

ABSTRACT
An operon comprising two genes, CA_P0037 and CA_P0036, that encode proteins of unknown function that were previously shown to be highly expressed in acidogenic cells and repressed in solventogenic and alcohologenic cells is located on the pSOL1 megaplasmid of Clostridium acetobutylicum upstream of adhE2. A CA_P0037::int (189/190s) mutant in which an intron was inserted at position 189/190 in the sense strand of CA_P0037 was successfully generated by the Targetron technique. The resultant mutant showed significantly different metabolic flux patterns in acidogenic (producing mainly lactate, butyrate, and butanol) and alcohologenic (producing mainly butyrate, acetate, and lactate) chemostat cultures but not in solventogenic or batch cultures. Transcriptomic investigation of the CA_P0037::int (189/190s) mutant showed that inactivation of CA_P0037 significantly affected the expression of more than 258 genes under acidogenic conditions. Surprisingly, genes belonging to the Fur regulon, involved in iron transport (CA_C1029-CA_C1032), or coding for the main flavodoxin (CA_C0587) were the most significantly expressed genes under all conditions, whereas fur (coding for the ferric uptake regulator) gene expression remained unchanged. Furthermore, most of the genes of the Rex regulon, such as the adhE2 and ldhA genes, and of the PerR regulon, such as rbr3A-rbr3B and dfx, were overexpressed in the mutant. In addition, the whole CA_P0037-CA_P0036 operon was highly expressed under all conditions in the CA_P0037::int (189/190s) mutant, suggesting a self-regulated expression mechanism. Cap0037 was shown to bind to the CA_P0037-CA_P0036 operon, sol operon, and adc promoters, and the binding sites were determined by DNA footprinting. Finally, a putative Cap0037 regulon was generated using a bioinformatic approach.
IMPORTANCE
Clostridium acetobutylicum is well-known for its ability to produce solvents, especially n-butanol. Understanding the regulatory network of C. acetobutylicum will be crucial for further engineering to obtain a strain capable of producing n-butanol at high yield and selectivity. This study has discovered that the Cap0037 protein is a novel regulator of C. acetobutylicum that drastically affects metabolism under both acidogenic and alcohologenic fermentation conditions. This is pioneering work for further determining the regulatory mechanism of Cap0037 in C. acetobutylicum and studying the role of proteins homologous to Cap0037 in other members of the phylum Firmicutes.
INTRODUCTION
Clostridium acetobutylicum is a Gram-positive, strictly anaerobic, spore-forming bacterium now considered the model organism for the study of solventogenic clostridia (1, 2). The superiority of butanol over ethanol as an alternative biofuel has attracted research interest in C. acetobutylicum and other recombinant bacteria producing butanol as a major product (3). Understanding the complex regulatory network of C. acetobutylicum metabolism is crucial for further manipulating the genotype to obtain industrial strains, but our understanding of cellular functioning remains very limited. Some global regulators have already been studied. A peroxide repressor (PerR)-homologous protein was identified to be a key repressor that plays an important role in defense against oxidative stress (4, 5). In the same family as PerR, a ferric uptake regulator (Fur), which helps C. acetobutylicum sense and respond to iron availability at multiple levels using a sophisticated system, was identified and well characterized (6). Spo0A was found to directly control the solventogenic switch, expression of the adc and ptb genes (7), and activation of the sporulation program in C. acetobutylicum (8). The carbon storage regulator (CsrA) was also shown to directly or indirectly regulate multiple pathways, including flagellum assembly, oligopeptide transport, iron uptake, phosphotransferase systems (PTS), synthesis of riboflavin, stage III sporulation, and central carbon metabolism (9). Recently, a redox-sensing transcriptional repressor (Rex) has been found to modulate its DNA binding activity in response to the NADH/NAD+ ratio and plays a role in the alcohologenic shift of C. acetobutylicum (10, 11).
The regulatory network of C. acetobutylicum remains largely unexplored. In this study, we found Cap0037 to be a novel self-regulated protein that globally affects C. acetobutylicum metabolism. CA_P0037 is the first gene of a polycistronic operon with CA_P0036 (Fig. 1). This operon is located upstream of adhE2 on the pSOL1 megaplasmid, and it was significantly expressed in acidogenic cells and repressed in solventogenic and alcohologenic cells (12, 13). Furthermore, the two proteins were present at a 1:1 molar ratio and were among the 30 most abundant proteins in the cell (around 50,000 molecules per cell) (12). Another study found that CA_P0037-CA_P0036 expression was slightly downregulated in an adc mutant, clearly upregulated during solventogenesis in a pta mutant and an adc pta double mutant, and strongly repressed in a ptb mutant independent of the cultivation conditions (14). Interestingly, Cap0037 and Cap0036 were found to appear in multiple spots (up to five in the case of Cap0037 and up to at least four in the case of Cap0036) in a two-dimensional (2-D) gel analysis of acidogenic cells, suggesting posttranslational modifications, such as phosphorylation, acetylation, glycosylation, or methylation (13).
FIG 1 .

Construction of CA_P0037::int mutants. (A) DNA configuration of mutant with a group II intron inserted at position 189/190 on the sense strand of CA_P0037. (B) PCR on genomic DNA of CA_P0037::int mutant (MT) and wild-type (WT) strains with different pairs of primers (intron probe-F and CAP0037-Fb primers and CAP0037-Rb and CAP0037-Fb primers) to confirm intron insertion and primers LtrA 5D and LtrA 3R to demonstrate the loss of pCUI1-cap37(189s). PCR was performed on genomic DNA of complemented mutant (MTC) strain with primers pthla-MF and cap37-sfo1-R to demonstrate the presence of the pSOS95-Cap0037 plasmid. (C) Detection of the Cap0037 protein in the wild type (WT), CA_P0037::int mutant (MT), and CA_P0037::int complemented mutant (MTC). The Western blot was generated using polyclonal antibodies raised against purified His-tagged Cap0037. The positions of molecular mass markers (in kilodaltons) are shown to the left of the Marker blot. (D) Southern blot to demonstrate single intron insertion in the CA_P0037::int mutant. The intron probe was DIG labeled and hybridized to EcoRI-digested genomic DNA of the CA_P0037::int mutant (lane 1) and to the AgeI-SbfI-digested pCUI1-cap37(189s) plasmid as a positive control (lane 2) with expected sizes of 4,388 bp and 1,718 bp, respectively.
In order to investigate the roles of these proteins in the acidogenic state of C. acetobutylicum, we inactivated CA_P0037 via the mobile group II intron. The regulatory role of Cap0037 and the observed phenotypes will be discussed in light of the different -omic data sets. DNA footprinting was used to determine DNA binding motifs of Cap0037 on its own promoter and in the upstream region of selected genes shown to be up- or downregulated. Putative Cap0037 binding sites across C. acetobutylicum genomes were analyzed via a bioinformatic approach. This study builds a foundation to further investigate the regulatory mechanism of this novel protein in C. acetobutylicum, which should be beneficial for understanding the role of homologous proteins in other members of the phylum Firmicutes.
RESULTS AND DISCUSSION
Phylogenetic tree and bioinformatic analysis of Cap0037/Cap0036.
Cap0037 and Cap0036 are proteins of unknown function. They are well conserved in other Firmicutes such as Clostridium, Bacillus, and Geobacillus (see Fig. S1 in the supplemental material). The functions of these two conserved proteins have not been characterized.
Cap0036 was found to be homologous to some cytoplasmic proteins of the Bacillus species and also to some bactofilin family proteins from the Geobacillus species (see Fig. S1A in the supplemental material). However, no transmembrane domains could be identified in Cap0036 using TMpred (15) and OCTOPUS (16), indicating that Cap0036 is most probably a cytoplasmic protein, rather than a membrane protein of the bactofilin family.
Noticeably, Cap0037 is homologous to the YgaS-like protein in Bacillus cereus G9241 (a putative DNA-binding protein), although the relationship is distant (see Fig. S1B in the supplemental material). A helix-turn-helix DNA binding motif (ISKKELLEITHISYGQLYRWKR) was found in the N terminus of the Cap0037 protein sequence (by the NPS@ web-based biotool [17]). As a result, Cap0037 is likely a novel transcriptional regulator. The 1:1 expression ratio of Cap0037 and Cap0036 (12) suggested an interaction of these two proteins that forms a system acting as an unknown regulatory mechanism in C. acetobutylicum. Interestingly, a transmembrane domain (LIRKLGIAICFLMLIPNEIYI) and an outer membrane region (ENTASLVLKVNARECLEELKSKLSL) were found at the C terminus of Cap0037 (OCTOPUS program [16]).
In this study, CA_P0037 was inactivated, and the effect of CA_P0037 disruption was elucidated at the system level.
Disruption of CA_P0037 by pCUI-Cap0037 (189s).
The ClosTron technique was first applied (18, 19) to inactivate CA_P0037 targeting an insertion into the 277/278 antisense of the CA_P0037 coding sequence (CDS), but targeted intron insertion was not detected in all of the erythromycin-resistant clones obtained. Therefore, we used another intron-based method developed in our laboratory (20) for knocking out CA_P0037 based on the pCUI-cap0037 vector as mentioned in Materials and Methods. The mutant was successfully obtained with the use of plasmid pCUI-Cap0037: a 1-kbp-long intron was inserted at position 189/190 in the sense strand of CA_P0037. PCR using two external primers flanking the targeted position confirmed the insertion of the intron into the gene (Fig. 1B). After selecting for the CA_P0037::int mutant, the strain was subjected to 5′-fluorouracil (5′-FU) selection to cure the pCUI-Cap0037 plasmid to avoid intron splicing by LtrA (the LtrA-encoding gene is present on the plasmid backbone). Loss of the plasmid was validated by sensitivity to erythromycin and by PCR (Fig. 1B). Southern blotting was conducted to confirm a single intron insertion into the genome (Fig. 1D).
Quantitative transcriptional analysis of the CA_P0037::int mutant under acidogenic chemostat culture conditions showed that the numbers of CA_P0037 and CA_P0036 mRNA molecules per cell (88 and 76, respectively) were similar to those in the wild-type (WT) strain (78 for both), indicating that the intron insertion has no polar effect and does not affect the stability of the transcript. Such results were expected as, in contrast to the ClosTron, the short intron that was inserted does not contain any terminator of transcription. On the other hand, when the level of Cap0037 protein was measured by Western blotting under acidogenesis conditions, no protein was detected for the CA_P0037::int mutant (Fig. 1C). However, when the CA_P0037::int mutant was transformed with the pSOS95-Cap0037 plasmid to express the CA_P0037 gene under control of the thlA promoter under acidogenesis conditions, both the production of Cap0037 and the phenotype (as will be seen below) were restored.
Carbon and electron fluxes of the CA_P0037::int mutant under different physiological conditions.
Acidogenesis and alcohologenesis were the two states that were significantly changed in the CA_P0037::int mutant. The normalized metabolic fluxes were compared to previously published data for the control strain (12).
In the acidogenic condition, the mutant strain underwent a profound change in its metabolism: the normalized fluxes of butyrate and acetate decreased 2.5- and 5.7-fold, respectively, while the normalized fluxes of lactate and butanol increased 85- and 9.7-fold, respectively (Fig. 2A). The normalized fluxes of ethanol, on the other hand, remained unchanged. In a complementation experiment, the CA_P0037::int mutant was transformed with the pSOS95-Cap0037 plasmid to express the CA_P0037 gene under control of the thlA promoter. When this strain was evaluated in acidogenic chemostat culture in the presence of clarithromycin (40 µg/ml) for the maintenance of the plasmid, the normalized metabolic fluxes (Fig. 2A) were not significantly different from those of the wild-type strain (12), demonstrating that the CA_P0037::int mutant phenotype was attributable only to CA_P0037 inactivation. The production of butanol by the CA_P0037::int strain under acidogenesis conditions is explained by the higher adhE2 expression (~50-fold higher than the control strain, with 21 mRNA molecules/cell) (12) (see Data Set S1 in the supplemental material), and although the expression of the sol operon (ctfA, ctfB, and adhE1) was increased ~6-fold, the number of mRNA molecules remained low (0.5 mRNA molecule/cell) compared to adhE2 (21). The increase in lactate formation was associated with an 83-fold increase in ldhA expression (12) (Data Set S1). For the CA_P0037::int mutant, the acetate flux decreased 5.7-fold compared to the control strain, although pta-ack (CA_C1742–CA_C1743) expression was unchanged (Data Set S1). Thus, flux is controlled at the enzyme level via a decrease in the acetyl coenzyme A (acetyl-CoA) pool, probably due to the twofold-higher expression of most of the genes (CA_C2708–CA_C2712) coding for the enzymes converting acetoacetyl-CoA to butyryl-CoA.
FIG 2 .

Metabolic fluxes of the CA_P0037::int mutant and complementary strain versus control strain under acidogenesis conditions. (A) Carbon fluxes; (B) electron flux. All values are normalized to the flux of the initial carbon source (millimoles per gram [dry cell weight] per hour). Glucose flux is normalized and set at 100. The values of the different strains are shown in color as follows: the corresponding mutant in blue, the complementary strain in purple, and the control strain in green. The control data were from reference 12. Metabolic fluxes of the CA_P0037::int mutant versus control strain under alcohologenesis and solventogenesis conditions can be found in the supplemental material. Abbreviations: Fdox and Fdred, oxidized and reduced ferredoxin, respectively; OAA, oxaloacetate; αKG, α-ketoglutarate.
The alcohologenesis state (pH 6.3, in a mixture of glucose and glycerol) was the second most affected state following the disruption of CA_P0037 (see Fig. S2 in the supplemental material). Although all of the glucose provided in the medium was consumed, the normalized flux of glycerol consumption was reduced more than 15-fold compared to the control strain. While the control strain produced mainly ethanol (normalized flux of 8.6%), butyrate (normalized flux of 25.1%), and butanol (normalized flux of 61.1%), the mutant produced mainly acids: butyrate (normalized flux of 63.6%), acetate (normalized flux of 21.9%), and lactate (normalized flux of 11.8%) (Fig. S2A). The ethanol and butanol fluxes were 1.8-fold and 9.8-fold lower than those in the parental strain, respectively, suggesting that, in the mutant, the alcohologenesis state was barely induced, and it behaved like the control strain in the acidogenesis state. The lower glycerol consumption of the CA_P0037::int strain is explained by the lower expression (four- to sixfold decrease) of the gene cluster coding for glycerol transport and utilization (CAC1319–CAC1323). The increase in lactate formation was associated with a 32-fold increase in ldhA expression, and the lower production of butanol was associated with 10-fold-lower adhE2 expression. On the other hand, the butyrate flux increase was not associated with a change in the expression of ptb-buk (CA_C3074–CA_C3075), which remained unchanged (12) (Data Set S1). Thus, flux is controlled at the enzyme level via an increase in the butyryl-CoA pool, probably due to the 10-fold-lower level of expression of adhE2.
The carbon flux of the mutant strain under solventogenesis conditions was compared to previously published data for the control strain. The CA_P0037::int mutant was not able to consume all glucose, and therefore, the titers of products were lower, but the fluxes were not significantly different from those of the wild-type strain (see Fig. S2B in the supplemental material). The level of expression of both the sol operon and the adhE2 gene were higher in the mutant (two- and fourfold, respectively), although the number of mRNA molecules for adhE1 (12 molecules/cell) was much higher than for adhE2 (~1 molecule/cell). Surprisingly, the expression of adc was downregulated threefold in the mutant, although the number of mRNA molecules per cell (4.3) remained high.
The normalized electron fluxes were analyzed for the CA_P0037::int mutant under three different conditions. In acidogenesis, the primary use of reduced ferredoxin was switched from hydrogen to NADH production in response to the high expression of adhE2 and ldhA in the mutant (Fig. 2B). In this metabolic state, the normalized hydrogen production fluxes decreased ~3-fold, while the normalized fluxes of NADH production from reduced ferredoxin increased 2.4-fold. The decrease in hydrogen production was not attributable to the expression of hydA (CA_C0028), which remained unchanged, but to the lower flux of reduced ferredoxin production, as due to high ldhA expression, lactate became the major product. The 2-fold increase in the normalized ferredoxin-NAD+ reductase fluxes might be explained by the 500-fold-higher expression of CA_C0587 encoding the major flavodoxin and the 1.5-fold-lower expression of CA_C0303 encoding the major ferredoxin, as well as the possibility that reduced flavodoxin might be a better substrate than ferredoxin for the ferredoxin-NAD+ reductase. In solventogenesis, the normalized electron fluxes were not significantly changed in agreement with the conserved product pattern (see Fig. S2D in the supplemental material). In alcohologenesis, butyrate was the main fermentation product of the CA_P0037::int mutant, and the normalized hydrogenase flux increased 1.4-fold, while the normalized ferredoxin-NAD+ reductase fluxes decreased 2.6-fold (Fig. S2C). The high normalized hydrogen production and low normalized ferredoxin-NAD+ reductase fluxes and normalized butanol fluxes can be explained by the fivefold downregulation of CA_C3486 encoding a potential multimeric flavodoxin and the proposed physiological role (12) in alcohologenic culture of the wild-type strain, providing a better substrate for the ferredoxin NAD+ reductase than for hydrogenase.
Determination of the Cap0037 DNA binding site.
To study the possible regulatory function of Cap0037 in central metabolism, we analyzed the ability of Cap0037 to bind to the promoter regions of several genes coding for key enzymes of the central metabolism of C. acetobutylicum that were either upregulated (CA_P0037/CA_P0036, ldhA, the sol operon, and fld1 [CA_C0587]) or downregulated (adc) in the CA_P0037::int mutant under acidogenesis conditions. A shift was observed only for the promoter regions of CA_P0037/CA_P0036 (Fig. 3A), the sol and adc operon (see Fig. S3 in the supplemental material), indicating that the upregulation of ldhA and fld1 (CA_0587) was an indirect effect of CA_P0037 inactivation (data for ldhA and fld1 promoters are not shown).
FIG 3 .
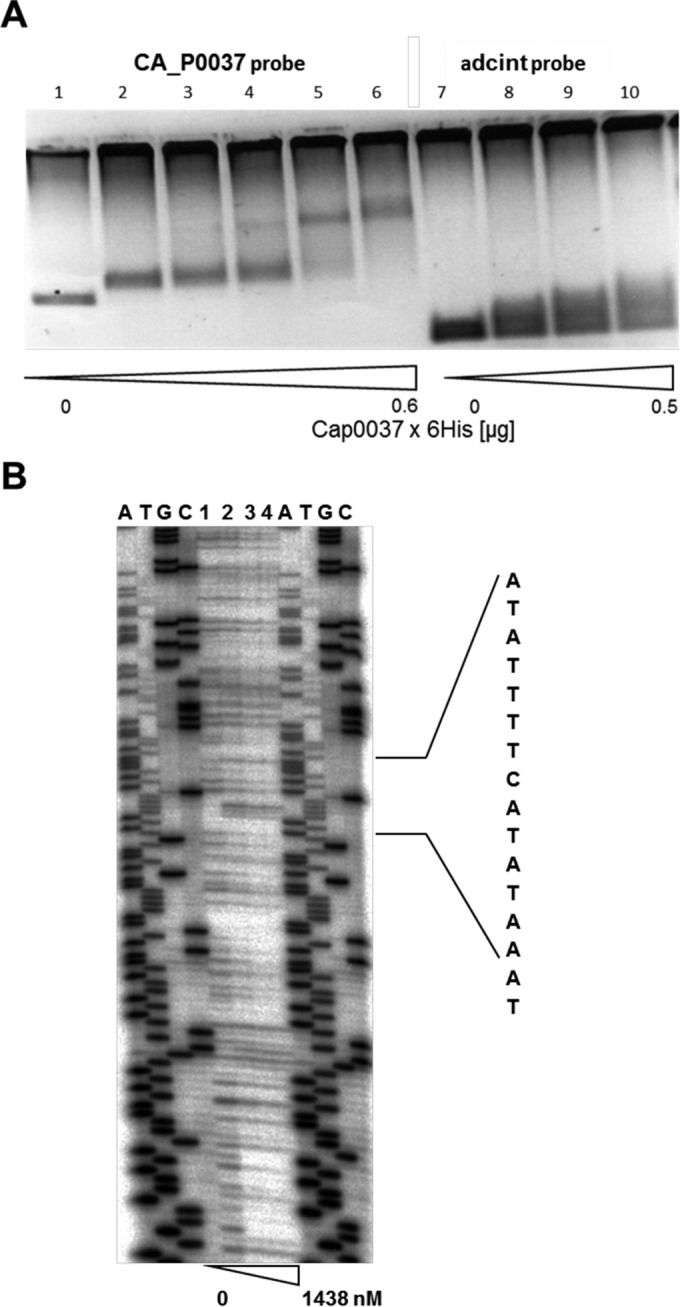
Analysis of Cap0037 binding to the promoter region of the CA_P0037-CA_P0036 operon. (A) EMSAs using the promoter region of the CA_P0037-CA_P0036 operon and Cap0037 protein. Lanes 1 to 6, 0, 0.1, 0.2, 0.3, 0.5, and 0.6 µg protein, respectively; lanes 7 to 10, 0, 0.1, 0.2, and 0.5 µg protein, respectively. (B) DNase I protection assay (DNA footprinting) of Cap0037 interacting with the CA_P0037/CA_P0036 promoter region (probe 85). End-labeled DNA fragment carrying the promoter region of the CA_P0037-CA_P0036 operon was incubated with different concentrations of Cap0037, subjected to DNase I cleavage, and analyzed on a sequencing gel. Sequencing reaction was performed with plasmid pDrive_86kurz. Lanes 1 to 4 contain 0 to 1,438 nM Cap0037. The assigned region on the right side indicates the region protected by Cap0037 and the respective sequence.
We used DNA footprinting to determine the DNA binding sites (BSs) of Cap0037 in the promoter regions of CA_P0037 (Fig. 3B) and in the sol operon and adc gene (see Fig. S4 in the supplemental material). Interestingly, Cap0037 was found to bind to the 5′-ATATTTTCATATAAAT-3′ sequence which overlaps the putative −10 region of the CAP0037-CAP0036 operon promoter (determined by the BPROM tool [21]), suggesting that Cap0037 might repress its own transcription. This hypothesis is in agreement with the derepression (50.7- and 68.6-fold higher than the levels of expression of the control) of the CA_P0037-CA_P0036 operon observed under alcohologenesis and solventogenesis conditions of the CA_P0037::int mutant. Two binding sites were found in the adc promoter region (Fig. S4A and B). The first was 5′-ATAAGTTTATATAAAT-3′, located upstream of −35, which contains three mismatches compared to the binding site (Fig. 4) found in the CA_P0037 promoter (Fig. 5). Cap0037 could potentially bind to this sequence to activate the expression of adc, which fit with the transcriptomic data for the CA_P0037::int mutant, in which adc was downregulated under all conditions. The second binding site in the adc promoter region, 5′-TAATGTAAATATAAAT-3′, is less conserved compared to the CAP0036-CAP0037 operon promoter binding with five mismatches, and it is located between the −10 and +1 promoter region (Fig. 5), suggesting that it might also be involved in the repression of adc under conditions that have not been characterized yet. It is worth noting that a Spo0A~P binding box was found upstream of the −35 region of the adc promoter (7). This box is only 6 bp downstream of the first Cap0037 binding site (Fig. 5). Hence, there might be some synergy between Spo0A~P and Cap0037 for the activation of adc.
FIG 4 .
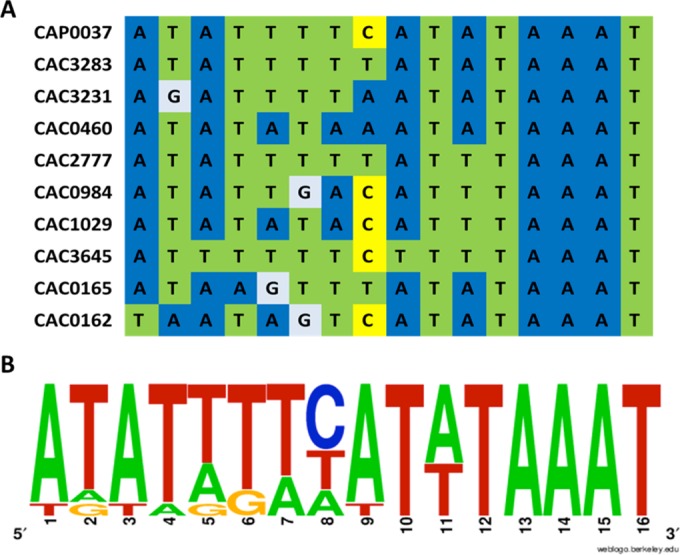
Alignment of putative Cap0037 binding sequence (A) and sequence logo showing the frequencies of residues (B).
FIG 5 .

DNA sequence of the 5′ flanking region of CA_P0037 (A), adc (B), and sol operon (C). Cap0037 binding sites (Cap37 BS) determined in this study are shown in red boxes. Spo0A binding sites (Spo0A BS) are shown in orange boxes (7, 32). Promoter −35 and −10 elements are shown in blue (7, 33). Putative −35 and −10 sequences of the CA_P0037-CA_P0036 operon were analyzed by the BPROM tool. Three imperfect repeats in the sol promoter are shown in green letters (7). Coding sequences (CDS) are shown in brown.
In the sol promoter region, Cap0037 binds to the 5′-TAAATATACTGATAAT-3′ sequence located upstream of the Spo0A~P binding box and the −35 region of the sol operon promoter (Fig. 5). Interestingly, this Cap0037 binding site overlapped the R1 sequence (5′-TCTAAATATA-3′) that has been proposed (22) to be the binding site of an additional activator protein, which acts in concert with Spo0A~P to activate the transcription of the sol operon. Binding of Cap0037 would prevent the binding of this activator and would explain the upregulation of the sol operon in the CA_P0037::int mutant in acidogenesis and solventogenesis (see Data Set S1 in the supplemental material).
Using these experimentally validated binding sequences, we can search for putative Cap0037 binding sites across the genome using the RegPredict tool.
Determination of putative Cap0037 binding sites across the C. acetobutylicum genome.
The 5′-ATATTTTCATATAAAT-3′ sequence and the conserved motif were used as references for the construction of a putative Cap0037 regulon by whole C. acetobutylicum genome analysis using RegPredict. The sequences with scores higher than 5.0 (which allow three mismatches or less) were selected and further analyzed for their relative position to the CDS of the corresponding gene. In addition to the three Cap0037 binding sites previously identified in the electrophoretic mobility shift assays, eight other putative Cap0037 binding sites were discovered (see Table S2 in the supplemental material). Of these eight sites, the transcription of the genes potentially regulated by Cap0037 binding was analyzed for the CA_P0037::int mutant and compared to the wild type in the three metabolic states.
The first putative Cap0037 binding site was located between the −35 and −10 promoter regions of an operon (CA_C1029-CA_C1032) encoding an iron transport system of the Feo family. This operon was strikingly upregulated under all conditions in the CA_P0037::int mutant. Previously, this operon was shown to be controlled by Fur (encoded by CA_C1682), the ferric uptake regulator, and was found to be highly induced by iron limitation or in the fur::int mutant (6). There is one Fur box located downstream of the −10 promoter region of CA_C1029-CA_C1032 (6). However, expression of the Fur-encoding gene was unchanged in the CA_P0037::int mutant under all continuous culture conditions. Thus, Cap0037 might be the second repressor of this iron transport system, raising the question of how Cap0037 and/or Cap0037 together with Fur carry out a regulatory role.
The second putative Cap0037 binding site was located 7 bp upstream of the −35 promoter region of an operon (CA_C2777-CA_C2778) encoding a glutaredoxin and a rubredoxin. This operon was upregulated under all conditions in the CA_P0037::int mutant. Previously, this operon was shown to be controlled by PerR (encoded by CA_C1682), the peroxide repressor, and was found to be induced by oxygen exposure or in a ΔperR mutant (5). There is one PerR box located downstream of the −10 promoter region of the CA_C2277-CA_C2278 operon. Thus, in addition to the iron transport discussed above, which could be regulated by the Cap0037 and Fur proteins, we have potentially identified another operon that can be controlled by two regulators, Cap0037 and PerR.
The third and fourth putative Cap0037 binding sites were in the promoter regions of two genes encoding potential transcriptional regulators of the MarR family for CA_C3283 and of a CRO repressor-like DNA-binding protein for CA_C3645. The Cap0037 binding site was located downstream of the transcription start point (TSP) of CA_C3283, which is part of a three-gene operon (CA_C3283-CA_C3281) including two genes encoding a potential ABC transporter. All three genes were upregulated in the Cap0037::int mutant under acidogenesis and alcohologenesis conditions. The regulatory effects of Cac3283 on the transcription of the two genes encoding the ABC transporter and the role of this ABC transporter are still unknown. CA_C3645 is expressed as a monocistronic operon, and the Cap0037 binding site was located downstream of the TSP. Its transcription was upregulated under acidogenesis conditions only in the CA_P0037::int mutant.
The last four putative Cap0037 binding sites were located in the promoter region of a three-gene operon (CA_C0984-CA_C0986) encoding membrane proteins of unknown function, a three-gene operon (CA_C0460-CA_C0462) involved in butanoate metabolism, a gene (CA_C3231) encoding a potential HAD (haloacid dehalogenase-like) type of phosphatase, and the CA_C0091 gene (encoding ketol-acid reductoisomerase, involved in valine, leucine, and isoleucine biosynthesis). However, the transcription levels of all of these genes were poorly affected in the CA_P0037::int mutant (see Table S2 in the supplemental material).
CA_P0037 inactivation affects the expression of the Rex, Fur, and PerR regulons.
The global effects of CA_P0037 inactivation on C. acetobutylicum metabolism are presented in the supplemental material, and we will concentrate here on how CA_P0037 inactivation affects the expression of the Rex, Fur, and PerR regulons. The redox-sensing transcriptional repressor (Rex) has been found to modulate its DNA binding activity in response to the NADH/NAD+ ratio and plays a role in the alcohologenic shift of C. acetobutylicum. Rex binding sites were determined in the promoters of different operons, the adhE2, ldhA, crt, thlA, asrT, ptb, and nadA operons (10, 11). Those operons were upregulated in the Rex mutant. The ldhA gene was the most induced gene, followed by adhE2 (11). In the CA_P0037::int mutant, Rex expression was only 1.4-fold higher in acidogenesis and was unchanged in alcohologenesis and solventogenesis. However, except for the thlA gene, all of the other genes of the Rex regulon, such as adhE2, ldhA, crt-bcd-etfAB-hbd, nadABC, and ptb-buk, were upregulated to various degrees (see Table S3 in the supplemental material). As there are no putative Cap0037 binding sites in the promoter regions of all the genes or operons shown to be controlled by Rex, it is tempting to speculate that the effect of CA_P0037 inactivation might be indirect and occur through an increase in the NADH/NAD+ ratio. The fact that thlA was unchanged or slightly downregulated in the CA_P0037::int mutant but highly upregulated in the Rex mutant suggests a more complex regulatory mechanism of thlA expression.
In a perR mutant or after the wild-type strain is exposed to oxygen, the genes encoding proteins involved in oxygen and reactive oxygen species (ROS) detoxification, including the reverse rubrerythins (rbr3A-rbr3B), desulfoferrodoxin (dfx), glutaredoxin (CA_C2777), rubredoxin (rd and CA_C2778), NADH-dependent rubredoxin oxidoreductase (nror), the oxygen-reducing flavoproteins (fprA1 and fprA2), and a flavodoxin (CA_C2452), were upregulated (5). The CA_P0037::int mutant behaved quite similarly to the perR mutant under acidogenic and alcohologenic conditions (see Table S4 in the supplemental material). As shown above, a putative Cap0037 binding site was located 7 bp upstream of the −35 promoter region of CA_C2777-CA_C2778 encoding a glutaredoxin and a rubredoxin, respectively. However, no putative Cap0037 binding sites were identified in the promoter regions of all of the other genes or operons shown to be controlled by PerR, and it is probable that the effect of CA_P0037 inactivation is indirect.
Interestingly, we also found a common regulatory effect of Cap0037 with the fur mutant. Table S4 in the supplemental material compares the CA_P0037::int mutant with the fur mutant and the wild-type strain under iron limiting conditions. One of the most critical similarities is the overexpression of genes involved in iron transport and metabolism. As discussed above, the ferrous transport system FeoA (encoded by CA_C1029-CA_C1032), which contains a putative Cap0037 box and a Fur box in the promoter, was strikingly overexpressed in the CA_P0037::int mutant, the Fur mutant, and in the iron-limited WT. The second putative ferrous iron uptake system FeoB (encoded by CA_C0447-CA_C0448) for Fe2+ and the ferrichrome-dependent system (encoded by CA_C0788-CA_C0791) were also highly upregulated in all cases, although less significantly in the iron-limited WT. A flavodoxin (fld1)-encoding gene (CA_C0587) was strikingly upregulated in the iron-limited WT, the fur mutant, and the CA_P0037::int mutant (Table S4). A Fur box was found in the promoter of fld1 (CA_C0587), but no putative Cap0037 box was identified. It has been well documented that the ferredoxin concentration is reduced dramatically in clostridia under low iron conditions, fld1 is induced, and flavodoxin replaces ferredoxin as a single electron carrier. However, in the CA_P0037::int mutant, the expression level of fdx (CA_C0303), encoding the main ferredoxin, was not reduced. In iron deficiency or in a fur mutant or CA_P0037 inactivation, the levels of genes belonging to the ribGBAH operon, which is involved in riboflavin synthesis, are strongly induced. Riboflavin biosynthesis genes were previously shown to be upregulated only by butyrate stress and not by acetate or butanol stress (23). Riboflavin is a precursor of the flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD) coenzymes. The expression of the rib operon was shown to be regulated by an FMN-sensing riboswitch element (24) in Bacillus subtilis, but the regulatory mechanism in C. acetobutylicum has yet to be characterized.
To summarize, the CA_P0037::int mutant cells behave as if they are Fe2+ limited, although they are in excess iron conditions. However, the upregulated Fur regulon in the mutant is an indirect regulatory effect rather than reflecting a real shortage in the intracellular Fe2+ concentration, and except for the FeoA-encoding operon (CA_C1029-CA_C1032), no Cap0037 box was identified in the promoter regions of the upregulated genes.
Conclusion.
Collectively, these results demonstrate that in the strict anaerobe C. acetobutylicum, Cap0037, beside controlling its own gene, directly or indirectly controls a large set of genes belonging to the Rex, PerR, and Fur regulons in addition to the sol operon and adc gene that encode genes directly involved in solvent formation. These controls drastically affect the primary metabolism of C. acetobutylicum. However, more experiments will be needed to understand the mechanism(s) behind the global regulation by Cap0037.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture media.
The bacterial strains, plasmids, and primers used in this study are listed in Table S1 in the supplemental material. C. acetobutylicum Δcac1502 Δupp and its derivative mutants were grown in clostridial growth medium (CGM) for routine manipulation and in synthetic medium (SM) for sporulation and the production of solvents. A reinforced clostridial agar (RCA) plate with antibiotic at a pH of 5.8 was used after electroporation, and a pH of 6.5 was used for the replication of transformants. The Escherichia coli strain was grown in Luria-Bertani (LB) medium at 37°C. Antibiotics were used at the following concentrations: erythromycin (Erm) and clarithromycin at 40 µg/ml and thiamphenicol (Tm) at 10 µg/ml for C. acetobutylicum and ampicillin at 50 µg/ml for E. coli.
Plasmid construction.
The CA_P0037::int mutant was successfully generated by using pCUI-Cap0037 (189s). This plasmid is derived from the pCUI plasmid, which was developed by our lab (20). The intron in this vector is smaller than in the pMTL007C-E2 system (18) because it does not contain a marker gene. The pCUI-Cap0037 (189s) plasmid was transformed into Clostridium acetobutylicum Δcac1502 Δupp by electroporation without prior methylation (25), and the transformants were selected on an RCA plate at a pH of 6.5 containing thiamphenicol at 40 µg/ml. In order to select for mutants, screening by PCR with two external primers must be performed. In the plasmid pCUI-Cap0037 (189s), the retargeting region was designed via the Perutka algorithm for the position 189/190 sense strand. In the presence of the ltrA gene, the integrants could lose the intron due to mRNA maturation. As a result, after screening and selecting for insertion, the plasmid pCUI-Cap0037 (189s) in the mutant was cured by spreading the cells on RCA at a pH of 6.5 containing 5′-fluorouracil (5-FU) at 1 mM (25). The colonies were then checked for plasmid loss by testing their sensitivity to Tm, Erm, and PCR using LtrA 5D and LtrA 3R primers that are specific for the vector backbone. The mutant CA_P0037::int was successfully generated by this second strategy.
For the complementation experiment, CA_P0037 CDS and its ribosomal binding site (RBS) were amplified by Cap0037-F-RBS BamHI and Cap0037-R-SfoI and cloned into a pSOS95 shuttle vector (26) by two restriction sites, BamHI and SfoI, to obtain the plasmid pSOS95-Cap0037. This pSOS95 vector allows the expression of CA_P0037 under the constitutive thlA promoter. The plasmid pSOS95-Cap0037 was then electroporated into the CA_P0037::int mutant and selected on an RCA plate with Erm at 40 µg/ml. The complementary strain carrying the plasmid was maintained in liquid SM culture under pressure from clarithromycin at 40 µg/ml.
Plasmid pBS2 was used for heterologous production of Cap0037. For construction of plasmid pBS2, the 630-bp CA_P0037 open reading frame was amplified by PCR using primers ygaS1hinCl and yghSrückCl with C. acetobutylicum WT genomic DNA as the template. The PCR product was digested with NdeI and HindIII and ligated into digested vector pET29a(+). Vector pET29a(+) contains a His tag sequence that is added to the C terminus of the produced protein.
Plasmids pDrive_86kurz, pDrive_144, pDrive_adcStart, and pDrive_sol were used for sequencing reaction of DNase I protection assay (DNA footprinting). For construction of pDrive_86kurz, pDrive_144, pDrive_adcStart, and pDrive_sol, probes 85 (256 bp), 144 (249 bp), adcStart (263 bp), and sol (245 bp) were amplified by PCR using genomic DNA from C. acetobutylicum WT as the template and primers 86 fwd (fwd stands for forward) and 85 rev (rev stands for reverse), 144 rev and 145 fwd, adcstart FP EcoRI fwd and adcstart FP EcoRV rev, and sol FP EcoRI fwd and sol FP EcoRV rev, respectively. PCR products were ligated into pDrive cloning vector. The generated PCR probes, probes 85 (256 bp), 144 (249 bp), adcStart (263 bp), and sol (245 bp), which contain parts of respective promoter regions, were used as probes for DNase I protection assay (DNA footprinting).
Continuous culture.
Continuous fermentation was performed as previously described (12). The cultures were maintained under acidogenesis, solventogenesis and alcohologenesis conditions, fed with 995 mM total carbon, and maintained at a dilution rate of 0.05 h−1. Samples for the production profile analysis and mRNA extraction were prepared under steady-state conditions. Results for the Clostridium acetobutylicum Δcac1502 Δupp Cap0037::int mutant were compared with the control strain Clostridium acetobutylicum Δcac1502 Δupp (12).
For the complementary strain, continuous culture under acidogenic conditions was performed in the presence of clarithromycin at 40 µg/ml for a short period to avoid losing the plasmid. The production profile was analyzed and compared with the CA_P0037::int mutant.
Isolation of total mRNA and microarray.
Total RNA was isolated from continuous cultures using the RNeasy kit protocol (Qiagen, Hilden, Germany). The protocol and microarray slides used in this study were the same as described elsewhere (12) to obtain comparable results.
Analytical methods.
The concentrations of the substrate and fermentation products were measured by high-pressure liquid chromatography (HPLC) (Agilent 1200 series, Massy, France). The separation was obtained with an Aminex HPX-87H column (300 by 7.8 mm) (Bio-Rad), at 14°C with 0.5 mM H2SO4 at a flow rate of 0.5 ml/min. Detection was achieved by reading the refractive index and UV absorbance (210 nm).
Southern blot analysis.
Genomic DNA of the CA_P0037::int mutant was digested with EcoRI restriction enzyme. The probe for hybridization was prepared by PCR using Intron Probe-F (F stands for forward) and Intron Probe-R (R stands for reverse) primers and was then labeled using the DIG High Prime DNA labeling and detection starter kit II (Roche). The protocols for hybridization and detection were performed according to the Roche instructions.
Expression and purification of His-tagged protein.
E. coli BL21(DE3) cells (Novagen R and D Systems) containing pBS2 was used for overproduction of protein Cap0037. The cells were grown aerobically at 37°C in LB medium supplemented with either kanamycin (50 µg/ml) or ampicillin (100 µg/ml) to an optical density at 600 nm (OD600) of 0.7 and induced by the addition of 1 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) at 37°C until stationary phase was reached. The cells were then harvested by centrifugation (4,300 × g, 10 min, 4°C) and washed twice with buffer containing imidazole (10 mM). The weight of the cell pellet (wet weight) was determined and solubilized in 3 volumes of the buffer mentioned above. The cells were disrupted using a French pressure cell (4 MPa) (French Pressure Cell/Press, G. Heinemann Ultraschall und Labortechnik, Schwäbisch Gmünd, Germany). The fusion protein was purified by nickel-nitrilotriacetic acid (NTA) affinity chromatography using Ni-NTA agarose (Qiagen GmbH, Hilden, Germany). For elution, imidazole buffers with 75, 100, 200, and 250 mM imidazole were used. The purity of the protein was analyzed by SDS-polyacrylamide gel electrophoresis, and protein concentration was quantified by Pierce BCA protein assay kit (Pierce, Rockford, IL, USA) according to the instructions provided by the manufacturer.
Electrophoretic mobility shift assays (EMSAs).
For electrophoretic mobility shift assays (EMSAs) of the adc, CA_P0037/CA_P0036, and sol operon, the respective DNA probes were amplified by PCR using genomic DNA from C. acetobutylicum ATCC 824 as the template. For amplifying the 5′ untranslated region of the adc gene, the adc probe (318 bp), primers adc_forward and adc_reverse (see Table S1 in the supplemental material) were used. For EMSAs to the 5′ untranslated region of the CA_P0037-CA_P0036 operon, the CAP0037 probe (539 bp), primers 94_fwd and PEX1_kurz were used. For EMSAs to the 5′ untranslated region of the sol operon, the sol probe (219 bp) and primers sol_frag_F and sol_frag_R were used. The adcint probe (233 bp) was used as a negative control and was amplified out of the internal region of the adc gene, using primers adcmit1 and adcmit2. In the binding assays, DNA probes were mixed with increasing amounts of the respective fusion proteins in 20-µl reaction mixtures with 0.5 µl poly(dI-dC) (2 µg/µl) and 2-µl band shift buffer (200 mM Tris, 500 mM Na glutamate, 100 mM MgCl2 ⋅ 6H2O, 63.6 mM EDTA, 0.5% Nonidet P-40 [vol/vol], 50% glycerol [vol/vol]). After incubation for 20 min at room temperature, samples were loaded on a 2% agarose gel in 1× TAE buffer (200 mM Tris, 100 mM acetate, 5 mM EDTA [pH 7.5]) and separated by electrophoresis (70 V, 50 min). The gel was stained using ethidium bromide.
For electrophoretic mobility shift assays of ldhA and fld1, PCR DNA fragments of ldhA and fld1 promoters were generated by two pairs of primers (ldh-prom-F [prom stands for promoter] and ldh-prom-R primers and fld1-prom-F and fld1-prom-R primers) and DIG labeled (with DIG High Prime DNA labeling and detection starter kit II [Roche]). Binding assays were done as described above. The detection of DNA shifts was performed according to Roche protocol.
DNase I protection assays (DNA footprinting).
To determine the exact binding sites of protein Cap0037 at the 5′ untranslated regions of the CA_P0037-CA_P0036 operon, adc operon, and sol operon, different DNA fragments (probes) were amplified by PCR (see “Plasmid construction” above3.2). The primers generated restriction sites at both ends of the probes (see “Plasmid construction” above3.2). The probes were radioactively 5′ labeled using [γ-32P]ATP. Labeling was performed in a 25-µl reaction mixture with 20 pmol DNA, 2 µl polynucleotide kinase (10 U/µl) (Thermo Scientific, St. Leon-Rot, Germany), 2.5 µl reaction buffer (10× binding buffer [see below] and 2 µl bovine serum albumin [BSA] 100 mg/ml [wt/vol]), and 4 pmol [γ-32P]ATP (20 µCi). The reaction mixture was incubated at 37°C for 1 h. One end of the DNA was digested using EcoRV, removing a 5′-labeled phosphate. Purification of the labeled DNA was carried out by Microspin G-25 columns (GE Healthcare, Buckinghamshire, United Kingdom) according to the instructions of the manufacturer. For protein binding, the respective radiolabeled DNA probe was mixed with increasing amounts of Cap0037 in a 20-µl reaction mixture together with 0.5 µl poly(dI-DC) (2 µg/µl) and 2 µl of10× binding buffer (500 mM Tris, 500 mM KCl, 50 mM MgCl2 ⋅ 6H2O, 10 mM EDTA). After incubation for 30 min at 37°C, 1.1 µl buffer R1 (120 mM MgCl2 ⋅ 6H2O, 120 mM CaCl2 ⋅ 2H2O) and 0.1 U DNase (Thermo Scientific, St. Leon-Rot, Germany) were added. The reaction was stopped after just 1 min by adding 1 µl EDTA (0.5 M, pH 8) and placing the reaction mixture on ice. The sample was purified by phenol chloroform extraction, and the DNA was ethanol precipitated and suspended in 3 µl of sequencing gel loading buffer (30 mg bromophenol blue, 30 mg xylene cyanole, 200 mM EDTA [pH 8], and 10 ml formamide). After an incubation at 80°C for 2 min, the DNase I digestion product was loaded onto a 7 M urea−6% polyacrylamide gel and separated at 1,500 V, 46 mA, and 60 W for 2 h. The dideoxy sequencing reaction was performed using the respective plasmid (see “Plasmid construction” above3.2) and T7 sequencing kit (Affymetrix, Santa Clara, CA, USA), and run in the same gel as the footprint sample. The gel was vacuum dried, exposed on Hyperfilm MP (Amersham Biosciences, Europe GmbH, Freiburg, Germany) for 24 h and developed with VCURIX 60V (AGFA Graphics GmbH & Co. KG, Düsseldorf, Germany).
Bioinformatic tools. (i) BLAST searches, amino acid sequence alignments, and phylogenetic analysis.
Identification of orthologs was performed using the BLAST tool provided by NCBI (27). Amino acid sequence alignment and the phylogenetic tree were processed using the phylogeny program (28).
(ii) Transmembrane protein prediction.
Cap0036 amino acid sequence was used as an input to find possible transmembrane regions using two bioinformatic web-based tools, TMpred (15) and OCTOPUS (16).
(iii) DNA binding motif search and promoter element prediction.
Amino acid sequences of Cap0037 were subjected to a search for the helix-turn-helix DNA binding motif using the NPS@ web-based biotool (https://npsa-prabi.ibcp.fr/) (17). To predict the promoters of different genes, BPROM web-based tool was used (21).
(iv) Putative Cap0037 regulon.
Construction of putative Cap0037 regulon was performed using an established comparative genomics method (29) implemented in the RegPredict webserver (http://regpredict.lbl.gov) (30) and Artemis Java Webstart (31). Genes with potential upstream binding sites that had high scores and/or were conserved were rebuilt to improve search accuracy. Genome sequences and annotations of C. acetobutylicum were obtained from GenBank (http://www.ncbi.nlm.nih.gov/GenBank/).
Western blot analysis.
Aliquots containing 100 µg of purified His-tagged Cap0037 were mixed with an equal amount of adjuvant (Specoll) and injected into a New Zealand White rabbit. Antiserum was collected 9 weeks after the first immunization. Western blot analyses were carried out with antiserum diluted 1,000-fold by using a standard protocol (32).
Microarray data accession number.
The microarray data can be accessed at GEO through accession numbers GSE81273 and GSE69973 for the CA_P0037::int mutant and the control strain (12), respectively.
SUPPLEMENTAL MATERIAL
Transcriptomic and proteomic data Download
Phylogenetic trees of Cap0036 (A) and Cap0037 (B) sequences from Clostridium acetobutylicum and their neighbor proteins from other bacteria. The neighbor proteins were selected by running BLAST on the NCBI database. Cap0036 and Cap0037 proteins from C. acetobutylicum are shown in blue. Download
Metabolic fluxes of mutant CA_P0037::int versus control strain in different metabolic states, such as alcohologenesis (AL) and solventogenesis (SO). (A and B) Carbon fluxes; (C and D) electron fluxes. All values are normalized to the flux of the initial carbon source (millimoles per gram [dry cell weight] per hour). Glucose flux is normalized and set at 100 for acidogenesis and solventogenesis, and the sum of glucose and half of the glycerol normalized as 100 for alcohologenesis. The values of the corresponding mutant are shown in blue, and those of the control strain are shown in green. The control data were from reference 12. Download
(A) EMSAs using the promoter region of adc and the Cap0037 protein. Lanes 1 to 8, 0, 0.2, 0.3, 0.5, 0.6, 0.7, 0.8, and 0.9 µg protein, respectively; lanes 9 to 12, 0, 0.2, 0.6, and 0.9 µg protein, respectively. (B) EMSAs using the promoter region of the sol operon and the Cap0037 protein. Lanes 1 to 5, 0, 0.28, 0.42, 0.7, and 1 µg protein, respectively; lanes 6 to 8, 0, 0.42, and 1 µg protein, respectively. Download
(A) DNase I protection assay I (DNA footprinting) of Cap0037 interacting with the adc promoter region (probe 144). End-labeled DNA fragment carrying the promoter region of the adc operon was incubated with different concentrations of Cap0037, subjected to DNase I cleavage, and analyzed on a sequencing gel. The sequencing reaction was performed with plasmid pDrive_144. (B) DNA footprinting of Cap0037 interacting with the adc promoter region (transcription start) (probe adcStart). End-labeled DNA fragment carrying the promoter region of the adc operon (transcription start) was incubated with different concentrations of Cap0037, subjected to DNase I cleavage, and analyzed on sequencing gels. The sequencing reaction was performed with plasmid pDrive_adcStart. (C) DNA footprinting of Cap0037 interacting with the sol promoter region (probe sol). End-labeled DNA fragment carrying the promoter region of the sol operon was incubated with different concentrations of Cap0037, subjected to DNase I cleavage, and analyzed on sequencing gels. The sequencing reaction was performed with plasmid pDrive_sol. Lanes 1 to 7 contain 0 to 2,308 nM Cap0037. The assigned region on the right side indicates the region protected by Cap0037 and the respective sequence. Download
Bacterial strains, plasmids, and primers used in this study
Prediction of the putative Cap0037 regulon and the corresponding relative transcript levels of those genes of the CA_P0037::int mutant in the three metabolic states, acidogenesis (AC), alcohologenesis (AL), and solventogenesis (SO). Orange letters are mismatched nucleotides compared to the binding box ATATTTTCATATAAAT in the CA_P0037/CA_P0036 promoter.
Relative transcript levels of genes belonging to the Rex regulon of the C. acetobutylicum CA_P0037::int mutant in the three metabolic states, acidogenesis (AC), alcohologenesis (AL), and solventogenesis (SO), and of the rexA mutant (data from reference 11). n.a., not available; n.d., not detected.
Relative transcript levels of selected genes of the CA_P0037::int mutant in the three metabolic states (acidogenesis [AC], alcohologenesis [AL], and solventogenesis [SO]). The transcript levels in the oxygen-exposed WT (+O2 WT), ΔperR mutant, iron-limited WT (−Fe WT), and fur::int mutant are shown. Data for the oxygen-exposed WT and the ΔperR mutant are from reference 5, and data for iron-limited WT and fur::int mutant are from reference 6. n.a., not available.
ACKNOWLEDGMENTS
We thank Sophie Lamarre and Lidwine Trouilh for help with the data analysis.
This work was financially supported by the European Community’s Seventh Framework Program “CLOSTNET” (PEOPLE-ITN-2008-237942) to Ngoc-Phuong-Thao Nguyen.
Footnotes
Citation Nguyen N-P-T, Linder S, Flitsch SK, Schiel-Bengelsdorf B, Dürre P, Soucaille P. 2016. Cap0037, a novel global regulator of Clostridium acetobutylicum metabolism. mBio 7(5):e01218-16. doi:10.1128/mBio.01218-16.
REFERENCES
- 1.Lütke-Eversloh T, Bahl H. 2011. Metabolic engineering of Clostridium acetobutylicum: recent advances to improve butanol production. Curr Opin Biotechnol 22:634–647. doi: 10.1016/j.copbio.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 2.Nair RV, Bennett GN, Papoutsakis ET. 1994. Molecular characterization of an aldehyde/alcohol dehydrogenase gene from Clostridium acetobutylicum ATCC 824. J Bacteriol 176:871–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atsumi S, Liao JC. 2008. Metabolic engineering for advanced biofuels production from Escherichia coli. Curr Opin Biotechnol 19:414–419. doi: 10.1016/j.copbio.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hillmann F, Fischer R-JJ, Saint-Prix F, Girbal L, Bahl H. 2008. PerR acts as a switch for oxygen tolerance in the strict anaerobe Clostridium acetobutylicum. Mol Microbiol 68:848–860. doi: 10.1111/j.1365-2958.2008.06192.x. [DOI] [PubMed] [Google Scholar]
- 5.Hillmann F, Döring C, Riebe O, Ehrenreich A, Fischer R-J, Bahl H. 2009. The role of PerR in O2-affected gene expression of Clostridium acetobutylicum. J Bacteriol 191:6082–6093. doi: 10.1128/JB.00351-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vasileva D, Janssen H, Hönicke D, Ehrenreich A, Bahl H. 2012. Effect of iron limitation and fur gene inactivation on the transcriptional profile of the strict anaerobe Clostridium acetobutylicum. Microbiology 158:1918–1929. doi: 10.1099/mic.0.056978-0. [DOI] [PubMed] [Google Scholar]
- 7.Ravagnani A, Jennert KCB, Steiner E, Grünberg R, Jefferies JR, Wilkinson SR, Young DI, Tidswell EC, Brown DP, Youngman P, Morris JG, Young M. 2000. Spo0A directly controls the switch from acid to solvent production in solvent-forming clostridia. Mol Microbiol 37:1172–1185. doi: 10.1046/j.1365-2958.2000.02071.x. [DOI] [PubMed] [Google Scholar]
- 8.Harris LM, Welker NE, Papoutsakis ET. 2002. Northern, morphological, and fermentation analysis of spo0A inactivation and overexpression in Clostridium acetobutylicum ATCC 824. J Bacteriol 184:3586–3597. doi: 10.1128/JB.184.13.3586-3597.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan Y, Liu ZZ-Y, Zheng H-J, Li F-L. 2015. Comparative transcriptome analysis between csrA-disruption Clostridium acetobutylicum and its parent strain. Mol Biosyst 11:1434–1442. doi: 10.1039/c4mb00600c. [DOI] [PubMed] [Google Scholar]
- 10.Wietzke M, Bahl H. 2012. The redox-sensing protein Rex, a transcriptional regulator of solventogenesis in Clostridium acetobutylicum. Appl Microbiol Biotechnol 96:749–761. doi: 10.1007/s00253-012-4112-2. [DOI] [PubMed] [Google Scholar]
- 11.Zhang L, Nie X, Ravcheev DA, Rodionov DA, Sheng J, Gu Y, Yang S, Jiang W, Yang C. 2014. Redox-responsive repressor Rex modulates alcohol production and oxidative stress tolerance in Clostridium acetobutylicum. J Bacteriol 196:3949–3963. doi: 10.1128/JB.02037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoo M, Bestel-Corre G, Croux C, Riviere A, Meynial-Salles I, Soucaille P. 2015. A quantitative system-scale characterization of the metabolism of Clostridium acetobutylicum. mBio 6:e01808-15. doi: 10.1128/mBio.01808-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janssen H, Döring C, Ehrenreich A, Voigt B, Hecker M, Bahl H, Fischer RJ. 2010. A proteomic and transcriptional view of acidogenic and solventogenic steady-state cells of Clostridium acetobutylicum in a chemostat culture. Appl Microbiol Biotechnol 87:2209–2226. doi: 10.1007/s00253-010-2741-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hönicke D, Lütke-Eversloh T, Liu Z, Lehmann DD, Liebl W, Ehrenreich A. 2014. Chemostat cultivation and transcriptional analyses of Clostridium acetobutylicum mutants with defects in the acid and acetone biosynthetic pathways. Appl Microbiol Biotechnol 98:9777–9794. doi: 10.1007/s00253-014-6040-9. [DOI] [PubMed] [Google Scholar]
- 15.Hofmann K, Stoffel W. 1993. TMbase - a database of membrane spanning protein segments. Biol Chem Hoppe Seyler 374:166. [Google Scholar]
- 16.Viklund H, Elofsson A. 2008. OCTOPUS: improving topology prediction by two-track ANN-based preference scores and an extended topological grammar. Bioinformatics 24:1662–1668. doi: 10.1093/bioinformatics/btn221. [DOI] [PubMed] [Google Scholar]
- 17.Dodd IB, Egan JB. 1990. Improved detection of helix-turn-helix DNA-binding motifs in protein sequences. Nucleic Acids Res 18:5019–5026. doi: 10.1093/nar/18.17.5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heap JT, Kuehne SA, Ehsaan M, Cartman ST, Cooksley CM, Scott JC, Minton NP. 2010. The ClosTron: mutagenesis in Clostridium refined and streamlined. J Microbiol Methods 80:49–55. doi: 10.1016/j.mimet.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 19.Kuehne SA, Minton NP. 2012. ClosTron-mediated engineering of Clostridium. Bioengineered 3:247–254. doi: 10.4161/bioe.21004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soucaille P. December 2010. Process for the stable gene interruption in clostridia. US provisional application no. 61/220,606. [Google Scholar]
- 21.Solovyev V, Salamov A. 2011. Automatic annotation of microbial genomes and metagenomic sequences, p 61–78. In Li RW. (ed), Metagenomics and its applications in agriculture, biomedicine and environmental studies. Nova Science Publishers, Hauppauge, NY. [Google Scholar]
- 22.Thormann K, Feustel L, Lorenz K, Dürre P, Nakotte S. 2002. Control of butanol formation in Clostridium acetobutylicum by transcriptional activation. J Bacteriol 184:1966–1973. doi: 10.1128/JB.184.7.1966-1973.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alsaker KV, Paredes C, Papoutsakis ET. 2010. Metabolite stress and tolerance in the production of biofuels and chemicals: gene-expression-based systems analysis of butanol, butyrate, and acetate stresses in the anaerobe Clostridium acetobutylicum. Biotechnol Bioeng 105:1131–1147. doi: 10.1002/bit.22628. [DOI] [PubMed] [Google Scholar]
- 24.Mironov AS, Gusarov I, Rafikov R, Lopez LE, Shatalin K, Kreneva RA, Perumov DA, Nudler E. 2002. Sensing small molecules by nascent RNA. Cell 111:747–756. doi: 10.1016/S0092-8674(02)01134-0. [DOI] [PubMed] [Google Scholar]
- 25.Croux C, Nguyen NP, Lee J, Raynaud C, Saint-Prix F, Gonzalez-Pajuelo M, Meynial-Salles I, Soucaille P. 2016. Construction of a restriction-less, marker-less mutant useful for functional genomic and metabolic engineering of the biofuel producer Clostridium acetobutylicum. Biotechnol Biofuels 9:23. doi: 10.1186/s13068-016-0432-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raynaud C, Sarçabal P, Meynial-Salles I, Croux C, Soucaille P. 2003. Molecular characterization of the 1,3-propanediol (1,3-PD) operon of Clostridium butyricum. Proc Natl Acad Sci U S A 100:5010–5015. doi: 10.1073/pnas.0734105100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard J-F, Guindon S, Lefort V, Lescot M, Claverie J-M, Gascuel O. 2008. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36:W465–W469. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodionov DA. 2007. Comparative genomic reconstruction of transcriptional regulatory networks in bacteria. Chem Rev 107:3467–3497. doi: 10.1002/chin.200746276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Novichkov PS, Laikova ON, Novichkova ES, Gelfand MS, Arkin AP, Dubchak I, Rodionov DA. 2010. RegPrecise: a database of curated genomic inferences of transcriptional regulatory interactions in prokaryotes. Nucleic Acids Res 38:D111–D118. doi: 10.1093/nar/gkp894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. 2000. Artemis: sequence visualization and annotation. Bioinformatics 16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 32.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 33.Thormann K, Feustel L, Lorenz K, Nakotte S, Dürre P. 2002. Control of butanol formation in Clostridium acetobutylicum by transcriptional activation. J Bacteriol 184:1966–1973. doi: 10.1128/JB.184.7.1966-1973.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gerischer U, Dürre P. 1992. mRNA analysis of the adc gene region of Clostridium acetobutylicum during the shift to solventogenesis. J Bacteriol 174:426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transcriptomic and proteomic data Download
Phylogenetic trees of Cap0036 (A) and Cap0037 (B) sequences from Clostridium acetobutylicum and their neighbor proteins from other bacteria. The neighbor proteins were selected by running BLAST on the NCBI database. Cap0036 and Cap0037 proteins from C. acetobutylicum are shown in blue. Download
Metabolic fluxes of mutant CA_P0037::int versus control strain in different metabolic states, such as alcohologenesis (AL) and solventogenesis (SO). (A and B) Carbon fluxes; (C and D) electron fluxes. All values are normalized to the flux of the initial carbon source (millimoles per gram [dry cell weight] per hour). Glucose flux is normalized and set at 100 for acidogenesis and solventogenesis, and the sum of glucose and half of the glycerol normalized as 100 for alcohologenesis. The values of the corresponding mutant are shown in blue, and those of the control strain are shown in green. The control data were from reference 12. Download
(A) EMSAs using the promoter region of adc and the Cap0037 protein. Lanes 1 to 8, 0, 0.2, 0.3, 0.5, 0.6, 0.7, 0.8, and 0.9 µg protein, respectively; lanes 9 to 12, 0, 0.2, 0.6, and 0.9 µg protein, respectively. (B) EMSAs using the promoter region of the sol operon and the Cap0037 protein. Lanes 1 to 5, 0, 0.28, 0.42, 0.7, and 1 µg protein, respectively; lanes 6 to 8, 0, 0.42, and 1 µg protein, respectively. Download
(A) DNase I protection assay I (DNA footprinting) of Cap0037 interacting with the adc promoter region (probe 144). End-labeled DNA fragment carrying the promoter region of the adc operon was incubated with different concentrations of Cap0037, subjected to DNase I cleavage, and analyzed on a sequencing gel. The sequencing reaction was performed with plasmid pDrive_144. (B) DNA footprinting of Cap0037 interacting with the adc promoter region (transcription start) (probe adcStart). End-labeled DNA fragment carrying the promoter region of the adc operon (transcription start) was incubated with different concentrations of Cap0037, subjected to DNase I cleavage, and analyzed on sequencing gels. The sequencing reaction was performed with plasmid pDrive_adcStart. (C) DNA footprinting of Cap0037 interacting with the sol promoter region (probe sol). End-labeled DNA fragment carrying the promoter region of the sol operon was incubated with different concentrations of Cap0037, subjected to DNase I cleavage, and analyzed on sequencing gels. The sequencing reaction was performed with plasmid pDrive_sol. Lanes 1 to 7 contain 0 to 2,308 nM Cap0037. The assigned region on the right side indicates the region protected by Cap0037 and the respective sequence. Download
Bacterial strains, plasmids, and primers used in this study
Prediction of the putative Cap0037 regulon and the corresponding relative transcript levels of those genes of the CA_P0037::int mutant in the three metabolic states, acidogenesis (AC), alcohologenesis (AL), and solventogenesis (SO). Orange letters are mismatched nucleotides compared to the binding box ATATTTTCATATAAAT in the CA_P0037/CA_P0036 promoter.
Relative transcript levels of genes belonging to the Rex regulon of the C. acetobutylicum CA_P0037::int mutant in the three metabolic states, acidogenesis (AC), alcohologenesis (AL), and solventogenesis (SO), and of the rexA mutant (data from reference 11). n.a., not available; n.d., not detected.
Relative transcript levels of selected genes of the CA_P0037::int mutant in the three metabolic states (acidogenesis [AC], alcohologenesis [AL], and solventogenesis [SO]). The transcript levels in the oxygen-exposed WT (+O2 WT), ΔperR mutant, iron-limited WT (−Fe WT), and fur::int mutant are shown. Data for the oxygen-exposed WT and the ΔperR mutant are from reference 5, and data for iron-limited WT and fur::int mutant are from reference 6. n.a., not available.