

Abstract
The risk of recurrence following radiation therapy remains high for a significant number of prostate cancer patients. The development of in vitro isogenic models of radioresistance through exposure to fractionated radiation is an increasingly used approach to investigate the mechanisms of radioresistance in cancer cells and help guide improvements in radiotherapy standards. We treated 22Rv1 prostate cancer cells with fractionated 2 Gy radiation to a cumulative total dose of 60 Gy. This process selected for 22Rv1-cells with increased clonogenic survival following subsequent radiation exposure but increased sensitivity to Docetaxel. This RR-22Rv1 cell line was enriched in S-phase cells, less susceptible to DNA damage, radiation-induced apoptosis and acquired enhanced migration potential, when compared to wild type and aged matched control 22Rv1 cells. The selection of radioresistant cancer cells during fractionated radiation therapy may have implications in the development and administration of future targeted therapy in conjunction with radiation therapy.
Following a prostate cancer diagnosis, approximately 50 percent of men will receive radiation therapy. Patients with PSA >20 ng/ml or biopsy Gleason score 8–10 or T2-3N0M0 localised prostate carcinoma are recognised as high risk1. The optimal management of these patients remains unclear. Randomized control trials recommend the combination of external beam radiotherapy with androgen deprivation therapy to improve overall survival2, but recurrence rates in these patients remain high and are associated with a limited chance of cure3. The characterisation of the radiobiological properties of prostate tumours, increasingly related to the eight cancer hallsmarks4, is essential to guide the evaluation of current as well as novel therapeutic options. It may also provide a means to select patients most likely to benefit from these strategies.
Modifications in the radiobiological properties of tumours can take several forms. Most likely, it results in an increased capacity of irradiated clonogens to overcome the anti-proliferative effects of radiation, evidenced by a quantifiable change in the relationship between clonogenic survival and radiation dose5. This change can be attributed to the capacity for these cells to overcome the induction and repair radiation damage6, ignore pro-apoptotic signals7 and avoid the transition to a senescent state8,9. But other factors complicate this relationship. First, tumour microenvironmental factors and the tumour vasculature10 may also reduce response to radiotherapy11. Second, rapidly accumulating evidence identifies the number of uncontrolled cancer stem cells following a radiotherapy regimen as a key to local tumour control probability12,13,14.
Exposure of cancer cells to fractionated radiation schedules can select a cancer subpopulation with modified cell fate in response to subsequent radiation exposure and affect tumour control probablity15. This selection process is increasingly reproduced in vitro to investigate the molecular response of cancer cells and guide the development of novel biomarkers of radiotherapy failure (reviewed in16). Few of these isogenic models currently exist for prostate cancer.
This study aimed to generate and characterise an isogenic model of radioresistant prostate cancer. Of the four commonly used prostate cancer cell lines, only 22Rv1 cells may be representative of primary disease17. This non-metastatic prostate cancer cell line was exposed to a fractionated radiation protocol. The resulting subline was evaluated for modification in radiation response and oncogenic properties. Our data suggests that this newly established radioresistant model has the potential to support discovery of novel biomarkers predictive of radiotherapy success.
Results
Selection of radioresistant 22Rv1 cells by fractionated irradiation
Wild type 22Rv1 (WT-22Rv1) were either exposed to 2-Gy fractionated radiation to a cumulative dose of 60Gy (RR-22Rv1) or mock irradiated (age matched controls AMC-22Rv1). At the end of this process, the proliferative potential following increasing radiation doses (2–10Gy) was measured in RR-22Rv1, AMC-22Rv1 and WT-22Rv1 using clonogenic assays. The individual experiments were used to define an average survival curve, with the deviation defined by summing the variance for each individual experiment at the corresponding dose and taking the square root (Fig. 1A). The surviving fraction of RR-22Rv1 cells was significantly higher than that of the WT-22Rv1 cell line at all doses tested. This increase was sustained one month later (RR-22Rv1-1M). Each experimental repeat was fitted with a linear curve between 0–6 Gy. The slope was then calculated to assess the decrease in survival. Wild-type 22RV1 cell survival decreased 15.24%/Gy (±0.6%), age-matched control cell survival decreased 15.20%/Gy (±0.37%), and radiation resistant cell survival decreased 13.72%/Gy ± (0.38%). There was no significant difference in the survival of wild type and age-matched controls (p > 0.05), but there was a significant difference between wild type and radiation resistant (p < 0.001) and between age-matched control and radiation resistant (p < 0.001) cells. The selection of radioresistant 22Rv1 cells was further evidenced by an increase in the area under the curve of the radiation survival curve from 2.3 (WT-22Rv1) to 3.14 (RR-22Rv1 cells). Mock-irradiated, aged-matched controls (AMC-22Rv1) exhibited an intermediate area under the curve of 2.7 and significantly reduced clonogenic survival compared to that of RR-22Rv1 cells at all doses tested, but 2 Gy (p > 0.05).
Figure 1. Generation of a stable isogenic radiation resistant prostate cancer cell line.

(A) Surviving fraction, after 60Gy cumulative dose of X-radiation, of the radiation resistant (RR-22RV1) isogenic cell line model compared with 22RV1 wild type (WT-22RV1) control and 22RV1 age matched control (AMC-22RV1). The isogenic cell line shows consistent resistance to radiation after one month of growth with no treatment (RR-22RV1 + 1M). Survival of irradiated cells was measured by clonogenic cell survival assay after X-radiation with 0–10 Gy. Points are measured as means ± SEM of at least five individual experiments. (B) Surviving fraction of aerobic and hypoxic (0.5% O2, 24 hrs) WT-22Rv1 and RR-22Rv1 cells. (C) Clonogenic survival of 22RV1-WT, 2RRv1-AMC and 22RV1-RR cells treated with 0.01, 0.5 and 0.1 nM Docetaxel for 48 hours. Points are measured as means ± SEM of at least five individual experiments. Mean ± SEM, *refers to significant differences in the RR cell line vs WT control. †refers to significant differences between RR and AMC cells. Δ refers to significant differences between AMC and WT control cells. *P ≤ 0.05, **P ≤ 0.01.
Hypoxic response of selected radioresistant 22Rv1 cells
To evaluate whether the radioresistance of RR-22Rv1 could be further increased, the clonogenic survival of WT-22Rv1 and RR-22Rv1 was measured following exposure to 0.5% oxygen for 24 h prior to irradiation (Fig. 1B). Hypoxic treatment increased the survival of WT-22Rv1 cells by up to 4-fold following irradiation (ANOVA, p < 0.0001) with an oxygen enhancement ratio of ~2.2 at 50% survival. The clonogenic survival of RR-22Rv1 cells was significantly higher than that of hypoxic WT-22Rv1 cells over the 0–6 Gy dose range (mean difference in survival of 18% at 2 Gy (p < 0.001), 10% at 4 Gy (p < 0.05) and 11% at 6 Gy (p < 0.05)). Hypoxia further protected RR-22Rv1 from the effect of radiation with a significant increase in clonogenic survival at all doses tested with an oxygen enhancement ratio of ~1.2.
Sensitivity of selected radioresistant 22Rv1 cells to Docetaxel
The response of RR-22Rv1, AMC-22RV1 and WT-22RV1 cells to docetaxel was investigated using clonogenic assays (Fig. 1C). Clonogenic survivals following docetaxel treatment (0.01, 0.05, 0,1nM, 48 h) were significantly reduced in 22Rv1-RR and 22Rv1-AMC, when compared to 22Rv1-WT cells at all concentrations tested (p < 0.005).
S-phase cell cycle fraction of radioresistant 22Rv1 cells
Cells cycle distributions were next measured by PI staining and flow cytometry (Fig. 2A). The % of cells in the S-phase was significantly higher in RR-2Rv1 (43.2 ± 1.3%) than in WT-22Rv1 (20.9 ± 3.3%, p = 0.0007) but not AMC-22Rv1 (34.6 ± 5.1%, p = 0.11). This pattern was maintained following irradiation (2-Gy) (ANOVA, p = 0.002). Radiation exposure did not increase the % of S-phase cells in all three lines (WT-22Rv1, p = 0.42; AMC-22Rv1, p = 0.76; RR-22Rv1, p = 0.99). The % of cells in the G2 phase were not significant different in both untreated (ANOVA, p = 0.23) and irradiated cell lines (ANOVA, p = 0.11).
Figure 2. Fractionated radiation exposure selects for 22Rv1 cells with enriched S-phase fraction and apoptosis resistance.

(A) Cell cycle analysis of WT-22RV1, RR-22RV1 and AMC-22RV1 cells untreated and four hours post irradiation with 2 Gy. (B) Radiation induced apoptosis measured using Annexin V and PI staining in WT-22RV1, RR-22RV1 and AMC-22RV1, 6 hours after treatment with 2 Gy and 4 Gy X-irradiation. (C) SA-β-gal activity measured with C12FDG and flow cytometry, with and without treatment of 2 Gy, 4 Gy and 8 Gy of ionising radiation. n = 2 *refers to significant differences in the RR cell line vs WT control. †refers to significant differences between RR and AMC cells. Δrefers to significant differences between AMC and WT control cells. *P ≤ 0.05, **P ≤ 0.01.
Apoptosis sensitivity of radioresistant 22Rv1 cells
Apoptotic cell death following exposure to radiation (2, 4 Gy) was quantified in all three lines using annexin V-FITC/PI staining and flow cytometry and compared to that of aged matched controls (Fig. 2B). The % of cells in early apoptosis was significantly reduced in RR-22Rv1 when compared to WT-22Rv1 and AMC-22Rv1 at both doses. The baseline % of apoptotic cells were similar across the three cell lines (ANOVA, p = 0.77). Following exposure to radiation (2, 4 Gy), the % of apoptotic cells increased in WT-22Rv1 (2 Gy: 3.58 ± 0.15 to 6.50 ± 0.65. p = 0.024; 4G y: 3.58 ± 0.15 to 7.5 ± 0.83, p = 0.03) and AMC-22Rv1 (2 Gy: 4.59 ± 0.67 to 4.34 ± 1.17, p = 0.86; 4 Gy: 4.59 ± 0.67 to 8.22 ± 0.95, p = 0.03) but not in RR-22Rv1 cells (2 Gy: 3.79 ± 1.05 to 2.51 ± 0.77, p = 0.38; 4 Gy: 3.79 ± 1.05 to 2.25 ± 0.7, p = 0.28).
Susceptibility to radiation-induced senescence of radioresistant 22Rv1 cells
The levels of senescence-associated β-galactosidase (SA-β-Gal) were next measured by flow cytometry18 (Fig. 2C). A non-significant trend towards elevated β-galactosidase levels was seen in untreated RR-22Rv1 cells, when compared to AMC- and WT-22Rv1 cells (ANOVA, p = 0.14). A similar trend was observed in irradiated cell lines (ANOVA, 2 Gy, p = 0.18; 4 Gy p = 0.17, 8 Gy p = 0.06). Radiation exposure appeared to increase senescence levels within each of the three lines, but the differences did not reach statistical significance (ANOVA, WT-22Rv1 p = 0.32. AMC-22Rv1, p = 0.22. RR-22Rv1, p = 0.56).
DNA repair capacity of radioresistant 22Rv1 cells
The alkaline comet assay (ACA) was used to quantify immediate levels of induced DNA damage (% Tail DNA) in the three cell lines following irradiation (0–10 Gy) (Fig. 3A). The level of initial DNA damage was significantly lower in RR-22Rv1 cells when compared to both AMC-22Rv1 and WT-22Rv1 in unirradiated cells and sustained across all doses tested (ANOVA, p < 0.0001). Initial DNA damage formation increased exponentially with increasing radiation dose in all three lines. At 10 Gy, the absolute increase in % Tail DNA compared to unirradiated control was highest in 48.5 ± 0.9% in WT-22Rv1 (0 Gy: 13.3 ± 0.55% to 10 Gy 61.6 ± 0.7%) 30.6 ± 0.68% in AMC-22Rv1 (0 Gy: 10.3 ± 0.36% to 10 Gy: 40.9 ± 0.6%) and lowest in 25.47 ± 0.5% in RR-22Rv1 (0 Gy: 7.03 ± 0.32% to 10Gy: 32.5 ± 0.44%). A strong correlation between the % tail DNA and clonogenic survival was identified in all three lines (22Rv1-RR, r = −0.79, p = 0.0028; 22Rv1-AMC, r = −0.94, p = 0.01; 22Rv1-WT, r = −0.94, p = 0.01) (Fig. 3B). ACA was next used to assess the extent of DNA damage repair for up to 50 min following 8Gy exposure (Fig. 3C). The DNA damage levels decreased rapidly within the first 15 min in all three lines. The reduction was greatest in RR-22Rv1 (2.37-fold), when compared to AMC-22Rv1 (1.81-fold) and WT-22Rv1 (1.3-fold) (p < 0.0001). For RR-22Rv1, beyond this time point, the % amount of DNA damage continued to decrease, albeit at a slower pace. Fifty minutes post exposure, the initial % tail DNA was reduced 3.9-fold in RR-22Rv1 cells (p < 0.0001) compared to 2.3- (p < 0.0001) and 1.12-fold (p = 0.005) in AMC-22Rv1 and WT-22Rv1 cells, respectively.
Figure 3. Increased proliferative potential following radiation exposure is associated with reduced levels of DNA damage.

(A) DNA damage in response to treatment with a range of radiation doses (0–10 Gy) in each of the three cell lines as measured by alkaline comet assay. N = 3 independent experiments. (B) Correlation between clonogenic survival and % tail DNA damage in 22Rv1-RR cells. (C) DNA repair capabilities of the 22Rv1-RR, WT and AMC cell lines. Initial damage and repair up to 50 min post irradiation with 8 Gy is measured using the alkaline comet assay. n = 5 independent experiments. DNA damage was calculated using % tail DNA for each comet, 400 comets were scored on two slides/4 gels per replicate and SEM calculated. *refers to significant differences in the RR cell line vs WT control. †refers to significant differences between RR and AMC cells. Δ refers to significant differences between AMC and WT control cells. *P ≤ 0.05, **P ≤ 0.01.
ROS levels in radioresistant 22Rv1 cells
The levels of reactive oxygen species (ROS) in the live and dead cell subpopulations of irradiated (2 Gy, 4 Gy) and unirradiated cells were measured using CM-H2DCFDA staining analysed by flow cytometry. In the live cells (Fig. 4A), the measures of ROS appeared elevated in RR-22Rv1 and AMC-22Rv1 cells but were not significantly different between the three cell lines for the unirradiated (ANOVA, p = 0.092) and the irradiated cells (ANOVA, 2 Gy, p = 0.07; 4 Gy, p = 0.16). Similarly, exposure to radiation did not significantly change the amounts of ROS within each of the three lines (ANOVA, WT-22Rv1, p = 0.12 ; AMC-22Rv1, p = 0.74, RR-22Rv1, p = 0.68). In the dead/dying subpopulation of cells (Fig. 4B), the measures of ROS were significantly different between the three cell lines in the unirradiated (ANOVA, p = 0.003) and irradiated cells (ANOVA, 2 Gy, p = 0.001; 4 Gy, p = 0.002) and highest in the AMC-22Rv1 and RR-22Rv1 cells. However, exposure to radiation did not significantly change the levels of ROS within all three lines (ANOVA, WT-22Rv1, p = 0.92; AMC-22Rv1, p = 0.09, RR-22Rv1, p = 0.10). Catalase activity was measured in irradiated (4 Gy) and unirradiated cells (Fig. 4C). At a cell number of 5 × 106, catalase activity was significantly different between the three cell lines in unirradiated (ANOVA, p = 0.01) and irradiated cells (ANOVA, p = 0.01). Subgroup comparisons indicate that catalase activity is significantly lower in AMC-22Rv1 cells, when compared to WT-22Rv1 under both conditions. In response to radiation exposure, catalase activity was not statistically different in WT-22Rv1 (p = 0.5), AMC-22Rv1 (p = 0.85) and RR-22Rv1 (p = 0.78), when compared to unirradiated controls. Finally, the radiation clonogenic survival curve of both WT-22Rv1 and RR-22-Rv1 cells was not modified following treatment with the anti-oxidant epigallocatechin-3-gallate (EGCG) (Fig. 4D).
Figure 4. Fractionated radiation exposure selects for cells with high reactive oxygen species levels.

(A) Reactive Oxygen species levels in the live cell population (isolated using PI) following treatment with radiation (2/4 Gy) compared to untreated controls as measured by Median Fluorescence Intensity (MFI) of DCFDA. Mean ± SD for 4 experiments. (B) Levels of reactive oxygen species in the measured in the dead/dying cell population (isolated using PI uptake) following treatment with radiation (2/4 Gy) compared to untreated controls as measured by Median Fluorescence Intensity (MFI) of DCFDA. Mean ± SD for 4 experiments. (C) Catalase activity of isogenic radiation resistant cell lines and controls before and after treatment with 4 Gy x-irradiation. Points mean of 4 independent experiments ± SEM. (D) Radiation clonogenic survival curve of 22Rv1-WT and 22Rv1-RR cells treated with 10 uM ECGC and untreated controls. Mean ± SD for 4 experiments. *refers to significant differences in the RR cell line vs WT control, †refers to significant differences between RR and AMC cell lines and Δ refers to significant differences between AMC and WT control cells. Φ differences between WT 4 Gy vs AMC 4 GY, ψ differences between WT 4 Gy and RR 4 Gy. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Migration capacity of 22Rv1 cells
The CD44+ fraction was determined in each cell line by flow cytometry (Fig. 5A). The fraction of CD44+ cells was significantly reduced in the radioresistant RR-22RV1 (52%), when compared to WT-22RV1(91%) and AMC-22RV1 cell populations (95%) (ANOVA, p < 0.0001). There was no significant difference between the fractions of CD44+ cells in the WT-22RV1 cell population when compared to the AMC-22RV1. A wound healing assay was performed on RR-22Rv1, AMC-22Rv1 and WT-22Rv1 cells to assess cellular motility (Fig. 5B)19. Over a 48 h period, wound healing was most pronounced in 22Rv1-RR, as evidenced with significantly decreased wound area, when compared to 22Rv1-WT cells (p = 0.02). This effect was prevented by treatment with the stem cell inhibitor salinomycin in all three lines (Fig. 5B).
Figure 5. Exposure to fractionated radiation selects for 22Rv1 cells with reduced CD44 expression and enhanced migration capacity.
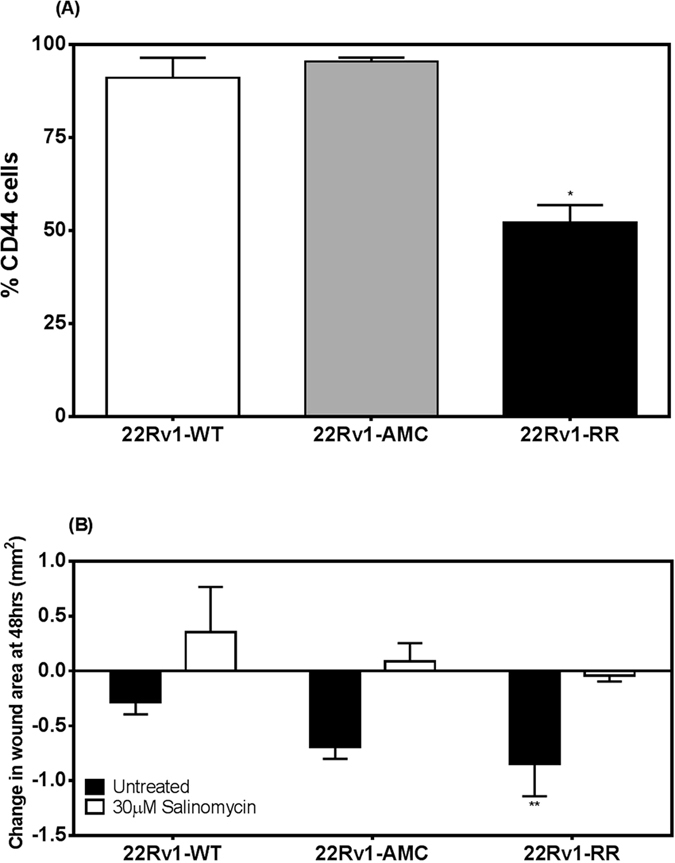
(A) The %CD44 positive cells was determined by flow-cytometry from the fluorescence intensities measured in 22Rv1-RR, AMC, and WT cells stained with FITC-conjugated CD44 antibody (B) The change in wound area was measured 48 hours following injury in 22Rv1-WT, AMC and RR cells in the absence or presence of 30 μM salinomycin (24 hours). n = 3; *p < 0.05.
Discussion
Isogenic models of radioresistance have been generated through exposure of cancer cell lines to a variety of fractionation schedules with total doses within a 40–60 Gy range20,21,22 and overall treatment times varying from 5 days23 to 6 years16,24. In prostate cancer, isogenic models of LncaP, PC-3 and DU145 cells were generated through 2 Gy daily exposure over 5 consecutive days and associated with a 1.6, 1.5 and 1.5 fold increase in the radiation dose needed to induce 0.1% survival (dose modifying factor), when compared to wild type cells23. The radiation-surviving cell population of DU145, PC-3, LnCaP and 22Rv1 cells following exposure to 35 doses of 2 Gy was isolated to examine effects on plasticity25 and neuroendocrine differentiation26. A number of limitations prevent the reproduction of a clinical radiotherapy delivery under experimental conditions such as cell ageing and the necessity for recovery periods16.
In this study, 22Rv1 cells were exposed to repeated 2 Gy-dose fractions and allowed to recover to a set confluence of ~70–80% in between fractions. The cumulative exposure of 22Rv1 cells to 60 Gy-fractionated radiation resulted in the generation of a sub-line with a significantly increased clonogenic survival potential following radiation exposure, when compared to wild type and mock irradiated, aged-matched cells. Increased colony forming ability is one of the most commonly reported consequences of protracted fractionated radiation exposure and is often associated with a modification of cell cycle distribution16,22. The resulting RR-22Rv1 cell line was enriched in S-phase cells, when compared to WT-22Rv1 cells. Enrichment in this radioresistant cell cycle phase has been reported in other radioresistant cell models16. In prostate cancer specimens, a larger S-phase fraction has been associated with more aggressive tumours27,28 and reduced local tumour control probability following radiotherapy29. Further evaluation of the underlying mechanisms for the amplification of the S-phase cell population is warranted.
The microtubule targeting agent Docetaxel is the standard of care first line chemotherapeutic drug for the treatment of hormone refractory prostate cancer30. Its effect is however limited by the poorly understood development of taxane-refractory tumours31. RR-22Rv1 and AMC-22Rv1 cells were more sensitive to Docetaxel than WT-22Rv1, suggesting distinct, possibly age-related, mechanisms of resistance.
Evaluations of the volume of prostate tumours suggest that 20% is exposed to less than 5 mmHg (0.7%) oxygen and prostate tumours are therefore considered hypoxic in nature32. Clinical evidence and modelling studies have demonstrated that tumour cell proliferation rates, DNA repair capacity and cellular hypoxia collectively modulate tumour control probability in response to radiation therapy33. The resulting RR-22Rv1 line was significantly more resistant to radiation than hypoxic WT-22Rv1 cells. Further enhancement of the radioresistance of RR-22Rv1 cells by hypoxia was small, suggesting that levels of radioresistance may have peaked in these cells.
Formation of DNA damage is the key event for cell killing by ionizing radiation34 and a cell’s ability to repair such DNA damage is key to determining a cell’s fate following irradiation35. Analysis of % Tail DNA immediately and up to 50 min after radiation exposure indicated that RR-22Rv1 cells were significantly less sensitive to initial damage induction than WT-22Rv1 and AMC-22Rv1 cells and repaired damage more effectively. This characteristic of radioresistant cells was documented in other isogenic cell models16,36,37. In prostate cancer patients receiving androgen deprivation therapy, the induction of reduced DNA repair capacity was proposed as a possible underlying mechanism for the improved response of these patients to subsequent radiotherapy treatment38. The % tail DNA damage peaked at 30 min post-irradiation in WT-22Rv1 cells but not in AMC-22Rv1 or RR-22Rv1 cells. The mechanistic interpretation of this peak should be further investigated. It may reflect induction of DNA damage by secondary radiation-induced reactive oxygen species, such as those released by the mitochondria39.
Mitochondrial respiration is increasingly documented to contribute to tumor cell survival and proliferation40. Mitochondrial dysfunction has been associated with cellular fate following irradiation41 and a direct role in radiation-induced G2/M arrest39 and apoptosis induction has been proposed42. Following irradiation, WT-22Rv1 and AMC-22Rv1 cells were more susceptible to apoptosis induction than RR-22Rv1 cells. Modification in radiation-induced apoptosis sensitivity has been reported in some43 but not other21 isogenic models. The prognostic potential of apoptosis sensitivity in prostate radiotherapy patients has been proposed44 but remains unconfirmed. Nonetheless, the pharmacologic radio sensitization of prostate cancer cells is often achieved through induction of apoptosis45. Further evaluation of the underlying mechanisms for this reduced susceptibility to radiation-induced apoptosis is required.
The induction of permanent growth arrest through senescence was proposed as an alternative mechanism in the therapeutic response to therapy in cancer cells with deregulated apoptotic signalling46. The induction of senescence in response to elevated DNA damage has been associated with increased therapeutic sensitivity47. In prostate cancer patients, increased expression of β-galactosidase (GLB1) was associated with a reduced risk of recurrence48. A proportion of surviving DU145 and PC3 cells exposed to 35 fractions of 2 Gy developed a senescent-like morphology associated with elevated mRNA expression of key senescence markers25. Our RR-22Rv1 cells however failed to show statistically significant evidence for a possible reduction in the induction of therapeutic senescence. Our analysis of β-galactosidase activity would not detect other permanent growth arrests such as the induction of neuroendocrine differentiation (NED). Therapeutic NED has also been identified as a cause of radioresistance that may be limited to CD44+ prostate cancer stem cells49. The evaluation of the stemness of our model is pending, but our preliminary analysis of CD44 expression suggests that exposure to fractionated radiation reduces the fraction of CD44-expressing 22Rv1 cells. Downregulation of CD44 was associated with increased radiosensitivity in DU145 and PC3 cells50, but increased migration potential51. Wound closure was more efficient in CD44-deprived RR-22Rv1 than WT-22Rv1 or AMC-22Rv1 cells. This process was inhibited by salinomycin, an anti-coccidial drug increasingly tested for its potential anti-cancer and anti-cancer stem cell properties52,53. This sodium ionophore monensin similarly inhibited proliferation and migration of LnCaP cells51. Further investigation into the role of CD44 in the selection of a radioresistant subpopulation of cells is warranted.
The ability of prostate cancer cells to manage oxidative stress plays an important role in cell signalling and their response to therapeutic injury54. In particular, reduced susceptibility to ROS-induced cellular damage and efficient repair of radiation-induced DNA damage was proposed as an underlying mechanisms to the observed radioresistance of prostate cancer stem cells55. Lower ROS basal levels were associated with increased Nfr2 levels and radiosensitivity in prostate cancer cells56. Basal ROS levels were lower in WT-22Rv1, than in AMC-22Rv1 and RR-22Rv1 cells. This reduction was associated with increased catalase activity in WT-22Rv1 cells. ROS levels were elevated in dying RR-22Rv1 cells. This suggests that cells with high ROS levels are preferentially eliminated in this cell population or reflects the likely release of ROS in dying cells. However, the sensitivity of cells to the anti-oxidant epigallocatechin-3-gallate (EGCG), whose therapeutic potential in prostate cancer was proposed previously57, remained unchanged in both WT- and RR-22Rv1 cells.
Conclusion
Exposure to fractionated radiation progressively selected for 22Rv1 cells with enhanced oncogenic properties protective against radiation injury and supportive of tumour invasion. The characterisation of the radioresistance of these cells using in vivo dose response assays is required to account for the likely effect of spontaneous (non-radiation) death rates58 and the tumour microenvironment59 on tumour control probability in a clinical setting15. This data must be considered within the context of one cell line, and the limitations of in vitro models, but the phenotyping modifications observed support the clinical relevance of this model to enable further study of the mechanisms of radioresistance in prostate cancer cells.
The progressive selection of radioresistant cells throughout a protracted radiation therapy protocol may have clinical implications. In the era of personalised medicine, concurrent targeted therapies may require careful timing and perhaps may be more effective when administered toward the end of radiotherapy delivery.
Materials and Methods
Cell lines and culture
The human prostate cancer cell line 22Rv1 was obtained from American Type Culture Collection. Cells were maintained as monolayers in RPMI cell culture medium containing l-glutamine (Lonza, Castleford, UK) with 10% foetal bovine serum (Gibco, Dublin, Ireland) and 1% pen/strep (Lonza). The cells were maintained at 37 °C in 95% humidified air containing 5% CO2 and sub-cultured once to twice weekly to maintain exponential growth unless otherwise stated. Cells were grown to approximately 70–80% confluency in vented 75 cm2 culture flasks prior to irradiation. Each irradiation consisted of 2 Gy X-rays dose (250 keV, 15 mA) using an RS225 cabinet irradiator (Gulmay Medical, Surrey, UK). This process was repeated weekly until the cells received to a total cumulative of 60 Gy. Mock irradiated cells were cultured in parallel as age-matched controls (AMC-22Rv1). Hypoxia (0.5% O2, pO2 < 2 mmHq) was achieved by exposing cells in a 1000 in vivo hypoxic chamber (BioTrace, Bracknell, UK) to a mixture of nitrogen, CO2 (5%) and compressed air to achieve a 0.5% oxygen concentration.
Clonogenic Assay
Cell survival was evaluated using a standard colony forming assay. Cells in exponential growth phase (approx. 70% confluent) were harvested by trypsinisation (Lonza) and counted in order to obtain a single cell suspension of 1 × 106 cells/ml. Cells were seeded into six well plates at a density of 1 × 103 −1 × 104 cells/well and allowed to adhere to plates overnight in the incubator at 37 °C in 95% humidified air containing 5% CO2. Cells were then irradiated (2, 4, 6, 8 or 10 Gy single doses) and incubated for 7–10 days under the conditions previously described. Following incubation, colonies were fixed and stained using 0.05% crystal violet in 70% methanol solution. Plates were dried overnight and colonies containing >50 cells counted using the ColCount instrument (Oxford Optronix Ltd, Oxford, UK). Surviving fraction was calculated as no. colonies/(no. cells seeded X PE). The platting efficiency PE was calculated using the no. colonies/no. cells seeded in the unirradiated cells.
Cell Cycle Analysis
Cell cycle analysis was performed using Propidium Iodide (PI) staining and flow cytometry as previously described60. Cells were harvested at 4 h, 8 h, 12 h, 24 h, 30 h post irradiation, using trypsin-EDTA(Lonza), and the pellet fixed and permeabilised by dropwise addition of 4 ml ice cold 70% EtOH (Sigma-Aldrich, Wicklow, Ireland). Cells were fixed for a minimum of 2 h at 4 °C. Fixed cells were centrifuged at 180 g × 3 min and the supernatant decanted, samples were then washed in PBS and centrifuged as before. Each sample was resuspended in 0.5 ml Triton X-100 (0.1% v/v in PBS)(Sigma-Aldrich) containing PI (0.02 mg/ml PI)(Sigma-Aldrich) and RNase A (0.2 mg/ml) (Sigma-Aldrich). Unstained fixed controls were resuspended in PBS. Samples were incubated at 37 °C for 30 min and analysed immediately using CyAnTM ADP Analyzer (Beckman Coulter, Clare, Ireland.). Cell cycle phase was determined using flowJo software (FlowJo LLC, Or, USA).
Apoptosis Assay
Apoptotic cells were detected using annexin V-FITC/PI staining and flow cytometry as previously described61. Following treatment with 0, 2 or 4 Gy of X-irradiation, cells were trypsinised and combined with supernatant containing non-adherent cells. Cell pellets were resuspended in ice cold 1X binding buffer (0.1M Hepes (Sigma-Aldrich) 1.4M NaCl (Sigma-Aldrich) and 25 mM CaCl2, at pH 7.4 (Sigma-Aldrich). Cells were stained with 3 μl Annexin V-FITC (IQ Products, Groningen, Netherlands) and incubated at 4 °C. Cells were washed in ice cold PBS and resuspended in 1X binding buffer containing PI (0.05 μg/ml) (Invitrogen, Dublin, Ireland). Cell were analysed using the CyAnTM ADP Analyser (Beckman Coulter) and apoptotic cells were detected using the FlowJo software (FLowJo LLC, Or, USA).
Senescence
β-Galactosisdase activity was measured by flow cytometry as previously reported18. Cells were seeded in 6 well plates at a density of 1–2 × 105 cells/well and allowed adhere overnight prior to irradiation (2 Gy, 4 Gy or 8 Gy) and returned to the incubator along with mock irradiated controls for 24 h. Cells were washed with PBS and incubated for 1 h at 37 °C, 5% CO2 with Bafilomycin A1 (Sigma-Aldrich) supplemented growth media to adjust lysosomal pH to pH6. 2 mM 5-Dodecanoylaminofluorescein di-β-D-galactopyranoside (C12FDG) (Sigma-Aldrich) was added and samples were incubated for 1 h before analysis. Cells were then trypsinised and resuspended in 1 ml PBS and analysed immediately using a CyAnTM ADP Analyser (Beckman Coulter). Mean fluorescence intensity was calculated using FlowJo software.
Alkaline comet assay
A previously reported version of the alkaline comet assay (ACA)62 adapted from the original protocol by Singh et al.63 was used to determine levels of DNA damage and repair capability after irradiation. All reagents used were obtained from Sigma-Aldrich, Dorset, UK. Briefly, 4 × 104 cells resuspended in low melting point agar (0.6% in PBS) were layered onto agar-coated slides (1% in ddH2O) prior to irradiation on ice and in the dark. Following this slides for the repair study were incubated at 37 °C, 5% CO2, 95% humidified air in RPMI media containing 20% FBS and 5% pen/strep for up to 50 min. All slides were then incubated overnight in ice cold lysis buffer (100 mM NA2EDTA, 2.5M NaCL, 10 mM Tris-HCL, pH 10 with 1% Triton X added immediately before use). Following electrophoresis (30 V, 30 mA, 20 min in 300 mM NaOH, 1 mM Na2EDTA, pH 13), the slides were incubated in neutralization buffer (0.4M Tris-HCL, pH7.5), washed and allowed to dry for 2 h at 37 °C prior to PI staining (2.5 μg/ml). Comets were visualised and measured using Komet 5.0 software (Kinetics imaging software ltd, Brombourough UK) and fluorescent microscope at a magnification of 20X. 50 comets were scored/gel. The mean % tail DNA, calculated as the percentage of DNA present in the comet tail (measured as an intensity) compared to total comet intensity), was deduced from three (dose response) or four (repair study) independent experiments and presented as mean ± SEM.
Oxidative Stress
Intracellular oxygen species were detected using Di(Acetoxymethyl Ester) (6-Carboxy-2′,7′-Dichlorodihydrofluorescein Diacetate) (CM-H2DCFDA)(Life Technologies, Dublin, Ireland) and flow cytometry. Exponentially growing cells were collected by trypsinisation. Samples were incubated for 30 min in the dark at 37 °C, 5% CO2, 95% humidified air in PBS containing 10 μM CM-H2DCFDA, with controls incubated in PBS. After incubation cells were pelleted at 180 × g for 3 min and the supernatant decanted. Samples were resuspended in PBS and irradiated with 2 Gy or 4 Gy X-rays, controls were mock irradiated and placed directly on ice. PI (1 mg/ml) was added to the samples except appropriate controls. Samples were analysed immediately using CyAnTM ADP Analyser (Beckman Coulter). Data was analysed using FlowJo software.
Catalase Activity
Catalase activity was measured using a visual method as previously reported64. Cells harvested as previously described were resuspended in pyrex tubes (Corning, Flintshire, UK) in varying concentrations (105–1.57 cells) in 100 μl of PBS (Lonza) prior to mock or 4-Gy irradiation. Catalase powder (Sigma-Aldrich) was dissolved in 100 μl distilled pure water to generate a calibration curve for catalase activity (0–200units). 100 μl1% Triton X-100 and 100 μl undiluted hydrogen peroxide (30%) were added to the solutions, mixed thoroughly and incubated at room temperature for 15 min before the height of O2-forming foam was measured with a ruler.
Migration Assay
A wound healing assay was performed with the Ibidi Wound Healing 6 Well Plate Assay Kit (Ibidi, GmbH, Martinsried, Germany). Cells were seeded into Ibidi insert wells at 500,000 cells/well with total volume 70 μl, allowed to adhere for 24 h and incubated in 30 μM salinomycin (Sigma-Aldrich) treated media for 24 h. After incubation, the insert was removed and images were taken every at 24 and 48 h. Image analysis and migration rate determination was performed using ImageJ Analysis software (Bethesda, USA).
CD44 Expression
Cell pellets were resuspended in staining buffer containing CD44 (FITC-conjugated mouse monoclonal anti-human, BD Pharminogen) and incubated for 20 min at 4 °C in the dark. Cells were washed and fluorescence intensity was measured using CyAnTM ADP Analyser (Beckman Coulter).
Docetaxel treatment
Cells were seeded into 6 well plates at a density of 750 cells/well for control and vehicle control (1X DMSO) and 3 × 103 cells/well for treated. Cells were treated with 0.01 nM, 0.05 nM, 0.1 nM, 0.25 nM, 0.5 nM and 1 nM of Docetaxel (Sigma Aldrich) for 48 h or left untreated as controls.
Statistical Analysis
All statistical analysis was carried out using Prism (version 6 for mac OS X and windows 7, Graphpad software La Jolla California USA, www.graphpad.com). The analysis of differences in the means of continuous data was performed with unpaired two tailed Student t-Tests, under the assumption that the data is Normally distributed. A One-way ANOVA with Bonferroni correction was used when the means of more than two groups were compared. A spearman correlation was used to quantify the non-linear relationship between the % tail DNA and clonogenic surviving fractions. All experiments were performed in triplicates unless otherwise stated. A p-value < 0.05 was considered statistically significant.
Additional Information
How to cite this article: McDermott, N. et al. Fractionated radiation exposure amplifies the radioresistant nature of prostate cancer cells. Sci. Rep. 6, 34796; doi: 10.1038/srep34796 (2016).
Acknowledgments
We would like to acknowledge support from the Irish Cancer Society (grant code PCA12MAR). Gabrielle Nortey sand Christopher Hernandez were Mount Sinai International Exchange Program minority student participants. Their work was supported in part by grant MD001452 from the National Center on Minority Health and Health Disparities of the National Institutes of Health. Their work was supported in part by grant MD 001452 from the National Center on Minority Health and Health Disparities of the National Institutes of Health, Dr. Luz Claudio, Principal Investigator. Sarah Hurley was supported by a grant from the M.Sc. in Molecular Medicine, Department of Clinical Medicine at Trinity College Dublin. We would also like to acknowledge the help and support of Dr. Eamon Breen in the Institute of Molecular Medicine Flow Cytometry Facility.
Footnotes
Author Contributions N.M.D. contributed to all aspects of the manuscript. A.M. all experimental aspects of the study. N.G. conducted the CD44 and cell migration assays. C.H. conducted the senescence study. S.H. generated the docetaxel data. K.B. and S.B. assisted with the comet assays. B.M. contributed to the ROS experiments. B.M. performed the survival curve data analysis. N.L. assisted with the generation of the isogenic cell line. G.D.D.J. assisted with the comet assay and preparation of the manuscript. L.M. contributed to all aspects of the manuscript. All authors reviewed the manuscript.
References
- Heidenreich A. et al. EAU guidelines on prostate cancer. part 1: screening, diagnosis, and local treatment with curative intent-update 2013. Eur Urol 65, 124–137, doi: 10.1016/j.eururo.2013.09.046 (2014). [DOI] [PubMed] [Google Scholar]
- Cooperberg M. R., Broering J. M. & Carroll P. R. Time trends and local variation in primary treatment of localized prostate cancer. J Clin Oncol 28, 1117–1123, doi: 10.1200/JCO.2009.26.0133 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y. H. et al. Contemporary risk profile of prostate cancer in the United States. J Natl Cancer Inst 101, 1280–1283, doi: 10.1093/jnci/djp262 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good J. S. & Harrington K. J. The hallmarks of cancer and the radiation oncologist: updating the 5Rs of radiobiology. Clin Oncol (R Coll Radiol) 25, 569–577, doi: 10.1016/j.clon.2013.06.009 (2013). [DOI] [PubMed] [Google Scholar]
- Franken N. A., Rodermond H. M., Stap J., Haveman J. & van Bree C. Clonogenic assay of cells in vitro. Nat Protoc 1, 2315–2319, doi: 10.1038/nprot.2006.339 (2006). [DOI] [PubMed] [Google Scholar]
- Barendsen G. W., Van Bree C. & Franken N. A. Importance of cell proliferative state and potentially lethal damage repair on radiation effectiveness: implications for combined tumor treatments (review). Int J Oncol 19, 247–256 (2001). [DOI] [PubMed] [Google Scholar]
- Brown J. M. & Attardi L. D. The role of apoptosis in cancer development and treatment response. Nat. Rev. Cancer 5, 231–237 (2005). [DOI] [PubMed] [Google Scholar]
- Fang Y., DeMarco V. G. & Nicholl M. B. Resveratrol enhances radiation sensitivity in prostate cancer by inhibiting cell proliferation and promoting cell senescence and apoptosis. Cancer Sci 103, 1090–1098, doi: 10.1111/j.1349-7006.2012.02272.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day R. M., Snow A. L. & Panganiban R. A. Radiation-induced accelerated senescence: a fate worse than death? Cell Cycle 13, 2011–2012, doi: 10.4161/cc.29457 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J. M. Vasculogenesis: a crucial player in the resistance of solid tumours to radiotherapy. Br J Radiol 87, 20130686, doi: 10.1259/bjr.20130686 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marignol L., Coffey M., Lawler M. & Hollywood D. Hypoxia in prostate cancer: a powerful shield against tumour destruction? Cancer Treat Rev 34, 313–327, doi: S0305-7372(08)00020-0 10.1016/j.ctrv.2008.01.006 (2008). [DOI] [PubMed] [Google Scholar]
- Krause M., Yaromina A., Eicheler W., Koch U. & Baumann M. Cancer stem cells: targets and potential biomarkers for radiotherapy. Clin Cancer Res 17, 7224–7229, doi: 10.1158/1078-0432.CCR-10-2639 (2011). [DOI] [PubMed] [Google Scholar]
- Baumann M., Krause M., Thames H., Trott K. & Zips D. Cancer stem cells and radiotherapy. Int J Radiat Biol 85, 391–402, doi: 910578057 10.1080/09553000902836404 (2009). [DOI] [PubMed] [Google Scholar]
- Baumann M., Krause M. & Hill R. Exploring the role of cancer stem cells in radioresistance. Nat. Rev. Cancer 8, 545–554, doi: nrc2419 10.1038/nrc2419 (2008). [DOI] [PubMed] [Google Scholar]
- Zaider M. & Hanin L. Tumor control probability in radiation treatment. Med Phys 38, 574–583, doi: 10.1118/1.3521406 (2011). [DOI] [PubMed] [Google Scholar]
- McDermott N., Meunier A., Lynch T. H., Hollywood D. & Marignol L. Isogenic radiation resistant cell lines: development and validation strategies. Int J Radiat Biol 90, 115–126, doi: 10.3109/09553002.2014.873557 (2014). [DOI] [PubMed] [Google Scholar]
- Sramkoski R. M. et al. A new human prostate carcinoma cell line, 22Rv1. In Vitro Cell Dev Biol Anim 35, 403–409 (1999). [DOI] [PubMed] [Google Scholar]
- Noppe G. et al. Rapid flow cytometric method for measuring senescence associated beta-galactosidase activity in human fibroblasts. Cytometry. Part A: the journal of the International Society for Analytical Cytology 75, 910–916, doi: 10.1002/cyto.a.20796 (2009). [DOI] [PubMed] [Google Scholar]
- Rodriguez L. G., Wu X. & Guan J. L. Wound-healing assay. Methods Mol Biol 294, 23–29 (2005). [DOI] [PubMed] [Google Scholar]
- Fukuda K. et al. Differential gene expression profiles of radioresistant oesophageal cancer cell lines established by continuous fractionated irradiation. Br J Cancer 91, 1543–1550, doi: 10.1038/sj.bjc.66021876602187 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynam-Lennon N. et al. Alterations in DNA repair efficiency are involved in the radioresistance of esophageal Adenocarcinoma. Radiat Res, doi: 10.1667/RR2295.1 (2010). [DOI] [PubMed] [Google Scholar]
- Henness S. et al. Changes in gene expression associated with stable drug and radiation resistance in small cell lung cancer cells are similar to those caused by a single X-ray dose. Radiat Res 161, 495–503 (2004). [DOI] [PubMed] [Google Scholar]
- Skvortsova I. et al. Intracellular signaling pathways regulating radioresistance of human prostate carcinoma cells. Proteomics 8, 4521–4533, doi: 10.1002/pmic.200800113 (2008). [DOI] [PubMed] [Google Scholar]
- Kuwahara Y. et al. Clinically relevant radioresistant cells efficiently repair DNA double-strand breaks induced by X-rays. Cancer Sci 100, 747–752, doi: 10.1111/j.1349-7006.2009.01082.x (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyjacova L. et al. Radiotherapy-induced plasticity of prostate cancer mobilizes stem-like non-adherent, Erk signaling-dependent cells. Cell Death Differ, doi: 10.1038/cdd.2014.97 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X. et al. Ionizing radiation induces prostate cancer neuroendocrine differentiation through interplay of CREB and ATF2: implications for disease progression. Cancer Res 68, 9663–9670, doi: 10.1158/0008-5472.CAN-08-2229 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Wahab M. et al. Androgen receptor antigen density and S-phase fraction in prostate cancer: a pilot study. Prostate Cancer Prostatic Dis 6, 294–300, doi: 10.1038/sj.pcan.4500672 (2003). [DOI] [PubMed] [Google Scholar]
- Nemoto R. et al. S-phase fraction of human prostate adenocarcinoma studied with in vivo bromodeoxyuridine labeling. Cancer 66, 509–514 (1990). [DOI] [PubMed] [Google Scholar]
- Centeno B. A. et al. Flow cytometric analysis of DNA ploidy, percent S-phase fraction, and total proliferative fraction as prognostic indicators of local control and survival following radiation therapy for prostate carcinoma. Int J Radiat Oncol Biol Phys 30, 309–315 (1994). [DOI] [PubMed] [Google Scholar]
- Niu G. et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 21, 2000–2008 (2002). [DOI] [PubMed] [Google Scholar]
- Mathew P. & Dipaola R. Taxane refractory prostate cancer. J Urol 178, S36–S41 (2007). [DOI] [PubMed] [Google Scholar]
- Chopra S. et al. Comparing oxygen-sensitive MRI (BOLD R2*) with oxygen electrode measurements: a pilot study in men with prostate cancer. Int J Radiat Biol 85, 805–813 (2009). [DOI] [PubMed] [Google Scholar]
- Nahum A. E., Movsas B., Horwitz E. M., Stobbe C. C. & Chapman J. D. Incorporating clinical measurements of hypoxia into tumor local control modeling of prostate cancer: implications for the alpha/beta ratio. Int J Radiat Oncol Biol Phys 57, 391–401 (2003). [DOI] [PubMed] [Google Scholar]
- Dugle D. L., Gillespie C. J. & Chapman J. D. DNA strand breaks, repair, and survival in x-irradiated mammalian cells. Proc Natl Acad Sci USA 73, 809–812 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bristow R. G., Ozcelik H., Jalali F., Chan N. & Vesprini D. Homologous recombination and prostate cancer: a model for novel DNA repair targets and therapies. Radiother Oncol 83, 220–230, doi: S0167-8140(07)00156-9 10.1016/j.radonc.2007.04.016 (2007). [DOI] [PubMed] [Google Scholar]
- de Llobet L. I. et al. Development and characterization of an isogenic cell line with a radioresistant phenotype. Clin Transl Oncol 15, 189–197, doi: 10.1007/s12094-012-0898-8 (2013). [DOI] [PubMed] [Google Scholar]
- Mihatsch J. et al. Selection of radioresistant tumor cells and presence of ALDH1 activity in vitro. Radiother Oncol 99, 300–306, doi: 10.1016/j.radonc.2011.06.003 (2011). [DOI] [PubMed] [Google Scholar]
- Tarish F. L. et al. Castration radiosensitizes prostate cancer tissue by impairing DNA double-strand break repair. Sci Transl Med 7, 312re311, doi: 10.1126/scitranslmed.aac5671 (2015). [DOI] [PubMed] [Google Scholar]
- Yamamori T. et al. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic Biol Med 53, 260–270, doi: 10.1016/j.freeradbiomed.2012.04.033 (2012). [DOI] [PubMed] [Google Scholar]
- Ward P. S. & Thompson C. B. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 21, 297–308, doi: 10.1016/j.ccr.2012.02.014 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bol V. et al. Reprogramming of tumor metabolism by targeting mitochondria improves tumor response to irradiation. Acta Oncol 1–9, doi: 10.3109/0284186X.2014.932006 (2014). [DOI] [PubMed] [Google Scholar]
- Wendt J. et al. TRAIL sensitizes for ionizing irradiation-induced apoptosis through an entirely Bax-dependent mitochondrial cell death pathway. Oncogene 24, 4052–4064 (2005). [DOI] [PubMed] [Google Scholar]
- Xie L. et al. Fractionated irradiation induced radio-resistant esophageal cancer EC109 cells seem to be more sensitive to chemotherapeutic drugs. Journal of Experimental & Clinical Cancer Research 28 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szostak M. J. & Kyprianou N. Radiation-induced apoptosis: predictive and therapeutic significance in radiotherapy of prostate cancer (review). Oncol Rep 7, 699–706 (2000). [DOI] [PubMed] [Google Scholar]
- Rae C., Haberkorn U., Babich J. W. & Mairs R. J. Inhibition of fatty acid synthase sensitizes prostate cancer cells to radiotherapy. Radiat Res 184, 482–493, doi: 10.1667/RR14173.1 (2015). [DOI] [PubMed] [Google Scholar]
- Schmitt C. A. et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 109, 335–346 (2002). [DOI] [PubMed] [Google Scholar]
- Ewald J. A., Desotelle J. A., Wilding G. & Jarrard D. F. Therapy-induced senescence in cancer. J Natl Cancer Inst 102, 1536–1546, doi: djq364 10.1093/jnci/djq364 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J. et al. Overexpression of the novel senescence marker beta-galactosidase (GLB1) in prostate cancer predicts reduced PSA recurrence. PLoS One 10, e0124366, doi: 10.1371/journal.pone.0124366 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C. D., Choo R. & Huang J. Neuroendocrine differentiation in prostate cancer: a mechanism of radioresistance and treatment failure. Frontiers in Oncology 5, 90, doi: 10.3389/fonc.2015.00090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W. et al. CD44 is a biomarker associated with human prostate cancer radiation sensitivity. Clin Exp Metastasis 29, 1–9, doi: 10.1007/s10585-011-9423-7 (2012). [DOI] [PubMed] [Google Scholar]
- Ketola K. et al. Salinomycin inhibits prostate cancer growth and migration via induction of oxidative stress. Br J Cancer 106, 99–106, doi: 10.1038/bjc.2011.530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi A. M., Tao Z. Q., Li H., Wang Y. Q. & Zhao J. Cancer stem cells targeting agents–a review. Eur Rev Med Pharmacol Sci 19, 4064–4067 (2015). [PubMed] [Google Scholar]
- Zhou S. et al. Salinomycin: a novel anti-cancer agent with known anti-coccidial activities. Curr Med Chem 20, 4095–4101 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta-Elera G., Garrett A. R., Robison R. A. & O’Neill K. L. The role of oxidative stress in prostate cancer. Eur J Cancer Prev 21, 155–162, doi: 10.1097/CEJ.0b013e32834a8002 (2012). [DOI] [PubMed] [Google Scholar]
- Jayakumar S., Kunwar A., Sandur S. K., Pandey B. N. & Chaubey R. C. Differential response of DU145 and PC3 prostate cancer cells to ionizing radiation: role of reactive oxygen species, GSH and Nrf2 in radiosensitivity. Biochim Biophys Acta 1840, 485–494, doi: 10.1016/j.bbagen.2013.10.006 (2014). [DOI] [PubMed] [Google Scholar]
- Liu M. et al. Nrf2 sensitizes prostate cancer cells to radiation via decreasing basal ROS levels. Biofactors 41, 52–57, doi: 10.1002/biof.1200 (2015). [DOI] [PubMed] [Google Scholar]
- Lecumberri E., Dupertuis Y. M., Miralbell R. & Pichard C. Green tea polyphenol epigallocatechin-3-gallate (EGCG) as adjuvant in cancer therapy. Clin. Nutr. 32, 894–903, doi: 10.1016/j.clnu.2013.03.008 (2013). [DOI] [PubMed] [Google Scholar]
- Hanin L. G. Iterated birth and death process as a model of radiation cell survival. Math Biosci 169, 89–107 (2001). [DOI] [PubMed] [Google Scholar]
- Shiao S. L., Chu G. C. & Chung L. W. Regulation of prostate cancer progression by the tumor microenvironment. Cancer Lett 380, 340–348, doi: 10.1016/j.canlet.2015.12.022 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishan A. Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. The Journal of cell biology 66, 188–193 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermes I., Haanen C., Steffens-Nakken H. & Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. Journal of immunological methods 184, 39–51 (1995). [DOI] [PubMed] [Google Scholar]
- Moneef M. A. et al. Measurements using the alkaline comet assay predict bladder cancer cell radiosensitivity. Br J Cancer 89, 2271–2276, doi: 10.1038/sj.bjc.6601333 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N. P., Stephens R. E. & Schneider E. L. Modifications of alkaline microgel electrophoresis for sensitive detection of DNA damage. Int J Radiat Biol 66, 23–28 (1994). [DOI] [PubMed] [Google Scholar]
- Iwase T. et al. A simple assay for measuring catalase activity: a visual approach. Sci. Rep. 3, 3081, doi: 10.1038/srep03081 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]