulting in the suppression of neurotransmitter release at both excitatory and inhibitory synapses, and they serve a critical role in the spatiotemporal regulation of both short- and long-term synaptic plasticity that supports adaptive learning of reward-motivated behaviors. However, mechanisms of eCB-mediated synaptic plasticity in reward areas of the brain are impaired following exposure to drugs of abuse. Because of this, it is theorized that maladaptive eCB signaling may contribute to the development and maintenance of addiction-related behavior. Here we review various forms of eCB-mediated synaptic plasticity present in regions of the brain involved in reward and reinforcement and explore the potential physiological relevance of maladaptive eCB signaling to addiction vulnerability.
Keywords: addiction, cocaine, drugs of abuse, endocannabinoid, nucleus accumbens, plasticity, reward, THC, ventral tegmental nucleus
Introduction
Emerging work has identified endocannabinoid (eCB) signaling as an important mediator of synaptic plasticity in mesocorticolimbic and corticostriatal pathways involved in the control of motivated behavior (Melis et al., 2014; Parsons and Hurd, 2015). The eCB system exploits a retrograde signaling mechanism that results in the suppression of neurotransmitter release at both excitatory and inhibitory synapses with both short- and long-lasting effects. Unlike other neuromodulators, eCBs integrate chemical signals from diverse neurotransmitter systems (e.g., GABA, glutamate, dopamine, acetylcholine) with changes in neuronal excitability, and their signaling represents a fundamental mechanism whereby a cell can control the gain of input from its own afferents (Alger, 2002). Through such neuromodulatory functions, eCBs play a vital role in the spatiotemporal regulation of synaptic plasticity that supports adaptive learning of reward-motivated behaviors and maintenance of affective homeostasis. However, the integrity of the eCB system can be compromised by repeated exposure to exogenous cannabinoids and other drugs of abuse, and abnormal eCB signaling has been identified throughout brain reward regions in the pathogenesis of addiction-related behavior (Gerdeman et al., 2003; Sidhpura and Parsons, 2011; Melis et al., 2014; Covey et al., 2015; Parsons and Hurd, 2015). Here we review various forms of eCB-mediated synaptic plasticity in regions of the brain involved in reward and reinforcement and explore the functional significance of maladaptive eCB signaling in drug-motivated behavior.
Overview of the eCB system
The eCB system encompasses several G-protein-coupled receptors (GPCRs) and lipid signaling molecules as well as their biosynthetic and metabolic machinery. There are two classical cannabinoid receptors: the cannabinoid type 1 (CB1) (Devane et al., 1988) and cannabinoid type 2 (CB2) (Munro et al., 1993) receptors. The CB1 receptor is found predominantly in the presynaptic compartment of neurons throughout the CNS (Herkenham et al., 1990) and is the most abundant GPCR in the brain (Mechoulam and Parker, 2013). Consistent with its role in reward and cognition, its regions of highest density include the hippocampus, amygdala, PFC, NAc, and caudate-putamen (Fig. 1) (Herkenham et al., 1990). Although the CB2 receptor is expressed primarily by immune cells in the periphery, recent evidence demonstrates that it is also present in neurons and glia (Van Sickle et al., 2005; Atwood and Mackie, 2010) and may functionally modulate neurotransmission in brain reward regions (Zhang et al., 2014, 2016). Sharing 48% sequence homology, both cannabinoid receptors couple to inhibitory Gi/o proteins and have the ability to activate several signal transduction mechanisms to inhibit adenylate cyclase activity and calcium influx through N-, P/Q-, and L-type calcium channels (Mackie and Hille, 1992; Twitchell et al., 1997; Gebremedhin et al., 1999) as well as stimulate inward rectifying potassium channels and the MAP kinase pathway (Mackie et al., 1995; Howlett, 2005). Through activation of these intracellular signaling cascades, presynaptic CB1 receptors directly reduce the probability of neurotransmitter release at both excitatory and inhibitory synapses and influence synaptic plasticity mechanisms throughout the brain.
Figure 1.

Density of CB1 receptor distribution across brain reward areas. Presynaptic CB1 receptors are Gi/o-coupled metabotropic receptors that are located throughout reward regions of the brain with varying levels of expression. Distribution of other components of the eCB system, such as the eCB synthetic enzymes N-arachidonoyl-phosphatidylethanolamine (NPLD) (Egertová et al., 2008) and DAGL (Suárez et al., 2011), follow a similar pattern, and mechanisms of CB1 receptor-mediated synaptic plasticity have been measured in mesocorticolimbic and corticostriatal pathways crucially involved in the pathophysiology of addiction.
Unlike GPCRs of other neurotransmitter systems, cannabinoid receptors have more than one endogenous agonist. The best-characterized eCB ligands are arachidonylethanolamide (AEA) or anandamide (from the Sanskrit word ananda, meaning “bliss”) (Devane et al., 1992) and 2-arachidonylglycerol (2-AG) (Fig. 2) (Mechoulam et al., 1995; Sugiura et al., 1995). Both AEA and 2-AG exert agonist activity at CB1 and CB2 receptors (Pertwee, 2010). AEA binds the CB1 receptor with higher affinity than the CB2 receptor but exhibits low efficacy at both receptors. However, 2-AG binds the CB1 and CB2 receptors with similar affinity and demonstrates greater potency and efficacy than AEA at both.
Figure 2.
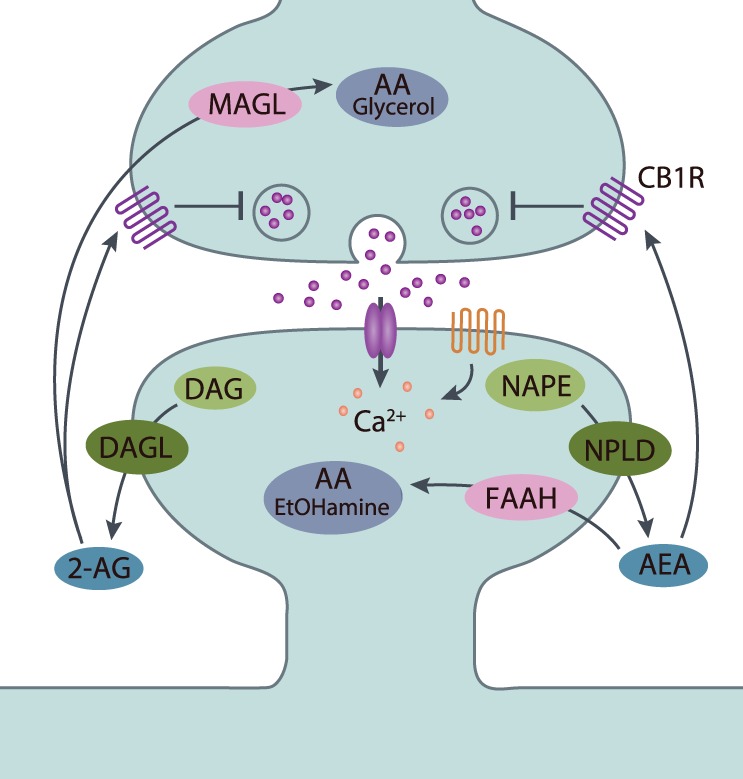
General mechanism of retrograde eCB signaling. Upon release of neurotransmitter (e.g., glutamate, GABA), postsynaptic depolarization results in elevations in intracellular calcium levels through activation of ionotropic receptors, Gq-coupled metabotropic receptors (e.g., Group I mGluRs, M1/M3 mAChRs, D2Rs), and/or voltage-gated calcium channels. The eCBs AEA and 2-AG are not stored in vesicles but instead are synthesized de novo from phospholipid precursors through calcium-dependent and -independent mechanisms. NAPE is hydrolyzed by N-arachidonoyl-phosphatidylethanolamine-specific phospholipase D (NPLD) to yield AEA, and DAG is converted to 2-AG by DAGL. Both eCB species traverse the synaptic cleft and activate presynaptic Gi/o-coupled CB1 receptors, thereby inhibiting adenylyl cyclase, regulating ion channels, and ultimately suppressing neurotransmitter release. eCB signaling is terminated following degradation by hydrolytic enzymes in the presynaptic and postsynaptic compartments. Primarily, AEA is converted to arachidonic acid (AA) and ethanolamine (EtOHamine) by fatty acid amide hydrolase (FAAH) localized to the postsynaptic cell, whereas 2-AG is hydrolyzed presynaptically into AA and glycerol by monacylglycerol lipase (MAGL).
Recent evidence also suggests that AEA and 2-AG are ligands of several other receptors that may be considered part of an “expanded” eCB system. Both molecules are functional agonists of the orphan GPCRs, GPR55 and GPR119 (Overton et al., 2006; Lauckner et al., 2008; Godlewski et al., 2009; Pertwee, 2010), and AEA in particular is an effective activator of the transient receptor potential vanilloid type 1 receptors (Zygmunt et al., 1999; Di Marzo and De Petrocellis, 2010), through which it may stimulate the presynaptic release of neurotransmitter (Musella et al., 2009). Therefore, eCBs may have significant impact on synaptic plasticity through additional mechanisms that are distinct from cannabinoid receptor-mediated signaling, and future work will be required to further characterize the influence of eCBs on these other signaling pathways during reward processing and motivated behavior.
In most instances, eCB ligands are generated and released on an ad hoc basis upon physiological or pathological neural activation of the postsynaptic neuron (Di Marzo et al., 1994; Cadas et al., 1996). As eCBs are membrane-diffusible lipid molecules, their physiochemical properties prohibit intracellular vesicle storage; therefore, they are synthesized “on demand” from cleavage of membrane phospholipids for immediate release. Activation of eCB biosynthetic machinery is generally calcium-dependent and occurs in response to sustained levels of neural stimulation (Freund et al., 2003), including depolarization as well as activation of ionotropic (Stella and Piomelli, 2001; Piomelli, 2003) or metabotropic (Giuffrida et al., 1999; Varma et al., 2001; Kim et al., 2002) receptors. Although several synthetic routes have been proposed (Sugiura et al., 1995; Leung et al., 2006; Liu et al., 2006; Simon and Cravatt, 2006), AEA is thought to primarily derive from the phospholipid precursor N-arachidonoyl-phosphatidylethanolamine (NAPE) that is released from the membrane by N-acyl-phosphatidylethanolamine-specific phospholipase D (NAPE-PLD) (Fig. 2) (Liu et al., 2006; but see Leung et al., 2006; Tsuboi et al., 2011). 2-AG, on the other hand, is synthesized from the hydrolytic metabolism of 1,2-diacylglycerol (DAG) by the sn-1-selective DAG lipases (DAGLs), DAGLα and DAGLβ (Bisogno et al., 2003; Jung et al., 2007; Murataeva et al., 2014; Shonesy et al., 2015).
eCB signaling is inactivated by cellular reuptake into both neurons and glia where eCBs are enzymatically degraded (Beltramo et al., 1997; Hillard and Jarrahian, 2000). While both AEA and 2-AG are synthesized and released from the postsynaptic compartment, there is spatial segregation of their catabolic enzymes (Fig. 2). AEA is hydrolyzed ultimately via fatty acid amide hydrolase located in the postsynaptic cell. In contrast, 2-AG undergoes predominant hydrolysis by monacylglycerol lipase (Blankman et al., 2007; Nomura et al., 2011; Chandra et al., 2013) located in the presynaptic cell (Gulyas et al., 2004; Ludányi et al., 2011), in addition to minor postsynaptic hydrolysis by α-β-hydrolase 6 (Marrs et al., 2010). This opposing organization of catabolic machinery is likely to underlie the differential physiological roles of AEA and 2-AG in eCB-mediated signaling (Gulyas et al., 2004; Kim and Alger, 2010; Sidhpura and Parsons, 2011). Although AEA can be released in an activity-dependent manner (Giuffrida et al., 1999), it is theorized to play a more general role in tonic eCB signaling at the CB1 receptor (Kim and Alger, 2010). The slow time course of AEA production (Giuffrida et al., 1999; Hohmann et al., 2005) and the location of its degradation machinery in the postsynaptic cell may help regulate interstitial levels of AEA (Sidhpura and Parsons, 2011). Furthermore, consistent with the presence of catabolic enzymes close to its site of action, 2-AG shapes phasic modulation of neurotransmission (Kim and Alger, 2010; Jung et al., 2012; Piomelli, 2014). Independent findings demonstrate a functional interaction between AEA and 2-AG (Maccarrone et al., 2008), and both eCB species can be recruited differentially from the same postsynaptic neuron with specific patterns of presynaptic activity (Puente et al., 2011; Lerner and Kreitzer, 2012). It is possible that each ligand participates in separate but overlapping forms of eCB-mediated synaptic plasticity. However, much is still unknown regarding the precise mechanisms underlying the production, transport, and degradation of eCBs, and additional work is essential for understanding their individual contributions to synaptic physiology.
Mechanisms of eCB-mediated synaptic plasticity
Short-term plasticity
Perhaps the best-studied forms of eCB-mediated plasticity are the transient or short-term mechanisms that result in brief (<1 min), stimulation-induced attenuation of neurotransmitter release during ongoing neurotransmission. These mechanisms have been termed depolarization-induced suppression of inhibition (DSI) or depolarization-induced suppression of excitation (DSE) at inhibitory (i.e., GABAergic) (Llano et al., 1991; Pitler and Alger, 1994; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001) or excitatory (i.e., glutamatergic) (Kreitzer and Regehr, 2001) synapses, respectively. These forms of eCB neuromodulation have been observed throughout brain regions critical to reward processing and the development of drug addiction, including the VTA (Melis et al., 2004; Riegel and Lupica, 2004), NAc (Hoffman and Lupica, 2001; Robbe et al., 2001), basolateral amygdala (Zhu and Lovinger, 2005), hippocampus (Isokawa and Alger, 2005), neocortex (Trettel and Levine, 2003; Bodor et al., 2005), and substantia nigra (Yanovsky et al., 2003). Induction of DSI/DSE requires specific patterns of afferent stimulation to depolarize the postsynaptic cell and activate voltage-gated calcium channels to elevate intracellular calcium levels and mobilize eCBs. Subsequently, eCBs activate CB1 receptors on the presynaptic cell and trigger intracellular signaling cascades that result in a decrement in neurotransmitter release for the length of CB1 receptor stimulation (Chevaleyre et al., 2006). Most evidence implicates 2-AG as the eCB species mediating DSI/DSE, as DSI is greatly reduced in DAGL knock-out mice (Gao et al., 2010), and inhibition of 2-AG clearance (Makara et al., 2005; Szabo et al., 2006; Pan et al., 2009), but not AEA clearance (Kim and Alger, 2004; Pan et al., 2009), prolongs both DSI and DSE.
An additional form of short-term eCB-mediated plasticity is driven by postsynaptic activation of Gq-coupled GPCRs (e.g., Group I mGluRs, M1/M3 mAChRs, and dopamine D2Rs) (Martin and Alger, 1999; Varma et al., 2001; Kim et al., 2002; Melis et al., 2004; Edwards et al., 2006; Uchigashima et al., 2007). Downstream of metabotropic receptor stimulation, 2-AG mobilization relies on phospholipase C and DAGL activation (Galante and Diana, 2004; Melis et al., 2004; Edwards et al., 2006) and, unlike DSI/DSE, is not calcium-dependent but can be potently augmented by coincident increases in intracellular calcium levels (Maejima et al., 2005; Hashimotodani et al., 2007; Kano et al., 2009). Through these short-term plasticity mechanisms, eCBs permit reversible and bidirectional modulation of synaptic strength at both excitatory and inhibitory synapses. This enables critical synaptic scaling and dynamic filtering of specific input frequencies during ongoing neural computation (Abbott et al., 1997; Abbott and Regehr, 2004; Klyachko and Stevens, 2006). Further, by markedly affecting the excitability of the postsynaptic cell (Wagner and Alger, 1996), eCB-mediated short-term synaptic transmission can influence the induction of long-term postsynaptic plasticity mechanisms (Carlson et al., 2002; Chevaleyre and Castillo, 2003, 2004; Zhu and Lovinger, 2007). Therefore, although transient, DSI/DSE and metabotropic receptor-mediated short-term plasticity can influence diverse neurotransmitter systems and represent important means by which eCBs make substantial contributions to network function.
Long-term plasticity
Transient eCB-mediated synaptic adaptations, such as DSI/DSE, persist for the duration of CB1 receptor activation, but eCBs can also establish changes in synaptic strength that are maintained beyond initial CB1 receptor stimulation (Chevaleyre et al., 2006). Sustained increases and decreases in synaptic strength that last for hours to weeks are referred to as LTP and LTD, respectively. eCB-mediated LTD (eCB-LTD) is the most well-characterized form of long-term presynaptic plasticity affected by eCBs. Although methods of induction at excitatory (eCB-LTDe) and inhibitory (eCB-LTDi) synapses differ somewhat by brain region, the predominant mechanism involves activity-dependent release of eCBs from the postsynaptic cell. eCB mobilization required for eCB-LTD is stimulated by neuronal depolarization or by activation of postsynaptic metabotropic receptors (Freund et al., 2003; Kano et al., 2009). Both high- and low-frequency patterns of neuronal stimulation elicit eCB release through calcium-dependent processes that do not rely on GPCRs (Calabresi et al., 2007; Heifets and Castillo, 2009; Lovinger, 2010). Instead, calcium influx through NMDA receptors and L-type and T-type voltage-gated calcium channels as well as calcium release from intracellular stores trigger postsynaptic eCB release (Beierlein and Regehr, 2006; Isokawa and Alger, 2006; Adermark and Lovinger, 2007; Ohno-Shosaku et al., 2007). In contrast, metabotropic receptor-mediated eCB mobilization is not calcium-dependent and elicits “on-demand” eCB mobilization through Gq (via Group I mGluRs, M1/M3 mAChRs, 5-HT2Rs, orexin receptors, and cholecystokinin receptors) or Gi/o (via D2Rs) protein signaling cascades (Chevaleyre et al., 2006; Heifets and Castillo, 2009).
In addition to activity-dependent release of eCBs, induction of eCB-LTD involves coincident presynaptic activity and prolonged CB1 receptor activation (Heifets and Castillo, 2009). The requirement for CB1 receptor binding at active synapses for generation of eCB-LTD imparts some degree of afferent specificity but also allows for both homosynaptic (i.e., target afferent) and heterosynaptic (i.e., nearby afferent) eCB-LTD. Furthermore, evidence suggests that the determining factor for the induction of DSI/DSE versus eCB-LTD may be the nominal time of CB1 receptor activation and subsequent differential recruitment of downstream effectors (Chevaleyre et al., 2006; Heifets and Castillo, 2009). Unlike DSI/DSE, eCB-LTD involves downregulation of the cAMP/PKA pathway, and this generates long-term interference with neurotransmitter release machinery (Lonart et al., 2003; Chevaleyre et al., 2007) and calcium influx via voltage-gated calcium channels (Robbe et al., 2002; Mato et al., 2005, 2008). Therefore, the extent of neural activity and eCB signaling within the local circuit may crucially determine whether CB1 receptor-mediated signaling results in brief or long-lasting depression of neurotransmission at the synapse. Long-term plasticity induced by eCB mechanisms has been measured in many brain regions, including those involved in addiction, such as the VTA, NAc, and others (Sidhpura and Parsons, 2011; Melis et al., 2014).
Effects of drugs of abuse on eCB-mediated synaptic plasticity in brain reward regions
VTA
The mesolimbic dopamine system is comprised of dopamine cells originating in the VTA of the midbrain that project rostrally to the NAc in the ventral forebrain and to the prefrontal cortex (Wise and Bozarth, 1985). A wide body of evidence supports a role for mesolimbic DA in reinforcement learning and motivation for incentive stimuli, such as reward-predictive cues (Wise and Rompre, 1989; Ikemoto, 2007; Berridge, 2009; Taber et al., 2012). Additionally, the rewarding properties of all known abused drugs are thought to arise, in part, from dopamine release in the NAc (Roberts et al., 1977; Wise and Bozarth, 1985; Di Chiara and Imperato, 1988; Volkow and Morales, 2015), and synaptic plasticity in the mesolimbic dopamine system is a critical target for drugs of abuse (Kauer, 2004). Importantly, administration of THC or cannabinoid agonists (French et al., 1997; Diana et al., 1998; Cheer et al., 2003) or cocaine (Sombers et al., 2009) increases the burst firing of VTA dopamine neurons as well as stimulates phasic dopamine release in the NAc in a CB1-dependent manner (Cheer et al., 2004, 2007; Wang et al., 2015). However, dopamine neurons do not express CB1 receptors (Herkenham et al., 1991; Julian et al., 2003), and current data support a CB1 receptor-mediated increase in dopamine neuron activity due to induction of local disinhibitory mechanisms, such as DSI (Lupica and Riegel, 2005) and eCB-LTDi (Pan et al., 2008a, b). During periods of burst firing, VTA dopamine neurons synthesize and release 2-AG predominantly onto presynaptic CB1-expressing GABAergic terminals originating from the globus pallidus, rostromedial tegmental nucleus, and local interneurons (Lecca et al., 2012). CB1 receptor binding transiently suppresses GABA-mediated inhibition and initiates a disinhibitory feedback loop that enhances dopamine cell firing (Melis et al., 2004; Riegel and Lupica, 2004; Lupica and Riegel, 2005; Alger and Kim, 2011). The long-term reduction in GABAergic inhibitory input to dopamine neurons via eCB-LTDi (Fig. 3a) also involves the coordination between D2 receptors and Group I metabotropic glutamate receptors (Pan et al., 2008a, b). Activation of postsynaptic mGluRs enhances the synthesis and release of eCBs and CB1 receptor activation, and presynaptic D2 receptor stimulation facilitates eCB-LTDi by augmenting the downstream effects of CB1 receptor signaling, such as inhibition of the cAMP/PKA pathway.
Figure 3.

Summary of drug-induced disruptions of eCB-mediated long-term plasticity in the VTA and NAc. a, b, Although the specific mechanism differs by brain region, eCB-LTD induction under normal conditions is primarily governed by activity-dependent release of eCBs from the postsynaptic cell and CB1 receptor stimulation on active afferents. a, Within the VTA, eCB-LTDi at GABAergic synapses permits adaptive dopamine cell firing, and induction involves additional coordination between presynaptic (e.g., D2 dopamine receptors) and postsynaptic (e.g., Group I mGluRs) metabotropic receptors. b, Control over accumbal glutamate release from cortical and limbic afferents through the induction of eCB-LTDe relies on activation of postsynaptic metabotropic receptors (e.g., mGluR5) and release of calcium from intracellular stores. c, d, Following exposure to drugs of abuse, such as THC or cocaine, parallel disturbances in eCB-LTD mechanisms that normally provide inhibitory control over VTA dopamine neuron activity and curb excitation of NAc MSNs instead promote activation of reward circuitry. c, Drug exposure facilitates the induction of eCB-LTDi in the VTA, removing GABA-mediated inhibition of dopamine neurons and enhancing their excitability. d, Drug-induced loss of eCB-LTDe at glutamatergic synapses in the NAc prevents control over excitation of MSNs.
Consistent with its role in promoting dopaminergic transmission (Cheer et al., 2007; Wang et al., 2015), cocaine interferes with both short-term (Lupica and Riegel, 2005; Wang et al., 2015) and long-term (Fig. 3a,c) (Liu et al., 2005; Pan et al., 2008a, b) eCB-mediated plasticity mechanisms in the VTA. Acute cocaine attenuates inhibition of dopamine neurons through facilitation of DSI at GABAergic synapses in the VTA (Wang et al., 2015). Specifically, cocaine mobilizes 2-AG via local inhibition of norepinephrine uptake and promotion of α1-adrenergic receptor stimulation of dopamine neurons. This results in a cocaine-mediated enhancement dopamine cell firing and dopamine release in the NAc.
Moreover, cocaine reduces inhibition of VTA dopamine neurons through facilitated induction of eCB-LTDi at GABAergic synapses (Liu et al., 2005; Pan et al., 2008a, b). In vivo cocaine exposure for 5–7 d occludes further induction of eCB-LTDi (Liu et al., 2005; Pan et al., 2008a, b), and cocaine's effects are blocked by pretreatment with D2, mGluR5, and CB1 receptor antagonists (Fourgeaud et al., 2004; Liu et al., 2005). Under many conditions, GABAergic inhibition suppresses LTP induction at excitatory synapses (Wigström and Gustafsson, 1983; Meredith et al., 2003). Accordingly, evidence suggests that cocaine-induced attenuation of LTD in GABAergic neurons in the VTA facilitates the induction of LTP at excitatory synapses (Liu et al., 2005). Therefore, through enhancement of eCB-LTDi induction, cocaine has the ability to increase the excitability of VTA dopamine neurons and may ultimately influence maladaptive drug-seeking behaviors. Other endogenous factors within the VTA, such as norepinephrine (Wang et al., 2015), brain-derived neurotrophic factor (Zhong et al., 2015), neurotensin-1 (Kortleven et al., 2012), and insulin (Labouèbe et al., 2013), also modulate eCB signaling and, in some cases, are involved in the effects of cocaine on dopamine neuron excitability (Wang et al., 2015; Zhong et al., 2015). Future work will determine the role these factors play in drug-mediated long-term plasticity mechanisms within the VTA.
NAc
The NAc is one of the major target regions of dopaminergic projections from the VTA as well as glutamatergic projections from cortical and limbic areas that are vital to the regulation of motivated behavior (Meredith et al., 2008; Sesack and Grace, 2010). Through convergent inputs from the VTA, PFC, amygdala, and hippocampus and output projections to motor regions, such as the ventral pallidum, the NAc enables adaptive behavioral responding to rewards and reward-predictive cues. Extensive work has identified long-term dysfunctional synaptic alterations in the NAc that promote vulnerability to relapse-related behavior (Gipson et al., 2014; Wolf, 2016). The majority of neurons (90%–95%) in the NAc are GABAergic medium spiny neurons (MSNs) that express either D1 or D2 dopamine receptors (Bock et al., 2013; MacAskill et al., 2014; Kupchik et al., 2015). Unlike projections of MSNs of the dorsal striatum, projections of D1- and D2-positive MSNs of the NAc do not segregate strictly via direct and indirect pathways to output nuclei (Kupchik et al., 2015). Instead, D1-positive MSNs project to the VTA and substantia nigra, whereas both D1- and D2-positive MSNs send projections to the ventral pallidum. Under many circumstances, these individual MSN populations are differentially impacted by exposure to drugs of abuse, and imbalance among their activity contributes to drug-induced behavior (Bock et al., 2013; MacAskill et al., 2014).
Like dopamine neurons of the VTA, MSNs participate in both short-term (Robbe et al., 2001) and long-term (Robbe et al., 2002) eCB-mediated synaptic plasticity (Fig. 3b). Although accumbal MSNs do not express CB1 receptors (Winters et al., 2012), CB1 receptors provide inhibitory control over local glutamate and GABA release in the NAc through their expression on cortical and limbic afferents as well as fast-spiking interneurons (Hoffman and Lupica, 2000, 2001; Robbe et al., 2001; Pistis et al., 2002; Winters et al., 2012). eCB-LTDe at glutamatergic synapses in the NAc is facilitated by postsynaptic activation of the Group I metabotropic receptor mGluR5 (Fig. 3b,d) (Robbe et al., 2001; Grueter et al., 2010) and release of calcium from intracellular ryanodine-sensitive stores (Robbe et al., 2001). Results suggest AEA as the eCB species mediating eCB-LTDe in the NAc, as preventing its degradation enhances LTD induction (Grueter et al., 2010). Moreover, accumbal eCB-LTDe manifests preferentially in D2-positive MSNs (Grueter et al., 2010). D2 receptor activation, however, is not necessary for eCB-LTDe induction in the accumbens (as opposed to the dorsal striatum) (Gerdeman et al., 2002) but can stimulate AEA production (Giuffrida et al., 1999). Thus, striatal dopamine and D2 receptors likely modulate fundamental eCB-LTDe induction mechanisms by facilitating eCB mobilization (Kreitzer and Malenka, 2005; Lerner and Kreitzer, 2012).
Studies from several laboratories have demonstrated disruption of eCB-LTDe in the NAc following exposure to drugs of abuse. Both single (Mato et al., 2004) and repeated (Hoffman et al., 2003; but see Mato et al., 2005) exposure to THC prevented eCB-LTDe in the NAc and reduced sensitivity to CB1 receptor agonists at the synapse (Hoffman et al., 2003), supporting pronounced cannabinoid-induced downregulation of the CB1 receptor. Additionally, accumbal eCB-LTDe was abolished 24 h after a single exposure to cocaine (Fourgeaud et al., 2004) and as long as 45 d after prior long-term cocaine self-administration (McCutcheon et al., 2011). However, in contrast to the effects of THC exposure, signaling at the CB1 receptor is not impaired following exposure to cocaine (Fourgeaud et al., 2004; McCutcheon et al., 2011). Instead, during extended withdrawal from cocaine self-administration, there is enhanced sensitivity to CB1 receptor agonists and an apparent uncoupling of mGluR5 with eCB synthetic machinery, suggesting a persistent attenuation of eCB tone (McCutcheon et al., 2011). Disabling of mGluR5-associated eCB-LTDe during protracted cocaine withdrawal is part of series of synaptic adaptations in the NAc that increase excitability of MSNs and promote susceptibility to relapse to drug-seeking behavior (McCutcheon et al., 2011; Wolf and Tseng, 2012; Wolf, 2016). More work will be needed to understand the full significance of drug-induced disruptions in accumbal eCB-LTD and motivated behavior, but present findings demonstrate significant abnormalities in eCB-mediated plasticity following exposure to drugs of abuse.
In conclusion, eCBs are key modulators of synaptic function and mediate mechanisms of both short- and long-term presynaptic plasticity in regions of the brain crucially involved in reward processing, such as the VTA and NAc. Accumulating evidence demonstrates perturbations in normal eCB-mediated synaptic plasticity in these brain areas following experience with drugs of abuse. Specifically, drug-induced disruptions in eCB plasticity result in concurrent loss of inhibitory control over dopamine neurons in the VTA and impairment in the regulation of excitatory signaling in the NAc. Together, these maladaptive synaptic alterations have the ability to potentiate reactivity of the mesocorticolimbic and corticostriatal pathways that are critical to drug-motivated behavior. Indeed, loss of accumbal eCB-LTD and related enhancement in excitability of MSNs are associated with heightened drug-seeking behavior (McCutcheon et al., 2011; Wolf, 2016). These findings suggest that therapeutic approaches aimed at restoring normal eCB-mediated synaptic plasticity might have significant impact on the treatment of addiction. Therefore, additional work is necessary to further characterize mechanisms of eCB-mediated plasticity throughout reward circuitry and determine their contribution to the pathophysiology and treatment of drug addiction.
Footnotes
Supported by National Institutes of Health Grants DA022340 and DA042595 to J.F.C.
The authors declare no competing financial interests.
References
- Abbott LF, Regehr WG. Synaptic computation. Nature. 2004;431:796–803. doi: 10.1038/nature03010. [DOI] [PubMed] [Google Scholar]
- Abbott LF, Varela JA, Sen K, Nelson SB. Synaptic depression and cortical gain control. Science. 1997;275:220–224. doi: 10.1126/science.275.5297.221. [DOI] [PubMed] [Google Scholar]
- Adermark L, Lovinger DM. Combined activation of L-type Ca2+ channels and synaptic transmission is sufficient to induce striatal long-term depression. J Neurosci. 2007;27:6781–6787. doi: 10.1523/JNEUROSCI.0280-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alger BE. Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol. 2002;68:247–286. doi: 10.1016/S0301-0082(02)00080-1. [DOI] [PubMed] [Google Scholar]
- Alger BE, Kim J. Supply and demand for endocannabinoids. Trends Neurosci. 2011;34:304–315. doi: 10.1016/j.tins.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Mackie K. CB2: a cannabinoid receptor with an identity crisis. Br J Pharmacol. 2010;160:467–479. doi: 10.1111/j.1476-5381.2010.00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beierlein M, Regehr WG. Local interneurons regulate synaptic strength by retrograde release of endocannabinoids. J Neurosci. 2006;26:9935–9943. doi: 10.1523/JNEUROSCI.0958-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science. 1997;277:1094–1097. doi: 10.1126/science.277.5329.1094. [DOI] [PubMed] [Google Scholar]
- Berridge KC. “Liking” and “wanting” food rewards: brain substrates and roles in eating disorders. Physiol Behav. 2009;97:537–550. doi: 10.1016/j.physbeh.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams EJ, Gangadharan U, Hobbs C, Di Marzo V, Doherty P. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol. 2003;163:463–468. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock R, Shin JH, Kaplan AR, Dobi A, Markey E, Kramer PF, Gremel CM, Christensen CH, Adrover MF, Alvarez VA. Strengthening the accumbal indirect pathway promotes resilience to compulsive cocaine use. Nat Neurosci. 2013;16:632–638. doi: 10.1038/nn.3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodor AL, Katona I, Nyíri G, Mackie K, Ledent C, Hájos N, Freund TF. Endocannabinoid signaling in rat somatosensory cortex: laminar differences and involvement of specific interneuron types. J Neurosci. 2005;25:6845–6856. doi: 10.1523/JNEUROSCI.0442-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadas H, Gaillet S, Beltramo M, Venance L, Piomelli D. Biosynthesis of an endogenous cannabinoid precursor in neurons and its control by calcium and cAMP. J Neurosci. 1996;16:3934–3942. doi: 10.1523/JNEUROSCI.16-12-03934.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Picconi B, Tozzi A, Di Filippo M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 2007;30:211–219. doi: 10.1016/j.tins.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Carlson G, Wang Y, Alger BE. Endocannabinoids facilitate the induction of LTP in the hippocampus. Nat Neurosci. 2002;5:723–724. doi: 10.1038/nn879. [DOI] [PubMed] [Google Scholar]
- Chandra R, Lenz JD, Gancarz AM, Chaudhury D, Schroeder GL, Han MH, Cheer JF, Dietz DM, Lobo MK. Optogenetic inhibition of D1R containing nucleus accumbens neurons alters cocaine-mediated regulation of Tiam1. Front Mol Neurosci. 2013;6:13. doi: 10.3389/fnmol.2013.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheer JF, Kendall DA, Mason R, Marsden CA. Differential cannabinoid-induced electrophysiological effects in rat ventral tegmentum. Neuropharmacology. 2003;44:633–641. doi: 10.1016/S0028-3908(03)00029-7. [DOI] [PubMed] [Google Scholar]
- Cheer JF, Wassum KM, Heien ML, Phillips PE, Wightman RM. Cannabinoids enhance subsecond dopamine release in the nucleus accumbens of awake rats. J Neurosci. 2004;24:4393–4400. doi: 10.1523/JNEUROSCI.0529-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheer JF, Wassum KM, Sombers LA, Heien ML, Ariansen JL, Aragona BJ, Phillips PE, Wightman RM. Phasic dopamine release evoked by abused substances requires cannabinoid receptor activation. J Neurosci. 2007;27:791–795. doi: 10.1523/JNEUROSCI.4152-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron. 2003;38:461–472. doi: 10.1016/S0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Endocannabinoid-mediated metaplasticity in the hippocampus. Neuron. 2004;43:871–881. doi: 10.1016/j.neuron.2004.08.036. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Heifets BD, Kaeser PS, Südhof TC, Purpura DP, Castillo PE. Endocannabinoid-mediated long-term plasticity requires cAMP/PKA signaling and RIM1alpha. Neuron. 2007;54:801–812. doi: 10.1016/j.neuron.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covey DP, Wenzel JM, Cheer JF. Cannabinoid modulation of drug reward and the implications of marijuana legalization. Brain Res. 2015;1628:233–243. doi: 10.1016/j.brainres.2014.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devane WA, Dysarz FA, Johnson MR, Melvin LS, Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol. 1988;34:605–613. doi: 10.1016/j.brainres.2014.11.034. [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Diana M, Melis M, Gessa GL. Increase in meso-prefrontal dopaminergic activity after stimulation of CB1 receptors by cannabinoids. Eur J Neurosci. 1998;10:2825–2830. doi: 10.1111/j.1460-9568.1998.00292.x. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, De Petrocellis L. Endocannabinoids as regulators of transient receptor potential (TRP) channels: a further opportunity to develop new endocannabinoid-based therapeutic drugs. Curr Med Chem. 2010;17:1430–1449. doi: 10.2174/092986710790980078. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- Edwards DA, Kim J, Alger BE. Multiple mechanisms of endocannabinoid response initiation in hippocampus. J Neurophysiol. 2006;95:67–75. doi: 10.1152/jn.00813.2005. [DOI] [PubMed] [Google Scholar]
- Egertová M, Simon GM, Cravatt BF, Elphick MR. Localization of N-acyl phosphatidylethanolamine phospholipase D (NAPE-PLD) expression in mouse brain: a new perspective on N-acylethanolamines as neural signaling molecules. J Comp Neurol. 2008;506:604–615. doi: 10.1002/cne.21568. [DOI] [PubMed] [Google Scholar]
- Fourgeaud L, Mato S, Bouchet D, Hémar A, Worley PF, Manzoni OJ. A single in vivo exposure to cocaine abolishes endocannabinoid-mediated long-term depression in the nucleus accumbens. J Neurosci. 2004;24:6939–6945. doi: 10.1523/JNEUROSCI.0671-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French ED, Dillon K, Wu X. Cannabinoids excite dopamine neurons in the ventral tegmentum and substantia nigra. Neuroreport. 1997;8:649–652. doi: 10.1097/00001756-199702100-00014. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- Galante M, Diana MA. Group I metabotropic glutamate receptors inhibit GABA release at interneuron-Purkinje cell synapses through endocannabinoid production. J Neurosci. 2004;24:4865–4874. doi: 10.1523/JNEUROSCI.0403-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Vasilyev DV, Goncalves MB, Howell FV, Hobbs C, Reisenberg M, Shen R, Zhang MY, Strassle BW, Lu P, Mark L, Piesla MJ, Deng K, Kouranova EV, Ring RH, Whiteside GT, Bates B, Walsh FS, Williams G, Pangalos MN, et al. Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J Neurosci. 2010;30:2017–2024. doi: 10.1523/JNEUROSCI.5693-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebremedhin D, Lange AR, Campbell WB, Hillard CJ, Harder DR. Cannabinoid CB1 receptor of cat cerebral arterial muscle functions to inhibit L-type Ca2+ channel current. Am J Physiol. 1999;276:H2085–H2093. doi: 10.1152/ajpheart.1999.276.6.H2085. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Partridge JG, Lupica CR, Lovinger DM. It could be habit forming: drugs of abuse and striatal synaptic plasticity. Trends Neurosci. 2003;26:184–192. doi: 10.1016/S0166-2236(03)00065-1. [DOI] [PubMed] [Google Scholar]
- Gipson CD, Kupchik YM, Kalivas PW. Rapid, transient synaptic plasticity in addiction. Neuropharmacology. 2014;76:276–286. doi: 10.1016/j.neuropharm.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrida A, Parsons LH, Kerr TM, Rodríguez de Fonseca F, Navarro M, Piomelli D. Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat Neurosci. 1999;2:358–363. doi: 10.1038/7268. [DOI] [PubMed] [Google Scholar]
- Godlewski G, Offertáler L, Wagner JA, Kunos G. Receptors for acylethanolamides-GPR55 and GPR119. Prostaglandins Other Lipid Mediat. 2009;89:105–111. doi: 10.1016/j.prostaglandins.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueter BA, Brasnjo G, Malenka RC. Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat Neurosci. 2010;13:1519–1525. doi: 10.1038/nn.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyas AI, Cravatt BF, Bracey MH, Dinh TP, Piomelli D, Boscia F, Freund TF. Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. Eur J Neurosci. 2004;20:441–458. doi: 10.1111/j.1460-9568.2004.03428.x. [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Kano M. Ca2+-assisted receptor-driven endocannabinoid release: mechanisms that associate presynaptic and postsynaptic activities. Curr Opin Neurobiol. 2007;17:360–365. doi: 10.1016/j.conb.2007.03.012. [DOI] [PubMed] [Google Scholar]
- Heifets BD, Castillo PE. Endocannabinoid signaling and long-term synaptic plasticity. Annu Rev Physiol. 2009;71:283–306. doi: 10.1146/annurev.physiol.010908.163149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, Rice KC. Cannabinoid receptor localization in brain. Proc Natl Acad Sci U S A. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci. 1991;11:563–583. doi: 10.1523/JNEUROSCI.11-02-00563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillard CJ, Jarrahian A. The movement of N-arachidonoylethanolamine (anandamide) across cellular membranes. Chem Phys Lipids. 2000;108:123–134. doi: 10.1016/s0009-3084(00)00191-2. [DOI] [PubMed] [Google Scholar]
- Hoffman AF, Lupica CR. Mechanisms of cannabinoid inhibition of GABA(A) synaptic transmission in the hippocampus. J Neurosci. 2000;20:2470–2479. doi: 10.1523/JNEUROSCI.20-07-02470.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman AF, Lupica CR. Direct actions of cannabinoids on synaptic transmission in the nucleus accumbens: a comparison with opioids. J Neurophysiol. 2001;85:72–83. doi: 10.1152/jn.2001.85.1.72. [DOI] [PubMed] [Google Scholar]
- Hoffman AF, Oz M, Caulder T, Lupica CR. Functional tolerance and blockade of long-term depression at synapses in the nucleus accumbens after chronic cannabinoid exposure. J Neurosci. 2003;23:4815–4820. doi: 10.1523/JNEUROSCI.23-12-04815.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- Howlett AC. Cannabinoid receptor signaling. Handb Exp Pharmacol. 2005;168:53–79. doi: 10.1007/3-540-26573-2_2. [DOI] [PubMed] [Google Scholar]
- Ikemoto S. Dopamine reward circuitry: two projection systems from the ventral midbrain to the nucleus accumbens-olfactory tubercle complex. Brain Res Rev. 2007;56:27–78. doi: 10.1016/j.brainresrev.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isokawa M, Alger BE. Retrograde endocannabinoid regulation of GABAergic inhibition in the rat dentate gyrus granule cell. J Physiol. 2005;567:1001–1010. doi: 10.1113/jphysiol.2005.094219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isokawa M, Alger BE. Ryanodine receptor regulates endogenous cannabinoid mobilization in the hippocampus. J Neurophysiol. 2006;95:3001–3011. doi: 10.1152/jn.00975.2005. [DOI] [PubMed] [Google Scholar]
- Julian MD, Martin AB, Cuellar B, Rodriguez De Fonseca F, Navarro M, Moratalla R, Garcia-Segura LM. Neuroanatomical relationship between type 1 cannabinoid receptors and dopaminergic systems in the rat basal ganglia. Neuroscience. 2003;119:309–318. doi: 10.1016/S0306-4522(03)00070-8. [DOI] [PubMed] [Google Scholar]
- Jung KM, Astarita G, Zhu C, Wallace M, Mackie K, Piomelli D. A key role for diacylglycerol lipase-α in metabotropic glutamate receptor-dependent endocannabinoid mobilization. Mol Pharmacol. 2007;72:612–621. doi: 10.1124/mol.107.037796. [DOI] [PubMed] [Google Scholar]
- Jung KM, Sepers M, Henstridge CM, Lassalle O, Neuhofer D, Martin H, Ginger M, Frick A, DiPatrizio NV, Mackie K, Katona I, Piomelli D, Manzoni OJ. Uncoupling of the endocannabinoid signalling complex in a mouse model of fragile X syndrome. Nat Commun. 2012;3:1080. doi: 10.1038/ncomms2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Kauer JA. Learning mechanisms in addiction: synaptic plasticity in the ventral tegmental area as a result of exposure to drugs of abuse. Annu Rev Physiol. 2004;66:447–475. doi: 10.1146/annurev.physiol.66.032102.112534. [DOI] [PubMed] [Google Scholar]
- Kim J, Alger BE. Inhibition of cyclooxygenase-2 potentiates retrograde endocannabinoid effects in hippocampus. Nat Neurosci. 2004;7:697–698. doi: 10.1038/nn1262. [DOI] [PubMed] [Google Scholar]
- Kim J, Alger BE. Reduction in endocannabinoid tone is a homeostatic mechanism for specific inhibitory synapses. Nat Neurosci. 2010;13:592–600. doi: 10.1038/nn.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Isokawa M, Ledent C, Alger BE. Activation of muscarinic acetylcholine receptors enhances the release of endogenous cannabinoids in the hippocampus. J Neurosci. 2002;22:10182–10191. doi: 10.1523/JNEUROSCI.22-23-10182.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyachko VA, Stevens CF. Excitatory and feed-forward inhibitory hippocampal synapses work synergistically as an adaptive filter of natural spike trains. PLoS Biol. 2006;4:e207. doi: 10.1371/journal.pbio.0040207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortleven C, Bruneau LC, Trudeau LE. Neurotensin inhibits glutamate-mediated synaptic inputs onto ventral tegmental area dopamine neurons through the release of the endocannabinoid 2-AG. Neuropharmacology. 2012;63:983–991. doi: 10.1016/j.neuropharm.2012.07.037. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727. doi: 10.1016/S0896-6273(01)00246-X. [DOI] [PubMed] [Google Scholar]
- Kupchik YM, Brown RM, Heinsbroek JA, Lobo MK, Schwartz DJ, Kalivas PW. Coding the direct/indirect pathways by D1 and D2 receptors is not valid for accumbens projections. Nat Neurosci. 2015;18:1230–1232. doi: 10.1038/nn.4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labouèbe G, Liu S, Dias C, Zou H, Wong JC, Karunakaran S, Clee SM, Phillips AG, Boutrel B, Borgland SL. Insulin induces long-term depression of ventral tegmental area dopamine neurons via endocannabinoids. Nat Neurosci. 2013;16:300–308. doi: 10.1038/nn.3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauckner JE, Jensen JB, Chen HY, Lu HC, Hille B, Mackie K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc Natl Acad Sci U S A. 2008;105:2699–2704. doi: 10.1073/pnas.0711278105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecca S, Melis M, Luchicchi A, Muntoni AL, Pistis M. Inhibitory inputs from rostromedial tegmental neurons regulate spontaneous activity of midbrain dopamine cells and their responses to drugs of abuse. Neuropsychopharmacology. 2012;37:1164–1176. doi: 10.1038/npp.2011.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner TN, Kreitzer AC. RGS4 is required for dopaminergic control of striatal LTD and susceptibility to parkinsonian motor deficits. Neuron. 2012;73:347–359. doi: 10.1016/j.neuron.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung D, Saghatelian A, Simon GM, Cravatt BF. Inactivation of N-acyl phosphatidylethanolamine phospholipase D reveals multiple mechanisms for the biosynthesis of endocannabinoids. Biochemistry. 2006;45:4720–4726. doi: 10.1021/bi060163l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Wang L, Harvey-White J, Osei-Hyiaman D, Razdan R, Gong Q, Chan AC, Zhou Z, Huang BX, Kim HY, Kunos G. A biosynthetic pathway for anandamide. Proc Natl Acad Sci U S A. 2006;103:13345–13350. doi: 10.1073/pnas.0601832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QS, Pu L, Poo MM. Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature. 2005;437:1027–1031. doi: 10.1038/nature04050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano I, Leresche N, Marty A. Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synaptic currents. Neuron. 1991;6:565–574. doi: 10.1016/0896-6273(91)90059-9. [DOI] [PubMed] [Google Scholar]
- Lonart G, Schoch S, Kaeser PS, Larkin CJ, Südhof TC, Linden DJ. Phosphorylation of RIM1alpha by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell. 2003;115:49–60. doi: 10.1016/S0092-8674(03)00727-X. [DOI] [PubMed] [Google Scholar]
- Lovinger DM. Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology. 2010;58:951–961. doi: 10.1016/j.neuropharm.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludányi A, Hu SS, Yamazaki M, Tanimura A, Piomelli D, Watanabe M, Kano M, Sakimura K, Maglóczky Z, Mackie K, Freund TF, Katona I. Complementary synaptic distribution of enzymes responsible for synthesis and inactivation of the endocannabinoid 2-arachidonoylglycerol in the human hippocampus. Neuroscience. 2011;174:50–63. doi: 10.1016/j.neuroscience.2010.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupica CR, Riegel AC. Endocannabinoid release from midbrain dopamine neurons: a potential substrate for cannabinoid receptor antagonist treatment of addiction. Neuropharmacology. 2005;48:1105–1116. doi: 10.1016/j.neuropharm.2005.03.016. [DOI] [PubMed] [Google Scholar]
- MacAskill AF, Cassel JM, Carter AG. Cocaine exposure reorganizes cell type- and input-specific connectivity in the nucleus accumbens. Nat Neurosci. 2014;17:1198–1207. doi: 10.1038/nn.3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccarrone M, Rossi S, Bari M, De Chiara V, Fezza F, Musella A, Gasperi V, Prosperetti C, Bernardi G, Finazzi-Agrò A, Cravatt BF, Centonze D. Anandamide inhibits metabolism and physiological actions of 2-arachidonoylglycerol in the striatum. Nat Neurosci. 2008;11:152–159. doi: 10.1038/nn2042. [DOI] [PubMed] [Google Scholar]
- Mackie K, Hille B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci U S A. 1992;89:3825–3829. doi: 10.1073/pnas.89.9.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie K, Lai Y, Westenbroek R, Mitchell R. Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptor. J Neurosci. 1995;15:6552–6561. doi: 10.1523/JNEUROSCI.15-10-06552.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima T, Oka S, Hashimotodani Y, Ohno-Shosaku T, Aiba A, Wu D, Waku K, Sugiura T, Kano M. Synaptically driven endocannabinoid release requires Ca2+- assisted metabotropic glutamate receptor subtype 1 to phospholipase C β4 signaling cascade in the cerebellum. J Neurosci. 2005;25:6826–6835. doi: 10.1523/JNEUROSCI.0945-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makara JK, Mor M, Fegley D, Szabó SI, Kathuria S, Astarita G, Duranti A, Tontini A, Tarzia G, Rivara S, Freund TF, Piomelli D. Selective inhibition of 2-AG hydrolysis enhances endocannabinoid signaling in hippocampus. Nat Neurosci. 2005;8:1139–1141. doi: 10.1038/nn1521. [DOI] [PubMed] [Google Scholar]
- Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, Bodor AL, Muccioli GG, Hu SS, Woodruff G, Fung S, Lafourcade M, Alexander JP, Long JZ, Li W, Xu C, Möller T, Mackie K, Manzoni OJ, Cravatt BF, et al. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci. 2010;13:951–957. doi: 10.1038/nn.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LA, Alger BE. Muscarinic facilitation of the occurrence of depolarization-induced suppression of inhibition in rat hippocampus. Neuroscience. 1999;92:61–71. doi: 10.1016/S0306-4522(98)00745-3. [DOI] [PubMed] [Google Scholar]
- Mato S, Chevaleyre V, Robbe D, Pazos A, Castillo PE, Manzoni OJ. A single in-vivo exposure to δ9THC blocks endocannabinoid-mediated synaptic plasticity. Nat Neurosci. 2004;7:585–586. doi: 10.1038/nn1251. [DOI] [PubMed] [Google Scholar]
- Mato S, Robbe D, Puente N, Grandes P, Manzoni OJ. Presynaptic homeostatic plasticity rescues long-term depression after chronic delta 9-tetrahydrocannabinol exposure. J Neurosci. 2005;25:11619–11627. doi: 10.1523/JNEUROSCI.2294-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mato S, Lafourcade M, Robbe D, Bakiri Y, Manzoni OJ. Role of the cyclic-AMP/PKA cascade and of P/Q-type Ca2+ channels in endocannabinoid-mediated long-term depression in the nucleus accumbens. Neuropharmacology. 2008;54:87–94. doi: 10.1016/j.neuropharm.2007.04.014. [DOI] [PubMed] [Google Scholar]
- McCutcheon JE, Wang X, Tseng KY, Wolf ME, Marinelli M. Calcium-permeable AMPA receptors are present in nucleus accumbens synapses after prolonged withdrawal from cocaine self-administration but not experimenter-administered cocaine. J Neurosci. 2011;31:5737–5743. doi: 10.1523/JNEUROSCI.0350-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R, Parker LA. The endocannabinoid system and the brain. Annu Rev Psychol. 2013;64:21–47. doi: 10.1146/annurev-psych-113011-143739. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-D. [DOI] [PubMed] [Google Scholar]
- Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL. Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci. 2004;24:53–62. doi: 10.1523/JNEUROSCI.4503-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis M, Greco B, Tonini R. Interplay between synaptic endocannabinoid signaling and metaplasticity in neuronal circuit function and dysfunction. Eur J Neurosci. 2014;39:1189–1201. doi: 10.1111/ejn.12501. [DOI] [PubMed] [Google Scholar]
- Meredith GE, Baldo BA, Andrezjewski ME, Kelley AE. The structural basis for mapping behavior onto the ventral striatum and its subdivisions. Brain Struct Funct. 2008;213:17–27. doi: 10.1007/s00429-008-0175-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith RM, Floyer-Lea AM, Paulsen O. Maturation of long-term potentiation induction rules in rodent hippocampus: role of GABAergic inhibition. J Neurosci. 2003;23:11142–11146. doi: 10.1523/JNEUROSCI.23-35-11142.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- Murataeva N, Straiker A, Mackie K. Parsing the players: 2-arachidonoylglycerol synthesis and degradation in the CNS. Br J Pharmacol. 2014;171:1379–1391. doi: 10.1111/bph.12411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musella A, De Chiara V, Rossi S, Prosperetti C, Bernardi G, Maccarrone M, Centonze D. TRPV1 channels facilitate glutamate transmission in the striatum. Mol Cell Neurosci. 2009;40:89–97. doi: 10.1016/j.mcn.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MC, Ward AM, Hahn YK, Lichtman AH, Conti B, Cravatt BF. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science. 2011;334:809–813. doi: 10.1126/science.1209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29:729–738. doi: 10.1016/S0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Hashimotodani Y, Ano M, Takeda S, Tsubokawa H, Kano M. Endocannabinoid signalling triggered by NMDA receptor-mediated calcium entry into rat hippocampal neurons. J Physiol. 2007;584:407–418. doi: 10.1113/jphysiol.2007.137505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overton HA, Babbs AJ, Doel SM, Fyfe MC, Gardner LS, Griffin G, Jackson HC, Procter MJ, Rasamison CM, Tang-Christensen M, Widdowson PS, Williams GM, Reynet C. Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab. 2006;3:167–175. doi: 10.1016/j.cmet.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Pan B, Hillard CJ, Liu Q. D2 dopamine receptor activation facilitates endocannabinoid-mediated long-term synaptic depression of GABAergic synaptic transmission in midbrain dopamine neurons via cAMP-protein kinase A signaling. J Neurosci. 2008a;28:14018–14030. doi: 10.1523/JNEUROSCI.4035-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Hillard CJ, Liu QS. Endocannabinoid signaling mediates cocaine-induced inhibitory synaptic plasticity in midbrain dopamine neurons. J Neurosci. 2008b;28:1385–1397. doi: 10.1523/JNEUROSCI.4033-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Wang W, Long JZ, Sun D, Hillard CJ, Cravatt BF, Liu QS. Blockade of 2-arachidonoylglycerol hydrolysis by selective monoacylglycerol lipase inhibitor 4-nitrophenyl 4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate (JZL184) enhances retrograde endocannabinoid signaling. J Pharmacol Exp Ther. 2009;331:591–597. doi: 10.1124/jpet.109.158162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons LH, Hurd YL. Endocannabinoid signalling in reward and addiction. Nat Rev Neurosci. 2015;16:579–594. doi: 10.1038/nrn4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG. Receptors and channels targeted by synthetic cannabinoid receptor agonists and antagonists. Curr Med Chem. 2010;17:1360–1381. doi: 10.2174/092986710790980050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- Piomelli D. More surprises lying ahead: the endocannabinoids keep us guessing. Neuropharmacology. 2014;76:228–234. doi: 10.1016/j.neuropharm.2013.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistis M, Muntoni AL, Pillolla G, Gessa GL. Cannabinoids inhibit excitatory inputs to neurons in the shell of the nucleus accumbens: an in vivo electrophysiological study. Eur J Neurosci. 2002;15:1795–1802. doi: 10.1046/j.1460-9568.2002.02019.x. [DOI] [PubMed] [Google Scholar]
- Pitler TA, Alger BE. Depolarization-induced suppression of GABAergic inhibition in rat hippocampal pyramidal cells: G protein involvement in a presynaptic mechanism. Neuron. 1994;13:1447–1455. doi: 10.1016/0896-6273(94)90430-8. [DOI] [PubMed] [Google Scholar]
- Puente N, Cui Y, Lassalle O, Lafourcade M, Georges F, Venance L, Grandes P, Manzoni OJ. Polymodal activation of the endocannabinoid system in the extended amygdala. Nat Neurosci. 2011;14:1542–1547. doi: 10.1038/nn.2974. [DOI] [PubMed] [Google Scholar]
- Riegel AC, Lupica CR. Independent presynaptic and postsynaptic mechanisms regulate endocannabinoid signaling at multiple synapses in the ventral tegmental area. J Neurosci. 2004;24:11070–11078. doi: 10.1523/JNEUROSCI.3695-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Alonso G, Duchamp F, Bockaert J, Manzoni OJ. Localization and mechanisms of action of cannabinoid receptors at the glutamatergic synapses of the mouse nucleus accumbens. J Neurosci. 2001;21:109–116. doi: 10.1523/JNEUROSCI.21-01-00109.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ. Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci U S A. 2002;99:8384–8388. doi: 10.1073/pnas.122149199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DC, Corcoran ME, Fibiger HC. On the role of ascending catecholaminergic systems in intravenous self-administration of cocaine. Pharmacol Biochem Behav. 1977;6:615–620. doi: 10.1016/0091-3057(77)90084-3. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Grace AA. Cortico-basal ganglia reward network: microcircuitry. Neuropsychopharmacology. 2010;35:27–47. doi: 10.1038/npp.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shonesy BC, Winder DG, Patel S, Colbran RJ. The initiation of synaptic 2-AG mobilization requires both an increased supply of diacylglycerol precursor and increased postsynaptic calcium. Neuropharmacology. 2015;91:57–62. doi: 10.1016/j.neuropharm.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhpura N, Parsons LH. Endocannabinoid-mediated synaptic plasticity and addiction-related behavior. Neuropharmacology. 2011;61:1070–1087. doi: 10.1016/j.neuropharm.2011.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon GM, Cravatt BF. Endocannabinoid biosynthesis proceeding through glycerophospho-N-acyl ethanolamine and a role for alpha/beta-hydrolase 4 in this pathway. J Biol Chem. 2006;281:26465–26472. doi: 10.1074/jbc.M604660200. [DOI] [PubMed] [Google Scholar]
- Sombers LA, Beyene M, Carelli RM, Wightman RM. Synaptic overflow of dopamine in the nucleus accumbens arises from neuronal activity in the ventral tegmental area. J Neurosci. 2009;29:1735–1742. doi: 10.1523/JNEUROSCI.5562-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella N, Piomelli D. Receptor-dependent formation of endogenous cannabinoids in cortical neurons. Eur J Pharmacol. 2001;425:189–196. doi: 10.1016/S0014-2999(01)01182-7. [DOI] [PubMed] [Google Scholar]
- Suárez J, Ortíz O, Puente N, Bermúdez-Silva FJ, Blanco E, Fernández-Llebrez P, Grandes P, de Fonseca FR, Moratalla R. Distribution of diacylglycerol lipase alpha, an endocannabinoid synthesizing enzyme, in the rat forebrain. Neuroscience. 2011;192:112–131. doi: 10.1016/j.neuroscience.2011.06.062. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- Szabo B, Urbanski MJ, Bisogno T, Di Marzo V, Mendiguren A, Baer WU, Freiman I. Depolarization-induced retrograde synaptic inhibition in the mouse cerebellar cortex is mediated by 2-arachidonoylglycerol. J Physiol. 2006;577:263–280. doi: 10.1113/jphysiol.2006.119362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taber KH, Black DN, Porrino LJ, Hurley RA. Neuroanatomy of dopamine: reward and addiction. J Neuropsychiatry Clin Neurosci. 2012;24:1–4. doi: 10.1176/appi.neuropsych.24.1.1. [DOI] [PubMed] [Google Scholar]
- Trettel J, Levine ES. Endocannabinoids mediate rapid retrograde signaling at interneuron right-arrow pyramidal neuron synapses of the neocortex. J Neurophysiol. 2003;89:2334–2338. doi: 10.1152/jn.01037.2002. [DOI] [PubMed] [Google Scholar]
- Tsuboi K, Okamoto Y, Ikematsu N, Inoue M, Shimizu Y, Uyama T, Wang J, Deutsch DG, Burns MP, Ulloa NM, Tokumura A, Ueda N. Enzymatic formation of N-acylethanolamines from N-acylethanolamine plasmalogen through N-acylphosphatidylethanolamine-hydrolyzing phospholipase D-dependent and -independent pathways. Biochim Biophys Acta. 2011;1811:565–577. doi: 10.1016/j.bbalip.2011.07.009. [DOI] [PubMed] [Google Scholar]
- Twitchell W, Brown S, Mackie K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol. 1997;78:43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- Uchigashima M, Narushima M, Fukaya M, Katona I, Kano M, Watanabe M. Subcellular arrangement of molecules for 2-arachidonoyl-glycerol-mediated retrograde signaling and its physiological contribution to synaptic modulation in the striatum. J Neurosci. 2007;27:3663–3676. doi: 10.1523/JNEUROSCI.0448-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, Stella N, Makriyannis A, Piomelli D, Davison JS, Marnett LJ, Di Marzo V, Pittman QJ, Patel KD, Sharkey KA. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310:329–332. doi: 10.1126/science.1115740. [DOI] [PubMed] [Google Scholar]
- Varma N, Carlson GC, Ledent C, Alger BE. Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J Neurosci. 2001;21:RC188. doi: 10.1523/JNEUROSCI.21-24-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Morales M. The brain on drugs: from reward to addiction. Cell. 2015;162:712–725. doi: 10.1016/j.cell.2015.07.046. [DOI] [PubMed] [Google Scholar]
- Wagner JJ, Alger BE. Increased neuronal excitability during depolarization-induced suppression of inhibition in rat hippocampus. J Physiol. 1996;495:107–112. doi: 10.1113/jphysiol.1996.sp021577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Treadway T, Covey DP, Cheer JF, Lupica CR. Cocaine-induced endocannabinoid mobilization in the ventral tegmental area. Cell Rep. 2015;12:1997–2008. doi: 10.1016/j.celrep.2015.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigström H, Gustafsson B. Facilitated induction of hippocampal long-lasting potentiation during blockade of inhibition. Nature. 1983;301:603–604. doi: 10.1038/301603a0. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Winters BD, Krüger JM, Huang X, Gallaher ZR, Ishikawa M, Czaja K, Krueger JM, Huang YH, Schlüter OM, Dong Y. Cannabinoid receptor 1-expressing neurons in the nucleus accumbens. Proc Natl Acad Sci U S A. 2012;109:E2717–E2725. doi: 10.1073/pnas.1206303109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RA, Bozarth MA. Brain mechanisms of drug reward and euphoria. Psychiatr Med. 1985;3:445–460. [PubMed] [Google Scholar]
- Wise RA, Rompre PP. Brain dopamine and reward. Annu Rev Psychol. 1989;40:191–225. doi: 10.1146/annurev.ps.40.020189.001203. [DOI] [PubMed] [Google Scholar]
- Wolf ME. Synaptic mechanisms underlying persistent cocaine craving. Nat Rev Neurosci. 2016;17:351–365. doi: 10.1038/nrn.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME, Tseng KY. Calcium-permeable AMPA receptors in the VTA and nucleus accumbens after cocaine exposure: when, how, and why? Front Mol Neurosci. 2012;5:72. doi: 10.3389/fnmol.2012.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanovsky Y, Mades S, Misgeld U. Retrograde signaling changes the venue of postsynaptic inhibition in rat substantia nigra. Neuroscience. 2003;122:317–328. doi: 10.1016/S0306-4522(03)00607-9. [DOI] [PubMed] [Google Scholar]
- Zhang HY, Gao M, Liu QR, Bi GH, Li X, Yang HJ, Gardner EL, Wu J, Xi ZX. Cannabinoid CB2 receptors modulate midbrain dopamine neuronal activity and dopamine-related behavior in mice. Proc Natl Acad Sci U S A. 2014;111:E5007–E5015. doi: 10.1073/pnas.1413210111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HY, Gao M, Shen H, Bi GH, Yang HJ, Liu QR, Wu J, Gardner EL, Bonci A, Xi ZX. Expression of functional cannabinoid CB2 receptor in VTA dopamine neurons in rats. Addict Biol. 2016 doi: 10.1111/adb.12367. doi: 10.1111/adb.12367. Advance online publication. Retrieved Fe. 1, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong P, Liu Y, Hu Y, Wang T, Zhao YP, Liu QS. BDNF interacts with endocannabinoids to regulate cocaine-induced synaptic plasticity in mouse midbrain dopamine neurons. J Neurosci. 2015;35:4469–4481. doi: 10.1523/JNEUROSCI.2924-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PJ, Lovinger DM. Retrograde endocannabinoid signaling in a postsynaptic neuron/synaptic bouton preparation from basolateral amygdala. J Neurosci. 2005;25:6199–6207. doi: 10.1523/JNEUROSCI.1148-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PJ, Lovinger DM. Persistent synaptic activity produces long-lasting enhancement of endocannabinoid modulation and alters long-term synaptic plasticity. J Neurophysiol. 2007;97:4386–4389. doi: 10.1152/jn.01228.2006. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sørgård M, Di Marzo V, Julius D, Högestätt ED. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]