

Abstract
Tetrahydrobiopterin (BH4) is a regulator of endothelial nitric oxide synthase (eNOS) activity. Deficient levels result in eNOS uncoupling with a shift from nitric oxide (NO) to superoxide generation. The hph-1 mutant mouse has deficient GTP-cyclohydrolase 1 (GTPCH1) activity resulting in low BH4 tissue content. The adult hph-1 mouse has pulmonary hypertension, but whether such condition is present from birth is not known. Thus, we evaluated newborn animals’ pulmonary arterial medial thickness, biopterins content (BH4+BH2), H2O2 and eNOS, right ventricle-to-left-ventricle+septum ratio (RV/LV+septum) and near-resistance pulmonary arteries agonist-induced force and endothelium-dependent and-independent relaxation. The biopterins lung content was inversely related to age for both types, but significantly lower in hph-1 mice, as compared to wild-type animals. As judged by the RV/LV+septum newborn hph-1 mice have pulmonary hypertension and following a two-week 13% oxygen exposure the ratio was similar in both types. The pulmonary arterial agonist-induced force was reduced (P<0.01) in hph-1 animals and no type-dependent difference in endothelium-dependent or –independent vasorelaxation was observed. When compared to wild-type mice, the H2O2 lung content was increased, whereas the eNOS expression decreased (P<0.01) in hph-1 animals. The pulmonary arterial medial thickness, a surrogate marker of vascular remodeling was increased (P<0.01) in hph-1 compared to wild-type mice. In conclusion, our data suggest that pulmonary hypertension is present from birth in the GTPCH1 deficient mice, not as a result of impaired vasodilation, but secondary to vascular remodeling.
Keywords: Tetrahydrobiopterin, Pulmonary Hypertension, Endothelial Dysfunction
Graphical Abstract
Introduction
Tetrahydrobiopterin (BH4) is an essential co-factor in the regulation of nitric oxide (NO) production by all NO synthases (NOS) isoforms. In the absence of this cofactor, both neuronal and endothelial dimeric NOS generate superoxide anion, instead of NO. Low BH4 levels have been associated with systemic and pulmonary hypertension later in life and its supplementation shown to promote vasodilation and reduce systemic blood pressure when pathologically increased [1]. The beneficial response to BH4 supplementation supports the concept that NO production may be limited by BH4 availability.
BH4 deficiency is known to occur in neonates and is often diagnosed on the basis of screening for hyperphenylalaninemia [2–4]. Several of these kids benefit from BH4 treatment that normalizes their liver functions. The potential therapeutic effect of supplemental BH4 in neonates with pulmonary hypertension has not been adequately evaluated. The hph-1 mouse was generated by N-ethyl-N-nitrosourea-induced (ENU) mutagenesis as an attempt to create an animal model of hyperphenylalaninemia [5]. The mutated animals have defective guanosine triphosphate cyclohydrolase-1 (GTPCH1), the enzyme responsible for the first committed step in the BH4 synthesis pathway. The hph-1 mouse is congenitally GTPCH1 deficient and early in life, its liver BH4 content is negligible, [6].
Adult hph-1 mice have been reported to exhibit pulmonary and systemic hypertension [7–9]. Yet it is not known whether similar changes are present in these animals early in life. This observation, together with the fact that the newborn pulmonary vasculature has a lower potential for endothelial NOS (eNOS)-dependent relaxation [10] led us to speculate that pulmonary hypertension is not only expected, but likely more severe in the hph-1 newborn. Therefore, we evaluated the newborn hph-1 lung biopterin content, vascular remodeling, as well as pulmonary vasoconstriction and relaxation potential. We hypothesized that newborn hph-1 mice have pulmonary hypertension secondary to impaired pulmonary vasorelaxation. It was further hypothesized that chronic hypoxia exposure would result in pulmonary hypertension of greater severity in hph-1 pups, when compared with wild type animals.
Methodology
Chemicals and reagents
All chemicals and reagents were obtained from Sigma Aldrich (Oakville, Ontario Canada), unless otherwise indicated.
Animals
All procedures were conducted according to criteria established by the Canadian Council on Animal Care and were approved by the Animal Care Committees of The Hospital for Sick Children Research Institutes.
Adult hph-1 animals obtained from Dr. Un Jung Kang, University of Chicago were bred in-house and confirmed by genotyping as being GTPCH1 deficient (data not shown). C57BL/6 × CBA, the wild-type strain where the hph-1 mutated animals were obtained, served as controls and was purchased from a commercial supplier (Charles River, Ontario, Canada). The animals were housed in standard conditions of light and temperature and fed regular rodent pellets.
Mice of both sexes were studied between 0–7 days of life (newborn), or as adults (60–90 days). The following is the number of animals utilized: Wild-type: 0–1 day old (N=22) 5–7 days of age (N=20), 2 weeks old (N=26) and adults (N=4). hph-1: 0–1 day old (N=26) 5–7 days of age (N=38), 2 weeks old (N=22) and adults (N=4). The animals were sacrificed with an overdose of pentobarbital sodium (50 mg/kg ip) and the lungs and heart quickly removed.
Organ bath studies
The functional in-vitro evaluation of newborn pulmonary arteries has been previously described [11]. Briefly, third generation lung intralobar pulmonary artery ring segments (average diameter 80–100 μm and length = 2 mm) were dissected free and mounted in a wire myograph (Danish Myo Technology A/S, Denmark). Isometric changes were digitized and recorded online (Myodaq, Danish Myo Technology A/S, and Aarhus, Denmark). Tissues were bathed in Krebs-Henseleit buffer (NaCl, 115 mM; NaHCO3, 25 mM; NaHPO4, 1.38 mM; KCl, 2.51 mM; MgSO4 – 7 H2O, 2.46 mM; CaCl2, 1.91 mM; and dextrose, 5.56 mM) bubbled with air/6% CO2 and maintained at 37°C. After 1 h of equilibration, the optimal tissue resting tension was determined by repeated stimulation with 128 mM KCl until maximum active tension was reached. All subsequent force measurements were obtained at optimal resting tension.
The pulmonary vascular muscle contraction was induced with the thromboxane A2-mimetic U46619. Following precontraction with U46619 at concentrations equivalent to the EC75, the endothelium-dependent and -independent relaxation dose-response was obtained with acetylcholine and sodium nitroprusside respectively. The former was evaluated in the presence and absence of the NOS inhibitor N-(G)-nitro-L-arginine methyl ester (L-NAME; 100 μM) and PEG-catalase (200 u/ml). Contractile responses were normalized to the tissue cross-sectional area as follows: (width × diameter) × 2 and expressed as mN/mm2. The earliest age that it is possible to successfully study the mechanical properties of near-resistance pulmonary arteries (diameter 80 – 90 μm) is 5 days. As such, all myograph measurements were obtained in pulmonary arteries from 5–7 days old for newborn animals.
Histological studies
Animals were anesthetized with i.p. ketamine (80 mg/kg) and xylazine (20 mg/kg). Given their small size, it is technically impossible to perfuse newborn mouse lungs with a constant pressure before fixation. Randomly chosen animals either <24 h old, or 7 days of age from each type were prepared for lung histology. Immediately after death the chest cavity was opened and the trachea was cannulated with a 24 G plastic cannula (7 days old animals only, since this is not technically reproducible in the <24 h of age pups). The lungs were then inflated with 4% paraformaldehyde at a constant pressure (20 cm H2O) and paraffin-embedded.
A 4 μm section was cut and double-stained with hematoxylin-eosin and Masson trichrome to allow for clear demarcation of the internal and external elastic lamina, as well as muscle layer. With the aid of a computerized image analyzer system (Openlab Improvision) coupled with a fine resolution microscope, the external and internal perimeters of each identifiable pulmonary artery were measured. Pulmonary arteries were distinguished from veins on the basis of their greater muscle and connective tissue observed in the former, and on the more peripheral position in the acinus of the latter. Only arteries that were cross-sectionally cut were measured. The internal elastic lamina was utilized to demarcate the internal, whereas the external elastic lamina was chosen as the external arterial perimeter. The arterial muscle layer was quantified by measuring the medial area and the average medial thickness was calculated as previously described [12]. Results are shown as mean values from all suitable arteries (10–15 arteries per lung) from < 24 h of age (hph-1: N=4; wild-type: N=3) and 7 days old animals (hph-1: N=3; wild-type: N=4).
Cardiac ventricular weight
The total wet heart weight was measured, the right ventricle wall was dissected free and the right-to-left ventricular+septum weight ratio (RV/LV+septum) was generated and used as a surrogate marker for pulmonary hypertension-induced right ventricular hypertrophy, as previously reported [13].
BH4 and BH2 Measurement
Pterins concentrations were directly estimated in lung homogenates (50mM phosphate buffer, pH 2.6, with 0.1mM DTPA and 1mM DTE) using high pressure liquid chromatography (HPLC) with electrochemical detection and standards as described previously [14]. Results were normalized to protein content.
H2O2 microsensor
Lung homogenate supernatants were tested using a calibrated H2O2 specific sensor (World Precision Instruments, Saratosa, FL) attached to an Apollo 4000 Free Radical Analyzer. Results were measured as μmoles H2O2/g protein for lung and expressed as a percentage of the respective age wild-type values.
Western blot analysis
Lung tissue were homogenized in 10 mM Tris-HCl pH 7.4 lysis buffer containing 1% Triton X-100 and protease/phosphatase inhibitors (Roche), and centrifuged at 13,000 g for 30 min. Equivalent amounts of lysate proteins in Laemmli buffer were fractionated on 4–12% gradient SDS/PAGE and immunoblotted. The antibody against eNOS was used at 1:3000 (mouse IgG1, BD Biosciences, Mississauga, Ontario). Appropriate IgGs conjugated with horseradish peroxidase (1:20000 dilution) were used as secondary antibodies. Enhanced chemiluminescence (ECL, Perkin Elmer, Shelton, Connecticut, USA) reagent was used for detection. Band intensities were quantified and expressed relative to β-actin.
Hypoxia exposure
A sub-group of wild-type and hph-1 newborn mice were subjected to chronic hypoxia in order to evaluate whether BH4 deficiency potentiates the severity of pulmonary hypertension, as previously shown in adult hph-1 animals [8]. Each litter was nursed in either normoxia (21% O2) or hypoxia (13% O2) from birth for up to 14-d, as previously described in detail for newborn rats (33). In order to prevent a reduction in their breast milk supply, the mothers were rotated every 24 hrs between air and hypoxia. All pups tolerated this degree of chronic hypoxia well and no deaths were observed.
Data Analysis
Data were evaluated by one or two-way analysis of variance (ANOVA) with multiple comparisons obtained by the Tukey-Kramer test, or unpaired Student’s t-test, when appropriate. Statistical significance was accepted at P<0.05. All statistical analyses were performed with the Number Cruncher Statistical System (NCSS, Kaysville, Utah, USA). Data are presented as means±SEM.
Results
Lung biopterin content
The lung biopterin content was measured in newborn and adult wild-type and hph-1 mice. In both types the BH4 lung content was highest in the newborn, when compared with adult mice (Figure 1). The BH2 lung content was significantly different across ages for wild-type animals, but not hph-1 mice (Figure 1). Given their GTPCH1 deficiency, the BH4 lung content of the hph-1 mice was significantly lower (P<0.01), as compared with wild-type newborn and adult animals (Figure 1).
Figure 1.
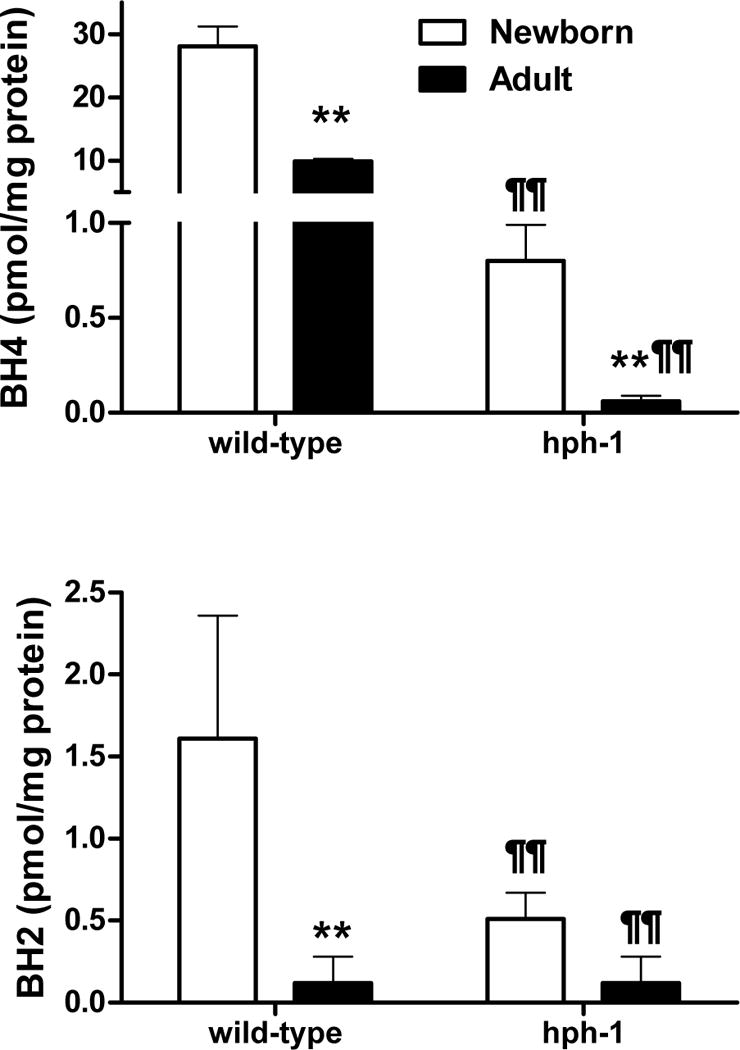
BH4 and BH2 lung content for newborn (3 days of age) and adult wild-type control and hph-1 mice. ** P<0.01 versus newborn values; P¶¶ < 0.01 versus wild-type respective values. N=4 per age and type group.
Body weight
Mutant and wild-type mice have litters of similar size (7–9 pups/litter). At birth, the hph-1 pups weighted 1.27±0.03 g which was significantly less (P=0.01), when compared with wild-type animals (1.43±0.05 g). The hph-1 pups did not show any obvious respiratory distress, but their weight gain during the first week of life was significantly lower (P<0.01) when compared with wild-type animals (Figure 2).
Figure 2.
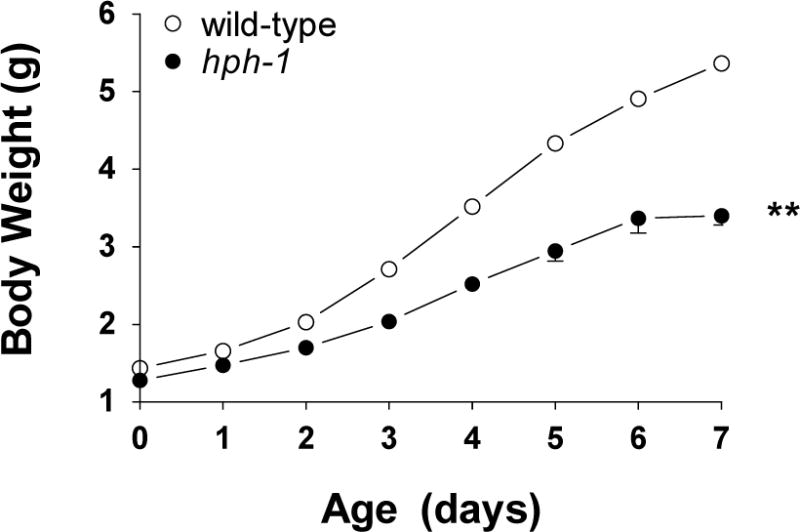
Body weight from birth to 7 days of age in hph-1 (N=14) and wild-type (N=16) mice. ** P<0.01 versus wild-type values by two-way ANOVA.
Evidence for pulmonary hypertension
As judged by the RV/LV+septum ratio data, the newborn hph-1 mice have a higher pulmonary vascular resistance at birth and at 7 days of age, when compared to age-matched wild-type animals (Figure 3). Chronic hypoxia induced a significant increase in the RV/LV+septum ratio of the wild-type newborn mice, but the degree of right ventricular hypertrophy was similar to the documented values for hph-1 animals (Figure 2). The hph-1 animals’ heart weight was significantly greater, when compared with age-matched wild-type animals, except following chronic hypoxia exposure. No difference in left ventricle+septum/total heart weight ratio was observed between wild-type and hph-1 newborn animals (N=10 per group; data not shown).
Figure 3.
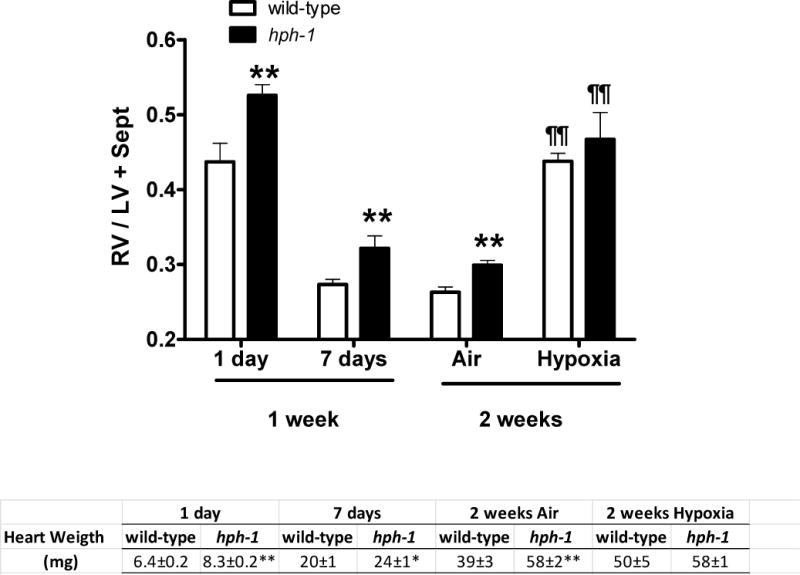
Right-to-left ventricle+septum (RV/LV+Sept) ratio at birth (N= 9 for wild-type and N=11 for hph-1 mice), at 7 days of age (N= 12 for wild-type and N=10 for hph-1 mice) and for two-week old animals. The two-week old animals were chronically exposed since day one of life to hypoxia (13% O2; N=18 wild-type and N=4 hph-1) or air (N=8 for wild-type and N=18 for hph-1). Data analyzed by Student’s t-test (one week) or two-way ANOVA with multiple comparisons evaluated by the Tukey-Kramer test (two-week). ** P<0.01 versus wild-type respective values; ¶¶ P< 0.01 versus respective air values.
H2O2 lung content
BH4 deficiency causes eNOS uncoupling, increased superoxide anion generation, oxidative injury to cells and impairment of organ functions is anticipated. Recent studies by others had shown increased oxidant-dependent loss of function in the adult hph-1 mice [15]. Thus, we further evaluated the newborn lung H2O2 content. The hph-1 H2O2 lung content was significantly higher when compared to age-matched wild-type controls (Figure 4).
Figure 4.
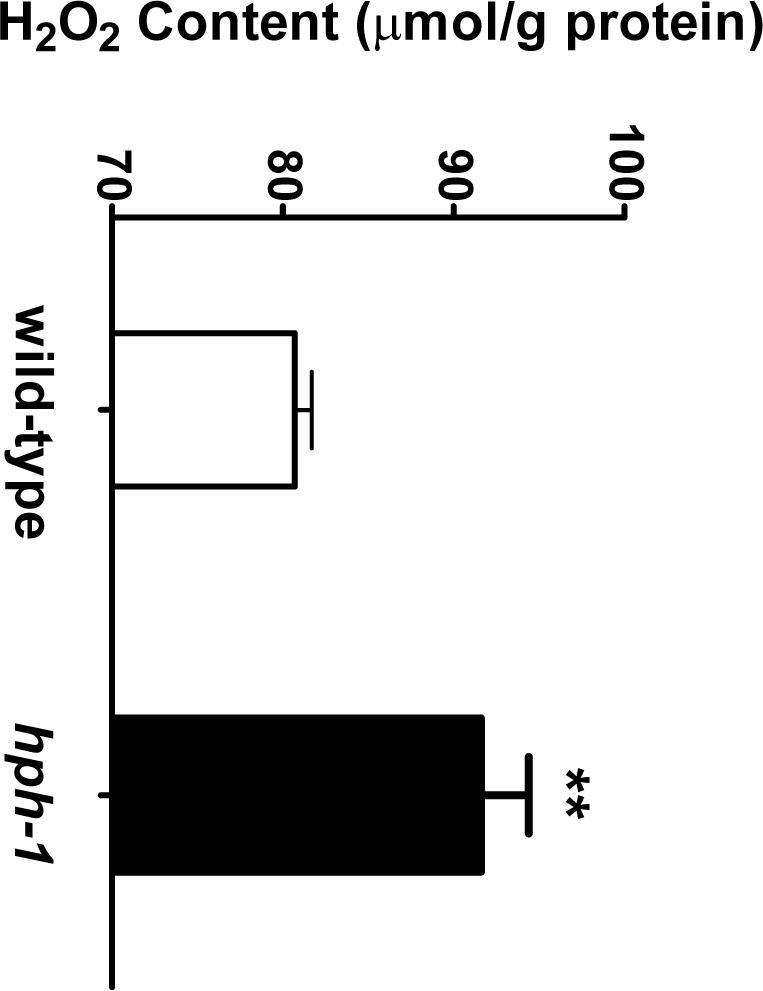
Newborn wild-type and hph-1 mouse lung H2O2 content measured by the H2O2 microsensor (N= 12 per group) ** P < 0.01 versus wild-type values by Student’s t-test.
Intrapulmonary arterial vasoconstriction and vasodilation potential
In order to evaluate the mechanism accounting for the pulmonary hypertension in the hph-1 newborn we measured the intrapulmonary arterial smooth muscle contraction response to the thromboxane A2 analogue U46619, as well as endothelium-dependent and independent-relaxation. In response to U46619, the newborn hph-1 mice pulmonary arterial muscle generated significantly less force, as compared to wild-type animals (Figure 5, panel A). PEG-catalase (200 units/ml) increased the U46619-induced force in the hph-1 pulmonary arteries, but not in wild-type animals, resulting in a similar dose-response curves in both mice types (Figure 5, panel B). These results suggest that an increased endogenous generation of H2O2 accounts for the reduced U46619 dose-response in hph-1 animals, when compared to wild-type pulmonary arteries.
Figure 5.
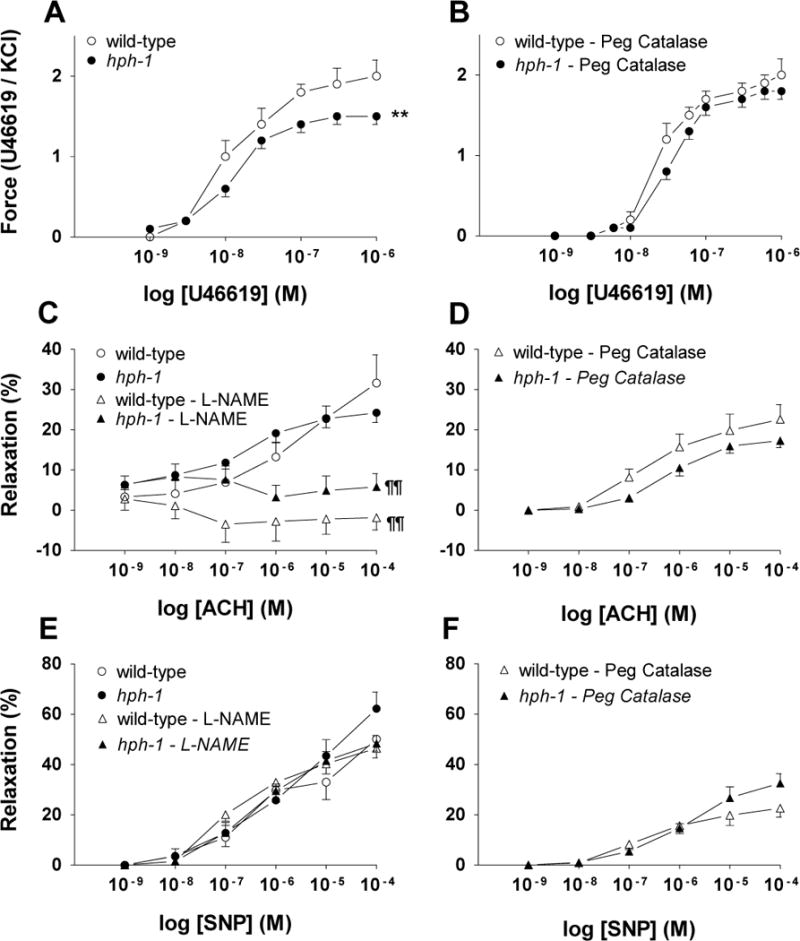
Wild-type and hph-1 mice pulmonary arteries contraction and relaxation. Thromboxane A2 analogue (U46619)-induced force generation in the absence (Panel A wild-type N=11; hph-1 N=20), or presence of PEG-catalase (200 u/ml) (Panel B, N=4 for both types). Panels C–F show endothelium-dependent (acetylcholine [ACH]) and endothelium-independent (sodium nitroprusside [SNP] dose-response relaxation obtained in the absence, or presence of the NOS inhibitor (L-NAME; 100 nM) or PEG-catalase (200 u/ml). Panel C (wild-type N=11; hph-1 N=20; N=4 per type for the L-NAME data). Panel E wild-type N=7; hph-1 N=16; N=4 per type for the L-NAME data). Panels D and F, N=4 per mouse type. For vasorelaxation measurements the pulmonary arteries were precontracted with the E75 concentration of U46619. ** P < 0.01 versus wild-type values by two-way ANOVA with Tukey-Kramer multiple comparisons test. ¶¶ P < 0.01 versus wild-type values by two-way ANOVA. Dose-response curves for ACH (Panel D) and SNP (Panel F) obtained in the presence of PEG-catalase were significantly (P<0.01) different than arteries evaluated in the absence of this H2O2 catalatic enzyme (Panels C and E) by two-way ANOVA.
Knowing that BH4 deficiency results in impaired eNOS function, we evaluated the newborn mice pulmonary arterial endothelium-dependent and -independent relaxation. No significant difference in the endothelium-dependent and -independent responses was observed in the pulmonary arteries (Figure 5, panels C and E). The NOS inhibitor L-NAME (10−4 M) suppressed the acetylcholine-induced pulmonary arterial vasorelaxation in both mouse types indicating that the pulmonary arteriolar response of hph-1 animals is eNOS-mediated (Figure 5, panel C and E). Whereas no statistically significant effect was seen in the acetylcholine-induced vasorelaxation (Figure 5, panel D) types, PEG-catalase reduced the SNP-induced relaxation for both mouse types (Figure 5, panels D and F as compared with panels C and E curves; P<0.01). This effect is likely secondary to the catalase-induced reduction in H2O2 given that others have shown a significant effect of this metabolite on the cGMP-dependent pulmonary arterial relaxation [16]
Pulmonary vascular remodeling
In spite of the evidence for an increased pulmonary vascular resistance in newborn and adult hph-1, compared to wild-type mice, neither increased pulmonary arterial muscle contraction potential, nor reduced vasodilation account for the hemodynamic changes. To further address the pathogenesis of pulmonary hypertension in hph-1 animals we evaluated the lung architecture looking for evidence of vascular remodeling. At birth, hph-1 animals’ resistance pulmonary arteries (internal diameter 15.2±0.6 μm) exhibited a significantly greater (P<0.01) medial thickness, when compared with age-matched wild-type controls, indicating vascular remodeling (Figure 6, panel A). At seven days of life (Figure 6, panel B), the hph-1 pups pulmonary arterial medial thickness remained increased (P<0.05) in small vessels (internal diameter <30 μm), whereas no mice type differences were observed for larger conductance pulmonary arteries (>30 μm).
Figure 6.
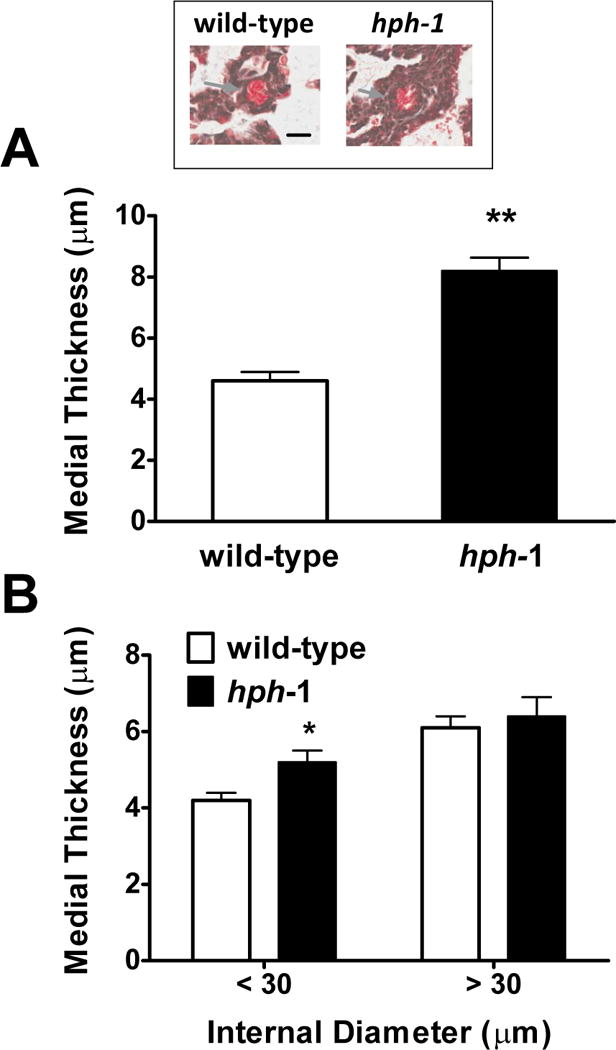
Wild-type control and hph-1 mice pulmonary arterial medial thickness for small (<30 μm; Panels A and B) and large (>30 μm, Panel B) pulmonary arteries, at <24 h of age (Panel A wild-type N=3; hph-1 N=4) and 7 days old (Panel B; wild-type; N=4; hph-1 N=3). * P < 0.05 ** P<0.01 versus wild-type respective values by Student’s t-test. N=3–6 animals per group. Insert shows representative small arteries from both types at < 24 h of age. Arrows point to the pulmonary artery. Bar=20 μm.
Lung eNOS expression
In order to further evaluate the eNOS pathway in hph-1 animals, we comparatively measured the lung eNOS expression. The eNOS/β-actin ratio was significantly reduced in hph-1 newborn animals (0.41±0.01; N=4), compared with wild-type mice (0.56±0.03; N=4; P<0.01). No statistically significant difference in β-actin densitometry values were observed between these mouse types.
Discussion
In this study we evaluated the newborn hph-1 mice pulmonary vasculature and demonstrated that in these BH4 deficient animals, right ventricular hypertrophy suggestive of pulmonary hypertension is present at birth. The pathogenesis of the higher pulmonary vascular resistance in hph-1 mice is not related to an abnormal vasoconstriction or vasorelaxation potential, but due to pulmonary vascular remodeling.
The hph-1 mutated mouse shows a 90% reduction in the activity of GTPCH1, the first step in the BH4 metabolic pathway resulting in an age-dependent lowered concentrations of this biopterin in several organs [17]. Although the hph-1 mouse has a high plasma phenylalanine level at birth, the levels progressively decrease with age in response to a developmental increase in BH4 accumulation in the liver. Yet, the adult hph-1 mouse liver BH4 content is significant reduced amounting to ~50% of wild-type levels [6].
In contrast to the age-dependent increase in liver and brain content [6] the present data demonstrated that the lung tissue BH4 content has a unique developmental pattern. As compared to adult animals, the lung BH4 content is highest in the newborn and this age-dependent pattern is present even in the hph-1 mice where pulmonary content of this biopterin remains low throughout life. The biochemical mechanism responsible for the developmental changes in BH4 tissue content in these mutated animals is unclear since they remain GTPCH1 deficient for life.
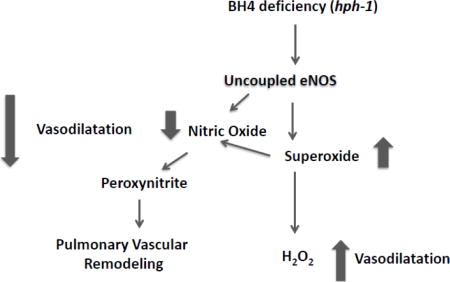
BH4 is a cofactor for NOS of which the endothelium-specific isoform plays a major role in pulmonary vascular NO generation. BH4 deficiency results in eNOS uncoupling. In the uncoupled state, eNOS cannot generate NO and releases superoxide anion instead from the decomposition of the heme-iron-dioxygen complex, an activity facilitated by L-arginine binding to the active site [18]. Following its diffusion, NO bioavailability rapidly decreases given its rate-limiting combination with the uncoupled NOS-generated superoxide resulting in the formation of peroxynitrite. Thus, eNOS uncoupling can theoretically result in pulmonary hypertension via two distinct, but complementary, pathways. Firstly, the reduced NO generation and bioavailability compromise vasodilation. Secondly, pulmonary hypertension can develop subsequent to reactive oxygen species-induced pulmonary vascular remodeling.
The adult hph-1 mouse is known to have pulmonary hypertension [8, 9]. In these animals a significant increase in the RV/LV+septum ratio has been documented in conjunction with pulmonary vascular remodeling and reduced serum NOx levels [9]. By cross breeding hph-1 with wild-type C57BL/6 mice Khoo et al [8] were able to generate adult animals with varying BH4 lung levels. The right ventricular systolic pressure was found to be inversely proportional to lung BH4 level in the crossbred mice and the tissue superoxide content was highest in the homozygous hph-1 adult animals [8]. Khoo et al further demonstrated that GTPCH1 overexpression in adult mice increased NOS activity, reduced superoxide generation and prevented pulmonary hypertension in the crossbred hph-1 adult mouse [8]. Lastly, the presence of lung vascular remodeling in adult hph-1 animals has been linked to eNOS dysfunction and reduced pulmonary vasodilatory potential [8, 19].
The hph-1 mouse has low-to-absent nonpulmonary tissue BH4 content at birth [5, 20, 21]. On that basis, we hypothesized that the newborn hph-1 mice not only would have pulmonary hypertension, but possibly a distinct lung phenotype, compared with adult animals. In the present study, we confirmed that pulmonary hypertension is present in the newborn hph-1 mouse at birth.
Pulmonary and systemic vascular eNOS uncoupling has been extensively documented in the adult hph-1 mouse [8, 9, 22, 23]. Yet, no significant difference in the endothelial-dependent pulmonary arterial vasorelaxation was observed in the newborn mutated mice in this study. H2O2 lung content, in this study was found to be significantly higher in hph-1 mice compared to the wild-type newborn animals. We previously showed that H2O2 is a pulmonary vasodilator in newborn and adult wild-type mice [11]. In addition, we recently reported the presence of pulmonary hypertension in adult mice heterozygous for endoglin (Eng+/− mice), where eNOS uncoupling is present [11, 24]. These mutated mice also show enhanced endothelium-dependent relaxation, increased lung H2O2 content and pulmonary vascular remodeling.
In addition, PEG-catalase reduced the NO donor-mediated relaxation response in wild-type and hph-1 mice suggesting that H2O2 plays a physiological role in the newborn regulation of pulmonary vasorelaxation. Previous studies have demonstrated H2O2 can directly activate cyclic GMP in pulmonary arteries via a compound I/H2O2 complex [25, 26] and is presently recognized as having prolonged signaling effects upon vascular tone regulation [27]
We propose that in the hph-1 mouse pulmonary arteries eNOS uncoupling results in H2O2 generation via superoxide radical anion dismutation. Thus, although eNOS uncoupling reduces NO generation, the enhanced eNOS-mediated H2O2 release offsets the reduced NO bioavailability and helps maintain the pulmonary endothelium-dependent vasorelaxation potential intact. That H2O2 plays a role in the regulation of pulmonary vascular tone under basal conditions in the hph-1 newborn animals is further demonstrated by the U46619 dose-response curves. The U46619-induced force is reduced in hph-1 pulmonary arteries and PEG-catalase, an H2O2 catalyst abolishes the dose-response differences amongst the mouse types.
Chronic hypoxia has been shown to potentiate the severity of pulmonary hypertension in the adult hph-1, compared to wild-type controls [8]. In the present study, as judged by the RV/LV+Sept ratio, chronic hypoxia does not potentiate the degree of pulmonary hypertension in hph-1 newborn animals. The reason for this age-dependent discrepancy is unclear, but it may pertain to developmental differences in the pathway responsible for oxidant-induced vascular remodeling. We have shown that peroxynitrite induces pulmonary vasoconstriction [28] and vascular remodeling in newborn rats [29, 30]. It is interesting to note that adult pulmonary hypertensive eNOS knockout mice do not show pulmonary vascular remodeling [31], unless exposed to chronic hypoxia [32].
eNOS-BH4 stoichiometry impacts on endothelium-dependent vasorelaxation potential and its disruptions leads to eNOS uncoupling in adult mice [22]. In the present study, we showed that the reduced lung BH4 content is associated with lower eNOS content in newborn hph-1 mice, compared to wild-type animals. Yet, our data indicate that whereas the lung BH4 content is reduced by 30-fold in hph-1 mice, compared to wild-type animals, the eNOS expression is decreased by only 25% in the mutated animals. Thus, it appears that it is the markedly reduced BH4, as opposed to eNOS-BH4 stoichiometry changes, that play a mechanistic role in the development of pulmonary hypertension in newborn hph-1 mice.
We speculate that the pulmonary hypertension observed in hph-1 mice is not related to enhanced vasoconstriction and/or reduced vasorelaxation, but eNOS uncoupling-induced pulmonary vascular remodeling. Others have shown pulmonary vascular remodeling is present in adult hph-1 mice [8] and the present study confirms that these changes are also evident at birth in these animals. This early occurrence indicates that the pulmonary vascular remodeling demonstrated here in the hph-1 newborn mice actually begins during the fetal period. The mechanisms accounting for such remodeling during fetal life are presently unclear, but likely related to BH-4 deficiency-induced reduction in lung NO generation prenatally. Nitric oxide is known to play an important role in the modulation of vascular remodeling and increased BH4 synthesis via endothelium-targeted overexpression of GTPCH1 inhibits vascular remodeling in mice [33].
The implications of the current findings to human physiology and the pathogenesis of pulmonary hypertension require further discussion. It is not known to what extent GTPCH1 is a rate-limiting enzyme in human lungs [34]. Neonates with GTPCH1 deficiency present mostly with neurological abnormalities and high phenylalanine plasma levels [35]. On the other hand, BH4 deficiency is associated with pulmonary hypertension [8, 9] and supplementation of this biopterin can reverse endothelial dysfunction [1]. The degree of pulmonary hypertension manifested in hph-1 mice does not shorten their life-span or activity but it negatively impacts on theirgrowth early in life. Thus, it is possible that newborns with BH4 deficiency have a rather clinically mild form of pulmonary hypertension that goes unrecognized until other additive factors are manifested.
In conclusion, we have demonstrated that BH4 deficiency induces pulmonary hypertension at birth in mice. The mechanism accounting for the increased pulmonary vascular resistance in GTPCH-1 mutated hph-1 mice relates to eNOS uncoupling-induced pulmonary vascular remodeling possibly mediated via increased peroxynitrite accumulation in the lung. Further understanding of the factors leading to pulmonary vascular remodeling in hph-1 animals may open new avenues for therapeutic intervention in pulmonary hypertension.
Highlights.
Tetrahydrobiopterin (BH4) is an endothelial nitric oxide synthase (eNOS) co-factor
BH4 deficiency results in eNOS uncoupling and reactive oxygen species generation
The hph-1 mutant mouse is congenitally BH4 deficient and pulmonary hypertensive at birth
Vascular remodeling is responsible for the high pulmonary vascular resistance in hph-1 mice
Acknowledgments
This work was funded by operating grants from the Canadian Institutes of Health and Research (MOP 93710; Jaques Belik) and The National Institutes of Health (HL67244; Jeannette Vasquez-Vivar).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Katusic ZS. Vascular protection by tetrahydrobiopterin: progress and therapeutic prospects. Trends Pharmacol Sci. 2009;30:48–54. doi: 10.1016/j.tips.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Furukawa Y, Nygaard TG, Gutlich M, Rajput AH, Pifl C, DiStefano L, et al. Striatal biopterin and tyrosine hydroxylase protein reduction in dopa-responsive dystonia. Neurology. 1999;53:1032–41. doi: 10.1212/wnl.53.5.1032. [DOI] [PubMed] [Google Scholar]
- 3.Longo N. Disorders of biopterin metabolism. J Inherit Metab Dis. 2009;32:333–42. doi: 10.1007/s10545-009-1067-2. [DOI] [PubMed] [Google Scholar]
- 4.Zurfluh MR, Giovannini M, Fiori L, Fiege B, Gokdemir Y, Baykal T, et al. Screening for tetrahydrobiopterin deficiencies using dried blood spots on filter paper. Mol Genet Metab. 2005;86(Suppl 1):S96–103. doi: 10.1016/j.ymgme.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 5.Bode VC, McDonald JD, Guenet JL, Simon D. hph-1: a mouse mutant with hereditary hyperphenylalaninemia induced by ethylnitrosourea mutagenesis. Genetics. 1988;118:299–305. doi: 10.1093/genetics/118.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hyland K, Gunasekera RS, Engle T, Arnold LA. Tetrahydrobiopterin and biogenic amine metabolism in the hph-1 mouse. J Neurochem. 1996;67:752–9. doi: 10.1046/j.1471-4159.1996.67020752.x. [DOI] [PubMed] [Google Scholar]
- 7.Du YH, Guan YY, Alp NJ, Channon KM, Chen AF. Endothelium-specific GTP cyclohydrolase I overexpression attenuates blood pressure progression in salt-sensitive low-renin hypertension. Circulation. 2008;117:1045–54. doi: 10.1161/CIRCULATIONAHA.107.748236. [DOI] [PubMed] [Google Scholar]
- 8.Khoo JP, Zhao L, Alp NJ, Bendall JK, Nicoli T, Rockett K, et al. Pivotal role for endothelial tetrahydrobiopterin in pulmonary hypertension. Circulation. 2005;111:2126–33. doi: 10.1161/01.CIR.0000162470.26840.89. [DOI] [PubMed] [Google Scholar]
- 9.Nandi M, Miller A, Stidwill R, Jacques TS, Lam AA, Haworth S, et al. Pulmonary hypertension in a GTP-cyclohydrolase 1-deficient mouse. Circulation. 2005;111:2086–90. doi: 10.1161/01.CIR.0000163268.32638.F4. [DOI] [PubMed] [Google Scholar]
- 10.Belik J, Stephens NL. Developmental differences in vascular smooth muscle mechanics in pulmonary and systemic circulations. J Appl Physiol. 1993;74:682–7. doi: 10.1152/jappl.1993.74.2.682. [DOI] [PubMed] [Google Scholar]
- 11.Belik J, Jerkic M, McIntyre BA, Pan J, Leen J, Yu LX, et al. Age-dependent endothelial nitric oxide synthase uncoupling in pulmonary arteries of endoglin heterozygous mice. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1170–L1178. doi: 10.1152/ajplung.00168.2009. [DOI] [PubMed] [Google Scholar]
- 12.Fornaro E, Li D, Pan J, Belik J. Prenatal exposure to fluoxetine induces fetal pulmonary hypertension in the rat. Am J Respir Crit Care Med. 2007;176:1035–40. doi: 10.1164/rccm.200701-163OC. [DOI] [PubMed] [Google Scholar]
- 13.Le Cras TD, Markham NE, Tuder RM, Voelkel NF, Abman SH. Treatment of newborn rats with a VEGF receptor inhibitor causes pulmonary hypertension and abnormal lung structure. Am J Physiol Lung Cell Mol Physiol. 2002;283:L555–L562. doi: 10.1152/ajplung.00408.2001. [DOI] [PubMed] [Google Scholar]
- 14.Vasquez-Vivar J, Whitsett J, Derrick M, Ji X, Yu L, Tan S. Tetrahydrobiopterin in the prevention of hypertonia in hypoxic fetal brain. Ann Neurol. 2009;66:323–31. doi: 10.1002/ana.21738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adlam D, Bendall JK, De Bono JP, Alp NJ, Khoo J, Nicoli T, et al. Relationships between nitric oxide-mediated endothelial function, eNOS coupling and blood pressure revealed by eNOS-GTP cyclohydrolase 1 double transgenic mice. Exp Physiol. 2007;92:119–26. doi: 10.1113/expphysiol.2006.035113. [DOI] [PubMed] [Google Scholar]
- 16.Burke-Wolin T, Wolin MS. H2O2 and cGMP may function as an O2 sensor in the pulmonary artery. J Appl Physiol. 1989;66:167–70. doi: 10.1152/jappl.1989.66.1.167. [DOI] [PubMed] [Google Scholar]
- 17.Hyland K, Gunasekara RS, Munk-Martin TL, Arnold LA, Engle T. The hph-1 mouse: a model for dominantly inherited GTP-cyclohydrolase deficiency. Ann Neurol. 2003;54(Suppl 6):S46–S48. doi: 10.1002/ana.10695. [DOI] [PubMed] [Google Scholar]
- 18.Vasquez-Vivar J, Martasek P, Whitsett J, Joseph J, Kalyanaraman B. The ratio between tetrahydrobiopterin and oxidized tetrahydrobiopterin analogues controls superoxide release from endothelial nitric oxide synthase: an EPR spin trapping study. Biochem J. 2002;362:733–9. doi: 10.1042/0264-6021:3620733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nandi M, Kelly P, Vallance P, Leiper J. Over-expression of GTP-cyclohydrolase 1 feedback regulatory protein attenuates LPS and cytokine-stimulated nitric oxide production. Vasc Med. 2008;13:29–36. doi: 10.1177/1358863X07085916. [DOI] [PubMed] [Google Scholar]
- 20.McDonald JD, Cotton RG, Jennings I, Ledley FD, Woo SL, Bode VC. Biochemical defect of the hph-1 mouse mutant is a deficiency in GTP-cyclohydrolase activity. J Neurochem. 1988;50:655–7. doi: 10.1111/j.1471-4159.1988.tb02961.x. [DOI] [PubMed] [Google Scholar]
- 21.McDonald JD, Bode VC. Hyperphenylalaninemia in the hph-1 mouse mutant. Pediatr Res. 1988;23:63–7. doi: 10.1203/00006450-198801000-00014. [DOI] [PubMed] [Google Scholar]
- 22.Bendall JK, Alp NJ, Warrick N, Cai S, Adlam D, Rockett K, et al. Stoichiometric relationships between endothelial tetrahydrobiopterin, endothelial NO synthase (eNOS) activity, and eNOS coupling in vivo: insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ Res. 2005;97:864–71. doi: 10.1161/01.RES.0000187447.03525.72. [DOI] [PubMed] [Google Scholar]
- 23.Cosentino F, Barker JE, Brand MP, Heales SJ, Werner ER, Tippins JR, et al. Reactive oxygen species mediate endothelium-dependent relaxations in tetrahydrobiopterin-deficient mice. Arterioscler Thromb Vasc Biol. 2001;21:496–502. doi: 10.1161/01.atv.21.4.496. [DOI] [PubMed] [Google Scholar]
- 24.Toporsian M, Jerkic M, Zhou YQ, Kabir MG, Yu LX, McIntyre BA, et al. Spontaneous adult-onset pulmonary arterial hypertension attributable to increased endothelial oxidative stress in a murine model of hereditary hemorrhagic telangiectasia. Arterioscler Thromb Vasc Biol. 2010;30:509–17. doi: 10.1161/ATVBAHA.109.200121. [DOI] [PubMed] [Google Scholar]
- 25.Iesaki T, Gupte SA, Kaminski PM, Wolin MS. Inhibition of guanylate cyclase stimulation by NO and bovine arterial relaxation to peroxynitrite and H2O2. Am J Physiol. 1999;277:H978–H985. doi: 10.1152/ajpheart.1999.277.3.H978. [DOI] [PubMed] [Google Scholar]
- 26.Wolin MS, Burke TM. Hydrogen peroxide elicits activation of bovine pulmonary arterial soluble guanylate cyclase by a mechanism associated with its metabolism by catalase. Biochem Biophys Res Commun. 1987;143:20–5. doi: 10.1016/0006-291x(87)90623-1. [DOI] [PubMed] [Google Scholar]
- 27.Cai H. Hydrogen peroxide regulation of endothelial function: origins, mechanisms, and consequences. Cardiovasc Res. 2005;68:26–36. doi: 10.1016/j.cardiores.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 28.Belik J, Jankov RP, Pan J, Tanswell AK. Peroxynitrite inhibits relaxation and induces pulmonary artery muscle contraction in the newborn rat. Free Radic Biol Med. 2004;37:1384–92. doi: 10.1016/j.freeradbiomed.2004.07.029. [DOI] [PubMed] [Google Scholar]
- 29.Belik J, Stevens D, Pan J, McIntyre BA, Kantores C, Ivanovska J, et al. Pulmonary vascular and cardiac effects of peroxynitrite decomposition in newborn rats. Free Radic Biol Med. 2010;49:1306–14. doi: 10.1016/j.freeradbiomed.2010.07.021. [DOI] [PubMed] [Google Scholar]
- 30.Masood A, Yi M, Lau M, Belcastro R, Shek S, Pan J, et al. Therapeutic effects of hypercapnia on chronic lung injury and vascular remodeling in neonatal rats. Am J Physiol Lung Cell Mol Physiol. 2009;297:L920–L930. doi: 10.1152/ajplung.00139.2009. [DOI] [PubMed] [Google Scholar]
- 31.Steudel W, Ichinose F, Huang PL, Hurford WE, Jones RC, Bevan JA, et al. Pulmonary vasoconstriction and hypertension in mice with targeted disruption of the endothelial nitric oxide synthase (NOS 3) gene. Circ Res. 1997;81:34–41. doi: 10.1161/01.res.81.1.34. [DOI] [PubMed] [Google Scholar]
- 32.Steudel W, Scherrer-Crosbie M, Bloch KD, Weimann J, Huang PL, Jones RC, et al. Sustained pulmonary hypertension and right ventricular hypertrophy after chronic hypoxia in mice with congenital deficiency of nitric oxide synthase 3. J Clin Invest. 1998;101:2468–77. doi: 10.1172/JCI2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ali ZA, Bursill CA, Douglas G, McNeill E, Papaspyridonos M, Tatham AL, et al. CCR2-mediated antiinflammatory effects of endothelial tetrahydrobiopterin inhibit vascular injury-induced accelerated atherosclerosis. Circulation. 2008;118:S71–S77. doi: 10.1161/CIRCULATIONAHA.107.753558. [DOI] [PubMed] [Google Scholar]
- 34.Thony B, Auerbach G, Blau N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem J. 2000;347(Pt 1):1–16. [PMC free article] [PubMed] [Google Scholar]
- 35.Jaggi L, Zurfluh MR, Schuler A, Ponzone A, Porta F, Fiori L, et al. Outcome and long-term follow-up of 36 patients with tetrahydrobiopterin deficiency. Mol Genet Metab. 2008;93:295–305. doi: 10.1016/j.ymgme.2007.10.004. [DOI] [PubMed] [Google Scholar]