

Abstract
Background. Ebola virus disease (EVD) is a severe viral illness caused by Ebola virus (EBOV). The 2013–2016 EVD outbreak in West Africa is the largest recorded, with >11 000 deaths. Development of the ReEBOV Antigen Rapid Test (ReEBOV RDT) was expedited to provide a point-of-care test for suspected EVD cases.
Methods. Recombinant EBOV viral protein 40 antigen was used to derive polyclonal antibodies for RDT and enzyme-linked immunosorbent assay development. ReEBOV RDT limits of detection (LOD), specificity, and interference were analytically validated on the basis of Food and Drug Administration (FDA) guidance.
Results. The ReEBOV RDT specificity estimate was 95% for donor serum panels and 97% for donor whole-blood specimens. The RDT demonstrated sensitivity to 3 species of Ebolavirus (Zaire ebolavirus, Sudan ebolavirus, and Bundibugyo ebolavirus) associated with human disease, with no cross-reactivity by pathogens associated with non-EBOV febrile illness, including malaria parasites. Interference testing exhibited no reactivity by medications in common use. The LOD for antigen was 4.7 ng/test in serum and 9.4 ng/test in whole blood. Quantitative reverse transcription–polymerase chain reaction testing of nonhuman primate samples determined the range to be equivalent to 3.0 × 105–9.0 × 108 genomes/mL.
Conclusions. The analytical validation presented here contributed to the ReEBOV RDT being the first antigen-based assay to receive FDA and World Health Organization emergency use authorization for this EVD outbreak, in February 2015.
Keywords: Ebola, point of care; diagnostic; lateral flow immunoassay
Ebola virus (EBOV; family Filoviridae) is one of the most lethal human pathogens known and is the causative agent of Ebola virus disease (EVD), which can be fatal in >50% cases. Filoviruses are enveloped, nonsegmented, negative-stranded RNA viruses. The RNA genome encodes 7 proteins, including nucleoprotein (NP), viral protein (VP) 24, VP30, VP35, VP40, glycoprotein (GP), and an RNA-dependent RNA polymerase (L) [1]. VP40 matrix protein is a peripheral membrane protein that is highly expressed in infected cells and free virions and mediates budding and viral particle release. EBOV now has a wide geographic distribution across sub-Saharan Africa. Updated risk mapping of environmental suitability for an EVD outbreak in West Africa suggests that 22.2 million people live in areas suitable for zoonotic transmission of EBOV [2].
Human exposure to EBOV from its animal reservoir, possibly fruit bats, or human exposure via intermediate vectors, such as nonhuman primates is a rare event. Following the initial animal-to-human transmission event, EBOV has demonstrated considerable capability for human-to-human transmission [3, 4]. EVD clinical manifestations can easily be confused with those of other endemic febrile illnesses, including malaria, typhoid fever, and Lassa fever, where misdiagnosis can lead to fatal consequences [5–8]. Advanced disease can result in spontaneous abortion, delirium, coma, or hemorrhage [8]. Hallmark features of VHF include many difficult-to-manage pathologies, including coagulation disorders, thrombocytopenia, vascular permeability, and hemorrhage, all of which can contribute to multiple organ failure and shock that can be fatal in >50% of cases [9, 10]. The rapid onset and progression of disease, coupled with a challenging diagnostic arena, highlight the critical need for rapid and accurate diagnostics for more-effective patient management.
Corgenix (Broomfield, Colorado) and its Viral Hemorrhagic Fever Consortium partners worked with the Food and Drug Administration (FDA) and World Health Organization (WHO) to expedite the validation of the ReEBOV Antigen Rapid Test (ReEBOV RDT; Corgenix, Broomfield, Colorado) to provide a point-of-care EVD screening. Through a collaborative effort with the FDA, the Draft Interactive Emergency Use Authorization (EUA) Review Template For Serological Assays–Antigen was issued in October 2014. During the same period, the WHO issued a separate guidance for point-of-care immunoassays (RDTs) [11]. Included in the FDA EUA Review Template were analytical performance guidelines, including limit of detection (LOD), specificity, interfering substance, and repeatability using contrived samples. Extensive specificity testing with nearest neighbor and cross-reactive pathogens in biosafety level 3 (BSL-3) and BSL-4 laboratories was also conducted. Herein, we describe the analytical performance of the ReEBOV RDT.
METHODS
Ebola Critical Reagent Development
To create viral antigen–specific assays, recombinant filovirus VP40 (rVP40) and NP (rNP) were prepared for antibody production and assay development [12–15]. Prior to the 2014 emergence of EVD in Guinea, ReEBOV RDT prototypes were developed using rVP40 and rNP antigens and respective polyclonal and monoclonal antibodies [16]. In June 2014, field testing of rVP40 and rNP enzyme-linked immunosorbent assays demonstrated strong reactivity of serum from patients with suspected EVD to these recombinant protein and antibody reagents, further strengthening confidence in the necessary cross-reactivity of the antibody reagents with the circulating strain of EBOV [17]. Ultimately, the decision was made to expedite RDT development toward detection of EBOV VP40 antigen, owing to reagent availability and promising feasibility studies.
ReEBOV RDT
The ReEBOV RDT is a diagnostic test that has been developed using caprine polyclonal antibodies (Autoimmune Technologies, New Orleans, LA) specific for EBOV rVP40 antigen (Zalgen Labs, Germantown, Maryland). The immunochromatographic dipstick design incorporates a plasma separator sample pad to separate plasma from whole blood or to allow the use of patient plasma or serum. The kit includes a lyophilized negative control composed of pooled negative human serum and a positive control composed of pooled negative human serum spiked with rVP40 antigen. The test is operated by introduction of 30 µL of whole blood, plasma, or serum onto the center of the sample pad. Inserting the dipstick into a culture tube containing 4 drops (approximately 200 µL) of sample buffer initiates the flow of sample and sample buffer. In the presence of EBOV antigen, VP40 specifically is absorbed by the colored nanoparticle reagent during flow through reagent pads. Capture of antigen-nanoparticle complexes by the EBOV VP40–specific antibodies composing the test line results in development of a faint pink to red signal that corresponds to the titer of EBOV VP40 antigen in the sample. Nanoparticles that are not complexed with EBOV VP40 antigen are captured by the control line, indicating a valid result. Visual interpretation of results is performed after a 15–25 minute incubation time. After signal development is complete, signal intensity may be scored with the use of visual aids included with the kit (Figure 1).
Figure 1.
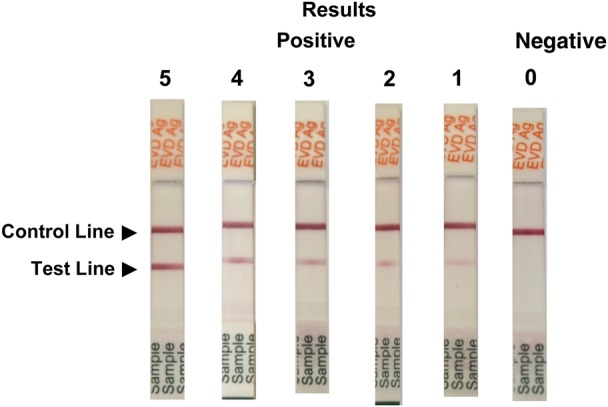
ReEBOV Antigen Rapid Test visual aid. The package insert visual aid provides the operator with a reproduction of valid positive and negative control results and a panel of invalid test results to aid in correct interpretation of the rapid test. A scale of test line signal intensity is also provided, to help guide results interpretation.
Validation Testing
The Galveston National Laboratory at the University of Texas Medical Branch (UTMB) was the site of validation studies where all BSL-4 work with live EBOV, VHF viral agent nearest neighbor, and nonhuman primate sample testing was performed. Infectious disease agent cross-reactive testing was performed at Tulane University (New Orleans, Louisiana). Analytical performance using rVP40 antigen was conducted at Corgenix. Whenever possible, a record of results was made by digital photography between 15 and 25 minutes after the test was initiated.
Donor Blood Collection
Fresh donor whole blood and serum was collected to evaluate specificity and contrived sensitivity. Donors completed donor forms, including Health Insurance Portability and Accountability Act acknowledgement, and were assigned random donor numbers. Serum and whole-blood specimens were collected into 5-mL tubes (BD) by an experienced phlebotomist. Serum tubes were incubated at room temperature for 30 minutes, followed by centrifugation at 400 g for 10 minutes. Initial testing was performed on the day of collection. Additional testing was performed within 48 hours of collection, using samples stored at 2°C–8°C. Tubes containing whole blood were inverted several times prior to preparation of spiked sample.
Range of Detection and LOD
The ReEBOV RDT assay range and LOD were determined for both rVP40 antigen and live Zaire EBOV (ZEBOV). The range of the ReEBOV RDT was determined in preliminary LOD testing. For rVP40 antigen, testing was conducted by serial dilution of rVP40 into negative human serum in a concentration range of 40.0 to 10 000 ng/mL. For live Zaire EBOV, testing was conducted by serial dilution of Zaire EBOV (Makona) in donor whole blood at a concentration range of 1.0 × 104 to 1.0 × 107 plaque-forming units (PFU)/mL. Each dilution level was tested with 5 replicates. Test strips were incubated a minimum of 15 minutes before visual scoring. The preliminary LOD was considered the lowest dilution in which all 5 test strips were positive by visual interpretation. Verification of the preliminary LOD was performed by testing 20 replicates of diluted rVP40 or Zaire EBOV at and near the preliminary LOD dilution levels.
Repeatability
Thirteen volunteers donated venous whole blood collected in ethylenediaminetetraacetic acid (EDTA)–lined vacutainers (BD Medicine). Randomized whole-blood samples were spiked with rVP40 antigen at a LOD of 9.4 ng/test and a 2-fold increases series up to 600 ng/test. Extra samples were spiked with rVP40 at the LOD and 2 times the LOD. A total of 75 replicates of unspiked whole blood were tested for assay specificity. All samples were blinded and tested according to instructions in the package insert. After incubation for 15 minutes, each dipstick was evaluated by 3 technicians. Scores were tabulated based on agreement of 3 technicians.
Interfering Substances
Interfering substances screening was conducted in accordance with Clinical Laboratory Standards Institute approved guideline EP7-A2 [18]. The rVP40 antigen was diluted in normal control serum at a concentration near the LOD of 0.2 µg/mL (6.0 ng/test). Substances and their concentrations to be tested were found in EP7-A2, appendix C or D [18]. For substances not listed, the equivalent of a 20X stock for the normal adult dose diluted in 5 L (blood volume) was prepared. Prior to testing, the 20X substance stock was diluted in an aliquot of the rVP40 sample and normal control serum without antigen. Each substance and solvent control was tested in 5 replicates. Additional levels of select substances were also requested by the FDA. A substance was considered noninterfering if the antigen-spiked median score did not differ from that of the solvent control or if it did not generate a false-positive signal in normal control serum.
Specificity and Sensitivity
To evaluate the specificity of the ReEBOV RDT, normal US serum panels developed from commercial donor serum were used (Plasma Services Group, Huntingdon Valley, Pennsylvania). A total of 4 panels representing 138 individual serum donors were tested. Each sample was tested according to its package insert and scored individually.
During fresh donor blood collection, 20 healthy US volunteers consented to finger-stick screening. Each volunteer provided duplicate finger sticks. One full drop (approximately 30 µL) was allowed to develop on the finger and then was transferred directly to the plasma separation sample pad by touching the dipstick directly to the blood drop. This was repeated to collect a second sample from each volunteer. Dipsticks were immediately inserted into test tubes containing sample buffer to initiate development of ReEBOV RDT. After incubation for 15 minutes, results were scored blinded by a second technician.
Thirteen donor whole-blood samples were collected in EDTA-lined vacutainers for ReEBOV RDT specificity and contrived sensitivity testing in a blinded, randomized design. Six replicates per whole-blood sample were evaluated independently by 3 technicians, yielding 234 observations for the specificity determination. Randomized whole-blood samples were spiked with rVP40 antigen at the whole-blood LOD of 9.4 ng/test and at 2-fold serial dilutions up to 600 ng/test. A minimum of 6 random spiked samples at each level were evaluated by 3 technicians, for a minimum of 18 observations per level. Extra samples were spiked with rVP40 at (117 observations) and 2-fold above (90 observations) the whole-blood LOD to verify the reproducibility of the previously described analytical LOD. A total of 303 observations were made with spiked whole-blood samples. All samples were blinded and tested according to guidance in the package insert. After incubation for 15 minutes, each dipstick was evaluated independently by 3 technicians.
Sixteen donor serum samples were collected for ReEBOV RDT specificity and contrived sensitivity testing in a blinded, randomized design. Six replicates per serum sample were tested and evaluated independently by 3 technicians, yielding 249 observations for the specificity determination. Randomized serum samples were spiked with rVP40 antigen at the serum LOD of 4.7 ng/test and at 2-fold increases up to 300 ng/test. A minimum of 5 random spiked samples at each level were evaluated by 3 technicians, for a minimum of 16 observations per level. Extra samples were spiked with rVP40 at (123 observations) and 2-fold above (99 observations) the serum LOD, to verify the reproducibility of the previously described analytical serum LOD. A total of 323 observations were made with spiked whole-blood samples. All samples were blinded and tested according to instructions in the package insert. After incubation for 15 minutes, each dipstick was evaluated independently by 3 technicians.
Cross-reactant and Nearest-Neighbor Screening
Nearest-neighbor and cross-reactant screening was conducted at multiple sites, depending on the required BSL for the agents (virus, bacteria, or parasite) tested. Testing was performed in a similar manner as in Interfering Substance section previously described. Normal control serum (ie, human serum) with or without rVP40 antigen diluted to 0.2 µg/mL (6.0 ng/test) was distributed or prepared on-site. BSL-4 viruses were tested at UTMB, BSL-3 vaccine virus strains were tested at the Centers for Disease Control and Prevention (Fort Collins), and remaining BSL-2 and BSL-3 pathogens were tested at the Tulane Health Science Center and the Tulane National Primate Research Center. The concentrations of pathogens tested fluctuated owing to available stocks. Every attempt was made to maintain a dilution factor of ≤5% of the antigen-spiked negative human serum or no-antigen controls to retain serum matrix integrity. All pathogens were tested in triplicate. Pathogens were considered non–cross-reactive if they did not produce false-positive or false-negative signals.
In cases of cross-reactive filovirus species, genomic equivalents of virus seed stocks used for analysis were determined by real-time quantitative polymerase chain reaction (qPCR). RNA was isolated from viral stocks by means of the Viral RNA mini-kit (Qiagen), using 100 µL of viral supernatant in 600 µL of buffer AVL containing 1% carrier RNA, per the manufacturer's instructions. The following primers/probe targeting the VP30 gene of ZEBOV-Makona and ZEBOV-Mayinga, the VP35-intergenic region of EBOV-Bundibugyo, and the L gene of EBOV-Sudan (Gulu) were used for real-time qPCR: for ZEBOV-Makona and ZEBOV-Mayinga, 6-carboxyfluorescein (6FAM)-5′ AGGCTTCCCTCGCTGCCGTTATG 3′-6 carboxytetramethylrhodamine (TAMRA); for EBOV-Bundibugyo, 6FAM-CGCAACCTCCACAGTCGCCT-TAMRA; for EBOV-Sudan (Gulu), 6FAM-CATCCAATCAAAGACATTGCGA-TAMRA; and for Marburg virus–Musoke, 6FAM-CGCGGCATTTCA-TAMRA (Life Technologies). Viral RNA was detected using the CFX96 detection system (BioRad Laboratories, Hercules, California) in One-Step probe real-time qPCR kits (Qiagen) with the following cycle conditions: for ZEBOV and EBOV-Bundibugyo, 50°C for 10 minutes, 95°C for 10 seconds, and 40 cycles of 95°C for 10 seconds and 57°C for 30 seconds; and for EBOV-Sudan (Gulu), 50°C for 10 minutes, 95°C for 10 seconds, and 40 cycles of 95°C for 10 seconds and 59°C for 30 seconds. Cycle thresholds representing viral genomes were analyzed with CFX Manager Software, and data are shown as genome equivalents (GEq) per milliliter. To create the GEq standard, RNA from viral stocks was extracted, and the number of strain-specific genomes was calculated using Avogadro's number and the molecular weight of each viral genome.
Nonhuman Primate Sampling
Animal studies were completed under BSL-4 biocontainment at the Galveston National Laboratory and were approved by the UTMB Institutional Laboratory Animal Care and Use Committee (IACUC) in accordance with state and federal statutes and regulations relating to experiments involving animals and with the Institutional Biosafety Committee. To assess feasibility of using the ReEBOV RDT to detect viral antigen in whole blood and plasma, biological samples were collected on days 4 and 6 after infection from ongoing pathogenesis studies involving serial necropsy over the course of disease (Geisbert et al, unpublished data). These experiments involved 4 healthy adult cynomolgus macaques (Macaca fascicularis) that were inoculated intramuscularly with 1000 PFU of EBOV strain Makona. All animals received physical examinations, and blood specimens were collected just prior to the time of challenge and on days 4 and 6 after challenge. Additionally, all animals were monitored daily and scored for disease progression, using scoring criteria approved by the UTMB IACUC. Criteria included posture/activity level, attitude/behavior, respiration, and hallmark filovirus disease features, including visible petechial rash, ecchymosis, and/or hemorrhage. Scores of ≥9 indicated that euthanasia criteria had been met. GEq per milliliter of each biological sample were determined as published elsewhere [19]. Plasma was processed from whole-blood samples, and both plasma and whole blood were tested without inactivation, using the ReEBOV RDT. Each sample was tested once and scored using the visual aid included with package insert.
Statistical Analysis
Data were analyzed using SAS, version 11.0 (SAS Institute), and GraphPad Prism, version 6.04 (GraphPad Software). Hypotheses involving dichotomous response variables were tested using the Student t test, the Fisher exact test, or logistic regression with receiver operating characteristics. Binomial proportion comparisons were used to establish significance across contingency estimates. Analyses were 2 tailed, with a significance threshold set at a P value of < .05. Confidence intervals (95%) [20].
RESULTS
LOD
The preliminary LOD for rVP40 spiked into human serum was equal to 160.0 ng/mL (4.7 ng/test). Verification of the spiked rVP40 LOD was conducted at 2.4 ng/mL and 4.7 ng/mL. At 4.7 ng/mL, 100% of the 20 replicates were positive and represented the LOD. LOD verification was repeated with rVP40 spiked into donor whole blood. The LOD concentrate was 2-fold higher at 9.4 ng/test (Table 1). The preliminary LOD for ZEBOV in whole blood was 1 × 106 PFU/mL. Verification of EBOV LOD in whole blood was confirmed by 100% positivity of the replicates (Table 2).
Table 1.
Limit of Detection (LOD) of Spiked Filovirus Recombinant Viral Protein 40 (VP40) Antigen in Serum and Whole-Blood Specimens
| VP40 Antigen Level |
Replicates, Proportiona | Result | |
|---|---|---|---|
| ng/mL | ng/Test | ||
| Preliminary | |||
| 10 000 | 300 | 5/5 | Positive |
| 5000 | 150 | 5/5 | Positive |
| 2500 | 75.0 | 5/5 | Positive |
| 1250 | 37.5 | 5/5 | Positive |
| 625 | 18.8 | 5/5 | Positive |
| 312 | 9.4 | 5/5 | Positive |
| 160 | 4.7 | 5/5 | LOD |
| 80 | 2.3 | 3/5 | Negative |
| 40 | 1.2 | 0/5 | Negative |
| Serum confirmation | |||
| 160 | 4.7 | 20/20 | LOD |
| 80 | 2.3 | 18/20 | … |
| Whole-blood confirmation | |||
| 312 | 9.4 | 18/20 | LOD |
| 160 | 4.7 | 13/20 | … |
| Blank control | 0/5 | Negative | |
a Data are no. of positive whole-blood specimens/no. tested.
Table 2.
Limit of Detection (LOD) of Zaire Ebola Virus (ZEBOV) in Whole-Blood Specimens
| ZEBOV Level |
Replicates, Proportiona | Result | |
|---|---|---|---|
| PFU/mL | PFU/test | ||
| Preliminary | |||
| 1 × 107 | 3 × 105 | 5/5 | Positive |
| 2 × 106 | 6 × 104 | 5/5 | Positive |
| 1 × 106 | 3 × 104 | 5/5 | LOD |
| 4 × 105 | 1.2 × 104 | 3/5 | Negative |
| 1 × 105 | 3 × 103 | 0/5 | Negative |
| 8 × 104 | 2.4 × 103 | 0/5 | Negative |
| 1 × 104 | 300 | 0/5 | Negative |
| Confirmation | |||
| 2 × 106 | 6 × 104 | 20/20 | Positive |
| 1 × 106 | 3 × 104 | 20/20 | LOD |
| Positive control | … | 2/2 | Positive |
| Negative control | … | 0/2 | Negative |
Abbreviation: PFU, plaque-forming units.
a Data are no. of positive whole-blood specimens/no. tested.
Repeatability
All 3 ReEBOV RDT evaluators agreed on unspiked sample scoring in 92% of cases (69 of 75), and 2 of 3 agreed in 100% of cases (75 of 75). For rNP-spiked samples, 2 of 3 evaluators agreed on 58% of occasions (21 of 36) for 9.4 ng/mL, which would correspond with the qualitative assay cutoff. At 18.8 ng/mL, all 3 evaluators agreed in 100% of cases (36 of 36), which would correspond with the LOD (Supplementary Table 1).
Interfering Substances
For the biologic substances, only hemoglobin and rheumatoid factor generated false results (Supplementary Table 2). Hemoglobin failed because of the interference with positive signal development from heavy background staining of the nitrocellulose membrane. Rheumatoid factor also failed because of false-positive signal above 39 IU/mL spiking concentration. Bilirubin also stained the nitrocellulose membrane yellow, but this did not interfere with negative or positive signal development. None of the drugs in common use interfered with positive or negative signal development or interpretation.
Mock Specificity and Sensitivity
Specificity of serum samples was evaluated using US normal serum panels developed from commercial donor serum. Four panels representing 138 individual serum donors were tested. Each sample was tested individually. A second technician scored the results independently (Supplementary Panel 1). The specificity of US donor panels was 94.9% (131 of 138; 95% CI, 89.8%–97.9%). Duplicate samples were collected by finger stick from 20 volunteers prior to attempting venous blood collection for mock clinical studies. The samples obtained by finger stick were blinded and evaluated by a single operator (Supplementary Panel 2). Thirty-nine of 40 were scored negative, and 1 duplicate generated a trace signal resulting in 97.5% specificity (95% CI, 86.8%–99.9%). The specificity estimate for donor whole-blood triplicate observations was 96.6% (226 of 234; 95% CI, 93.4%–98.5%). The effective LOD when antigen was spiked into whole blood was 18.8 ng/test. The first level of 9.4 ng/test was only detected in 60.7% of cases (71 of 117; 95% CI, 51.2%–69.6%), which would satisfy a qualitative assay cutoff (Table 3 and Supplementary Panel 3). The second level of 18.8 ng/test was detected in 97.8% of cases (88 of 90; 95% CI, 92.2%–99.7%) and would satisfy LOD criteria. The sensitivity estimate at ≥18.8 ng/mL spiked VP40 in triplicate was 93.0% (173 of 186; 95% CI, 88.3%–96.2%). The specificity estimate for donor serum triplicate observations was 80.3% (200/249; 95% CI, 74.8%–85.1%). The effective LOD when antigen was spiked into serum was 9.4 ng/test. The first level of 4.7 ng/test was detected in 87.0% of cases (107 of 123; 95% CI, 79.8%–91.9%), which would satisfy a qualitative assay cutoff. The second level of 9.4 ng/test was detected in 93.9% of cases (93 of 99; 95% CI, 87.2%–97.4%), which nearly satisfies the LOD criteria (Table 3 and Supplementary Panel 4). The sensitivity estimate at ≥9.4 ng/mL spiked VP40 in triplicate was 96.5% (193 of 200; 95% CI, 92.9%–98.6%).
Table 3.
Summary of Clinical Performance Specificity and Sensitivity
| Characteristic, Specimen | Negative | Positive | Negative Agreement |
|
|---|---|---|---|---|
| Samples, Proportion | Percentage (95% CI) | |||
| Specificity | ||||
| Normal serum panels | 131 | 7 | 131/138 | 94.9 (89.8–97.9) |
| Finger stick | 39 | 1 | 39/40 | 97.5 (86.8–99.9) |
| Whole-blood replicates | 226 | 8 | 226/234 | 96.6 (93.4–98.5) |
| Serum replicates | 200 | 49 | 200/249 | 80.3 (93.4–98.5) |
| Positive Agreement |
||||
| Samples, Proportion | Percentage (95% CI) | |||
| Spiked sensitivity, ng/test | ||||
| Whole blood | ||||
| 9.4 | 46 | 71 | 71/117 | 60.7 (51.2–69.6) |
| 18.8 | 2 | 90 | 90/92 | 97.8 (92.2–99.7) |
| Serum | ||||
| 4.7 | 16 | 123 | 107/123 | 92.9 (79.8–91.9) |
| 9.4 | 6 | 93 | 93/99 | 93.9 (87.2–97.4) |
Abbreviation: CI, confidence interval.
Cross-reactant and Nearest-Neighbor Specificity
None of the cross-reactants tested interfered with positive or negative signal development or interpretation (Supplementary Table 3). Paramyxoviridae were detected in 1 of 3 negative serum specimens, but the finding was recorded as nonreactive owing to the presence of 5 of 6 correct ReEBOV RDT results. Only negative human serum was spiked with filoviruses. The ReEBOV RDT was reactive to both Makona and Mayinga strains of ZEBOV. The ReEBOV RDT was also cross-reactive to Sudan and Bundibugyo EBOV species at 1.0 × 103–1.0 × 104 PFU/mL which equated to 2.2 × 109–2.5 × 1010 GEq/mL from in vitro viral stocks (Table 4). Reston EBOV and Marburg virus were nonreactive.
Table 4.
Nearest-Neighbor Filovirus Screening
| Filovirus | Type, Strain (Isolate) | Concentration, PFU/mL | Concentration, GEq/mL | RDT Score | Reactivity |
|---|---|---|---|---|---|
| Ebola virus | Zaire (Mayinga) | 3.3 × 106 | 2.55 × 1010 | +++ | Yes |
| Ebola virus | Zaire (Makona) | 1.3 × 105 | 1.78 × 1010 | +++++ | Yes |
| Ebola virus | Sudan, Gulu (200 011 676) | 3.75 × 105 | 1.63 × 1010 | +++ | Yes |
| Ebola virus | Sudan, Gulu (200 011 676) | 3.75 × 104 | 1.63 × 109 | +++ | Yes |
| Ebola virus | Bundibugyo (200 706 291) | 1.38 × 104 | 2.18 × 1010 | +++ | Yes |
| Ebola virus | Bundibugyo (200 706 291) | 1.38 × 103 | 2.18 × 109 | +++ | Yes |
| Ebola virus | Reston (AZ-1435) | 6.75 × 103 | ND | −−− | None |
| Marburg virus | Musoke | 2.2 × 105 | ND | −−− | None |
Abbreviations: GEq, genome equivalents; ND, no data; PFU, plaque-forming units; RDT, ReEBOV Antigen Rapid Test.
Nonhuman Primates
At day 4 after infection, all nonhuman primates exhibited fever and elevated clinical scores comparable to those during early stages of human EVD. All day 4 blood and plasma samples were scored as a 1 on scale of 1–5 for the ReEBOV RDT, and real-time qPCR determined a range of 3.0 × 105 to 1.0 × 107 EBOV genomes/mL, which equated to 6.5 × 104–5.2 × 105 PFU/mL. By day 6 after infection, nonhuman primates displayed clinical signs indicative of more-severe disease, including hallmark signs of EBOV infection—sustained fever, petechial rash, and marked changes in blood hematology and chemistry parameters—analogous to those seen in human disease. Day 6 samples were all scored as a 5 by the ReEBOV RDT, and real-time qPCR determined a range of 2.0 × 108 to 9.0 × 108 genomes/mL, which equated to 6.5 × 108–8.8 × 108 PFU/mL (Table 5).
Table 5.
In Vivo Sampling of Nonhuman Primates Challenged With Zaire Ebola Virus Strain Makona
| Day After Infection, Sample | Clinical Sign(s) | Positive By RDT | Viremia Level, PFU/mL | Real-time qPCR Result, GEq/mL |
|---|---|---|---|---|
| 4 | ||||
| Whole blood | Fever | 2/2 | … | 3.4 × 105 and 5.7 × 106 |
| Plasma | Fever | 2/2 | 6.5 × 104 and 5.23 × 105 | 2.4 × 106 and 1.4 × 107 |
| 6 | ||||
| Whole blood | Fever/hallmark | 2/2 | … | 2 × 108 and 3 × 108 |
| Plasma | Fever/hallmark | 2/2 | 3.05 × 107 and 7.25 × 107 | 6.6 × 108 and 8.8 × 108 |
Cynomolgus macaques were challenged with 1000 PFU/mL Ebola virus strain Makona. Sampling was performed on days 4 and 6 after challenge for RDT and real-time qPCR testing.
Abbreviations: GEq, genome equivalents; PFU, plaque-forming units; qPCR, quantitative polymerase chain reaction; RDT, ReEBOV Antigen Rapid Test.
DISCUSSION
The development of the ReEBOV RDT was expedited to provide a diagnostic test with a rapid time to result, to aid in EVD case triage and management. By building on the successful design of the ReLASV Antigen Rapid Test (Corgenix; unpublished data), the ReEBOV RDT provides a simple finger-stick test that could be administered in Ebola treatment unit triage and holding areas by staff members who have donned appropriate personal protective equipment. This diagnostic format would alleviate many of the concerns described by Perkins and Kessel, including delayed time to results of real-time qPCR and the logistics or biosecurity concerns in transporting samples, patients, and/or contacts to those facilities [21]. The regions affected by the current EVD outbreak are extremely challenging environments in which to provide accurate infectious disease diagnosis. Guinea, Liberia, and Sierra Leone (as well as Nigeria) are areas of endemicity for Lassa fever but also for more-common febrile illness, such as malaria, dengue, West Nile virus infection, and chikungunya [22–24]. The ReEBOV RDT successfully demonstrated specificity for EBOV in nearest-neighbor and cross-reactant tests and further demonstrated sensitivity to the EBOV strains known to cause EVD in humans. Sensitivity to Marburg virus was not expected, owing to low homology with the EBOV VP40 amino acid sequence. A variety of drugs in common use, including analgesics, antibiotics, antihelmitics, antimalarials, and antivirals, including ribavirin, demonstrated no interference with ReEBOV RDT performance.
The ReEBOV RDT specificity estimate ranged from 80.3% with replicate serum testing to as high as 97.5% for screening samples obtained by finger stick. This range in sensitivity has been independently observed in subsequent clinical studies [25, 26]. By use of experimental nonhuman primate infections to correlate disease presentation kinetics with ReEBOV RDT performance, the detection range of the ReEBOV RDT becomes clearer. Further, these studies highlight the value of using relevant animal models for the testing of human diagnostic tests against filoviruses. Large disparities of infectious units to genomic quantities present in repeatedly passaged viral stocks as compared to live infections may skew the interpretation of the infectious agent present if assessments of live virus within the sample are not also made. These data also suggest that genomic detection may be more sensitive when using in vitro–generated viral stocks but that this may not necessarily equate to the presence of similar levels of infectious units within the sample. Therefore, both values should be determined and presented in parallel.
The LOD of contrived virus spiked in whole blood was 1.0 × 106 PFU/mL in this study, compared with 5.9 × 104 PFU/mL in independent testing by the WHO. This WHO assessment was determined to be equivalent to 4.2 × 108 RNA copies/mL [26]. However, we observed that EBOV concentrations of 3.4 × 105 genomes/mL were present in experimentally infected nonhuman primates prior to the development of hallmark signs. In agreement with nonhuman primate testing, subsequent clinical testing demonstrated that the ReEBOV RDTs range of 1.0 × 105–1.0 × 109 genomes/mL correlated well with threshold cycle times observed in patients with EVD during the course of acute EVD [25, 27, 28]. The ReEBOV RDT analytical validation presented here for FDA and WHO applications demonstrated the capacity to accurately detect EVD and provide a rapid, point-of-care test for EVD case triage and management. Field trials and independent clinical studies further confirmed its potential clinical effectiveness (Boisen et al, unpublished data) [25, 26]. The ReEBOV RDT is the first point of care RDT to receive both FDA and WHO emergency use status for the ongoing 2013–2016 West African Ebola outbreak.
Supplementary Data
Supplementary materials are available at http://jid.oxfordjournals.org. Consisting of data provided by the author to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the author, so questions or comments should be addressed to the author.
Notes
Acknowledgments. We thank J. J. Laven (Arbovirus Diseases Branch, Centers for Disease Control and Prevention, Ft. Collins, CO), for her collaboration in the cross-reactant pathogen testing; Karla A. Fenton, for assistance with photography during the testing at the University of Texas Medical Branch; and the Bill and Melinda Gates Foundation and Paul Allen National Philanthropic Trust, for their generous support for the scale up and deployment of the ReEBOV Antigen Rapid Test to the Ebola-affected countries.
Financial support. This work was supported by the National Institute of Allergy and Infectious Diseases (Small Business Innovation Research Grant AI088843 and Challenge and Partnership grants AI067188 and AI082119) and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (physician scientist award 5K12HD043451), National Institute of Health, Department of Health and Human Services.
Potential conflicts of interest. The Viral Hemorrhagic Fever Consortium (available at: http://www.vhfc.org) is a public-private partnership of academic and industry scientists who are developing diagnostic tests, therapeutic agents, and vaccines for Lassa fever, Ebola, and other severe diseases. Tulane University (with which M. L. B., J. N. H., J. G. S., J. S. S., and R. F. G. are affiliated) and its various academic and industry partners (Corgenix [with which M. L. B., M. M. M., D. S. N., D. O., A. B. J., B. L. B., H. P., and K. R. P. are affiliated], Zalgen Labs [with which M. L. B., M. M. R., M. L. H., L. M. B., and R. F. G. are affiliated], and AutoImmune Technologies [with which P. C. K. and R. B. W. are affiliated]) have filed US and foreign patent applications on behalf of the Viral Hemorrhagic Fever Consortium for several technologies that have resulted from these efforts. Certain technical information may be keep as a trade secret. If commercial products are developed, consortium members may receive royalties or profits. This does not alter our adherence to all policies of the National Institutes of Health and The Journal of Infectious Diseases on sharing data and materials. All other authors report no potential conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Feldmann H, Geisbert TW. Ebola haemorrhagic fever. Lancet 2011; 377:849–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pigott DM, Golding N, Mylne A et al. Mapping the zoonotic niche of Ebola virus disease in Africa. Elife 2014; 3:e04395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ter Meulen J, Lukashevich I, Sidibe K et al. Hunting of peridomestic rodents and comsumption of their meat as possible risk factors for rodent-to-human transmission of Lassa virus in the Republic of Guinea. Am J Trop Med Hyg 1996; 55:661–6. [DOI] [PubMed] [Google Scholar]
- 4.Georges AJ, Leroy EM, Renaut AA et al. Ebola hemorrhagic fever outbreaks in Gabon, 1994–1997: epidemiologic and health control issues. J Infect Dis 1999; 179(suppl 1):S65–75. [DOI] [PubMed] [Google Scholar]
- 5.Johnson KM, McCormick JB, Webb PA, Smith ES, Elliott LH, King IJ. Clinical virology of Lassa fever in hospitalized patients. J infect Dis 1987; 155:456–64. [DOI] [PubMed] [Google Scholar]
- 6.Andersen KG, Shapiro BJ, Matranga CB et al. Clinical sequencing uncovers origins and evolution of Lassa virus. Cell 2015; 162:738–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shaffer JG, Grant DS, Schieffelin JS et al. Lassa fever in post-conflict Sierra Leone. PLoS Negl Trop Dis 2014; 8:e2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schieffelin JS, Shaffer JG, Goba A et al. Clinical illness and outcomes in patients with Ebola in Sierra Leone. N Engl J Med 2014; 371:2092–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yun NE, Walker DH. Pathogenesis of Lassa fever. Viruses 2012; 4:2031–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takada A, Kawaoka Y. The pathogenesis of Ebola hemorrhagic fever. Trends Microbiol 2001; 9:506–11. [DOI] [PubMed] [Google Scholar]
- 11.World Health Organization (WHO). Target product profile for Zaire ebolavirus rapid, simple test to be used in the control of the Ebola outbreak in West Africa. Geneva, Switzerland: WHO, 2014. http://www.who.int/medicines/publications/target-product-profile.pdf. Accessed 3 October 2014.
- 12.Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, Saphire EO. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 2008; 454:177–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirchdoerfer RN, Abelson DM, Li S, Wood MR, Saphire EO. Assembly of the ebola virus nucleoprotein from a chaperoned VP35 complex. Cell Rep 2015; 12:140–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hashiguchi T, Fusco ML, Bornholdt ZA et al. Structural basis for Marburg virus neutralization by a cross-reactive human antibody. Cell 2015; 160:904–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dias JM, Kuehne AI, Abelson DM et al. A shared structural solution for neutralizing ebolaviruses. Nat Struct Mol Biol 2011; 18:1424–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bornholdt ZA, Noda T, Abelson DM et al. Structural rearrangement of ebola virus VP40 begets multiple functions in the virus life cycle. Cell 2013; 154:763–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boisen ML, Oottamasathien D, Jones AB et al. Development of prototype filovirus recombinant antigen immunoassays. J Infect Dis 2015; 212(suppl 2):S359–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.CLSI. Interference testing in clinical chemistry. Approved guideline. Document EP07-A2 2nd ed Wayne, PA: CLSI, 2005. [Google Scholar]
- 19.Thi EP, Mire CE, Lee AC et al. Lipid nanoparticle siRNA treatment of Ebola-virus-Makona-infected nonhuman primates. Nature 2015; 521:362–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agresti A, Coull BA. Approximate is better than “exact” for interval estimation of binomial proportions. Amer Stat 1998; 52:119–26. [Google Scholar]
- 21.Perkins MD, Kessel M. What Ebola tells us about outbreak diagnostic readiness. Nat Biotechnol 2015; 33:464–9. [DOI] [PubMed] [Google Scholar]
- 22.Boisen ML, Schieffelin JS, Goba A et al. Multiple circulating infections can mimic the early stages of viral hemorrhagic fevers and possible human exposure to filoviruses in Sierra Leone prior to the 2014 outbreak. Viral Immunol 2015; 28:19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ansumana R, Jacobsen KH, Leski TA et al. Reemergence of chikungunya virus in Bo, Sierra Leone. Emerg Infect Dis 2013; 19:1108–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klempa B, Koulemou K, Auste B et al. Seroepidemiological study reveals regional co-occurrence of Lassa- and Hantavirus antibodies in Upper Guinea, West Africa. Trop Med Int Health 2013; 18:366–71. [DOI] [PubMed] [Google Scholar]
- 25.Broadhurst MJ. ReEBOV Antigen Rapid Test kit for point-of-care and laboratory-based testing for Ebola virus disease: a field validation study, 2015; 386:867–74. [DOI] [PubMed] [Google Scholar]
- 26.WHO. Public report for ReEBOV Antigen Rapid Test Kit (EA 0011-011-00), 2015. http://www.who.int/diagnostics_laboratory/procurement/150219_reebov_antigen_rapid_test_public_report.pdf. Accessed 21 March 2015.
- 27.Fitzpatrick G, Vogt F, Moi Gbabai OB et al. The contribution of Ebola viral load at admission and other patient characteristics to mortality in a Medecins Sans Frontieres Ebola case management centre, Kailahun, Sierra Leone, June-October 2014. J Infect Dis 2015; 212:1752–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crowe SJ, Maenner MJ, Kuah S et al. Prognostic indicators for Ebola patient survival. Emerg Infect Dis 2016; 22:217–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.