

Abstract
Background. The 2013–2016 West African Ebola virus disease (EVD) epidemic is the largest recorded. Triage on the basis of clinical signs had limited success, and the time to diagnosis by quantitative reverse transcription–polymerase chain reaction (qRT-PCR) could exceed 5 days. Here we describe the development and field validation of the ReEBOV Antigen Rapid Test (ReEBOV RDT) to aid triage of individuals with suspected EVD.
Methods. Samples from patients with suspected EVD were submitted to Kenema Government Hospital, Sierra Leone, for Lassa fever and EVD screening throughout 2014. Banked residual clinical samples were tested in November 2014 and January 2015 in a blinded field trial to estimate the clinical effectiveness of the ReEBOV RDT, compared with EBOV-specific qRT-PCR.
Results. Preliminary ReEBOV RDT performance demonstrated a positive percentage agreement (PPA) of 91.1% (195 of 214 results; 95% confidence interval [CI], 86.5%–94.6%) and a negative percentage agreement (NPA) of 90.2% (175 of 194; 95% CI, 85.1%–94.0%). The final estimates used by the Food and Drug Administration to determine whether to grant emergency use authorization for the test, which excluded a qRT-PCR reference method threshold cutoff, were a PPA of 62.1% (72 of 116 results; 95% CI, 52.6%–70.9%) and a NPA of 96.7% (58 of 60; 95% CI, 88.5%–99.6%), with a diagnostic likelihood of 18.6. A subsequent, independent evaluation by the World Health Organization generated results consistent with the preliminary performance estimates.
Conclusions. The ReEBOV RDT demonstrated the potential to provide clinically effective rapid and accurate point-of-care test results and, thus, to be a powerful tool for increasing triage efficiency.
Keywords: clinical, Ebola, point of care, lateral flow immunoassay, validation
Ebola virus (EBOV; family Filoviridae) is one of the most lethal human pathogens known. It is a causative agent of Ebola virus disease (EVD). EBOV is capable of significant human-to-human transmission after exposure to infected animals (eg, bats and nonhuman primates), which can also be considered food sources in EVD-endemic regions [1, 2]. EBOV has an overlapping geographic distribution across a wide region of sub-Saharan Africa, and there is a risk of coemergence with other viral hemorrhagic fever (VHF) agents (eg, Lassa virus, yellow fever virus, dengue virus, Crimean-Congo hemorrhagic fever virus, and Marburg virus) [3, 4]. VHFs share similarities in early clinical manifestations, which can lead to misdiagnosis and often-fatal consequences [5–7]. At the time of onset, patients with EVD will typically present with a nonspecific febrile illness, which commonly leads to misdiagnosis of other diseases, such as malaria, typhoid fever, and Lassa fever [8]. Advanced disease can result in spontaneous abortion, delirium, coma, or hemorrhage [7, 9–11]. Hallmark features of VHF disease, including coagulation disorders, hypotension, thrombocytopenia, vascular permeability, and hemorrhage, contribute to multiple organ failure, leading to shock, which can be fatal in >50% of cases [12]. After an incubation period of 1–3 weeks, VHFs can rapidly progress from onset of symptoms to death in 7–14 days. While some signs and symptoms may be associated with poor prognosis of EVD, individually or in combination they have limited sensitivity or specificity, owing to their nonspecific nature, resulting in weak diagnostic likelihood [13, 14].
The 2014 emergence of EVD in southern Guinea and its subsequent spread to Sierra Leone and Liberia required accelerated development of new therapeutics, vaccines, and diagnostic methods. In response to this unprecedented public health effort to contain the spread of EVD, the Viral Hemorrhagic Fever Consortium (VHFC) partners worked with the Food and Drug Administration (FDA) and World Health Organization (WHO) to expedite the validation of the ReEBOV Antigen Rapid Test (ReEBOV RDT) [15]. The goal was to provide a point-of-care test for EVD screening. The ReEBOV Antigen and immunoglobulin G (IgG) enzyme-linked immunosorbent assay (ELISA) were also developed to investigate the immune profile of suspected EVD cases. Herein, we describe the clinical performance of the ReEBOV RDT.
METHODS
The dynamic course of the 2013–2016 EVD outbreak compromised the full implementation of a ReEBOV RDT clinical study design [16]. Because of the magnitude of the caseload at Kenema Government Hospital (KGH) during June through September 2014, the hospital was no longer treating EVD cases. However, a holding center at KGH was still accepting individuals with suspected EVD for transport to the nearby Ebola treatment unit (ETU). The VHF Lab at KGH continued to provide diagnoses for patients and maintain the EVD sample bank. By November 2014, the EVD outbreak in Kenema District had subsided. We proceeded with field trials of the ReEBOV RDT, using the available surplus banked serum and plasma EVD samples, in November 2014 and January 2015.
Whole-blood samples from cases suspected on the basis of the WHO EVD case definition [17] were collected at the KGH ETU and other ETUs in the country and delivered to the VHF Lab. All samples were deidentified and assigned a case series number. All manipulations were undertaken using WHO recommendations for personal protective equipment and biosafety practices [18]. An aliquot of each sample was collected for viral RNA extraction from whole blood. The remaining sample was centrifuged to separate plasma and then aliquoted for sample banking and additional screening of plasma by the RDT and ELISA. Previously banked samples were blinded and randomized for RDT and qRT-PCR testing. US normal serum panels or Sierra Leone asymptomatic controls were included in sample screening for internal reference during ELISA testing.
This study was approved by the Tulane University Human Research Protection Program, the Sierra Leone Ethics and Scientific Review Committee, and the Ministry of Health and Sanitation of Sierra Leone. All sample collection was performed for patient care purposes. A waiver of consent was granted for testing banked excess clinical samples collected from patients with suspected EVD. Informed consent was obtained from all asymptomatic controls.
ReEBOV RDT
The ReEBOV RDT (Corgenix, Broomfield, Colorado) is a rapid diagnostic test that was developed using affinity-purified caprine polyclonal antibodies (Autoimmune Technologies, New Orleans, Louisiana) specific for EBOV VP40 antigen. The immunochromatographic dipstick design incorporates a plasma separator sample pad to separate plasma from whole-blood specimens (obtained via capillaries or veins) or allow the use of processed plasma and serum (Supplementary Figure 1). The test is operated by introduction of 30 µL of whole blood, plasma, or serum to the sample pad. Insertion of the dipstick into a culture tube containing 4 drops (approximately 200 µL) of sample buffer initiates the flow of sample and sample buffer. In the presence of EBOV antigen, VP40 is specifically absorbed by the colored nanoparticle reagent during flow through reagent pads. Capture of antigen-nanoparticle complexes by the EBOV VP40–specific test line results in development of a faint pink-to-red signal that corresponds to the titer of EBOV VP40 antigen in the sample. Nanoparticles that are not complexed with VP40 antigen are captured by the control line, which indicates a valid result. Visual interpretation of the result is performed after incubation for 15–25 minutes. Signal intensity was scored on a scale of 1–5, which represents a faint pink to red line. A visual aid is included in each kit to assist in signal interpretation (Figure 1).
Figure 1.

ReEBOV Antigen Rapid Test Kit results card. The results card visual aid was developed in collaboration with the Food and Drug Administration and the World Health Organization to aid ReEBOV rapid diagnostic test operators in test results interpretation. The card provides a full-scale image of a developed rapid test and hole to align the capped tube. Correct alignment allows inexperienced operators to correctly identify the control and test line signals. A scale of test line signals aids in test interpretation.
After RDT results were recorded, the dipstick was removed from the test tube for measuring the reflectance (in mV) of the test signal, using an ESEQuant Lateral Flow Reader (LFR; Qiagen Lake Constance). Each test, visual score, and reflectance were recorded on ReEBOV RDT worksheets, and a digital photograph was taken. RDT measurements were also transferred to secured laboratory databases.
ReEBOV IgG ELISA
The ReEBOV IgG ELISA test (Corgenix, Broomfield, Colorado) uses 96-well plates coated with a mixture of recombinant EBOV VP40 and NP antigens (Zalgen Labs, Germantown, Maryland). A lyophilized negative control (human serum) and reference (EBOV VP40– and EBOV NP–specific bioconjugates in human serum) are reconstituted with laboratory-grade water. Reference, negative control, and samples (serum or plasma) are diluted 1:100 in the supplied sample buffer and added to coated microwell plate wells (100 µL/well). Following incubation at ambient temperature for 30 minutes, microwells are washed 4 times with a phosphate-buffered saline (PBS)–Tween solution. Human IgG–specific horseradish peroxidase (HRP) conjugate is added (100 µL/well). Following incubation at ambient temperature for another 30 minutes, the wash step is repeated. TMB substrate is added (100 µL/well) and incubated at ambient temperature for 15 minutes, followed by addition of stop solution (100 µL/well). Microwells are read at A450 nm (with A600 nm subtraction). IgG ELISA responded to titered serum from EBOV-challenged NHPs (Supplementary Figure 2). The OD450 nm positive cutoff of 0.320 for IgG was established by comparison of US control serum to samples from individuals with suspected or confirmed EVD (Supplementary Figure 3).
ReEBOV Antigen ELISA
The ReEBOV Antigen ELISA (Corgenix, Broomfield, Colorado) uses 96-well plates coated with a mixture of EBOV VP40– and EBOV NP–specific caprine polyclonal antibody (Autoimmune Technologies, New Orleans, Louisiana). A lyophilized negative control (human serum) and reference (recombinant EBOV VP40 and NP antigens in human serum) are reconstituted with laboratory-grade water. Reference, negative control, and samples (serum or plasma) are diluted 1:10 in the supplied sample buffer and added to coated microwell plate wells (100 µL/well). Following incubation for 60 minutes at 35°C, microwells are washed 4 times with a PBS-Tween solution. EBOV VP40– and EBOV NP–specific caprine polyclonal antibody HRP conjugate is added (100 µL/well). Following incubation for 30 minutes at ambient temperature, the wash step is repeated. TMB substrate is added (100 µL/well) and incubated at ambient temperature for 15 minutes, followed by addition of stop solution (100 µL/well). Microwells are read at A450 nm (with A600 nm subtraction). Potential clinical utility was observed by comparison of US control serum to samples from patients with suspected or confirmed EVD (Supplementary Figure 4). Clinical performance was established concurrent with EBOV RDT testing versus the qRT-PCR reference method. ReEBOV Ag ELISA demonstrated significant reactivity (P < .0001) to confirmed EVD and a positive OD450nm cutoff of > 0.120 established by logistic fit analysis (Supplementary Figure 5). Clinical performance estimate was a positive percentage agreement (PPA) of 81.9% and a negative percentage agreement of 97.4% (Supplementary Table 1).
EBOV qRT-PCR
Viral RNA was extracted from 140 µL of clinical samples (banked serum and plasma), using the QIAmp Viral RNA Mini Kit (Qiagen, Germany) as per the manufacturer's recommended protocol. RNA was eluted in 80 µL of AVE buffer and stored at −20°C until analyzed. qRT-PCR reactions were set up as previously described [19]. qRT-PCR was performed using the SuperScript III One-Step RT-PCR with Platinum Taq DNA Polymerase (Life Technologies). Forward and reverse primers were used for Zaire EBOV GP (F2000 and R2079) and NP (F565 and R640) with a FAM probe for both [19]. qRT-PCR samples were analyzed on an Applied Biosystems StepOnePlus RT-PCR System. All samples were tested blindly and independently of any other diagnostic information. The EBOV qRT-PCR negative cutoff was ≥37 cycles, as established by nonlinear regression analysis of an EBOV RNA standard (Primer Design, United Kingdom). The standard curves generated were used to estimate a cycle threshold (Ct) near the lower limit of detection. A Ct of 37 was the lowest Ct with a mean of >10 genome equivalents (Supplementary Figure 6). A reverse logistic fit of qRT-PCR Ct values versus RDT results was conducted to determine the Ct for optimal agreement of both methods (mean Ct, 34.5) at which the PPA was 90.2% and the NPA was 92.8% (Supplementary Figure 7).
Statistical Analysis
Evaluation of clinical performance followed Clinical Laboratory Standards Institute and FDA guidance for qualitative diagnostic tests [20, 21]. Data were analyzed using v11.0 (SAS Institute) and GraphPad Prism v. 6.04 (GraphPad Software). Hypotheses involving dichotomous response variables were tested using the Student t test, the Fisher exact test, or logistic regression with receiver operating characteristics. Binomial proportion comparisons were used to establish significance across contingency estimates. Analyses were 2 tailed, with a significance threshold set at P < .05.
RESULTS
The field trial examined 408 samples, comprising 196 plasma and 212 serum samples, with paired RDT and qRT-PCR data (cutoff Ct, ≥ 37). Age and sex distribution data were not available. Overall, the PCR-confirmed EVD incidence was 52.5% (214 of 408 samples). The PCR-confirmed EVD incidence for plasma samples was 61.32% (130 of 212) versus 42.86% (84 of 196) for serum, which was significantly different (P = .0002). A bias was observed, with a higher PPA for serum (94.6%) and a higher NPA for plasma (97.3%; Table 1). The preliminary RDT performance estimate of the combined plasma and serum data set was a PPA of 91.1% (195 of 214 samples; 95% CI, 86.5%–94.6%) and a NPA of 90.2% (175 of 194; 95% CI, 85.1%–94.0%), with a diagnostic likelihood of 9.3 (Supplementary Table 2). For the FDA emergency use authorization application, the performance estimate was limited to plasma samples, and qRT-PCR results did not include the Ct cutoff. The final clinical performance estimate was a PPA of 62.1% (72 of 116 samples; 95% CI, 52.6%–70.9%) and a NPA 96.7% (58 of 60; 95% CI, 88.5%–99.6%), with a resulting diagnostic likelihood of 18.6 (Table 2).
Table 1.
ReEBOV Rapid Diagnostic Test (RDT) Performance Bias by Sample Type
| Sample Type | RDT Positive Agreementa |
RDT Negative Agreement |
||||
|---|---|---|---|---|---|---|
| Proportion | Percentage (95% CI) | P Value | Proportion | Percentage (95% CI) | P Value | |
| Serum | 123/130 | 94.6 (89.2–97.8) | .0254 | 66/82 | 80.5 (70.3–88.4) | <.0001 |
| Plasma | 72/84 | 85.7 (76.4–92.4) | 109/112 | 97.3 (92.4–99.4) | ||
Abbreviation: CI, confidence interval.
a Quantitative reverse transcription–polymerase chain reaction analysis had a cycle threshold cutoff of ≥ 37.
Table 2.
Clinical Performance of the ReEBOV Antigen Rapid Test
| Clinical Performance | qRT-PCR Positivea | qRT-PCR Negative | Total |
|---|---|---|---|
| RDT positive | 72b | 2 | 74 |
| RDT negative | 44c | 58 | 102 |
| Total | 116 | 60 | 176 |
| Proportion | Percentage (95% CI) |
||
| PPA | 72/116 | 62.1 (52.6–70.9) | |
| NPA | 58/60 | 96.7 (88.5–99.6) | |
| PPV | 72/74 | 97.3 (90.6–99.7) | |
| NPV | 58/102 | 56.9 (46.7–66.6) | |
| Accuracy | 130/176 | 73.9 (66.9–79.8) | |
The diagnostic likelihood was 18.6 (P < .0001).
Abbreviations: CI, confidence interval; Ct, cycle threshold; NPA, negative percentage agreement; NPV, negative predictive value; PPA, positive percentage agreement; PPV, positive predictive value; qRT-PCR, quantitative reverse transcription–polymerase chain reaction; RDT, rapid diagnostic test.
a A Ct value of <45 was considered positive.
b RDT true-positive Ct range, 19.6–29.1.
c RDT false-negative Ct range, 33.6–40.2.
The qRT-PCR Ct interquartile range (IQR) was similar between sample types (Figure 2), so a comparison of means was performed. Means testing confirmed no significant difference in the Ct values of the qRT-PCR reference method between plasma and serum for RDT-positive samples (P = .6164) and RDT-negative samples (P = .7478). Based on this finding, we examined the combined data set for additional performance characteristics. This larger sample size exceeded the minimum sample size estimate for PPA or NPA within a 0.05 margin of error for a 95% CI [16].
Figure 2.

Comparison of mean quantitative reverse transcription–polymerase chain reaction (qRT-PCR) values of plasma and serum specimens testing positive or negative by rapid diagnostic test (RDT) There were no significant differences in mean values and comparable interquartile ranges for RDT-positive and RDT-negative samples. Bias in PCR-confirmed incidence may be attributed to temporal prevalence of serum sample collection during the early stage of the EVD outbreak as compared to later stages, when whole-blood (plasma) samples were predominately collected for standardized qRT-PCR confirmation protocols. The dashed line denotes the qRT-PCR cycle threshold (Ct) cutoff. ****P < .0001, by the Fisher test. Abbreviation: NS, not significant.
The visual scoring of the ReEBOV RDT revealed a trend of qRT-PCR Ct and RDT from negative (scored 0) to moderate (scored 3) signals. The qRT-PCR maintains a Ct IQR of 20.8–26.5 for RDTs that scored ≥2 (Figure 3). Means comparison for qRT-PCR CT within the RDT scoring range was significant. For instance, a RDT visual score of 2 versus 4 corresponded to a mean Ct of 27.9 versus 22.3 (P = .0002). However, given the subjective nature of the RDT scoring, attempting to assign a linear correlation between a qRT-PCR Ct representing viral RNA and a RDT signal representing viral antigenemia is not appropriate. Instead, the relationship of ReEBOV RDT scoring to EVD qRT-PCR Ct demonstrates a high positive predictive value (PPV) when the visual signal score was ≥2 (Table 3), based on the visual aid. The combined PPV for the moderate (2–3) and high (4–5) visual scoring is 97.0% (161 of 166 specimens; 95% CI, 93.1%–99.0%) with a margin of error of 0.03.
Figure 3.
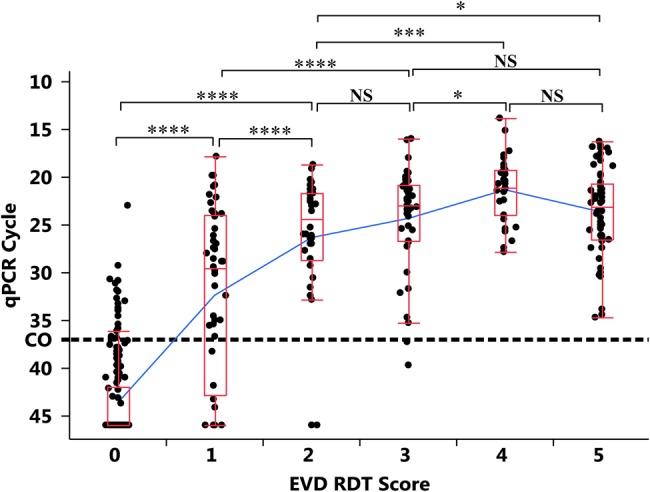
Comparison of mean quantitative reverse transcription–polymerase chain reaction (qRT-PCR) cycle threshold (Ct) values, by visual rapid diagnostic test score. There is a significant relationship between the negative signal (0) and the development of low-to-moderate signal (1–3). However, at a score of ≥2, the PCR Ct values remain positive, with an interquartile range of 20.8–26.5. qRT-PCR–nonreactive samples were assigned a Ct value of 46 for distribution analysis. Quantile box plots are shown in red. Mean values are connected by a blue line. qRT-PCR cutoff Ct values are shown by a dotted line. *P < .05, **P < .01, ***P < .005, and ****P < .0001, by the Fisher test. Abbreviations: EVD, Ebola virus disease; NS, not significant; RDT, rapid diagnostic test.
Table 3.
Positive Agreement of Stratified ReEBOV Rapid Diagnostic Test Visual Scores
| Visual Score | qRT-PCR Positive | qRT-PCR Negative | PPV (95% CI) |
|---|---|---|---|
| 5 | 56 | 0 | 100 (96.3–100) |
| 4 | 31 | 0 | 100 (88.8–100) |
| 3 | 39 | 2 | 95.1 (83.5–99.4) |
| 2 | 35 | 3 | 92.1 (78.6–98.3) |
| 1 | 34 | 14 | 70.8 (56.7–81.9) |
A total of 161 of 166 specimens had a visual signal score of ≥2, for a PPV of 97.0% (95% CI, 93.1%–99.0%).
Abbreviations: CI, confidence interval; PPV, positive predictive value; qRT-PCR, quantitative reverse transcription–polymerase chain reaction.
The correlation of antigen ELISA and qRT-PCR CT was R2 = 0.628 (Figure 4A), while no correlation (R2 = 0.044) with IgG ELISA was observed (Figure 4B). Quantile density contours elucidate the indeterminate range in qRT-PCR results (Ct, 30–40) in which the transition from acute EVD (antigenemia) and seropositive EVD lies. Most reported qRT-PCR Ct cutoffs lie within this range, including the threshold proposed for this study [22–25].
Figure 4.

Correlation of Ebola virus (EBOV) load, antigenemia, and immunoglobulin G (IgG) seropositivity. Correlation charts display quantile density heat maps of data distribution and linear regression. A, Correlation between ReEBOV antigen enzyme-linked immunosorbent assay (ELISA) (antigenemia) and quantitative reverse transcription–polymerase chain reaction (qRT-PCR) cycle threshold (Ct) values (R2 = 0.628). B, Lack of correlation between ReEBOV IgG seropositivity and qRT-PCR positivity (R2 = 0.044), revealing possible IgG-positive, PCR-negative EVD survivors. qRT-PCR-nonreactive samples were assigned a Ct of 46 for distribution analysis. Dashed lines denote qPCR Ct and ELISA OD450 cutoffs. There were 396 observations.
DISCUSSION
The field trial results of the ReEBOV RDT were included in the FDA emergency use authorization application as requested in light of the fact that a prospective study would delay the submission. As previously stated, the final application data set was restricted to plasma samples, and qRT-PCR confirmation of EVD excluded the use of a cutoff, such that a Ct value of <45 was considered a confirmed positive result. Clinical data were not available for the banked samples, so morbidity and mortality covariates could not be examined. We consider the preliminary performance estimate to be justified, based on the analysis of the KGH qRT-PCR results and covariate relationship to ELISA results. However, we also stand by the final performance estimate authorized by the FDA.
The WHO also conducted an independent clinical study (in January 2105) of the ReEBOV RDT at 2 sites in the Freetown area of Sierra Leone. This study tested fresh whole-blood specimens collected from veins and banked plasma samples. The reference method was the RealStar Filovirus Screen RT-PCR Kit 1.0 (Altona Diagnostics), with a Ct cutoff of <40. The WHO clinical performance evaluated yielded a PPA of 91.8% (89 of 97 samples; 95% CI, 84.4%–96.4%) and a NPA of 84.6% (165 of 195; 95% CI, 78.8%–89.4%), with a diagnostic likelihood 5.96. Compared to the preliminary performance at KGH, there was not a significant difference in PPA (P = .855) or NPA (P = .0966).
A second independent study (performed in February 2015) was conducted jointly by Partners In Heath (Boston, Massachusetts) and Public Health England (Porton Down, United Kingdom) [24]. For this study, the primary site was the Public Health England field reference laboratory in Port Loko, Sierra Leone. Samples enrolled in the study were referrals from several sites and clinics in western Sierra Leone. The reference method was the RealStar Filovirus Screen RT-PCR kit 1.0 (Altona Diagnostics). Importantly, this was the first study to fully model the point-of-care test concept by evaluating blood collected by finger sticks, as well as screening of fresh venous whole-blood specimens. The clinical performances for both finger-stick and whole-blood specimens were identical, with a PPA of 100% and a NPA of 92.2%. Agreement between evaluations of finger-stick and whole-blood specimens was 93.0%, with all discordant results being weak (score 1) false-positive results. The diagnostic likelihoods were 12.8 for finger-stick specimens and 14.3 for fresh whole-blood specimens.
Based on the VHFC analytical validation, the KGH field trial, and the WHO independent clinical study, the WHO and FDA granted emergency use authorization to the ReEBOV RDT for in vitro diagnostic use, in February 2015. Approved sample types were limited to whole-blood specimens obtained from finger stick or veins and plasma. These are the most appropriate samples since the RDT is design for point-of-care testing of finger-stick specimens or venous whole-blood specimens, which has been standardized for use in qRT-PCR confirmation testing. The authorized intended use of the ReEBOV RDT is for the presumptive detection of EBOV in individuals with signs and symptoms of EBOV infection in conjunction with epidemiological risk factors, including geographic location with a high prevalence of EBOV infection. Use is intended for circumstances when an authorized EBOV nucleic acid test is not available. The ReEBOV RDT is not intended for general EVD screening of nonfebrile cases, such as via airport screening or contact tracing. The ReEBOV RDT is authorized for use in laboratories or facilities adequately equipped, trained, and capable of such testing.
In the wake of the West African EVD outbreak, studies have reviewed the use of qRT-PCR to confirm the triage of patients with suspected EVD, based on the EVD clinical case definition. One study at the MSF case management center (CMC) site in Kailahun, Sierra Leone, evaluated the performance of the triage system based on the EVD case definition while awaiting PCR test results [13]. Patients with suspected EVD were separated on the basis of symptoms at triage into isolation wards for patients with suspected EVD and those for patients with highly suspected EVD while they awaited RT-PCR results, often requiring an overnight stay or longer. Based on PCR, the PPV of symptom classification at triage was only 46% for suspected cases and 76% for highly suspected cases, resulting in a combined false-positive rate of 39% that, potentially, exposes those patients to nosocomial transmission of EVD. The readmission rate of non-EVD cases (ie, those with negative PCR results) at this site was observed at 11% (15 of 138), but transmission source could not be confirmed [26].
A second study at the MSF CMC in Kailahun analyzed the EBOV load based on qRT-PCR cycle time at admission during June–October 2014 [25]. The qRT-PCR implemented at the time used a Ct cutoff comparable to the 3 ReEBOV RDT validation studies and determined that a Ct of 25 was equivalent to 1.3 × 107 genomes/mL. The Ct IQRs at admission for both survivors (25–34) and fatal cases (20–25) were below the recommended Ct cutoff (≥37) of the KGH validation study. This would suggest that PCR-positive results would have correlated well with the detection range of the ReEBOV RDT and that its use during triage would be of benefit since the acceptable time to results of PCR was as much as 5 days for inclusion in this study. Another consideration is that the qRT-PCR Ct IQR for cases on both wards was similar, with Ct ranges of 25 to 32 for suspected cases and 21–32 for highly suspected cases, which suggests that qRT-PCR–positive cases on both holding wards were equally infectious prior to being moved to the isolation wards for patients with confirmed EVD.
The potential for the use of the EVD RDT in triage scenarios and surveillance programs was recently modeled [27]. The population-level transmission model analyzed RDT use based on the WHO performance estimate for the ReEBOV RDT (PPA, 91.8%; NPA, 84.6%) versus qRT-PCR with an assumed 100% sensitivity and 100% specificity. The population-wide transmission model was calibrated to actual incidence based on PCR-only testing. When wait times for PCR results were improved to <24 hours, a 30% reduction in transmission was predicted owing to reduced nosocomial transmission in holding areas. Under the scenario of caseload outpacing bed capacity, as experienced at the peak outbreak, the model suggests that RDT-only screening would lead to more-efficient use of bed capacity by turning away fewer EVD-positive cases, which could lead to further transmission within the population. The model suggested that combined use of RDTs for EVD triage, followed by EVD confirmation with qRT-PCR, could have reduced the size of the epidemic by 32%, owing to a reduction in the number of patients with nosocomial EVD infections who are discharged back into the population.
The ReEBOV RDT met the challenge to fulfill the need for low-cost, minimal-resource, point-of-care RDTs to screen for and triage individuals with EVD. Its expedited development and validation was intended to contribute to EVD case management during the peak of the EVD transmission, but fortunately the caseload began declining in 2015. The studies discussed above suggest that implementation of this EVD RDT for screening could have contributed to improved case management efforts in areas where EVD transmission was still active and may be of great utility for future EVD outbreaks in otherwise resource-constrained regions.
Supplementary Data
Supplementary materials are available at http://jid.oxfordjournals.org. Consisting of data provided by the author to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the author, so questions or comments should be addressed to the author.
Notes
Acknowledgments. We thank the Office of the President of Sierra Leone (His Excellency President Ernest Bai Koroma and Marty Jones), the Sierra Leone Ministry of Health and Sanitation (Hon. Minister A. B. Fofonah and A. Jambai), and the Kenema District Health Management Team.
Financial support. This work was supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH; Small Business Innovation Research Grant AI088843 and Challenge and Partnership grants AI067188 and AI082119), the Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH (Physician Scientist Award 5K12HD043451-43 14); the Bill and Melinda Gates Foundation (grant OOP1123820); and the Paul Allen National Philanthropic Trust.
Potential conflicts of interest. The Viral Hemorrhagic Fever Consortium (VHFC; available at: http://www.vhfc.org) is a public-private partnership of academic and industry scientists who are developing diagnostic tests, therapeutic agents, and vaccines for Lassa fever, Ebola, and other severe diseases. Tulane University and its various academic and industry partners (Corgenix Medical Corporation; Zalgen Labs; and AutoImmune Technologies) have filed US and foreign patent applications on behalf of the VHFC for several technologies that have resulted from these efforts. Certain technical information may be kept as a trade secret. If commercial products are developed, consortium members may receive royalties or profits. This does not alter our adherence to all policies of the National Institutes of Health and The Journal of Infectious Diseases on sharing data and materials. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Georges AJ, Leroy EM, Renaut AA et al. Ebola Hemorrhagic Fever Outbreaks in Gabon, 1994–1997: Epidemiologic and Health Control Issues. J Infect Dis 1999; 179(suppl 1):S65–75. [DOI] [PubMed] [Google Scholar]
- 2.Leroy EM, Epelboin A, Mondonge V et al. Human Ebola outbreak resulting from direct exposure to fruit bats in Luebo, Democratic Republic of Congo, 2007. Vector Borne Zoonotic Dis 2009; 9:723–8. [DOI] [PubMed] [Google Scholar]
- 3.Mylne AQ, Pigott DM, Longbottom J et al. Mapping the zoonotic niche of Lassa fever in Africa. Trans R Soc Trop Med Hyg 2015; 109:483–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pigott DM, Golding N, Mylne A et al. Mapping the zoonotic niche of Ebola virus disease in Africa. Elife 2014; 3:e04395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson KM, McCormick JB, Webb PA, Smith ES, Elliott LH, King IJ. Clinical virology of Lassa fever in hospitalized patients. J Infect Dis 1987; 155:456–64. [DOI] [PubMed] [Google Scholar]
- 6.Shaffer JG, Grant DS, Schieffelin JS et al. Lassa Fever in Post-Conflict Sierra Leone. PLoS Negl Trop Dis 2014; 8:e2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schieffelin JS, Shaffer JG, Goba A et al. Clinical illness and outcomes in patients with Ebola in Sierra Leone. N Engl J Med 2014; 371:2092–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yun NE, Walker DH. Pathogenesis of Lassa fever. Viruses 2012; 4:2031–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hunt L, Gupta-Wright A, Simms V et al. Clinical presentation, biochemical, and haematological parameters and their association with outcome in patients with Ebola virus disease: an observational cohort study. Lancet Infect Dis 2015; 15:1292–9. [DOI] [PubMed] [Google Scholar]
- 10.Arwady MA, Bawo L, Hunter JC et al. Evolution of ebola virus disease from exotic infection to global health priority, Liberia, mid-2014. Emerg Infect Dis 2015; 21:578–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petti S, Messano GA, Vingolo EM, Marsella LT, Scully C. The face of Ebola: changing frequency of haemorrhage in the West African compared with Eastern-Central African outbreaks. BMC Infect Dis 2015; 15:564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takada A, Kawaoka Y. The pathogenesis of Ebola hemorrhagic fever. Trends Microbiol 2001; 9:506–11. [DOI] [PubMed] [Google Scholar]
- 13.Vogt F, Fitzpatrick G, Patten G et al. Assessment of the MSF triage system, separating patients into different wards pending Ebola virus laboratory confirmation, Kailahun, Sierra Leone, July to September 2014. Euro Surveill 2015; doi:10.2807/1560-7917.ES.2015.20.50.30097. [DOI] [PubMed] [Google Scholar]
- 14.Lado M, Walker NF, Baker P et al. Clinical features of patients isolated for suspected Ebola virus disease at Connaught Hospital, Freetown, Sierra Leone: a retrospective cohort study. Lancet Infect Dis 2015; 15:1024–33. [DOI] [PubMed] [Google Scholar]
- 15.WHO. Target Product Profile for Zaire ebolavirus rapid, simple test to be used in the control of the Ebola outbreak in West Africa 2014.
- 16.Schieffelin J, Moses LM, Shaffer J, Goba A, Grant DS. Clinical validation trial of a diagnostic for Ebola Zaire antigen detection: Design rationale and challenges to implementation. Clin Trials 2016; 13:66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.WHO. EVD Case definition, 2014.
- 18.WHO. Personal protective equipment in the context of filovirus disease outbreak response, 2014. [PubMed]
- 19.Trombley AR, Wachter L, Garrison J et al. Comprehensive panel of real-time TaqMan polymerase chain reaction assays for detection and absolute quantification of filoviruses, arenaviruses, and New World hantaviruses. Am J Trop Med Hyg 2010; 82:954–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.FDA U. Statistical Guide on Reporting Results from Studies Evaluating Diagnostic Tests, 2007.
- 21.CLSI. User Protocol for Evaluation of Evaluation of Qualitative Test Performance: Approved Guideline - Second Edition, 2008; EP12-A2.
- 22.Crowe SJ, Maenner MJ, Kuah S et al. Prognostic indicators for Ebola patient survival. Emerg Infect Dis 2016; 22:217–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de La Vega MA, Caleo G, Audet J et al. Ebola viral load at diagnosis associates with patient outcome and outbreak evolution. J Clin Invest 2015; 125:4421–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Broadhurst MJ. ReEBOV Antigen Rapid Test kit for point-of-care and laboratory-based testing for Ebola virus disease: a field validation study, 2015. [DOI] [PubMed]
- 25.Fitzpatrick G, Vogt F, Moi Gbabai OB et al. The contribution of Ebola viral load at admission and other patient characteristics to mortality in a Medecins Sans Frontieres Ebola case management centre, Kailahun, Sierra Leone, June-October 2014. J Infect Dis 2015; 212:1752–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fitzpatrick G, Vogt F, Moi Gbabai OB et al. Describing readmissions to an Ebola case management centre (CMC), Sierra Leone, 2014. Euro Surveill 2014; 19:pii:20924. [PubMed] [Google Scholar]
- 27.Nouvellet P, Garske T, Mills HL et al. The role of rapid diagnostics in managing Ebola epidemics. Nature 2015; 528:S109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.